

Abstract
Background
The main goal of successful gene selection for microarray data is to find compact and predictive gene subsets which could improve the accuracy. Though a large pool of available methods exists, selecting the optimal gene subset for accurate classification is still very challenging for the diagnosis and treatment of cancer.
Results
To obtain the most predictive genes subsets without filtering out critical genes, a gene selection method based on least absolute shrinkage and selection operator (LASSO) and an improved binary particle swarm optimization (BPSO) is proposed in this paper. To avoid overfitting of LASSO, the initial gene pool is divided into clusters based on their structure. LASSO is then employed to select high predictive genes and further calculate the contribution value which indicates the genes’ sensitivity to samples’ classes. With the second-level gene pool established by double filter strategy, the BPSO encoding the contribution information obtained from LASSO is improved to perform gene selection. Moreover, from the perspective of the bit change probability, a new mapping function is defined to guide the updating of the particle to select the more predictive genes in the improved BPSO.
Conclusions
With the compact gene pool obtained by double filter strategies, the improved BPSO could select the optimal gene subsets with high probability. The experimental results on several public microarray data with extreme learning machine verify the effectiveness of the proposed method compared to the relevant methods.
Keywords: Binary particle swarm optimization, Gene selection, LASSO, Extreme learning machine
Background
DNA microarray datasets have been used to identify the optimal gene subset and perform sample classification between different disease phenotypes, for diagnostic and prognostic purposes [1]. However, many computational methods have difficulties in selecting the optimal set of genes as a result of the small number of samples compared to the huge number of genes, irrelevant genes, and noisy genes [2], which leads poor generalization in the classification process. As a data preprocessing technique, gene selection is a key step for classification [3]. Selecting a critical gene subset could not only decrease the computational complexity and gene redundancy, but also increase the classification accuracy. However, gene selection is a tough task for the high-dimensional microarray data.
Fortunately, the development of swarm intelligence optimization algorithm offers great advantages for microarray data [4]. Due to its simple operation, fast convergence, good global search ability, the swarm intelligence optimization algorithm has been widely accepted and successfully applied to solve a lot of problems.
As an efficient global search technique, particle swarm optimization (PSO) [5, 6] has been widely applied to microarray data. Precisely because of its fast convergence speed and good convergence accuracy, PSO has attracted much more attention [7, 8] in gene selection. In [9], a combination of teaching learning-based optimization (TLBO) and particle swarm optimization was proposed to find the small optimal gene subset. In [10], the binary PSO (BPSO) coupled with filter method was implemented in searching optimal gene subsets. Sahu et al. [11] proposed a novel feature selection algorithm using PSO for microarray data, which used filtering technique such as signal-to-noise ratio (SNR) score combined with PSO to select key genes for classification and achieved a better classification accuracy than other non-PSO algorithms. In the Kmeans-PSO-ELM [12], the initial gene pool was firstly grouped into several clusters by the the K-means method, and then a compact set of informative genes were obtained after combining the standard PSO with extreme learning machine. These hybrid methods mentioned above had the ability of searching a small predictive gene subset for sample classification. However, the genes selected by these methods were not easily interpretable. Moreover, despite the fact that PSO shows superior performance for selecting optimal feature subsets, it still suffers from the drawback that it is easy to converge to local minima and lead to premature convergence. To overcome the deficiencies of the above PSO based gene selection methods, a modified discrete PSO combined support vector machines (SVM) was proposed in [13] for tumor classification, which verified that the modified PSO was a useful tool for gene selection. In [14], an improved PSO (PSO-RG) with a new global best position (gbest) updating mechanism was proposed to avoid being trapped in a local optimum and achieved superior classification performance. In [15], a gene selection method based on hybrid model BPSO and Bayesian linear discriminant analysis (BLDA) was proposed to select genes with lower redundancy and high classification accuracy. Although the method could relieve the premature problem of PSO and select compact gene subsets, the proposed method selects genes that are not easily interpretable and it may also filtering out some critical genes.
To obtain predictive genes with more interpretability, two gene selection methods based on binary PSO and gene-to-class sensitivity (GCS) information were proposed in [16, 17]. In the KMeans-GCSI-MBPSO-ELM [16], a modified BPSO coupling GCS information (GCSI) combined with ELM was used to select smallest possible gene subsets. Although it could obtain predictive genes with lower redundancy and better interpretability, it might filter out a few critical genes highly related to sample classification in some cases and thus lead into worse classification accuracy.
Least absolute shrinkage and selection operator (LASSO) is a regression analysis method that performs both variable selection and regularization in order to enhance the prediction accuracy and interpretability of the statistical model it produces [18]. Since it can typically extremely sparse, leading to interpretable models with only very few predictor variables, LASSO become another powerful feature selection method [19]. In [20], LASSO was used to select the top key variables in the regression process and achieved a superior performance in gene selection. However, the limitation of the method is that the gene structure in microarray data is not taken into account enough. Furthermore, it is computational costly and may cause overfitting problem.
According to the above analysis, some current PSO-based gene selection methods lack interpretability as well as filtering out some key genes. Some LASSO-based methods do not consider gene structure and have overfitting problem with high computational cost. To overcome those deficiencies, an efficient gene method combining LASSO with an improved BPSO is proposed in this paper. Firstly, the signal-to-noise ratio (SNR) filter method is employed to filter out some genes in order to establish the initial gene pool. The genes in initial pool are divided into different clusters based on their true geometric structure. Then, LASSO is conducted to select the top contributing genes in each cluster individually to establish the second level gene pool. Finally, an improved BPSO is proposed to select the optimal gene subset. In the improved BPSO, to obtain predictive genes with better interpretability, the contribution values from the LASSO process, indicating the genes’ sensitivity to samples’ classes, are encoded into the initial and update process of the BPSO. Moreover, from the perspective of the bit change probability, a new mapping function is defined to guide the updating of the particle in order that the swarm can converge to the global optimum with high possibility. With the compact gene pool obtained by double filter strategies, the improved BPSO could select the optimal gene subsets with high probability. Experimental results on several public microarray data verify the effectiveness and efficiency of the proposed hybrid gene selection method.
The remainder of this paper is organized as follows. The related preliminaries and the proposed gene selection method are described in “Methods” section. Simulations are carried out and results are analyzed in “Results” section. Finally, the concluding remarks are offered in “Conclusions” section.
Methods
Binary particle swarm optimization
Particle swarm optimization (PSO) is a population-based optimization algorithm in search for the best solution by simulating the movement of flock of birds [6]. The binary PSO [21] which is used for discrete problem was proposed. Its general steps are described as follows.
The velocity of the i − th particle is represented by vi = (vi1, vi2, ..., viD) and the position of the i − th particle is represented by xi = (xi1, xi2, ..., xiD), i = 1, 2, ..., n, where n is the size of population. Once the adaptive values personal best position (pbest) and gbest are obtained, the features of the pbest and gbest particle can be traced with regard to their position and velocity. Each particle is updated according to the following equation:
| 1 |
| 2 |
| 3 |
where j = (1, 2, ..., D); pbestij = (pbesti1, pbesti2, ..., pbestiD) is the personal best position of the i − th particle and gbest(t) = (gbest1, gbest2, ..., gbestD) is the global best position in the t-th iteration; ω is the inertial weight of BPSO; t denotes the iteration number; c1 and c2 are two acceleration factors which can balance the impact of pbest and gbest; r1 and r2 are two numbers randomly generated in [0, 1].
Least absolute shrinkage and selection operator
To improve variable selection, Tibshirani [18] developed the least absolute shrinkage and selection operator (LASSO). LASSO is a combination of ridge regression. It can automatically select a set of informative variables through the regression coefficients in the linear regression model shrinking to zero [22].
Suppose that the data (xi, yi) contains n samples and m features, x = (x1, x2, ..., xm),where xj = (x1j, x2j, ...xnj)T are the predictor variables, y = (y1, y2, ...ynj)T and yi is the responses. Assume that the xij are standardized and the yi are centralization, there is:
| 4 |
Letting regression coefficients β = (β1, β2, ...βm), the LASSO estimate is defined as follows:
| 5 |
where t ≥ 0 is a tuning parameter.
Extreme learning machine
To solve the problem of gradient-based learning algorithms, a learning algorithm for single-hidden layer feedforward neural networks (SLFNs) called extreme learning machine (ELM) was proposed in [23]. In ELM, the input weights and hidden biases are randomly selected, and then the output weights are calculated by generalized inverse of hidden output matrix. ELM has much better generalization performance with much faster learning speed than gradient-based algorithms [24, 25, 26]. For N arbitrary distinct samples (xi, ti) (i = 1, 2, ..., N), where xi = [xi1, xi2, ..., xin] ∈ Rn, ti = [ti1, ti2, ..., tim] ∈ Rm. A SLFN with NH hidden nodes and activation function g( ) can approximate these N samples with zero error. This means that:
| 6 |
where T represents the target matrix vectors, H is the hidden output matrix:
| 7 |
where the ωhi = [ωhi1, ωhi2.., ωhin]T is the input weight vector connecting the i − th hidden neuron and input neurons, the ωoi = [ωoi1, ωoi2.., ωoim] is the output weight vector connecting the i − th hidden neuron and the output neurons.
In the process of learning, the input weight and the hidden biases are arbitrarily chosen and need not be adjusted at all. Secondly, the smallest norm least-squares solution of the Eq. (6) is obtained as follows:
| 8 |
where H+ is the Moore-Penrose inverse of H.
The proposed gene selection method based on LASSO and BPSO
The proposed method is aimed to deal with the two problems on how to take advantage of intrinsic gene structure information, avoid overfitting with less complexity and how to select the optimal gene subsets to improve the classification accuracy without filtering out key genes. In the selection process, geodesic distance is calculated as the measurement between genes, which can preserve the intrinsic geometry of high dimensional microarray data. To decrease the complexity of the model and avoid overfitting of LASSO, the initial gene pool data are divided into clusters by using the K-medoids approach. The clustering process based on geodesic distance could solve the former problem. To solve the latter problem, the improved BPSO is proposed to select the possible gene subsets, including encoding gene contribution information and defining a new mapping function, which could help particles converge to the optimal with higher possibility. The gene contribution information and the improved BPSO are depicted in detail in the following subsections.
The contribution value of each gene obtained by LASSO
As mentioned above, the candidate elite gene pool is established by LASSO. However, LASSO is an ordinary least squares cost function extended with a L1 penalty on the regression coefficients. Since the interval of parameter t is crucial for LASSO and hard to be determined in practical, in this study, Least angle regression (LAR) is used for LASSO. It would return the entire solution path directly for any fixed sample set. So the LASSO process can be described as follows:
Step 1: Normalize all the gene variables and centralize all the predictors:
Set residual,the coefficients β = (β1, β2, ..., βm) are initialized to 0, where ;
Step 2: Find the xj which is most correlated with r, the current correlation coefficient ;
Step 3: Move βj from 0 towards the inner product of the xj and r until some other variable xk has as much correlation with the current residual r;
Step 4: Move βj and βk the inner product of the α = (xj, xk) and r until some other variable xp has as much correlation with the current residual r; If the coefficient βm is decreased to 0, then delete the corresponding variable xm and recalculate the r;
Step 5: Repeat step 2 to step 4 until all variables have been calculated by the model.
After the LASSO method, top contributing genes are selected and the corresponding regression coefficients are obtained. Furthermore, the value of the regression coefficients β = (β1, β2, ...βm) are the contribution values of genes to the class.
The improved BPSO
In this study, BPSO is modified from two aspects. One is to encode the contribution value to the BPSO to select those genes which are much related to samples’ classes. The other is to modify the mapping function of velocity in BPSO for increasing the probability of finding the optimal with fewer iterations.
Generally, a variable with larger coefficient value makes more contribution to regression model. Consequently, it is convincing that gene with a large contribution value contributes more to samples’ classification than one with small contribution value, so it is reasonable to select those genes with large contribution values to achieve higher classification accuracy. In this study, the contribution value is encoding into the BPSO in the swarm initialization and update.
It is possible that the particles initialized by the traditional BPSO randomly are far from the global minima or near some local minima, which may lead to slow convergence and premature. In this study, to make the initial swarm near the global minima with high probability, the particles are initialized according to the contribution values of genes selected. Hence, the top twenty percentages of the genes with the largest contribution values are randomly initialized as 1 s or 0 s after all genes sorted in descending order according to their contribution values. The rest components related to the eighty percentages of the genes with the smallest contribution values are initialized as 0, which indicates that those genes are not selected.
In the swarm update process, given that the high contribution value indicating the high possibility to be selected, the formula of updating the particles is modified according to the contribution value as follows:
| 9 |
where Contribution(j) is the contribution value of the j-th gene. s( ) is the mapping function and avg(Contribution) is the average contribution value of all genes. xij and vij are the j-th component of the position and velocity respectively of the i-th particle. Under the effective guidance it directly searches the optimal gene subset sensitive to the class from the candidate elite gene pool.
To make sure the particle could converge to the global best position with higher possibility, the mapping function in BPSO is defined as follows:
| 10 |
where T is the maximum iteration number.
The interpretation for the new mapping function can be described as follows:
In binary PSO, each particle consists of binary code, bit change probability is first proposed in [21], it represent the change probability of every bit in binary code. According to the analysis in [27], if the bit in (t-1)-th iteration is 0, the changing probability of bit in t-th iteration is s(vij(t)); Similarly, if bit is 1 in (t-1)-th iteration, the changing probability of bit in t-th iteration is (1 − s(vij(t))). Thus, the change probability of bit in t t-th iteration is calculated as follows:
| 11 |
The relation of the bit change probability to vij can be simply characterized in Fig. 1. As can be seen, when particle xij converge to global location gbestj, the change rate is 0.5 which is up to maximum. That is, if BPSO converges to global optimal particle, its velocity is 0 which means that the rate of bit changing is maximum, so BPSO is more stochastic and lacks search directionality thus it may can not converge to the global best position.
Fig. 5.

The heatmap of expression levels based on the top ten frequently selected genes on the five data. (a) Colon (b) Brain cancer (c)Leukemia. (d) Lymphoma (e) LUNG
From the above idea, different from the mapping function in [27], the new mapping function differs from the original sigmoid in two respects. First consideration is the difference between the probability mapping function and the sigmoid function on velocity. The purpose is to make the probability function value is 0 when the speed is 0. Second consideration is from the iteration number aspect to make sure the BPSO can convergence to global optimal. The function curve is shown in Figs. 2 and 3.
Fig. 6.

The comparison of the 5-fold CV accuracy on the test data versus the iteration number between the original BPSO and the improved BPSO on the five data. (a) Colon (b) Brain cancer (c) Leukemia (d) Lymphoma (e) LUNG
Fig. 7.

The contribution value of the selected genes versus iteration number of IBPSO on the five data. (a) Colon (b) Brain cancer (c) Leukemia (d) Lymphoma (e) LUNG
From the Fig. 2, under the new mapping function, the mapping value is 0 when the velocity is 0 so the change rate is 0 which meets the requirements. Besides, as can be seen in Fig. 3, in addition to the different mappings of bit velocity, the mapping function also take the iteration number into consideration, with the iteration number increasing, the mapping value is more closer to 1, which means the gene selected with higher probability.
The steps of the proposed gene selection method
Since the proposed method combines the LASSO with BPSO based on K-medoids and ELM to perform gene selection, it is referred to as the KL-IBPSO-ELM method. Figure 4 depicts the frame of the proposed gene selection method, and the detailed steps are described as follows:
Fig. 8.
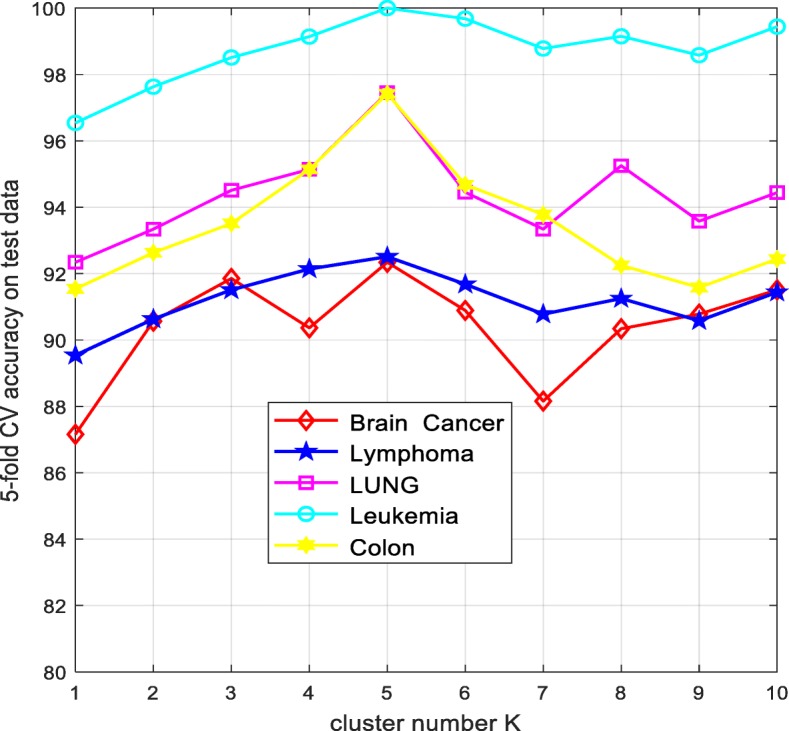
The number of the clusters versus the 5-fold CV accuracy on the test data obtained by ELM
Step 1: Form an initial gene pool. The dataset is divided into training and testing datasets. Selecting 200 genes from all original genes by using SNR method [28] on the training data. Then, the training dataset is further divided into the training and validation datasets.
Step 2: Establish the candidate elite gene pool. First, calculate the geodesic distance between every two genes in initial pool. The geodesic distance can reflect flow structure of the high dimensional gene data more precisely [29]. Then, employ the K-medoids to cluster the genes. The purpose of clustering before LASSO is to give full consideration of gene structure as well as decrease the computational complexity. Finally, the top contributing genes are obtained by LASSO selecting in every clusters. Moreover, the contribution values of the elite genes are gained.
