

Abstract
Sepsis is an often-deadly complication of infection in which systemic inflammation damages the vasculature, leading to tissue hypoperfusion and multiple organ failure. Currently, the standard of care for sepsis is predominantly supportive, with few therapeutic options available. Because of increased sepsis incidence worldwide, there is an urgent need for discovery of novel therapeutic targets and development of new treatments. The recently discovered function of the endoplasmic reticulum (ER) in regulation of inflammation offers a potential avenue for sepsis control. Here, we identify the ER-resident protein Sigma-1 receptor (S1R) as an essential inhibitor of cytokine production in a preclinical model of septic shock. Mice lacking S1R succumb quickly to hypercytokinemia induced by a sub-lethal challenge in two models of acute inflammation. Mechanistically, we find that S1R restricts the endonuclease activity of the ER stress sensor IRE1 and cytokine expression, but does not inhibit the classical inflammatory signaling pathways. These findings could have substantial clinical implications, as we further find that fluvoxamine, an anti-depressant therapeutic with high affinity for S1R, protects mice from lethal septic shock and dampens the inflammatory response in human blood leukocytes. Our data reveal the contribution of S1R to the restraint of the inflammatory response, and place S1R as a possible therapeutic target to treat bacterial-derived inflammatory pathology.
Single sentence summary:
Sigma-1 receptor is a critical inhibitor of endoplasmic reticulum-driven inflammation and a potential therapeutic target in septic shock.
Introduction:
The endoplasmic reticulum (ER) is increasingly recognized as a powerful controller of inflammatory signaling (1, 2) and the response of immune cells to diverse stimuli (3, 4). Among the major ER stress sensors, Inositol-Requiring Enzyme 1α (IRE1) is selectively activated by the TLR4 ligand lipopolysaccharide (LPS) (3). IRE1 regulates inflammatory cytokine production via both its endonuclease activity and transcriptional regulation (2, 3) and the control of cellular signaling pathways (5). Given the considerable potential of IRE1 to modulate inflammation, there is interest in targeting IRE1 for therapeutic benefit (2, 6). However, caution should be applied, as IRE1 function is also critical during homeostasis, including in the liver and pancreas (7, 8). Therefore, to take full advantage of this potent inflammatory mediator, it is essential to identify alternative methods for targeting IRE1 signaling.
Sigma-1 receptor (S1R) is a ubiquitously expressed ER resident chaperone protein that associates with IRE1 during ER stress (9). S1R function is well described in the central nervous system (10), where it has been implicated in the regulation of neurodegenerative diseases (11, 12), cell fate control, and immune activity of microglia (13, 14). Targeting S1R has been reported to influence immune cells and cytokine production in vitro (15), with many well-tolerated S1R ligands currently in clinical use, placing S1R as an attractive therapeutic target (16).
In this work, we identify S1R as a critical regulator of IRE1-driven inflammation. S1R deficiency potently enhances inflammatory cytokine production in a manner dependent on IRE1 activity and reduces survival during models of hyper-inflammation and septic shock in mice. Conversely, forced expression of S1R can dampen the inflammatory response to LPS. Further, we show that the S1R ligand fluvoxamine can enhance survival in mouse models of inflammation and sepsis and can inhibit the inflammatory response in human peripheral blood cells. Collectively, our data show that S1R is uniquely poised to sensitively control IRE1 activity during inflammation.
Results:
S1R controls LPS-induced IRE1 activity in macrophages
S1R has been shown to interact with IRE1 under strong ER stress-inducing conditions (9). Given the role for IRE1 during the inflammatory response (2, 3), we wanted to test if S1R participates in ER-mediated inflammation. We first used the BirA proximity ligation assay to test if S1R interacts with IRE1 during LPS challenge in vitro. For this experiment, we used HEK293 cells that express mTLR4/MD2/CD14 and therefore respond to LPS (17). Cells were transfected with S1R conjugated to the bifunctional ligase/repressor BirA (BirA), or BirA alone as control, resulting in the biotinylation of proteins that are in close proximity to S1R (Fig. 1A) (18). We observed IRE1 biotinylation during homeostasis that was enhanced following LPS treatment (Fig. 1B-C), indicating proximity and possible association (direct or indirect) between S1R and IRE1.
Figure 1: S1R is an inhibitor of IRE1 during inflammation.

(A) Experimental design and principle of proximity ligation assay. HA: Hemagglutinin. (B) Western blots on input lysates and biotinylated (streptavidin pulldown) proximity ligation samples of HEK293 transfected with BirA or S1R-BirA, then stimulated for 24 hours with 100 ng/mL LPS in the presence of 80μM biotin. (C) Densitometric quantification of B (N=4, *P<0.05, repeated measures one-way ANOVA with post-hoc Sidak test). (D) Activity modulators of IRE1 and experimental parameters used in the study. XBP1 (US): Unspliced XBP1 transcript; XBP1 (S): Spliced XBP1 transcript. (E) XBP1 splicing ratio (i.e. GAPDH-normalized spliced XBP1 transcript/GAPDH-normalized unspliced XBP1 transcript) in S1R WT or KO BMDM stimulated for 6 hours with DMSO, 100 ng/mL LPS or 100 ng/mL LPS + IRE1 inhibitor (5μM 4μ8C) (N= 3, n.s. not significant, *P<0.05, two-way ANOVA with post-hoc Sidak test). (F) XBP1 splicing ratio in S1R WT or KO BMDM stimulated with DMSO or IRE1 activator (5μM or 10μM APY29) for 6 hours (N= 3, each dot represents one individual experiment, n.s. not significant, two-way ANOVA with post-hoc Sidak test).
Upon activation with LPS, IRE1 endonuclease activity is triggered and splices the mRNA that encodes the transcription factor X-box binding protein-1 (XBP1) (Fig. 1D), resulting in expression of active XBP1 protein. We found increased LPS-induced XBP1 splicing in mouse bone marrow derived macrophages (BMDM) lacking S1R, indicating elevated inducible, but not basal, IRE1 endonuclease activity in S1R knockout (KO) macrophages (Fig. 1E). To confirm that XBP1 splicing was mediated by IRE1 endonuclease activity, the selective IRE1 endonuclease inhibitor 4μ8C was tested (19). Treatment with 4μ8C abolished LPS-induced XBP1 splicing in both genotypes, ruling out IRE1-independent XBP1 splicing (Fig. 1E). Importantly, we ruled out the presence of a larger pool of IRE1 in S1R KO cells by treating cells with APY29, which forces IRE1-dependent XBP1 splicing (20). In this IRE1 stimulation paradigm, XBP1 splicing amounts were equal in both genotypes (Fig. 1F), indicating that S1R KO affects IRE1 activity, and not IRE1 protein abundance or substrate availability.
S1R critically regulates inflammatory cytokine production via IRE1
Because IRE1 activity is required for cytokine production (2, 3, 5), likely via XBP1 mediated transactivation of IL-6 and TNF-α, we next asked if S1R deficiency alters macrophage cytokine expression upon exposure to LPS. We found that S1R KO BMDM had elevated expression of IL-6 and pro-IL-1β transcripts and secreted higher amounts of IL-6 protein, when compared to wild type (WT) cells (Fig. 2A-B and fig. S1A). However, S1R deficiency does not result in a global increase of cytokine production, as the anti-inflammatory cytokine IL-10 expression was unaffected in S1R KO BMDM (fig. S1B). Having established that deletion of S1R leads to an increased inflammatory response, we examined whether overexpression of S1R could be anti-inflammatory. We overexpressed S1R in HEK293 that express mTLR4/MD2/CD14 and monitored expression of IL-8 following LPS treatment (17). Relative to control transfected cells, overexpression of S1R resulted in a significant decrease in IL-8 production after LPS stimulation (p<0.05, Fig. 2C). These data collectively suggest that overexpression of S1R can dampen inflammation, whereas S1R deficiency contributes to an enhanced inflammatory response.
Figure 2: S1R controls the production of inflammatory cytokines by inhibiting IRE1.

(A) Relative Quantity (RQ) of IL-6 was determined by qPCR on S1R WT or KO BMDM stimulated for 6 hours with vehicle (NT) or 100ng/mL LPS (Representative of 4 independent experiments, one mouse is shown, **P<0.01, two-way ANOVA with post-hoc Tukey test). (B) IL-6 ELISA on supernatant from BMDM stimulated for 6 hours with 1μg/mL LPS (Representative of 4 independent experiments, one mouse is shown, ***P<0.001, t-test). (C) qPCR on TLR4-MD2-CD14-expressing HEK293 cells that were transfected with empty vector (EV) or S1R and stimulated for 6 hours with 100ng/mL LPS (N=3, each dot pair represents one independent experiment, *p<0.05, paired t-test). (D) qPCR on BMDM stimulated for 6 hours with vehicle (DMSO), 100 ng/mL LPS, or 100 ng/mL LPS + IRE1 inhibitor (5μM 4μ8C) (Representative of 3 independent experiments, one mouse is shown, n.s. not significant, ***P<0.001, two-way ANOVA with post-hoc Tukey test). (E) IL-6 ELISA on supernatant from BMDM stimulated for 6 hours with 1μg/mL LPS with either <1% DMSO, NF-κB inhibitor (20μM JSH-23), ERK inhibitor (20μM PD98059), JNK inhibitor (1μM SP600125), or IRE1 inhibitor (5μM 4μ8C) (Representative of 3 independent experiments, one mouse is shown, n.s. not significant, **P<0.01, ***P<0.001, two-way ANOVA with post-hoc Tukey test).
We next tested whether IRE1 endonuclease activity is responsible for the increase in pro-inflammatory cytokine expression in S1R KO cells. Pro-inflammatory cytokines, including IL-6, are rapidly induced by LPS in mice and humans, and correlate with poor prognosis in sepsis (21, 22). We treated WT and S1R KO BMDM with LPS in the presence or absence of 4μ8C and analyzed IL-6 expression by qPCR. Inhibition of IRE1 endonuclease activity reduced IL-6 expression in KO cells to the amount observed in WT BMDM (Fig. 2D). Because S1R is an ER-resident protein, we wanted to rule out that deletion of S1R might result in global ER dysfunction, which could lead to the observed increase in IRE1 activation. To test this, we performed an immunoblot for ER-resident proteins that become upregulated during ER stress (23): Protein kinase R-like Endoplasmic Reticulum Kinase (PERK), binding immunoglobulin protein (BiP) and protein disulfide isomerase (PDI). We found comparable amounts of protein expression of all three proteins in S1R KO BMDM at baseline, with no change elicited by stimulation of BMDM with LPS (fig. S1C). Therefore, we conclude that global ER stress does not drive IRE1 activity in S1R KO BMDM. Cell surface expression of TLR4 was unaffected by S1R deletion ruling out differential expression of the LPS receptor (fig. S2A-B). Activation of NF-κB (fig. S2C), MAP kinase (ERK1/2), and the c-Jun N-terminal kinase (JNK) (fig. S2D) was identical between WT and S1R KO cells after LPS treatment. Finally, when we tested selective pharmacologic inhibitors of NF-κB, JNK, ERK1/2, and IRE1 for the ability to normalize LPS-induced IL-6 secretion in S1R KO BMDM, only the IRE1 inhibitor was effective at blunting the augmented inflammatory response of S1R KO cells (Fig. 2E). It is important to note that although NF-κB, JNK, and ERK1/2 inhibitors suppressed IL-6 production in both genotypes, S1R KO BMDM still produced elevated IL-6 relative to WT BMDM (Fig. 2E), indicating that these pathways are functional in S1R KO BMDM. We also cultured primary lung fibroblasts from the S1R KO and WT mice and tested their response to LPS stimulation. Fibroblasts also presented with enhanced LPS-induced XBP1 splicing and inflammatory cytokine production that can be corrected by IRE1 inhibition (fig. S3A-B). Taken together, these findings indicate that IRE1 signaling is selectively perturbed in S1R KO cells, and that the pro-inflammatory effects of S1R deletion likely depend on the endonuclease activity of IRE1.
S1R-deficient mice display increased mortality in sub-lethal models of sepsis
To test the function of S1R in vivo, we subjected S1R KO mice to LPS injection, an animal model to study the inflammatory response to endotoxin (24). S1R KO mice and WT littermate controls were injected with a sub-lethal dose of LPS (5 mg/kg) and survival was monitored for 6 days (Fig. 3A). WT animals experienced very low mortality (9%), whereas 62% of S1R KO mice succumbed to LPS-induced death (Fig. 3B), suggesting that S1R potently inhibits systemic inflammation. We next analyzed the concentration of pro-inflammatory cytokines TNF-α and IL-6 in serum at their reported peak expression, as these cytokines have been extensively shown to correlate with LPS-induced mortality (21, 25). Peak serum TNF-α and IL-6 were significantly increased in LPS-challenged S1R KO mice, when compared to controls (p<0.05, Fig. 3C-D), whereas neither cytokine was detectable in the serum of unchallenged mice. To test if the increase in TNF-α and IL-6 in S1R KO mice was due to baseline differences in the composition of immune cells, we performed an immunophenotyping analysis of blood (fig. S4), the peritoneal cavity (fig. S5) and immune organs (spleen and lymph nodes; fig. S6). Our flow cytometry analyses revealed no significant differences in the innate and adaptive cell numbers and frequency, suggesting that S1R-deficient mice do not have an overt immune defect.
Figure 3: S1R is protective in murine models of inflammation and septic shock.

(A) Experimental design. (B) Survival curve of WT and S1R KO mice following LPS administration (n=11-13 mice/ group, LPS=5mg/kg, **P<0.01, log-rank test) (C) ELISA for TNF-α in serum 1.5 hours after LPS injection (each dot represents one mouse, *P<0.05, t-test). (D) ELISA for IL-6 in serum collected 3 hours after LPS injection (each dot represents one mouse, *P<0.05, t-test). (E) Survival curve of WT and S1R KO mice following administration of fecal content (n=10-13 mice/ group, fecal slurry =1g/kg, *P<0.05, log-rank test). (F) ELISA for IL-6 in serum collected 3 hours after fecal slurry injection (each dot represents one mouse, *P<0.05, t-test). (G) Rectal temperature of animals presented in E (n= 10-13 mice/group, **p<0.01, ***p<0.001, two-way repeated measures ANOVA with post-hoc Sidak test). (H-K) Mice were injected with 5mg/kg LPS or 1mg/kg fecal slurry and, 24 hours later, serum was analyzed for amount of (H) alanine aminotransferase (ALT) (I) aspartate aminotransferase (AST) (J) creatinine (K) and creatine kinase (CK) (each dot represents one mouse, n=10-12 per group. *p<0.05, **p<0.01, ***p<0.001, two-way ANOVA. Outliers have been removed from visualization and are available in Data file S1.).
Although LPS injection is a convenient model for study of endotoxin-mediated inflammation, the use of a single pathogen-associated molecular pattern (PAMP) does not fully recapitulate the biological complexity of sepsis. Therefore, we tested S1R KO mice in fecal-induced peritonitis (FIP, Fig. 3E), a model of sepsis that involves injection of fecal material containing live bacteria (26). Similar to our LPS challenge finding, WT mice receiving a sub-lethal dose of fecal slurry (1g/kg of body weight) did not succumb to septic shock induced by FIP, whereas S1R KO mice experienced significant mortality (p<0.05, Fig. 3E). This increased mortality correlated with increased serum IL-6 (p<0.05, Fig. 3F) and significantly lowered core body temperature in S1R KO mice (p<0.01, Fig. 3G). S1R deficiency was also associated with elevated markers of organ failure, as revealed by serum chemistry analysis performed 24h after the initiation of septic shock. Two indicators of impaired liver function alanine transferase (ALT, p<0.05, Fig. 3H) and aspartate aminotransferase (AST, p<0.001, Fig. 3I), an indicator of kidney dysfunction, creatinine (p<0.001, Fig. 3J), and an indicator of heart dysfunction, creatine kinase (CK, p<0.01, Fig. 3K), were all significantly elevated in S1R-deficient animals in both LPS and FIP models. Taken together, our data demonstrate increased susceptibility to models of sepsis and inflammation in S1R deficiency, characterized by elevated cytokines and multi-organ dysfunction.
S1R activation and IRE1 inhibition are protective in an animal model of inflammation
To test if LPS-challenged S1R KO mice have increased IRE1 activity, we first examined XBP1 splicing in the liver, a key organ in the pathological progression of sepsis. LPS-challenged S1R KO mice had increased hepatic XBP1 splicing when compared to WT mice (Fig. 4A). This finding suggests that similar IRE1-dependent inflammatory mechanisms we identified in cultured macrophages may be at work in vivo. If increased IRE1 activity is responsible for reduced survival of S1R KO mice during LPS challenge, then IRE1 inhibition should protect S1R KO mice subjected to LPS challenge (Fig. 4B). Due to the reported short half-life of the IRE1 inhibitor 4μ8C in vivo (19), we selected instead to use STF 083010 (herein referred to as STF), an effective IRE1 inhibitor (fig. S7A) that has been used in in vivo studies (19, 27). Again, LPS-challenged S1R KO mice that received vehicle control experienced rapid mortality (Fig. 4C). Remarkably, STF administration (30 mg/kg at 0h and 24h) spared S1R KO mice from LPS-induced mortality (Fig. 4C), whereas it did not significantly affect the survival of WT mice.
Figure 4: Pharmacological modulation of S1R and IRE1 function in sepsis models.

(A) XBP1 splicing ratio from liver homogenate of mice challenged with 5mg/kg LPS for 3 hours. Data shown are ratio of XBP1 spliced transcript/XBP1 unspliced transcript (each dot represents one mouse, **P<0.01, two-way ANOVA with post-hoc Sidak test). (B) Experimental design. (C) Survival curve of WT and S1R KO mice treated with vehicle (33% Kolliphor in saline) or STF (30mg/kg) following administration of LPS as indicated in B (n=15-16 mice/ group, LPS=2mg/kg, **P<0.01, ***P<0.001, log-rank test). (D) IL-6 peritoneal exudate 3 hours after LPS injection in mice (each dot represents one mouse, *P<0.05, two-way ANOVA with post-hoc Sidak test. (E) Survival curve of WT and S1R KO mice treated with vehicle or fluvoxamine (20mg/kg) following administration of LPS as indicated in E (n=13-17 mice/group, LPS = 6mg/kg *P<0.05, **P<0.01, log-rank test). (F) Serum IL-6 ELISA 3 hours after LPS injection in mice challenged as shown in E (each dot represents one mouse, n.s. not significant, **P<0.01, two-way ANOVA with post-hoc Sidak test. Genotype-treatment interaction: *P<0.05, two-way ANOVA).
The finding that an IRE1 inhibitor rescues S1R KO mice in a model of endotoxemia is in agreement with our hypothesis that cytokine production and LPS-induced mortality in S1R KO mice require excessive IRE1 endonuclease activity. Further supporting this hypothesis, we detected significantly higher IL-6 after 3h in the peritoneal exudate in LPS-challenged vehicle-treated S1R KO mice compared to WT controls, which was corrected by STF treatment (p<0.05, Fig. 4D). IL-6 in the serum following STF treatment was not significantly different (fig. S7B). In this treatment paradigm, we noted that vehicle (Kolliphor) treatment significantly increased LPS-induced IL-6 in the serum when compared to LPS alone (p<0.01, fig. S7C). Because Kolliphor exacerbates LPS-induced inflammation, a lower dose of LPS was selected than in other experiments. Importantly, injection of Kolliphor alone did not result in a detectable concentration of serum IL-6, nor did it cause any mortality in WT or KO mice, suggesting that vehicle treatment exacerbates LPS-induced inflammation, but is not inflammatory on its own (fig. S7D). Collectively, these findings suggest that cytokine production and LPS-induced mortality in S1R KO mice require excessive IRE1 endonuclease activity. We next aimed to directly assess whether S1R function might be manipulated for benefit in an in vivo inflammatory context. We selected fluvoxamine (FLV), an antidepressant drug with low-nanomolar affinity for S1R, which has also been reported to have anti-inflammatory properties (28). In order to elicit higher mortality in WT mice, we selected a higher dose of LPS (6 mg/kg) for this experiment, and first administered fluvoxamine (20mg/kg) at the same time as LPS (Fig. 4B). Fluvoxamine treatment significantly protected WT mice from mortality and reduced serum IL-6 whereas, as expected, no significant effect was observed in S1R KO animals (p<0.05, Fig. 4E-F). These results indicate that the anti-inflammatory effect of fluvoxamine is mediated by S1R.
Therapeutic administration of a S1R ligand is beneficial in preclinical models of sepsis and inflammation.
We next tested whether fluvoxamine could be therapeutically administered to protect C57BL/6J from LPS administration or ongoing FIP sepsis model. Fluvoxamine was administered as indicated in Fig. 5A (90 min post LPS challenge) and Fig. 5B (30 min post FIP induction), after animals presented with a significant sickness behavior characterized by a decrease in body temperature (p<0.001, Fig. 5C-D) and a clinical presentation of sepsis signs (p<0.01, Fig. 5E-F). Therapeutic administration of fluvoxamine improved the clinical score (Fig. 5E-F) and temperature (Fig. 5G-H) of challenged animals. The treatment also significantly enhanced survival in both animal models (p<0.01, Fig. 5I-J). Remarkably, fluvoxamine treatment was also beneficial in the FIP model when administered at an even later time point post FIP induction (90 min instead of 30 min, Fig. 5K-L). To directly compare the effectiveness of fluvoxamine to the currently available therapeutics, we also administered Ceftriaxone, an antibiotic currently used as a standard of care for sepsis patients (29), 90 min post-FIP induction. Fluvoxamine administration was as efficacious in enhancing survival as Ceftriaxone (CRO, 100mg/kg, Fig. 5L), and the combination of fluvoxamine and ceftriaxone did not further improve survival when compared to single treatment (Fig. 5L).
Figure 5: Therapeutic administration of the S1R agonist fluvoxamine is protective during models of inflammation and sepsis.

(A-B) Experimental design for (A) LPS challenge or (B) FIP with therapeutic S1R agonist treatment. (C) Rectal temperature of mice measured immediately before LPS injection and one hour after (each dot represents one mouse, ***p<0.001, paired t-test). (D) Rectal temperature of mice measured immediately before FIP induction and 0.5 hour after (each dot represents one mouse, ***p<0.001, paired t-test). (E) Clinical score, expressed as total murine sepsis score, of mice treated as in A, **p<0.01, ***p<0.001, repeated measures two-way ANOVA with post-hoc Sidak test. (F) Clinical score, expressed as total murine sepsis score, of mice treated as in B, (n=14 mice/group, ***p<0.001, two-way ANOVA). (G) Rectal temperatures of mice 24 hours after I.P. LPS injection, treated with saline vehicle or FLV as indicated in A (each dot represents one mouse, ***p<0.05, t-test). (H) Survival curve of mice challenged with 6mg/kg LPS and given therapeutic FLV or saline as indicated in A (n=16-20, ***p<0.001, log-rank test). (I) Rectal temperatures of mice 24 hours after FIP induction, treated with saline vehicle or FLV as indicated in B (each dot represents one mouse, **p<0.01, t-test). (J) Survival curve of mice challenged with 1.5 g/kg fecal slurry and given therapeutic FLV or saline as indicated in I (n=14 mice/group **p<0.01, log-rank test). (K) Experimental design for FIP challenge with FLV and the antibiotic ceftriaxone (CRO) treatment. (L) Survival of C57/Bl6 mice treated with fluvoxamine (20mg/kg) i.p. and/or ceftriaxone (100mg/kg s.c.) following administration of fecal slurry at 2 g/kg (n=10-12 mice/group, **p<0.01, ***p<0.001, log-rank test).
Fluvoxamine is anti-inflammatory in human cells.
To assess whether targeting S1R can dampen inflammation in human cells, heparinized peripheral blood from healthy donors was stimulated ex vivo with LPS (10ng/ml) in the presence or absence of fluvoxamine (20 μM) and the production of inflammatory mediators was measured by multiplex analysis. Fluvoxamine significantly reduced LPS-induced IL-6 (p<0.01, Fig. 6A), IL-1β (p<0.05, Fig. 6B), and IL-12 p40 (p<0.01, Fig. 6C), and decreased IL-8 (Fig. 6D) production in cells from all donors analyzed. These data indicate that the anti-inflammatory action of this S1R ligand is likely conserved across species. Importantly, modulation of S1R during LPS treatment was limited to a subset of inflammatory mediators (fig. S8) and was not the result of global cytokine suppression. Taken together, our data show that fluvoxamine can influence the inflammatory response in murine and human cells in a S1R dependent manner and suggest that therapeutic exploitation of S1R targeting might hold promise for the control of inflammatory insults.
Figure 6: The S1R agonist fluvoxamine is anti-inflammatory in human cells.
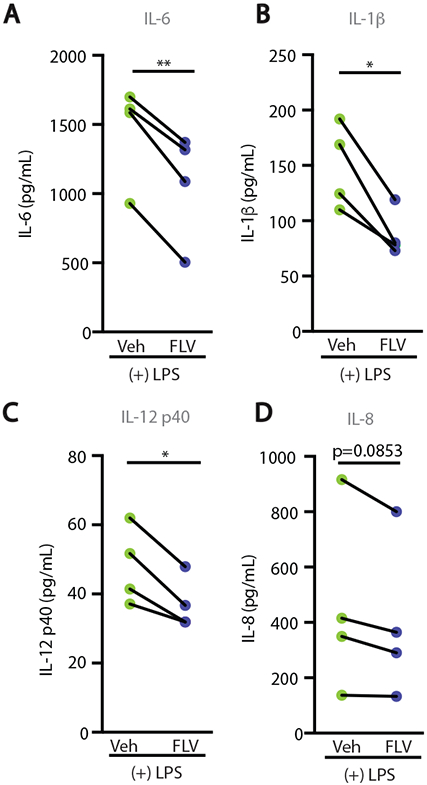
(A-D) Multiplex ELISA on serum from human blood. Heparinized whole blood was stimulated ex vivo with 10ng/mL LPS and vehicle (RPMI) or 20μM FLV for 4 hours (n=4, each dot pair represents serum from one participant, *p<0.05, **p<0.01, paired t-test).
Discussion:
The ER stress sensing protein IRE1α (as well as the closely related protein IRE1β) is able to powerfully affect the inflammatory behavior of both immune and non-immune cells in numerous contexts (1, 30). However, little is yet known about the factors that modulate the extent of IRE1 signaling during inflammation. Here we identify S1R as a regulator of IRE1 endonuclease function during LPS-induced inflammation (fig. S9). S1R and IRE1 may associate both basally and after LPS stimulation, suggesting that S1R is uniquely poised to sensitively control IRE1 activity. This finding is particularly exciting, since the physiological and pathophysiological relevance of IRE1 is well-established (30). However, direct therapeutic targeting of IRE1 has been met with substantial challenges (7, 8), and alternative routes toward IRE1 signaling modulation are sought after. We propose that S1R, which can be targeted by several drugs that are already in clinical use, might represent one such option.
One important caveat of our study is that, although preclinical models of septic shock are convenient for the discovery of new therapeutic treatments for sepsis, they incompletely replicate human sepsis and translational efficacy of preclinical findings in human patients is difficult to predict. There are likely many factors contributing to this challenge, including diversity of predisposition and underlying physiological features, heterogeneous pathophysiology, and variability of causative infectious agents (31). Clinically defining such a broad array of processes as one syndrome, sepsis, complicates the application of novel therapeutic approaches. Work to more effectively design clinical sepsis studies to properly apply preclinical findings is an active area of the sepsis field (32). At the same time, novel preclinical sepsis models and methods are arising that may allow for experimentation in a wider range of conditions associated with sepsis (33). Despite these challenges, preclinical sepsis models have shown promise, and more precise classification of sepsis conditions may lead to efficacious application of interventions that have been identified in animal models.
Here we show that S1R deficiency appears to selectively enhance activity of IRE1 and does not influence other inflammatory pathways, including NF-κB, JNK, and ERK. The ability of the S1R-IRE1 interaction to influence immune and non-immune cell activity may prove to be of importance in inflammatory and degenerative diseases in which S1R and IRE1 dysfunction have been implicated, including Alzheimer’s disease (34, 35) and amyotrophic lateral sclerosis (11, 36). Additionally, although our study focused on XBP1 splicing as an indicator of IRE1 endonuclease activity, IRE1 can also cleave other RNA species (in a process called regulated IRE1-dependent decay, or RIDD), which may drive inflammation as well (1). Some of our observed findings may be a result of RIDD and may not depend on XBP1.
Another potential mechanism is that S1R may be altering calcium signaling, as S1R has been shown to modulate the conductivity of inositol triphosphate receptor (IP3R) (37). However, several lines of evidence suggest that changes in calcium signaling do not produce the observed inflammatory effects. First, we show that the cytosolic inflammatory signaling proteins NF-κB, JNK, and ERK1/2, all of which are sensitive to changes in calcium, are not affected by S1R deletion. Additionally, calcium dysregulation might inhibit protein folding, but we do not observe changes in ER chaperone abundance basally or after LPS stimulation of S1R KO BMDM, again suggesting that S1R does not strongly perturb calcium homeostasis in mammalian cells. Further studies examining calcium flux in S1R KO BMDM might reveal additional important features of S1R function, such as during prolonged ER stress, but calcium flux does not appear to be central to the pathway described in this study.
One limitation of our study is that we have not fully elucidated the mechanism by which S1R controls IRE1. Although we have demonstrated with our proximity ligation approach that S1R and IRE1 were in close proximity and that interaction was further promoted upon LPS stimulation, our study did not address if intermediates were necessary for the formation of the putative S1R-IRE1 complex. Our attempts at co-immunoprecipitation did not convincingly demonstrate a strong association between endogenous S1R and IRE1, unlike what has been described with over-expressed proteins (9). This suggests that the interaction might be transient and/or require intermediates. Furthermore, our study did not explore what are the signaling events that control S1R activity in the context of inflammation. To date, only one endogenous S1R ligand has been identified, N,N-Dimethyl tryptamine (DMT), a tryptophan metabolite, known for its psychedelic activity (38). Limited evidence suggests that DMT-S1R interactions can influence the activation of immune cells (15). Perhaps understanding the interaction of S1R and its endogenous ligands that are regulated by inflammation could help answer this open question.
Although we found that the anti-inflammatory action of fluvoxamine depends on S1R in our animal models of sepsis and inflammation, there is still much to be learned about fluvoxamine and other Selective Serotonin Reuptake Inhibitors (SSRIs) in inflammation. Some SSRIs that do not have affinity for S1R also have reported anti-inflammatory properties, linking serotonin signaling in immune cells to inflammation (39). Furthermore, it remains unknown whether the requirement for S1R in fluvoxamine efficacy is due to direct binding of fluvoxamine to S1R. Nonetheless, it will be important to consider S1R when studying SSRIs in inflammation.
Materials and Methods
Study design
The goal of our study was to identify the role of S1R during LPS-mediated inflammation. Using animal models of inflammation and sepsis, we demonstrated that S1R is an inhibitor of cytokine production. We elucidated the mechanism by which S1R controls the inflammatory response via IRE1 with primary BMDM and HEK-293. Using pharmacological inhibitors, we used two in vivo models of sepsis to validate our in vitro findings showing the mechanism of action of S1R and IRE1. We finally confirm our results obtained with fluvoxamine using human blood samples. In all experiments, animals were randomly assigned to treatment groups and researchers were blinded during treatment and data collection. Group and sample size for each experiment are indicated in each figure legend. No statistical methods were used to predetermine sample sizes for in vitro experiments. Sample sizes for in vivo and ex vivo experiments were predetermined using G*Power, with 1-β≥0.85. Post-hoc power calculations were performed on in vitro studies (except where representative data are shown) using G*Power to ensure that 1-β ≥0.85. Primary data are reported in data file S1.
Mice
C57BL/6J (8 weeks old) were purchased from The Jackson Laboratory. The S1R knockout mouse strain was acquired from The Mutant Mouse Resource and Research Centers and bred to C57BL/6J at the University of Virginia to generate WT and KO mice used in the study (40, 41). All animal experiments were approved and complied with regulations of the Institutional Animal Care and Use Committee at University of Virginia (#3918).
Tissue culture conditions and Reagents
HEK293 mTLR4/MD2/CD14 (Invivogen, 293-mtlr4md2cd14), primary lung fibroblasts, and BMDM were isolated and maintained as described (42, 43). Cells and animals were treated with LPS (Sigma, L4391), 4μ8C (Tocris, 4479), APY-29 (Medchem Express HY-17537), PD98059 (Medchem Express HY-12028), JSH-23 (Medchem Express, HY-13982), SP600125 (Medchem Express, HY-12041), STF 083010 (Medchem Express, HY-15845), Fluvoxamine (Medchem Express, HY-B0103A), and Ceftriaxone (Hospira, NDC: 0409–7337-01), as described in the text.
LPS challenge
In vivo LPS challenge was performed on adult mice (8–12 weeks of age). LPS from E. coli 0111:B4 (Sigma-Aldrich, L2630) was injected intraperitoneally, as described in the text. STF083010 (Medchem Express, HY-15845) was resuspended in 33% Kolliphor-EL (Sigma, C5135) and administered intraperitoneally at 30mg/kg immediately after and again 24 hours after LPS injection. Fluvoxamine was resuspended in saline and administered at 20mg/kg as indicated in the text. Blood for serum ELISA was collected from facial vein at the predicted peak serum concentration of TNF-α and IL-6 (21).
Fecal-induced peritonitis
Fecal material was isolated from the caecum of age- and sex- matched WT animals coming from the UVA vivarium for Fig. 3 or from The Jackson Laboratory for Fig. 5, resuspended in saline and passed through a 70μM strainer to remove large particles. The slurry was prepared fresh for each experiment and administered intraperitoneally. Core body temperature was measured and mice were scored with murine sepsis severity scale by two independent, blinded researchers (26). Blood for serum ELISA was collected from facial vein at 3 hours post FIP induction. Fluvoxamine in saline was administered intraperitoneally at a dose of 20mg/kg at the same time as FIP, 30 minutes later, or 90 minutes later, as indicated in schematic figure panels, and ceftriaxone in saline was given at a dose of 100mg/kg subcutaneously as indicated in the text.
Serum preparation
Serum was collected 24 hours after injection of LPS or fecal slurry. Serum chemistry analysis was performed by Comparative Clinical Pathology Services LLC. ELISA was performed on serum as described below.
ELISA
ELISA for IL-6 and TNF-α were performed as previously described (25). Antibodies used were: anti-mouse IL-6 MP5–20F3 (Biolegend, 504501) 0.5μg/mL; biotin anti-mouse IL-6 MP5–32C11 (Biolegend, 504601) 1μg/mL; anti-mouse TNF-α (R&D systems, AF-410-NA) 0.5μg/mL; biotin anti-mouse TNF-α (R&D systems, BAF410) 0.25μg/mL.
Peritoneal exudates collection
Peritoneal cavities content were collected 3h after LPS injection in PBS + 5mM EDTA, then centrifuged to pellet cells. Supernatants were collected for ELISA, and cells were washed and stored as previously described (44).
Western blot
Protein extraction and western blot were performed as previously described (44). Antibodies were used according to manufacturer’s instruction: Actin (Sigma Aldrich, A2228) 1:5000; BiP (BD Biosciences, 610798) 1:1000; total ERK1/2 (CST, 9102) 1:1000; phospho-ERK1/2 (CST, 4370) 1:1000; total IRE1α (CST, 3294) 1:1000; 1:1000; total JNK (CST, 9252) 1:1000; phospho-JNK (CST, 9251) 1:1000; total p65 NF-κB (CST, 8242) 1:1000; phospho p65 NF-κB (CST, 3033) 1:1000; PDI (Abcam, 2792) 1:1000; total PERK (CST, 3192) 1:1000. Linear level adjustments were applied to entire images to enhance visualization.
Cloning and transfection
Plasmids used were: MGC Mouse Sigmar1 cDNA (GE Life Sciences, MMM1013–202768624) and pcDNA3.1 MCS-BirA(R118G) HA (addgene#36047) (18). S1R-BirA HA construct was generated by cloning Murine S1R ORF upstream of BiRA into pcDNA3.1 MCS-BirA HA. HEK293 mTLR4/MD2/CD14 were transfected using X-tremeGENE HP transfection reagent (Roche, 06366244001) according to the manufacturer’s instructions.
Proximity biotinylation
Culture medium was supplemented with 80μM biotin (Research Products International, B40040) and LPS 18 hours after transfection. Biotinylated proteins were purified as described (44).
cDNA synthesis and quantitative PCR
Total RNA was extracted using an ISOLATE II RNA kit (Bioline, 52073) and cDNA synthesis was performed with the SensiFAST cDNA synthesis kit (Bioline, BIO-65054). TaqMan Probes were obtained from Thermo Fisher (GAPDH: Mm99999915_g1; IL-6: Mm00446190_m1; pro-IL-1β: Mm00434228_m1; IL10: Mm004396). Primers for the detection of XBP1, IL-8, and actin were previously published (45-47). qPCR was performed as described previously (44).
Flow cytometry
Flow cytometric analyses were performed as described (48). The following Abs were used: TLR4/MD-2 Complex APC (Thermo Fisher Scientific, 17–9924-82), F4/80 Antigen PE (Thermo Fisher Scientific, 12–4801-80), F4/80 Antigen PE Cy7 (Thermo Fisher Scientific, 25–4801-82), CD11b PE Cy7 (Biolegend, 101215), CD11b eFluor 450 (Thermo Fisher Scientific, 48–0112-82), CD11b APC (Thermo Fisher Scientific, 17–0112-82), CD45 PerCP Cy5.5 (Thermo Fisher Scientific, 45–0451-82), CD45 APC (Thermo Fisher Scientific, 17–0451-82), Ly6G APC Cy7 (Tonbo Biosciences, 25–1276), CD19 PE Cy7 (Thermo Fisher Scientific, 25–0193-82), CD4 eFluor 450 (Thermo Fisher Scientific, 48–0042-82), TCRβ (Thermo Fisher Scientific, 12–5961-83), CD8 Alexa Fluor 488 (Thermo Fisher Scientific, 53–0081-82), CD11c PerCP Cy5.5(Thermo Fisher Scientific, 45–0114-82), CD115 APC (Thermo Fisher Scientific, 17–1152-82), Fc Block (Thermo Fisher Scientific, 14–9161-71) and a Zombie Aqua Fixable Viability kit (Biolegend, 423101).
Human whole blood stimulation
Study participants were healthy adults (ages 18–45). The study was approved by the Institutional Review Board (#13166) at the University of Virginia, and all participants signed informed consent before enrollment. Blood was collected into heparinized vacuum tube, then stimulated with 10ng/mL LPS +/− 20μM fluvoxamine for 4 hours, as described (49). Cytokine concentrations were determined by multiplex analysis.
Data analysis and statistics
Data are represented as mean +/− SEM. Densitometry was performed using ImageJ software. Statistical analyses, as indicated in each figure legend, were performed using GraphPad Prism 6. All t-tests were two-tailed. ROUT analysis was used to identify outliers, with Q = 1%, and outliers identified by this method were excluded from analysis. The D’Agostino & Pearson omnibus normality test was used to assess normality of data sets. Power analyses were performed with G*Power 3.1.
Supplementary Material
fig. S1. S1R deletion influences a subset of LPS-induced processes without causing global perturbation.
fig. S2. Canonical TLR4 signaling in macrophages is unperturbed by S1R deletion.
fig. S3. Gating strategy and quantification of immunophenotyping on blood.
fig. S4. Gating strategy and quantification of immunophenotyping on peritoneal contents.
fig. S5. Gating strategy and quantification of immunophenotyping on spleen and lymph node.
fig. S6. S1R controls inflammation in primary fibroblasts
fig. S7. STF affects cytokine production in vitro and in vivo.
fig. S8. Anti-inflammatory effect of fluvoxamine does not globally suppress cytokine production in human tissue.
fig. S9. Proposed mechanism of action of S1R during LPS mediated inflammatory response.
Data file S1: Primary data (Excel file)
Acknowledgements:
We thank Dr. Sanja Arandjelovic and the BIG center (University of Virginia) for critical reading of the manuscript.
Funding: The authors are supported by NIH grants R01 NS083542 (A.G.), R21 NS101281 (A.G.), the Owens Family Foundation (A.G.) and T32 GM007055 (D.A.R.).
Footnotes
Competing interests: D.A.R and A.G. are inventors on U.S. Provisional Patent Application No. 62/618,741 submitted by the University of Virginia entitled “Compositions and Methods for Regulating Inflammation”. There is no other competing interests.
Data and materials availability: All data related to this study can be found in the paper and Supplementary Materials.
References and Notes
- 1.Cho JA, Lee AH, Platzer B, Cross BC, Gardner BM, De Luca H, Luong P, Harding HP, Glimcher LH, Walter P, Fiebiger E, Ron D, Kagan JC, Lencer WI, The unfolded protein response element IRE1alpha senses bacterial proteins invading the ER to activate RIG-I and innate immune signaling. Cell host & microbe 13, 558–569 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 2.Qiu Q, Zheng Z, Chang L, Zhao YS, Tan C, Dandekar A, Zhang Z, Lin Z, Gui M, Li X, Zhang T, Kong Q, Li H, Chen S, Chen A, Kaufman RJ, Yang WL, Lin HK, Zhang D, Perlman H, Thorp E, Zhang K, Fang D, Toll-like receptor-mediated IRE1alpha activation as a therapeutic target for inflammatory arthritis. EMBO J 32, 2477–2490 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Martinon F, Chen X, Lee AH, Glimcher LH, TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol 11, 411–418 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cubillos-Ruiz JR, Silberman PC, Rutkowski MR, Chopra S, Perales-Puchalt A, Song M, Zhang S, Bettigole SE, Gupta D, Holcomb K, Ellenson LH, Caputo T, Lee AH, Conejo-Garcia JR, Glimcher LH, ER Stress Sensor XBP1 Controls Anti-tumor Immunity by Disrupting Dendritic Cell Homeostasis. Cell 161, 1527–1538 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Urano F, Wang X, Bertolotti A, Zhang Y, Chung P, Harding HP, Ron D, Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 287, 664–666 (2000). [DOI] [PubMed] [Google Scholar]
- 6.Abuaita BH, Burkholder KM, Boles BR, O'Riordan MX, The Endoplasmic Reticulum Stress Sensor Inositol-Requiring Enzyme 1alpha Augments Bacterial Killing through Sustained Oxidant Production. MBio 6, e00705 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fu S, Watkins SM, Hotamisligil GS, The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling. Cell metabolism 15, 623–634 (2012). [DOI] [PubMed] [Google Scholar]
- 8.Lipson KL, Fonseca SG, Ishigaki S, Nguyen LX, Foss E, Bortell R, Rossini AA, Urano F, Regulation of insulin biosynthesis in pancreatic beta cells by an endoplasmic reticulum-resident protein kinase IRE1. Cell metabolism 4, 245–254 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Mori T, Hayashi T, Hayashi E, Su TP, Sigma-1 receptor chaperone at the ER-mitochondrion interface mediates the mitochondrion-ER-nucleus signaling for cellular survival. PLoS One 8, e76941 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang B, Wang L, Chen T, Hong J, Sha S, Wang J, Xiao H, Chen L, Sigma-1 receptor deficiency reduces GABAergic inhibition in the basolateral amygdala leading to LTD impairment and depressive-like behaviors. Neuropharmacology 116, 387–398 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Mavlyutov TA, Guo LW, Epstein ML, Ruoho AE, Role of the Sigma-1 receptor in Amyotrophic Lateral Sclerosis (ALS). J Pharmacol Sci 127, 10–16 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maurice T, Goguadze N, Sigma-1 (sigma1) Receptor in Memory and Neurodegenerative Diseases. Handb Exp Pharmacol, (2017). [DOI] [PubMed] [Google Scholar]
- 13.Moritz C, Berardi F, Abate C, Peri F, Live imaging reveals a new role for the sigma-1 (sigma1) receptor in allowing microglia to leave brain injuries. Neurosci Lett 591, 13–18 (2015). [DOI] [PubMed] [Google Scholar]
- 14.Ha Y, Shanmugam AK, Markand S, Zorrilla E, Ganapathy V, Smith SB, Sigma receptor 1 modulates ER stress and Bcl2 in murine retina. