

Abstract
Background
Feed-forward loops (FFLs), consisting of miRNAs, transcription factors (TFs) and their common target genes, have been validated to be important for the initialization and development of complex diseases, including cancer. Esophageal Carcinoma (ESCA) and Stomach Adenocarcinoma (STAD) are two types of malignant tumors in the digestive tract. Understanding common and distinct molecular mechanisms of ESCA and STAD is extremely crucial.
Results
In this paper, we presented a computational framework to explore common and distinct FFLs, and molecular biomarkers for ESCA and STAD. We identified FFLs by combining regulation pairs and RNA-seq data. Then we constructed disease-specific co-expression networks based on the FFLs identified. We also used random walk with restart (RWR) on disease-specific co-expression networks to prioritize candidate molecules. We identified 148 and 242 FFLs for these two types of cancer, respectively. And we found that one TF, E2F3 was related to ESCA, two genes, DTNA and KCNMA1 were related to STAD, while one TF ESR1 and one gene KIT were associated with both of the two types of cancer.
Conclusions
This proposed computational framework predicted disease-related biomolecules effectively and discovered the correlation between two types of cancers, which helped develop the diagnostic and therapeutic strategies of Esophageal Carcinoma and Stomach Adenocarcinoma.
Keywords: Esophageal carcinoma, Stomach adenocarcinoma, Molecular mechanism, Feed-forward loop, Random walk with restart
Background
Esophageal Carcinoma (ESCA) and Stomach Adenocarcinoma (STAD) are two types of cancer in the digestive tract. ESCA ranks sixth in its cancer-related mortality rate [1, 2]. ESCA is classified histologically as esophageal adenocarcinoma (EAC) and esophageal squamous cell carcinoma (ESCC) [3]. Stomach Adenocarcinoma is one of common malignancies of digestive tract [4, 5]. Despite the advances in the treatment of STAD, the 5-year survival rate is 5~15% [6]. Both ESCA and STAD belong to digestive tract cancer, and the sites of their incidence are very close, so it is significant to explore the molecular mechanisms and the relationship between these two types of cancer.
Recently, comprehensive analysis of molecular characteristics of many types of cancer was performed, including STAD and ESCA. For example, Yin et al. conducted a case-control study based on their own patients and provided the first evidence that RANK rs1805034 T>C polymorphism was associated with susceptibility of ESCA [2]. A study by Pan et al. showed that lncRNA CASC9 in ESCA tissue was up-regulated [7]. SLC52A3 was proved to be useful for proliferation and colony formation of ESCA [8]. Baffa R et al. focused on loss of heterozygosity for chromosome 11 in STAD as early as 1996 [9]. An allelotype analysis was performed to identify chromosomal regions which were frequently deleted in STAD [10]. Korean researchers analyzed protein expression profiles of five STAD suppressor genes [11]. The Cancer Genome Atlas (TCGA) Research team performed a comprehensive molecular analysis of 559 patients of Stomach Adenocarcinoma and Esophageal Carcinoma, and found that EAC was closely resembled Stomach Adenocarcinoma by analyzing mRNA expression, DNA methylation and SCNA data [12]. In a recent study, the researchers questioned the use of PD-L1 as a biomarker in both of ESCA and STAD [13]. Most of the studies focused on ESCA or STAD separately, ignoring their potential common molecular characteristics, so it is of great importance to compare these two types of cancer.
Gene expression is regulated by many factors, among which TFs and miRNAs are two most important factors, and a feed-forward loop (FFL) consisting of two regulation factors and a common target gene plays an essential role in many biological processes [14]. FFLs were proved to be relevant to diseases, so some studies were performed to identify significant FFLs in complex diseases, including schizophrenia, Glioblastoma, T-cell acute lymphoblastic leukemia and so on [15–17]. There were also some studies identifying common FFLs in pan-cancer [18, 19]. Besides, TF-miRNA-lncRNA FFLs were identified [20]. However, FFLs in ESCA and STAD have not been studied yet as far as we know.
In this paper, we investigated the common and distinct regulatory properties of ESCA and STAD. Firstly, we identified miRNA-TF-gene FFLs by integrating gene/miRNA expression profiles and transcriptional/post-transcriptional regulation pairs. Then, we built and analyzed disease-specific co-expression networks based on the identified FFLs. Finally, we prioritized candidate disease-related biomolecules based on their scores.
Results
Overview of the proposed computational framework
The proposed computational framework consisted of the following five steps (Fig. 1), and we described each step briefly.
Step 1. Preprocessing of regulation pairs and expression profiles. We combined TF-target pairs and miRNA-target pairs from different algorithms and databases, and dealt with noisy data. For expression profiles, we filtered out the genes with low expression level and miRNAs with many missing values. We also calculated differentially expressed genes and miRNAs for ESCA and STAD using Limma [21] with adjusted p-value smaller than 0.05 and |log2FC| greater than 1.
Step 2. Construction of disease-specific regulatory networks. We constricted the target genes as differentially expressed genes and the miRNAs as differentially expressed miRNAs. And then we contained the regulation pairs whose spearman correlation coefficient (SCC) was greater than 0.3. As a result, the disease-specific regulatory networks for ESCA and STAD were constructed.
Step 3. Identification of 3-node FFLs. Three types of typical FFLs were identified from the disease-specific regulatory networks for ESCA and STAD.
Step 4. Construction of disease-specific co-expression networks. We calculated SCC for each pair of the molecules in the identified FFLs and then constructed diseased-specific co-expression networks using those pairs with coefficient absolutely greater than a predefined threshold.
Step 5. Prioritization of candidate molecules. Random walk with restart (RWR) was used to calculate the score of each biomolecule in the co-expression networks. The higher the score was, the more likely the biomolecule was a disease-related molecular.
Fig. 1.

Flowchart of the proposed computational framework. (1) Preprocessing of regulation pairs and expression profiles (2) Construction of disease-specific regulatory networks (3) Identification of 3-node FFLs (4) Construction of disease-specific co-expression networks (5) Prioritization of candidate molecules
Disease-specific regulatory network analysis
First of all, we combined differentially expressed molecules with preprocessed regulation pairs. And then we calculated SCC for each regulation pair. We chose the threshold with correlation as 0.3 and p-value as 0.05 so that we obtained the disease-specific regulatory networks for ESCA and STAD. The results were shown in Table 1.
Table 1.
Disease-specific regulatory networks for ESCA and STAD
| Cancer | Relationship | RegulationType | Pairs | miRNAs | TFs | Genes |
|---|---|---|---|---|---|---|
| ESCA | TF-gene | positive | 2096 | – | 154 | 1353 |
| negative | 1383 | – | 101 | 1032 | ||
| TF-miRNA | positive | 136 | 54 | 51 | – | |
| negative | 68 | 31 | 36 | – | ||
| miRNA-gene | negative | 1674 | 48 | – | 579 | |
| miRNA-TF | negative | 444 | 47 | 165 | – | |
| STAD | TF-gene | positive | 2454 | – | 199 | 1534 |
| negative | 1307 | – | 153 | 942 | ||
| TF-miRNA | positive | 154 | 70 | 51 | – | |
| negative | 165 | 78 | 45 | – | ||
| miRNA-gene | positive | 3689 | 80 | – | 847 | |
| miRNA-TF | negative | 1304 | 80 | 277 | – |
There were 79 miRNAs, 325 TFs, 1830 genes, and 5801 regulation pairs in ESCA-specific regulatory network (Fig. 2a). And the obtained STAD-specific regulatory network consisted of 116 miRNAs, 461 TFs, 2093 genes, and 9037 regulation pairs (Fig. 2b).
Fig. 2.

Disease-specific regulatory networks. a ESCA b STAD
MiRNA-TF-gene FFLs
We identified three categories of FFLs from the disease-specific regulatory networks for ESCA and STAD, respectively. And we named the FFLs identified from the ESCA-specific regulatory network as ESCA-specific FFL and the FFLs identified from the STAD-specific regulatory network as STAD-specific FFL. The results were summarized in Table 2.
Table 2.
The number of FFLs identified
| Cancer | FFL | FFLs | miRNAs | TFs | Genes |
|---|---|---|---|---|---|
| ESCA | TFP-FFL | 7 | 3 | 2 | 7 |
| TFN-FFL | 14 | 6 | 4 | 13 | |
| miRNAN-FFL | 127 | 19 | 8 | 46 | |
| STAD | TFP-FFL | 38 | 12 | 9 | 21 |
| TFN-FFL | 46 | 16 | 9 | 32 | |
| miRNAN-FFL | 158 | 38 | 21 | 48 |
There were 7 TFP-FFLs, 14 TFN-FFLs and 127 miRNAN-FFLs for ESCA, respectively. An ESCA-specific regulatory network was constructed based on the identified FFLs, which consisted of 26 miRNAs, 12 TFs, 60 genes and 240 regulation pairs (Fig. 3a).
Fig. 3.

Disease-specific FFL networks. a ESCA b STAD
What’s more, there were 38 TFP-FFLs, 46 TFN-FFLs and 158 miRNAN-FFLs for STAD. A STAD-specific FFL network was constructed based on the identified FFLs, which consisted of 47 miRNAs, 31 TFs, 87 genes and 401 regulation pairs (Fig. 3b).
For both of ESCA and STAD, the number of miRNAN-FFL was the largest one, which meant that this FFL model was the most common regulatory pattern, and the genes were mainly down-regulated.
We further investigated the common FFLs in the ESCA-specific FFL and STAD-specific FFL, and found that there were 3 TFP-FFLs, 8 TFN-FFLs, and 41 miRNAN-FFLs. It is exciting that STAD and ESCA shared so many FFLs, which provided a strong evidence for the potential closely relationship between STAD and ESCA. We further constructed a regulatory network for ESCA and STAD based on these common FFLs (Fig. 4), which was made up of three subnetworks. What’s more, there were 16 miRNAs, 6 TFs, 20 genes and 90 regulation pairs in this common network.
Fig. 4.
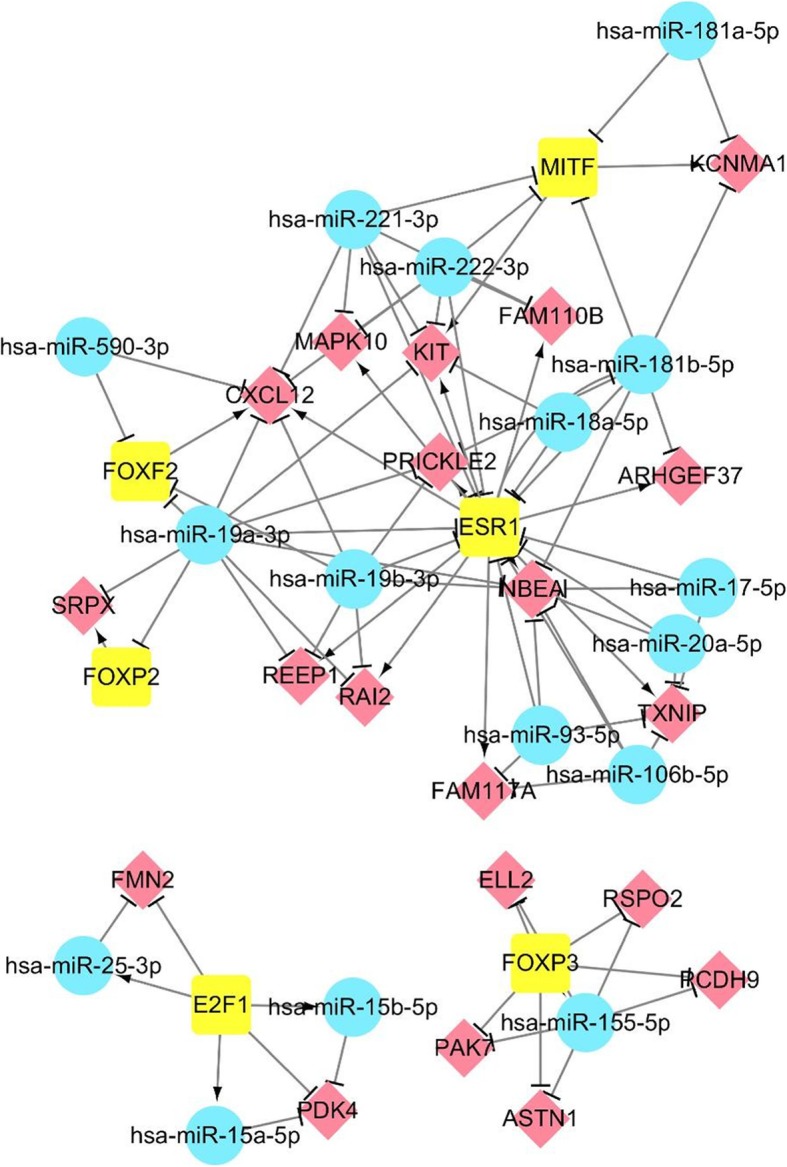
The common regulatory network for ESCA and STAD
We investigated the in-degree and out-degree properties of the regulatory network. Figure 5 showed the in-degree and out-degree distribution of this network. We found that the nodes which only had in-degree were all genes, and the number of nodes was 20. And there were 11 miRNAs and 2 TFs which only had out-degree in this network. 5 miRNAs and 4 TFs not only had in-degree but also had out-degree. Among these nodes, one TF ESR1 had highest in-degree and highest out-degree. We found it was related to both of ESCA and STAD. Genetic variations in ESR1 were associated with an increased risk of ESCA [22]. ESR1 regulated stomach-specific tumor suppressor gene TFF1, further influenced the development of STAD [23].
Fig. 5.

The in-degree and out-degree distribution of the regulatory network for ESCA and STAD
Gene set enrichment analysis
Gene set enrichment analysis is a meaningful way to understand the functions of genes in living cells. We applied the online tool DAVID [24] to perform gene set enrichment analysis for those genes. With the DAVID online tool, we set the threshold p-value as 0.05 and then obtained a list of entries.
For the STAD-specific FFL, there were 114 biomolecules, including 33 TFs and 81 genes, and they were enriched in a total of 191 annotation entries, including 162 GO terms and 29 BIOCARTA and KEGG pathways. For the ESCA-specific FFL, there were 72 biomolecules, including 12 TFs and 60 genes, and analyzed them with DAVID online tool. They were resulting in a total of 53 annotation entries, including 42 GO terms and 11 BIOCARTA and KEGG pathways. An additional file showed this in more detail [see Additional file 1].
We further found 32 common entries, including 27 GO terms and 5 KEGG pathways. We selected 10 enrichment entries for further analysis (Table 3). Among these entries, the disease-specific FFLs had similar number of genes. For biological processes, the genes in both types of disease-related FFLs were enriched in negative regulation of transcription from RNA polymerase II promoter, which also indicated that these genes played an important role in transcriptional regulation. For molecular components, the genes were enriched in the nucleoplasm. The nucleus was a necessary component in the cell, which also showed that these genes were vital and had an indispensable effect on the cell body and even the living body. For molecular function, the genes were enriched in transcription factor activity, sequence-specific DNA binding.
Table 3.
The common enrichment entries for ESCA and STAD
| Category | Term | Genes in ESCA | Genes in STAD |
|---|---|---|---|
| BP (GO:0000122) | Negative regulation of transcription from RNA polymerase II promoter | 12 | 27 |
| BP (GO:0045944) | Positive regulation of transcription from RNA polymerase II promoter | 11 | 28 |
| BP (GO:0009791) | Post-embryonic development | 3 | 5 |
| CC (GO:0005654) | Nucleoplasm | 18 | 28 |
| CC (GO:0005667) | Transcription factor complex | 5 | 11 |
| CC (GO:0043234) | Protein complex | 6 | 10 |
| MF (GO:0005515) | Protein binding | 42 | 72 |
| MF (GO:0003700) | Transcription factor activity, sequence-specific DNA binding | 12 | 26 |
| MF (GO:0019901) | Protein kinase binding | 6 | 7 |
| KEGG (hsa04110) | Cell cycle | 5 | 7 |
For biological pathways, genes were enriched in the cell cycle which was a continuous process passing from one generation to the next. The enriched members of this pathway for the ESCA-specific FFL were 4 TFs, E2F1, E2F3, MYC, TFDP1 and 1 gene GADD45B. And the enriched members of this pathway for the STAD-specific FFL were 4 TFs, E2F1, SMAD4, SMAD2, MYC and 3 genes which included CDKN1C, CDK1, GADD45B. The common members which were 2 TFs, E2F1, MYC and 1 gene GADD45B, were all related to both of two types of cancer. All these categories showed that these enriched genes may have a critical impact on the emergence and development of the disease.
Co-expression network analysis
We further focused on the molecules in the identified FFLs and investigated their SCC for all pairs of molecules to build disease-specific co-expression networks. We observed different sizes of co-expression network for different thresholds. Figure 6 showed the relationship between thresholds and the size of co-expression networks for STAD and ESCA. This relationship could be fitted to a cubic function. The cubic function’s inflection point is very meaningful. Before this point, the network size decreases sharply with the increase of threshold, and after this point, the network size decreases slowly with the increase of threshold. So the network at this point is more representative. The thresholds corresponding to the inflection points of the fitting functions of disease-specific co-expression networks for ESCA and STAD are about 0.6. Consequently, we chose 0.6 as the cut-off value in these two networks, and meanwhile p-value was less than 0.05. Finally, there were 98 nodes with 2666 pairs and 158 nodes with 5117 pairs in the disease-specific co-expression networks for ESCA and STAD, respectively.
Fig. 6.

The relationship between thresholds and edge numbers. a ESCA b STAD
Specifically, compared with the STAD-specific FFL, there were 3 molecules lost in the STAD-specific co-expression network. Because these 3 molecules had weak association with the other molecules.
Random walk with restart in co-expression network analysis
We investigated the molecules in the disease-specific co-expression networks for ESCA and STAD. We collected 15 and 39 disease-related molecules in disease-specific co-expression networks for ESCA and STAD, respectively, as we have mentioned in Methods. Taking these disease-related molecules as seed nodes, and the other 83 and 119 molecules as candidates, we ran RWR on the disease-specific co-expression networks for STAD and ESCA, respectively. As a result, we could obtained the scores for each candidate molecule. The higher the score was, the more relevant the candidate molecules were with the specific disease.
In order to evaluate and select the appropriate restart probability r, the AUC value of sorting correctness was calculated when the value r varies from 0.1 to 0.9 step by 0.1 using leave-one-out cross validation, following the method proposed by RWRMDA [25]. Table 4 shows the relationship between the restart probability and the corresponding AUC. And we found that the AUCs for both of two types of cancer were really great when r varied from 0.1 to 0.9. When the restart probability is 0.9, we obtained the highest AUC, so we assigned 0.9 to the restart probability.
Table 4.
The relationship between the restart probability and the corresponding AUC
| r | 0.1 | 0.2 | 0.3 | 0.4 | 0.5 |
|---|---|---|---|---|---|
| ESCA | 0.5550 | 0.6137 | 0.6442 | 0.6667 | 0.6796 |
| STAD | 0.6124 | 0.6660 | 0.7128 | 0.7468 | 0.7778 |
| r | 0.6 | 0.7 | 0.8 | 0.9 | |
| ESCA | 0.6996 | 0.7060 | 0.7181 | 0.7229 | |
| STAD | 0.7992 | 0.8214 | 0.8375 | 0.8500 |
We listed the top 20 candidate molecules for both ESCA and STAD (Tables 5 and 6). Also there were 12 out of 20 candidate molecules supported by literature in PubMed for ESCA (Table 5), and 13 out of 20 candidate molecules were supported by literature in PubMed for STAD (Table 6). And details of the all candidate molecules can be showed in additional file [see Additional file 2]. These results showed that our analysis was reliable in a certain degree. And the molecules un-supported by literature may be potential disease-related molecules.
Table 5.
Top 20 candidate molecules for ESCA
| Ranking | Molecule | Score(10−3) | PMID |
|---|---|---|---|
| 1 | RBPMS2 | 1.8594 | 29301256 |
| 2 | hsa-miR-15b-5p | 1.7756 | 25943911 |
| 3 | GADD45B | 1.7477 | 16026601 |
| 4 | hsa-miR-365a-3p | 1.6981 | – |
| 5 | SECISBP2L | 1.6595 | – |
| 6 | TFDP1 | 1.6582 | 14618416 |
| 7 | hsa-miR-222-3p | 1.6557 | 26258795 |
| 8 | AURKA | 1.6530 | 24953013 |
| 9 | SORCS1 | 1.6250 | – |
| 10 | hsa-miR-106b-5p | 1.6121 | 27619676 |
| 11 | hsa-miR-18a-5p | 1.5999 | 23643275 |
| 12 | TXNIP | 1.5953 | 29934340 |
| 13 | ARHGEF37 | 1.5939 | – |
| 14 | FBXL17 | 1.5910 | – |
| 15 | E2F3 | 1.5658 | 28751461 |
| 16 | hsa-miR-17-5p | 1.5597 | 28002789 |
| 17 | RAI2 | 1.5583 | – |
| 18 | ITPR1 | 1.5563 | – |
| 19 | SOX9 | 1.5302 | 29936467 |
| 20 | MITF | 1.5277 | – |
Table 6.
Top 20 candidate molecules for STAD
| Ranking | Molecules | Score(10−3) | PMID |
|---|---|---|---|
| 1 | CNN1 | 1.5602 | – |
| 2 | hsa-miR-15a-5p | 1.5177 | 26894855 |
| 3 | REEP1 | 1.2745 | – |
| 4 | DTNA | 1.2160 | 27858295 |
| 5 | FOXP2 | 1.1572 | 27382302 |
| 6 | hsa-miR-188-5p | 1.1426 | 29471891 |
| 7 | hsa-miR-590-3p | 1.1182 | 29516678 |
| 8 | DLG2 | 1.0800 | – |
| 9 | KCNMA1 | 1.0594 | 28231797 |
| 10 | RELN | 1.0589 | 19956836 |
| 11 | CFL2 | 1.0466 | 29342841 |
| 12 | hsa-miR-590-5p | 1.0191 | 27757042 |
| 13 | NECAB1 | 1.0111 | – |
| 14 | PRICKLE2 | 0.9895 | 16273260 |
| 15 | TUSC3 | 0.9824 | 22447362 |
| 16 | hsa-miR-424-5p | 0.9587 | 27655675 |
| 17 | GPRASP2 | 0.9453 | – |
| 18 | MEIS1 | 0.9435 | 28545608 |
| 19 | MAPK10 | 0.9298 | – |
| 20 | PCSK2 | 0.9209 | – |
Furthermore, we investigated the molecules in the ranking lists, and found four interesting genes RAI2, KCNMA1, NBEA and KIT. These four genes ranked relatively closely in their disease-specific ranking list, and ranked in the first half among the whole candidate molecules (Table 7). And KCNMA1, NBEA and KIT were all related with STAD supported by published literatures [26–28], and KIT was also related with ESCA [29]. According to the enrichment analysis results of ESCA and STAD, RAI2, KCNMA1 and KIT are involved in protein binding. NEBA is involved in protein kinase binding.
Table 7.
The common molecules supported by PubMed
| Candidate Molecule | ESCA_Ranking | STAD_Ranking | ESCA_PMID | STAD_PMID |
|---|---|---|---|---|
| RAI2 | 17 | 36 | – | – |
| KCNMA1 | 36 | 9 | – | 28231797 |
| NBEA | 37 | 52 | – | 28035468 |
| KIT | 41 | 50 | 21626441 | 25741136 |
Then we investigated the expression level of these 4 genes. And all these genes were down-regulated in these two types of cancer, as shown in Fig. 7, which showed that these two types of cancer shared similar molecular characteristics.
Fig. 7.

The expression levels of four genes in different samples. a RAI2 b NBEA c KCNMA1 d KIT
Discussion
We identified 148 and 242 FFLs for ESCA and STAD, respectively, and 52 FFLs were common for both of the two types of cancer, which meant that ESCA and STAD shared common regulatory properties. Gene set enrichment analysis for the genes in the FFLs also showed that they share many functional entries, including GO terms and biological pathways. For the top 20 candidate molecules in the ranking list, we validated 13 and 12 molecules in literature for ESCA and STAD, respectively, which also showed that our analysis is effective. We also investigated four genes, RAI2, KCNMA1, NBEA, and KIT, in the two ranking lists, and their potential functions for these two types of cancer. In all, we found that ESCA and STAD were close related with each other from the gene regulation prospect.
Conclusions
We proposed a computational framework to investigate the regulatory properties of ESCA and STAD. In detail, we integrated gene/ miRNA expression profiles and TF/miRNA-target pairs from different data sources. Then we constructed disease-specific regulatory networks for ESCA and STAD, respectively, and identified FFLs from these two regulatory networks. We further analyzed the molecules in the identified FFLs and built two disease-specific co-expression networks. Finally, we prioritized candidate disease molecules using random walk with restart in these two disease co-expression networks. The results showed that ESCA and STAD shared common gene regulatory properties and molecular characteristics.
In this study, we performed a systematic analysis of gene regulatory properties of two types of cancer in the digestive tract. We focused on three points: firstly, we compared the molecular mechanisms of these two types of cancer, ESCA and STAD. Secondly, we built disease-specific regulatory networks and identified FFLs. Thirdly, we built disease-specific co-expression networks and predicted candidate molecules with RWR.
However, there are some problems in our study. Firstly, our analysis was heavy influenced by the incomplete and noise public data. Secondly, more omics data should be included to provide a more comprehensive model for the complex biological system.
Methods
Data source and pre-processing
Transcriptional/post-transcriptional regulations
For transcriptional regulations, we obtained TF-target pairs from Transcriptional Regulatory Relationships Unraveled by Sentence-based Text mining (TRRUST) [30] and Human Transcriptional Regulation Interactions database (HTRIdb) [31] and obtained TF-miRNA pairs from mirTrans [32] and TransmiR [33]. As for the data in mirTrans, we reserved the pairs with affinity score no smaller than 1 and conservation score no smaller than 0.95. The TFs to be studied were derived from these four data sources. And then TF-target pairs were divided into TF-gene and TF-TF by the obtained TF, and the TF-TF pairs were removed.
For post-transcriptional regulations, we downloaded the miRNA-target pairs from miRanda [34], PITA [35], and TargetScan [36]. The pairs that appeared at least twice in these three databases were kept. Meanwhile, the miRNA-target pairs were divided into miRNA-TF pairs and miRNA-gene pairs.
Finally, there were 13,768 miRNA-TF pairs, 124,393 miRNA-gene pairs, 53,855 TF-gene pairs and 7036 TF-miRNA pairs, respectively.
Disease related genes and miRNAs
We collected disease-related genes from Online Mendelian Inheritance in Man (OMIM) [37] and the Catalogue Of Somatic Mutations In Cancer (COSMIC) [38]. OMIM is a database which collects data, including human genes, genetic phenotypes and the relationships between diseases and genes, while COSMIC is a database which explores the impact of somatic mutations in human cancer. We also collected disease-related miRNAs from miR2Disease [39], PhenomiR [40] and the Human microRNA Disease Database (HMDDv2.0) [41].
At last, we obtained 17 ESCA-related genes and 186 ESCA-related miRNAs, 30 STAD-related genes and 381 STAD-related miRNAs.
Gene and miRNA expression profiles
Clinical data and gene/miRNA expression profiles were downloaded from TCGA [42]. First, we retained the samples which satisfied the following three conditions. (1) They should be paired, i.e. there should be a corresponding normal sample for a tumor sample; (2) They should have gene expression profile; (3) They should have miRNA expression profile. We obtained 20 (10 tumor samples and 10 normal samples) and 64 (32 tumor samples and 32 normal samples) samples for ESCA and STAD, respectively.
We filtered the genes whose expression levels were less than 1 in half of the samples. And the miRNAs whose expression levels were missing in greater than 10% of samples were removed. For the remaining miRNAs, we retrieved miRNAs which were related to the specific disease, and then we deleted the miRNAs whose expression levels were missing in more than half of the samples.
After preprocessing, there were 17,150 genes and 471 miRNAs in gene/miRNA expression profiles for ESCA. And there were 17,059 genes and 477 miRNAs in gene/miRNA expression profiles for STAD.
The differential expression analysis is an important way to study the molecular mechanisms, which could help explain the mysteries of organisms. We can obtain differentially expressed molecules using the preprocessed expression data. We used the R package Limma [21] to calculate differentially expressed genes and differentially expressed miRNAs with adjusted p-value < 0.05 and |log2FC| >1.
For ESCA, we obtained 2769 differentially expressed genes and 105 differentially expressed miRNAs, which contained 1329 down-regulated genes, 1440 up-regulated genes, 17 down-regulated miRNAs, and 88 up-regulated miRNAs, respectively. For STAD, we obtained 3208 and 148 differentially expressed genes and miRNAs, which contained 1752 down-regulated genes, 1456 up-regulated genes, 14 down-regulated miRNAs and 134 up-regulated miRNAs, respectively.
MiRNA-TF-gene FFL
The FFL is one of the most important principles in regulating the responses of living cells, in which one TF A regulates another TF B, while A and B regulate their common target gene C [43]. In this study, we considered two kinds of regulation factors, TFs and miRNAs, so our FFL consists of three elements, one TF, one miRNA and one gene. Besides, we defined the molecule in one FFL regulating the other two molecules as the main regulation factor, and the expression level of the target gene depends on the main regulation factor. As a TF activates or regresses its target, while a miRNA regresses its target, and a TF and a miRNA may regulate mutually, these three elements may constitute multiple categories of FFL models. We focused on three models here (Fig. 8). These three models are really typical on the studies of molecular mechanisms of diseases [44].
Fig. 8.

Three categories of MiRNA-TF-gene FFL. a TFP-FFL b TFN-FFL c miRNAN-FFL
We named these three FFLs as TFP-FFL, TFN-FFL and miRNAN-FFL, respectively. As shown in Fig. 8, TFP-FFL describes that a TF inhibits its target miRNA and activates its target gene, and meanwhile the target miRNA inhibits the same target gene. In contrast, TFN-FFL describes that a TF activates its target miRNA and inhibits its target gene, and meanwhile the target miRNA inhibits the same target gene. Similarly, miRNAN-FFL describes that a miRNA inhibits its target gene and inhibits its target TF, meanwhile, the target TF activates the same target gene. The first two models take the TF as the main regulation factor, while the last one takes the miRNA as the main regulation factor. The final effect of TFP-FFL is to up-regulate the expression level of the target gene, while the other two FFLs will down-regulate the expression level of the target gene.
Random walk with restart
The random walk on a graph describes a walker walks from a current node to one of its neighbors randomly from a certain initial node s [45]. When a random walker is allowed to walk from the initial node s at each time step with a certain probability r, which is called random walk with restart [46].. Random walk with restart (RWR) has been successfully applied in ranking candidate disease genes by walking on biological molecular networks [46–48]. RWR with a restart probability r (0 < r < 1) is defined as Eq. (1).
| 1 |
W is a column-normalized adjacency matrix of the network, pl is a vector in which the i-th element holds the probability of being at node i at time step l [49]. p0 is an initial vector. Assuming we have m seed nodes, in p0, each seed node has a same initial probability which is 1/m, while each non-seed node has zero probability. The whole iteration process will stop when the difference between pl and pl + 1 is very small, say, less than 10− 6.
Supplementary information
Additional file 1. This .xls is a detail description of gene set enrichment analysis for STAD-specific FFL and ESCA-specific FFL
Additional file 2. This .xls is a detail description of all candidate molecules
Acknowledgements
Not applicable.
About this supplement
This article has been published as part of BMC Bioinformatics Volume 20 Supplement 22, 2019: Decipher computational analytics in digital health and precision medicine. The full contents of the supplement are available online at https://bmcbioinformatics.biomedcentral.com/articles/supplements/volume-20-supplement-22 .
Abbreviations
- ESCA
Esophageal Carcinoma
- FFL
Feed-forward loop
- RWR
random walk with restart
- SCC
spearman correlation coefficient
- STAD
Stomach Adenocarcinoma
- TF
transcription factor
Authors’ contributions
GQ, LY and YM designed the analysis pipeline, YM and JL preprocessed the data, and performed the analysis to generate presented results. GQ, LY, and QH prepared, revision and discussion the manuscript. All authors read and approved the final version of the manuscript.
Funding
This work was supported by the National Key Research and Development Program of China (2018YFC0116500) and the Natural Science Foundation of Shaanxi Province (No. 2017JM6038).
Publication costs were funded by the Natural Science Foundation of Shaanxi Province.
Availability of data and materials
Gene and miRNA datasets analysed during this study are included in TCGA (https://xenabrowser.net/datapages/) [42]. TF-target pairs were downloaded from TRRUST (https://www.grnpedia.org/trrust/) [30] and HTRIdb (http://www.lbbc.ibb.unesp.br/htri) [31]. TF-miRNA pairs were downloaded from mirTrans (http://mcube.nju.edu.cn/jwang/lab/soft/mirtrans/) [32] and TransmiR (http://cmbi.bjmu.edu.cn/transmir) [33]. MiRNA-target pairs were downloaded from miRanda (http://www.miranda.org/) [34], PITA (https://genie.weizmann.ac.il/pubs/mir07/mir07_dyn_data.html) [35] and TargetScan (http://www.tar-getscan.org/) [36]. Disease-related genes were downloaded from OMIM (http://www.omim.org/) [37] and COSMIC (http://cancer.sanger.ac.uk/cosmic) [38]. Disease-related mi-RNAs were downloaded from miR2Disease (http://www.mir2disease.org/) [39], PhenomiR (http://mips.helmholtz-muenchen.de/phenomir) [40] and HMDDV2.0 (http://210.73.221.6/hmdd) [41].
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12859-019-3230-6.
References
- 1.Pennathur A, Gibson MK, Jobe BA, Luketich JD. Oesophageal carcinoma. Lancet. 2013;381(9864):400–412. doi: 10.1016/S0140-6736(12)60643-6. [DOI] [PubMed] [Google Scholar]
- 2.Yin Jun, Wang Liming, Tang Weifeng, Wang Xu, Lv Lu, Shao Aizhong, Shi Yijun, Ding Guowen, Chen Suocheng, Gu Haiyong. RANK rs1805034 T>C Polymorphism Is Associated with Susceptibility of Esophageal Cancer in a Chinese Population. PLoS ONE. 2014;9(7):e101705. doi: 10.1371/journal.pone.0101705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siewert JR, Ott K. Are squamous and adenocarcinomas of the esophagus the same disease? Semin Radiat Oncol. 2007;17(1):38–44. doi: 10.1016/j.semradonc.2006.09.007. [DOI] [PubMed] [Google Scholar]
- 4.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65(2):87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 5.Gu J, Li Y, Fan L, Zhao Q, Tan B, Hua K, Wu G. Identification of aberrantly expressed long non-coding RNAs in stomach adenocarcinoma. Oncotarget. 2017;8(30):49201–49216. doi: 10.18632/oncotarget.17329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu J, Liu F, Shi Y, Tan H, Zhou L. Identification of key miRNAs and genes associated with stomach adenocarcinoma from The cancer Genome Atlas database. FEBS Open Bio. 2018;8(2):279–294. doi: 10.1002/2211-5463.12365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pan Z, Mao W, Bao Y, Zhang M, Su X, Xu X. The long noncoding RNA CASC9 regulates migration and invasion in esophageal cancer. Cancer Med. 2016;5(9):2442–2447. doi: 10.1002/cam4.770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Long L, Pang XX, Lei F, Zhang JS, Wang W, Liao LD, Xu XE, He JZ, Wu JY, Wu ZY, et al. SLC52A3 expression is activated by NF-kappaB p65/Rel-B and serves as a prognostic biomarker in esophageal cancer. Cell Mol Life Sci. 2018;75(14):2643–2661. doi: 10.1007/s00018-018-2757-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baffa R, Negrini M, Mandes B, Rugge M, Ranzani GN, Hirohashi S, Croce CM. Loss of heterozygosity for chromosome 11 in adenocarcinoma of the stomach. Cancer Res. 1996;56(2):268. [PubMed] [Google Scholar]
- 10.Tamura G, Sakata K, Nishizuka S, Maesawa C, Suzuki Y, Terashima M, Eda Y, Satodate R. Allelotype of adenoma and differentiated adenocarcinoma of the stomach. J Pathol. 1996;180(4):371–377. doi: 10.1002/(SICI)1096-9896(199612)180:4<371::AID-PATH704>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 11.Choi WH, Lee S, Cho S. Microsatellite alterations and protein expression of 5 major tumor suppressor genes in gastric adenocarcinomas. Transl Oncol. 2018;11(1):43–55. doi: 10.1016/j.tranon.2017.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Network CGAR Integrated genomic characterization of oesophageal carcinoma. Nature. 2017;541(7636):169–175. doi: 10.1038/nature20805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koemans WJ, Chalabi M, van Sandick JW, van Dieren JM, Kodach LL. Beyond the PD-L1 horizon: in search for a good biomarker to predict success of immunotherapy in gastric and esophageal adenocarcinoma. Cancer Lett. 2018;442:279–286. doi: 10.1016/j.canlet.2018.11.001. [DOI] [PubMed] [Google Scholar]
- 14.Kamapantula BK, Mayo ML, Perkins EJ, Ghosh P. The structural role of feed-forward loop motif in transcriptional regulatory networks. Mob Netw Appl. 2016;21(1):191–205. doi: 10.1007/s11036-016-0708-6. [DOI] [Google Scholar]
- 15.Ye H, Liu X, Lv M, Wu Y, Kuang S, Gong J, Yuan P, Zhong Z, Li Q, Jia H, et al. MicroRNA and transcription factor co-regulatory network analysis reveals miR-19 inhibits CYLD in T-cell acute lymphoblastic leukemia. Nucleic Acids Res. 2012;40(12):5201–5214. doi: 10.1093/nar/gks175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun J, Gong X, Purow B, Zhao Z. Uncovering MicroRNA and transcription factor mediated regulatory networks in glioblastoma. PLoS Comput Biol. 2012;8(7):e1002488. doi: 10.1371/journal.pcbi.1002488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo AY, Sun J, Jia P, Zhao Z. A novel microRNA and transcription factor mediated regulatory network in schizophrenia. BMC Syst Biol. 2010;4:10. doi: 10.1186/1752-0509-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yan Z, Shah PK, Amin SB, Samur MK, Huang N, Wang X, Misra V, Ji H, Gabuzda D, Li C. Integrative analysis of gene and miRNA expression profiles with transcription factor-miRNA feed-forward loops identifies regulators in human cancers. Nucleic Acids Res. 2012;40(17):e135. doi: 10.1093/nar/gks395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jiang W, Mitra R, Lin CC, Wang Q, Cheng F, Zhao Z. Systematic dissection of dysregulated transcription factor-miRNA feed-forward loops across tumor types. Brief Bioinform. 2016;17(6):996–1008. doi: 10.1093/bib/bbv107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jiang L, Yu X, Ma X, Liu H, Zhou S, Zhou X, Meng Q, Wang L, Jiang W. Identification of transcription factor-miRNA-lncRNA feed-forward loops in breast cancer subtypes. Comput Biol Chem. 2018;78:1–7. doi: 10.1016/j.compbiolchem.2018.11.008. [DOI] [PubMed] [Google Scholar]
- 21.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lagergren K, Ek WE, Levine D, Chow WH, Bernstein L, Casson AG, Risch HA, Shaheen NJ, Bird NC, Reid BJ, et al. Polymorphisms in genes of relevance for oestrogen and oxytocin pathways and risk of Barrett’s oesophagus and oesophageal adenocarcinoma: a pooled analysis from the BEACON Consortium. PLoS One. 2015;10(9):e0138738. doi: 10.1371/journal.pone.0138738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang WZ, Li Z, Wang JW, Du ML, Li BW, Zhang L, Li Q, Xu JH, Wang LJ, Li FY, et al. A functional polymorphism in TFF1 promoter is associated with the risk and prognosis of gastric cancer. Int J Cancer. 2018;142(9):1805–1816. doi: 10.1002/ijc.31197. [DOI] [PubMed] [Google Scholar]
- 24.Huang DW, Sherman BT, Tan Q, Collins JR, Alvord WG, Roayaei J, Stephens R, Baseler MW, Lane HC, Lempicki RA. The DAVID gene functional classification tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 2007;8(9):R183. doi: 10.1186/gb-2007-8-9-r183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen X, Liu M, Yan G-Y. RWRMDA: predicting novel human microRNA-disease associations. Mol BioSyst. 2012;8:2792–2798. doi: 10.1039/c2mb25180a. [DOI] [PubMed] [Google Scholar]
- 26.Wang YM, Gu ML, Ji F. Succinate dehydrogenase-deficient gastrointestinal stromal tumors. World J Gastroenterol. 2015;21(8):2303–2314. doi: 10.3748/wjg.v21.i8.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hou JY, Wang YG, Ma SJ, Yang BY, Li QP. Identification of a prognostic 5-Gene expression signature for gastric cancer. J Cancer Res Clin Oncol. 2017;143(4):619–629. doi: 10.1007/s00432-016-2324-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ma GX, Liu HT, Hua QH, Wang ML, Du ML, Lin YD, Ge YQ, Gong WD, Zhao QH, Qiang FL, et al. KCNMA1 cooperating with PTK2 is a novel tumor suppressor in gastric cancer and is associated with disease outcome. Mol Cancer. 2017;16. [DOI] [PMC free article] [PubMed]
- 29.Terada T. Primary esophageal small cell carcinoma with brain metastasis and with CD56, KIT, and PDGFRA expressions. Pathol Oncol Res. 2012;18(4):1091–1093. doi: 10.1007/s12253-011-9374-y. [DOI] [PubMed] [Google Scholar]
- 30.Han H, Cho JW, Lee S, Yun A, Kim H, Bae D, Yang S, Chan YK, Lee M, Kim E. TRRUST v2: an expanded reference database of human and mouse transcriptional regulatory interactions. Nucleic Acids Res. 2017;46(Database issue):D380–D386. doi: 10.1093/nar/gkx1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bovolenta Luiz A, Acencio Marcio L, Lemke Ney. HTRIdb: an open-access database for experimentally verified human transcriptional regulation interactions. BMC Genomics. 2012;13(1):405. doi: 10.1186/1471-2164-13-405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hua X, Tang R, Xu X, Wang Z, Xu Q, Chen L, Wingender E, Li J, Zhang C, Wang J. mirTrans: a resource of transcriptional regulation on microRNAs for human cell lines. Nucleic Acids Res. 2018;46(Database issue):D168–D174. doi: 10.1093/nar/gkx996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang J, Lu M, Qiu C, Cui Q. TransmiR: a transcription factor-microRNA regulation database. Nucleic Acids Res. 2010;38(Database issue):D119–D122. doi: 10.1093/nar/gkp803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Enright Anton J, John Bino, Gaul Ulrike, Tuschl Thomas, Sander Chris, Marks Debora S. Genome Biology. 2003;5(1):R1. doi: 10.1186/gb-2003-5-1-r1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Michael K, Nicola I, Ulrich U, Ulrike G, Eran S. The role of site accessibility in microRNA target recognition. Nat Genet. 2007;39(10):1278–1284. doi: 10.1038/ng2135. [DOI] [PubMed] [Google Scholar]
- 36.Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. 2015;4. 10.7554/eLife.05005http://www.targetscan.org/. Accessed on 16 Mar 2018. [DOI] [PMC free article] [PubMed]
- 37.Ada H, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33(1):514–517. doi: 10.1093/nar/30.1.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Forbes Simon A., Beare David, Gunasekaran Prasad, Leung Kenric, Bindal Nidhi, Boutselakis Harry, Ding Minjie, Bamford Sally, Cole Charlotte, Ward Sari, Kok Chai Yin, Jia Mingming, De Tisham, Teague Jon W., Stratton Michael R., McDermott Ultan, Campbell Peter J. COSMIC: exploring the world's knowledge of somatic mutations in human cancer. Nucleic Acids Research. 2014;43(D1):D805–D811. doi: 10.1093/nar/gku1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang QWY, Hao Y, Juan L, Teng M, Zhang X, Li M, Wang G, Liu Y. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009;37(1):D98–104. doi: 10.1093/nar/gkn714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ruepp Andreas, Kowarsch Andreas, Schmidl Daniel, Bruggenthin Felix, Brauner Barbara, Dunger Irmtraud, Fobo Gisela, Frishman Goar, Montrone Corinna, Theis Fabian J. PhenomiR: a knowledgebase for microRNA expression in diseases and biological processes. Genome Biology. 2010;11(1):R6. doi: 10.1186/gb-2010-11-1-r6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang L, Chengxiang Q, Jian T, Bin G, Jichun Y, Tianzi J, Qinghua C. HMDD v2.0: a database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 2014;42(Database issue):D1070. doi: 10.1093/nar/gkt1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tomczak K, Czerwińska P, Wiznerowicz M. The Cancer Genome Atlas (TCGA): an immeasurable source of knowledge. Contemp Oncol. 2015;19(1A):68–77. doi: 10.5114/wo.2014.47136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mangan S, Alon U. Structure and function of the feed-forward loop network motif. Proc Natl Acad Sci U S A. 2003;100(21):11980–11985. doi: 10.1073/pnas.2133841100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lai X, Wolkenhauer O, Vera J. Understanding microRNA-mediated gene regulatory networks through mathematical modelling. Nucleic Acids Res. 2016;44(13):6019–6035. doi: 10.1093/nar/gkw550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Can T, Ç Amo O, Glu, Singh AK. Analysis of protein-protein interaction networks using random walks; 2005.
- 46.Sebastian KH, Sebastian B, Denise H, Robinson PN. Walking the interactome for prioritization of candidate disease genes. Am J Hum Genet. 2008;82(4):949–958. doi: 10.1016/j.ajhg.2008.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huan T, Wu X, Bai Z, Chen JY. Seed-weighted random walk ranking for cancer biomarker prioritisation: a case study in leukaemia. Int J Data Min Bioinform. 2014;9(2):135–148. doi: 10.1504/IJDMB.2014.059064. [DOI] [PubMed] [Google Scholar]
- 48.Gutierrez-Arcelus M, Ongen H, Lappalainen T, Montgomery SB, Buil A, Yurovsky A, Bryois J, Padioleau I, Romano L, Planchon A, et al. Tissue-specific effects of genetic and epigenetic variation on gene regulation and splicing. PLoS Genet. 2015;11(1):e1004958. doi: 10.1371/journal.pgen.1004958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li L, Wang Y, An L, Kong X, Huang T. A network-based method using a random walk with restart algorithm and screening tests to identify novel genes associated with Menière’s disease. PLoS One. 2017;12(8):e0182592. doi: 10.1371/journal.pone.0182592. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. This .xls is a detail description of gene set enrichment analysis for STAD-specific FFL and ESCA-specific FFL
Additional file 2. This .xls is a detail description of all candidate molecules
Data Availability Statement
Gene and miRNA datasets analysed during this study are included in TCGA (https://xenabrowser.net/datapages/) [42]. TF-target pairs were downloaded from TRRUST (https://www.grnpedia.org/trrust/) [30] and HTRIdb (http://www.lbbc.ibb.unesp.br/htri) [31]. TF-miRNA pairs were downloaded from mirTrans (http://mcube.nju.edu.cn/jwang/lab/soft/mirtrans/) [32] and TransmiR (http://cmbi.bjmu.edu.cn/transmir) [33]. MiRNA-target pairs were downloaded from miRanda (http://www.miranda.org/) [34], PITA (https://genie.weizmann.ac.il/pubs/mir07/mir07_dyn_data.html) [35] and TargetScan (http://www.tar-getscan.org/) [36]. Disease-related genes were downloaded from OMIM (http://www.omim.org/) [37] and COSMIC (http://cancer.sanger.ac.uk/cosmic) [38]. Disease-related mi-RNAs were downloaded from miR2Disease (http://www.mir2disease.org/) [39], PhenomiR (http://mips.helmholtz-muenchen.de/phenomir) [40] and HMDDV2.0 (http://210.73.221.6/hmdd) [41].