Step 3: Use the improved BPSO to select the optimal gene subsets from the candidate elite gene pool. Initialize all particles according to the contribution Initialization rule. The position xij can be coded to 0 or 1. 1 means the i − th gene is selected and 0 means the i − th gene is not selected. Set the current position of each particle as its pbest, and compute the gbest. Update the particle according to contribution updating rule and new mapping function. Compute the fitness value of each particle. In this study, the selected gene number is not fixed so as to further avoid filter out key genes. Therefore, the fitness value of the i-th particle is adopted by the corresponding accuracy obtained by ELM denoted by the i-th particle.
The KL-IBPSO-ELM method firstly filter out the irrelevant genes by the SNR method. Then the LASSO selects the candidate elite genes in every clusters obtained through K-medoids method based on the geodesic distance. Finally, to obtain the optimal gene subsets, the BPSO is modified to improve its convergence by encoding the contribution value from LASSO and changing the new mapping function. It could select the most predictive gens with low redundancy effectively.
Additionally, the proposed gene selection method contains filtering irrelevant genes to establish the gene pool and using PSO to select functional gene subsets from the gene pool, and its computational complexity is at the same order of magnitude of that of the PSO-based [16, 17, 30] gene selection methods.
Results
Datasets
To verify the effectiveness and efficiency of the proposed method, we conduct experiments on the five open microarray datasets including Leukemia, Colon, Lymphoma, LUNG and Brain cancer data. The Leukemia, Colon, LUNG are available at: http://wwwgenome.wi.mit.edu/cgibin/cancer/datasets.cgi, http://microarray.princeton.edu/oncology/, http://www.biomedcentral.com/content/supplementary/1471-2105-7-228-S4.tgz, respectively [16, 17]. And the Brain and Lymphoma data are available at: http://linus.nci.nih.gov/~brb/DataArchive_New.html [16]. The detailed specification of the datasets is the same as in [30].
In the experiments on all data, the swarm size is 30, the maximum iteration number is selected as 100, the acceleration constants c1 and c2 are both selected as 1.49445, and the inertial weight varies from 0.9 to 0.4. The size of the second-level gene pool is 25 on all data. The parameter of cluster number is fixed as 5 on all data. The values of these parameters are determined by the cross-validation runs on the training datasets. All the experiments are run in MATLAB 8.1 environment.
The classification ability of the gene subsets selected by the proposed method
To verify the classification ability of the selected gene subsets, ELM is used to perform sample classification with some gene subsets selected by the KL-IBPSO-ELM method on the five datasets. The gene subsets which is selected by the proposed approach on five datasets are listed in Table 1. With the compact gene subsets selected by the proposed method, ELM obtains comparatively high prediction accuracies, which indicates that the KL-IBPSO-ELM method has the ability of selecting those predictive genes highly related to sample classes.
Table 1.
The classification accuracy obtained by ELM with different gene subsets selected by the KL-IBPSO-ELM method on the five microarray data
| Data | Selected gene subsets | 5-fold CV Accuracy Mean(%) ± std | Test Accuracy Mean(%) ± std |
|---|---|---|---|
| Colon | 493, 1902, 1060, 1346, 1982, 554, 1060 | 9307 ± 0.009 | 91.40 ± 0.011 |
| 377,1100,959, 475,1637,164,1764,304,60,897 | 95.81 ± 0.004 | 93.23 ± 0.012 | |
| 14,341,20,1886,164,1271,304,1136,165,1549,830,1897,1227,1042 | 97.42 ± 0.011 | 94.28 ± 0.022 | |
| 493,251,1346,377,554,1902 | 95.07 ± 0.009 | 92.16 ± 0.019 | |
| Brain cancer | 5202,3341,1243,1135,5051,30,4413,4935 | 91.63 ± 0.011 | 89.00 ± 0.012 |
| 18,3341,1582,2942,1198,6331,4917,724 | 92.67 ± 0.008 | 90.78 ± 0.011 | |
| 6429, 4917,6774,1975,587,2122,5051,6700,6828 | 92.36 ± 0.006 | 91.73 ± 0.011 | |
| 6429,4309,2304,3555,1975,3035,3341,1648,161, 724 | 91.15 ± 0.007 | 90.26 ± 0.012 | |
| Leukemia | 818,894,3135,3359,4653,4991,5094,5406,2356,445 | 100 ± 0.000 | 100 ± 0.000 |
| 3090,1694,1635,3276,1410,1523,1992,2659, | 100 ± 0.000 | 100 ± 0.000 | |
| 1268,3276,1523,1973,1855,2356,445,3150,818 | 100 ± 0.000 | 100 ± 0.000 | |
| 3276,1523,1973,1882,356,818,445,3135,2895,3082 | 100 ± 0.000 | 100 ± 0.000 | |
| Lymphoma | 4862,3589,3775,3356,343,962,3227,2666,2810,2734 | 90.09 ± 0.006 | 89.54 ± 0.011 |
| 4862,3589,3227,704,2810,4998 | 92.06 ± 0.008 | 90.63 ± 0.004 | |
| 4514,3589,5709,6172,2666,2810,3525 | 91.63 ± 0.006 | 90.24 ± 0.010 | |
| 3589,3775,5709,6565,5329,418,5818,4998 | 92.23 ± 0.005 | 91.63 ± 0.010 | |
| LUNG | 235,295,1411,1784,1921,1974,2264,2672,3187 | 91.64 ± 0.007 | 89.64 ± 0.013 |
| 1268,1523,1822,2356,445,2556,1318,1411,295,2005,1712 | 94.64 ± 0.007 | 90.64 ± 0.016 | |
| 2479,924,2969,1973,1822,580,2279,2128,1432,2005,414 | 95.81 ± 0.004 | 90.35 ± 0.011 | |
| 1268,3276,2969,441,295,2904,445,2895,2128,261,1028, 2005 | 91.35 ± 0.012 | 88.08 ± 0.021 |
The biological and functional analysis of the selected gene subsets
The experiments are carried out 500 times on each microarray data, and the top ten frequently selected genes by the proposed method are listed in Tables 2, 3, 4, 5, and 6. Many genes selected by the KL-IBPSO-ELM method were also selected by one or more methods proposed in [16, 17] [30–34].
Table 2.
The top ten frequently selected genes with the proposed method on the Brain cancer data
| Gene no. | Gene name | Description |
|---|---|---|
| 18 | AB000895 | Dachsous 1(Drosophila) #※ |
| 4917 | U65676 | Hermansky-Pudlak syndrome 1 |
| 4309 | U33849_at | Proprotein, convertase subtilisin/ kexin type 7 |
| 4413 | U39817 | Bloom syndrome ※ |
| 4657 | U51095 | caudal type homeo box transcription factor 1 |
| 4843 | U61262 | neogenin homolog 1 (chicken)#※ |
| 5931 | X58987 | dopamine receptor D1 #※ |
| 3041 | M64394 | Kell blood group #❖ |
| 6480 | X87159 | Sodium channel, nonvoltage-gated 1,beta (Liddle syndrome)# |
| 6429 | X83703 | ankyrin repeat domain 1 (cardiac muscle) |
Table 3.
The top ten frequently selected genes with the proposed method on the Colon data
| Gene no. | Gene name | Description |
|---|---|---|
| 493 | R87126 | Myosin heavy chain, nonmuscle (gallus gallus)✱ |
| 14 | H20709 | Myosin light chain alkali, smooth muscle isoform (human)#※❖✺✱ |
| 377 | Z50573 | H.sapiens mRNA for GCAP-II/uroguanylin precursor. #✺ |
| 251 | U37012 | Hunman cleavage and polyadenylation specificity factor mRNA,complete cds. ※ |
| 554 | H24401 | MAP KINASE PHOSPHATASE-1 (Homo sapiens) |
| 175 | T94579 | Human chitotriosidase precursor mRNA, complete cds #※✺ |
| 1346 | T62947 | 60S RIBOSOMAL PROTEIN L24 (Arabidopsis thaliana) |
| 1771 | J05032 | Human aspartyl-tRNA synthetase alpha-2 subunit mRNA, complete cds✺ |
| 765 | M76378 | Human cysteine-rich protein (CPR) gene, exons 5 and 6 #✺ |
| 1902 | U01038 | Human pLK mRNA, complete cds |
Table 4.
The top ten frequently selected genes with the proposed method on the LUNG data
| Gene no. | Gene name | Description |
|---|---|---|
| 580 | 39333_at | collagen, type IV, alpha 1# |
| 235 | 41,770 | Cluster Incl AA420624:nc61c12.r1 Homo sapiens cDNA |
| 295 | 36,681 | apolipoprotein D |
| 1411 | 37,954 | annexin A8 |
| 1784 | 35,874 | lymphoid-restricted membrane protein |
| 1921 | 32,748 | Cluster Incl AI557852:P6test.G05.r Homo sapiens cDNA |
| 1974 | 33,656 | ribosomal protein L37 |
| 2264 | 32,104 | calcium/calmodulin-dependent protein kinase (CaM kinase) II gamma |
| 2672 | 37,970 | mitogen-activated protein kinase 8 interacting protein 3 |
| 3178 | 38799_at | Cluster Incl AF068706:Homo sapiens gamma2-adaptin (G2 AD) mRNA, complete cds /cds = (763,3018) /gb = AF068706 /gi = 3,193,225 /ug = Hs.8991 ※ |
Table 5.
The top ten frequently selected genes with the proposed method on the Lymphoma data
| Gene no. | Gene name | Description |
|---|---|---|
| 806 | D86969 | PHD finger protein 16 ※# |
| 772 | D85423 | CDC5 cell division cycle 5-like (S. pombe) |
| 1703 | L06499 | ribosomal protein L37a |
| 2320 | M13207 | colony stimulating factor 2 (granulocyte-macrophage) |
| 3419 | S48983 | serum amyloid A4, constitutive |
| 3507 | S75213 | phosphodiesterase 4A, cAMP-specific (phosphodiesterase E2 dunce homolog, Drosophila) |
| 3755 | U07358 | mitogen-activated protein kinase kinase kinase 12 |
| 4998 | U69108 | TNF receptor-associated factor 5 |
| 5230 | U81600 | paired related homeobox 2 |
| 6651 | X97630 | MAP/microtubule affinity-regulating kinase 2 |
Table 6.
The top ten frequently selected genes with the proposed method on the Leukemia data
| Gene no. | Gene name | Description |
|---|---|---|
| 4847 | X95735 | Zyxin ※#⊙✱ |
| 894 | HG3162-HT3339 | Transcription Factor Iia |
| 2354 | M92287 | CCND3 Cyclin D3#⊙✱✺ |
| 4535 | X74262 | RETINOBLASTOMA BINDING PROTEIN P48 ✱ |
| 4991 | Y09615 | GB DEF = Mitochondrial transcription termination factor |
| 2642 | U05259 | MB-1 gene ※#⊙✱✺ |
| 818 | HG1879-HT1919 | Ras-Like Protein Tc10 |
| 6283 | Y00081 | IL6 Interleukin 6 (B cell stimulatory factor 2) |
| 6855 | M31523 | TCF3 Transcription factor 3 (E2A immunoglobulin enhancer binding factors E12/E47) ⊙✱✺ |
| 1882 | M27891 | CST3 Cystatin C (amyloid angiopathy and cerebral hemorrhage) ※#⊙✺ |
The heatmap with top ten frequently selected genes for the five data is shown in Fig. 5. From Fig. 5a, the expression levels of genes 765, 14, 493, 377, 175 are distinct in two classes. From Fig. 5b, the only expression level of gene 4309 has a distinct expression level in Desmoplastic class. From Fig. 5c, most of ten genes expression levels clearly differentiate between AML and ALL. From Fig. 5d, there has no single gene whose expression levels are distinct between the two classes. From Fig. 5e, The expression levels of genes 3178 are distinct from SQ and other classes, the ones of gene 2672,1974 and 580 are distinct from PC and other classes, the ones of gene 2264, 1974 and 235 are distinct from SM and other classes, and the gene 295 has distinct expression level in ADE. According to Table 4 and Fig. 5e, the gene 295, 235 and 1974 have distinct expression level and their frequency is higher than other crucial genes. Those genes are not selected by the relevant method, so it indicates that genes 295, 235 and 1974 could be the key genes which have been filtered out by other method. Similarly, on the Brain cancer data, the gene 4309 may be the new key genes to the class.
Fig. 1.
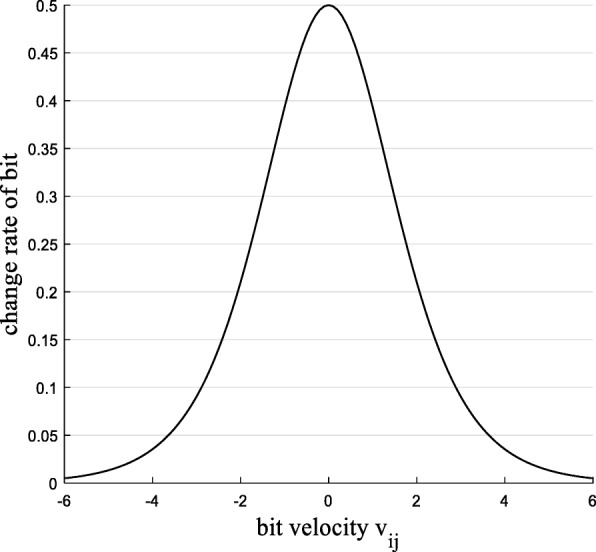
Relation between change rate and bit velocity
The comparison with other BPSO-based gene selection methods
To verify the efficiency of the proposed method, the KL-IBPSO-ELM is compared with BPSO-ELM, KMeans-BPSO-ELM, SC-IPSO-ELM and KMeans-GCSI-MBPSO-ELM. The parameters in all algorithms in all experiments are determined by trial and error. The mean results are listed in Table 7. The proposed mehod in this study outperform other four methods on the Brain cancer data. The SC-IPSO-ELM achives better performance than other methods on the Colon, Lymphoma and LUNG data, and the KMeans-GCSI-MBPSO-ELM method achieves better performance than the KL-IBPSO-ELM method on the Colon and LUNG data. On the Leukemia data, the KL-IBPSO-ELM achieves 100% 5-fold CV accuracy as well as the KMeans-GCSI-MBPSO-ELM and SC-IPSO-ELM methods. These results indicate that the KL-IBPSO-ELM is also capable of selecting those predictive genes highly related to samples’ classes.
Table 7.
The 5-fold CV classification accuracies of ELM based on the five gene selection methods on the five data
| Method | 5-fold CV Accuracy (Mean% ± std) and selected gene number | ||||
|---|---|---|---|---|---|
| Colon | Brain cancer | Leukemia | Lymphoma | LUNG | |
| BPSO-ELM | 93.34 ± 0.020(9) | 85.45 ± 0.023(7) | 98.50 ± 0.003(5) | 83.50 ± 0.027(8) | 94.80 ± 0.006(11) |
| KMeans-BPSO-ELM | 93.50 ± 0.020(9) | 87.23 ± 0.023(8) | 99.17 ± 0.010(4) | 85.14 ± 0.029(6) | 95.64 ± 0.006(12) |
| KMeans-GCSI-MBPSO-ELM | 97.61 ± 0.014(6) | 88.63 ± 0.022(6) | 100 ± 0.00(3) | 86.97 ± 0.024(8) | 97.10 ± 0.006(11) |
| SC-IPSO-ELM | 99.05 ± 0.011(13) | 91.88 ± 0.019(7) | 100 ± 0.00(3) | 93.79 ± 0.020(7) | 98.67 ± 0.019(11) |
| The proposed method | 97.42 ± 0.011(14) | 92.67 ± 0.008 (8) | 100 ± 0.000(8) | 92.23 ± 0.005(8) | 95.81 ± 0.004(11) |
The performance comparison between the original BPSO and the improved BPSO
To illustrate the performance of the improved BPSO, the experiments conducted by the KL + BPSO+ELM frame wtih the improved BPSO and original BPSO, respectively. Figure 6 shows the 5-fold CV accuracy on the test data on the five data versus the iteration number of the original BPSO compared with that of the improved BPSO. From Fig. 6, the improved BPSO finds the optimal gene subsets with only 30, 40, 38, 31 and 32 epochs on the Colon, Brain cancer, Leukemia, Lymphoma, LUNG data, respectively, whereas the original BPSO require 42, 50, 42, 38 and 40 epochs, on the above five data, respectively, which shows the improved BPSO could find the optimal with less iteration number than the original BPSO. Furthermore, on each specific epoch, the 5-fold CV accuracy of the improved BPSO is always higher than that of the original BPSO. These results indicate that the improved BPSO has the ability to converges slightly faster than the original BPSO and could selected the optimal gene subsets.
Fig. 2.
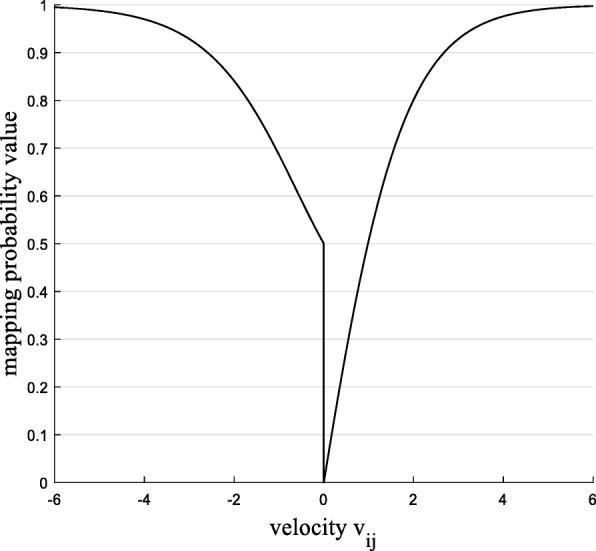
New curve of mapping function of probability
Figure 7 shows the contribution values of selected genes in every iteration of IBPSO on the five data. In the process of selecting the optimal gene subset, the KL-IBPSO-ELM is apt to select those genes with high contribution values, so the subsets’ contribution value has a increase trend as the iteration increases. The KL-IBPSO-ELM method does not always select those genes with the highest contribution values, and it also selects those critical genes with comparatively low contribution values to form the predictive gene subsets for achieving higher classification accuracy. Hence, the contribution curve fluctuates at the early iterations.
Fig. 3.
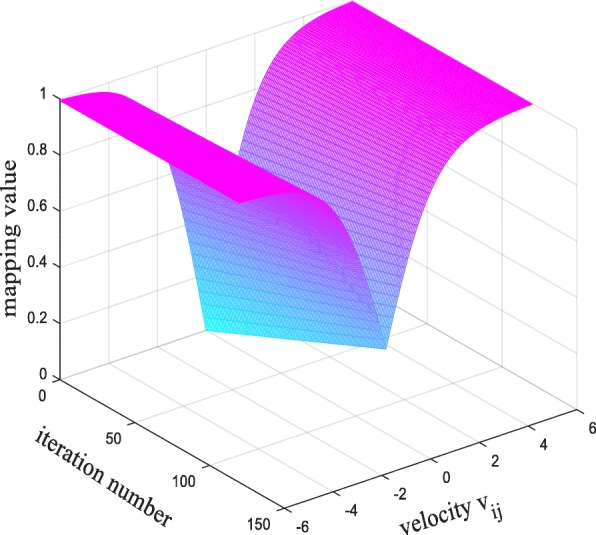
New mapping function curve in three-dimension
Discussion on the parameter selection
To cluster the genes in initial gene pool, it is critical to determine the number of the clusters. Fig. 8 shows the relationship between the classification accuracy on the test data obtained by ELM and the number of the clusters. From Fig. 8, the 5-fold CV accuracy does not have a specific trend as the values of the parameter k increases, and the accuracy is highest when the k is selected as 5 on the Colon, Brain cancer, Leukemia, Lymphoma and LUNG data. Thus the clusters number k is fixed as 5 in the experiments.
Fig. 4.

The frame of the proposed hybrid gene selection method
Conclusions
In this study, a gene selection method based on LASSO and BPSO was proposed to obtain the most predictive genes subsets. To give full consideration of gene structure as well as avoid LASSO overfitting, the candidate elite genes were selected by double filter method. Then by encoding the contribution value into the BPSO and defining a new mapping function, the improved BPSO was able to select a highly predictive and compact gene subset. Experimental results verified that the proposed method outperformed other PSO-based and GCSI-based gene selection methods. Although the proposed could avoid filter out some of the key genes and reduce the rate at which the selection of new important is ignored by other relevant method, the proposed method may increase the computational cost because of complex establishment process of the candidate elite gene pool. Future work will include how to simplify the model for gene selection and apply the new method to more complex microarray data including RNA-seq data.
Acknowledgments
The authors would like to thank the anonymous reviewers for their time and their valuable comments.
Abbreviations
- LASSO
least absolute shrinkage and selection operator
- PSO
particle swarm optimization.
- BPSO
binary particle swarm optimization
- ELM
extreme learning machine
- SLFNs
single-hidden layer feedforward neural network
- LAR
least angle regression
- TLBO
teaching learning-based optimization proposed in [9]
- SNR
signal-to-noise ratio
- PSO-RG
an improved PSO proposed in [14]
- BLDA
Bayesian linear discriminant analysis
- GCS
gene-to-class sensitivity information
- KL-IBPSO-ELM
the proposed gene selection method proposed in this study
- BPSO-ELM
a simple gene selection method combing BPSO with ELM
- KMeans-BPSO-ELM
a gene selection method proposed in [12]
- KMeans-GCSI-MBPSO-ELM
a gene selection method proposed in [16]
- SC-IPSO-ELM
a gene selection method proposed in [30]
Authors’ contributions
QLing proposed the frame. YX wrote the manuscript. YX, QLing, FH and QLiu conducted the experiments. All authors read and approved the final manuscript.
Funding
Publication costs are funded by the National Natural Science Foundation of China [Nos. 61976108 and 61572241], the National Key R&D Program of China [No. 2017YFC0806600], the Foundation of the Peak of Five Talents of Jiangsu Province [No. 2015-DZXX-024], the Fifth” 333 High Level Talented Person Cultivating Project” of Jiangsu Province [No. (2016) III-0845].
Availability of data and materials
Not applicable
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Maulik U. Analysis of gene microarray data in a soft computing framework. Appl Soft Comput. 2011;11:4152–4160. doi: 10.1016/j.asoc.2011.03.004. [DOI] [Google Scholar]
- 2.Mohamad MS, Omatu S, Deris S, et al. A modified binary particle swarm optimization for selecting the small subset of informative genes from gene expression data. IEEE Trans Inf Technol Biomed. 2011;15(6):813–822. doi: 10.1109/TITB.2011.2167756. [DOI] [PubMed] [Google Scholar]
- 3.Saeys Y, Inza I, et al. Review of feature selection techniques in bioinformatics. Bioinformatics. 2007;23(19):2507–2517. doi: 10.1093/bioinformatics/btm344. [DOI] [PubMed] [Google Scholar]
- 4.Karthikeyan T, Balakrishnan R. Swarm intelligence approach based on adaptive ELM classifier with ICGA selection for microarray gene expression and cancer classification. Res J Appl Sci Eng Technol. 2014;7(21):4445–4454. doi: 10.19026/rjaset.7.821. [DOI] [Google Scholar]
- 5.Kennedy J, Eberhart R. Particle swarm optimization, IEEE international conference on neural networks, 1995. Proc IEEE. 2002;4:1942–1948. [Google Scholar]
- 6.Sapiah S, Abdul RN, Muhammad ZZ. Particle swarm optimization feature selection for breast cancer recurrence prediction. IEEE Access. 2018;6:29637–29647. doi: 10.1109/ACCESS.2018.2843443. [DOI] [Google Scholar]
- 7.Mohamad MS, Omatu S, Deris S, et al. Particle swarm optimization for gene selection in classifying cancer classes. Artif Life Robot. 2009;14(1):16–19. doi: 10.1007/s10015-009-0712-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu L, Qi C, Chen S, et al. An improved heuristic optimization algorithm for feature learning based on morphological filtering and its application. IEEE Access. 2018;6:22754–22763. doi: 10.1109/ACCESS.2018.2827403. [DOI] [Google Scholar]
- 9.Shahbeig S, Helfroush MS, Rahideh A. A fuzzy multi-objective hybrid TLBO–PSO approach to select the associated genes with breast cancer. Signal Process. 2017;131:58–65. doi: 10.1016/j.sigpro.2016.07.035. [DOI] [Google Scholar]
- 10.Chuang LY, Yang CH, Wu KC, et al. A hybrid feature selection method for DNA microarray data. Comput Biol Med. 2011;41(4):228–237. doi: 10.1016/j.compbiomed.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 11.Sahu B, Mishra D. A novel feature selection algorithm using particle swarm optimization for cancer microarray data. Procedia Eng. 2012;38(5):27–31. doi: 10.1016/j.proeng.2012.06.005. [DOI] [Google Scholar]
- 12.Yang SX, Han F, Guan J. A hybrid gene selection and classification approach for microarray data based on clustering and PSO. Commun Comput Inf Sci. 2013;375:88–93. [Google Scholar]
- 13.Shen Q, Shi WM, Kong W, et al. A combination of modified particle swarm optimization algorithm and support vector machine for gene selection and tumor classification. Talanta. 2007;71(4):1679–1683. doi: 10.1016/j.talanta.2006.07.047. [DOI] [PubMed] [Google Scholar]
- 14.Chuang LY, Chang HW, et al. Improved binary PSO for feature selection using gene expression data. Comput Biol Chem. 2008;32:29–38. doi: 10.1016/j.compbiolchem.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 15.Joroughi M, Shamsi M, Saberkari H, et al. Gene selection and cancer classification based on microarray data using combined BPSO and BLDA algorithm. J Thorac Cardiovasc Surg. 2014;5(2):1931–1939. [Google Scholar]
- 16.Han F, Sun W, Ling QH. A novel strategy for gene selection of microarray data based on gene-to-class sensitivity information. PLoS One. 2014;9(5):e97530. doi: 10.1371/journal.pone.0097530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han F, Yang C, Wu YQ, Zhu JS, Ling QH, Song YQ, Huang DS. A gene selection method for microarray data based on binary pso encoding gene-to-class sensitivity information. IEEE/ACM Trans Comput Biol Bioinf. 2017;14(1):85–96. doi: 10.1109/TCBB.2015.2465906. [DOI] [PubMed] [Google Scholar]
- 18.Tibshirani RJ. Regression shrinkage and selection via the LASSO: a retrospective. J R Stat Soc. 1996;58(1):267–288. [Google Scholar]
- 19.Chrétien S, Guyeux C, Boyerguittaut M, et al. Using the LASSO for gene selection in bladder cancer data. Int Conf Comput Intell Methods Bioinf, 2015;1–6.
- 20.Zheng SF, Liu WX. Selecting informative genes by LASSO and Dantzig selector for linear classifiers. IEEE international conference on bioinformatics & biomedicine. 2011, pp. 677–680.
- 21.Kennedy J, Eberhart R. A discrete binary version of the particle swarm algorithm, IEEE international conference on systems, man, and cybernetics. Comput Cybern Simul. 2002;5:4104–4108. [Google Scholar]
- 22.Tibshirani RJ. The LASSO problem and uniqueness. Electron J Stat. 2013;7(1):1456–1490. doi: 10.1214/13-EJS815. [DOI] [Google Scholar]
- 23.Huang GB, Zhu QY, Siew CK. Extreme learning machine: a new learning scheme of feedforward neural networks. IEEE international joint conference on neural networks. 2004, pp. 985–990.
- 24.Shyamala N, Vijayakumar K. Microarray gene expression cancer diagnosis using modified extreme learning machine classification. Artif Intell Syst Mach Learn. 2014;6(8):293–296. [Google Scholar]
- 25.Yin Y, Zhao Y, Zhang B, et al. Enhancing ELM by Markov boundary based feature selection. Neurocomputing. 2017;261:57–69. doi: 10.1016/j.neucom.2016.09.119. [DOI] [Google Scholar]
- 26.Meng Y, et al. AGA-ELM algorithm for genetic data classification. J China Univ Metrol. 2017;28(1):97–102. [Google Scholar]
- 27.Liu J, Fan X. The analysis and improvement of binary particle swarm optimization. Int Conf Comput Intell Secur. 2009;1:254–258. [Google Scholar]
- 28.Paul TK, Iba H. Gene selection for classification of cancers using probabilistic model building genetic algorithm. Bio Syst. 2005;82(3):208–225. doi: 10.1016/j.biosystems.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 29.Yuan Y, Ji X, Sun Z, et al. Application of Isomap for cluster analyses of gene expression data. J Tsinghua Univ. 2004;44(9):1286–1289. [Google Scholar]
- 30.Han F, Tang D, Sun YWT, Cheng Z, Jiang J, Li QW. A hybrid gene selection method based on gene scoring strategy and improved particle swarm optimization. BMC Bioinf. 2019;20:289. doi: 10.1186/s12859-019-2773-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alon U, Barkai N, Notterman DA, Gish K, Ybarra S, Mack D, Levine AJ. Broad patterns of gene expression revealed by clustering analysis of tumor and normal colon tissues probed by oligonucleotide arrays. Proc Natl Acad Sci. 1999;96:6745–6750. doi: 10.1073/pnas.96.12.6745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang TM, Kecman V. Gene extraction for cancer diagnosis by support vector machines. Int Conf Artif Neural Netw. 2005;35:185–194. doi: 10.1016/j.artmed.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 33.Tong DL. Hybridising Genetic algorithm-neural network (GANN) in marker genes detection International Conference on Machine Learning & Cybernetics. 2009, pp. 12–15.
- 34.Lee KE, Sha N, Dougherty ER, et al. Gene selection: a Bayesian variable selection approach. Bioinformatics. 2003;19(1):90–97. doi: 10.1093/bioinformatics/19.1.90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable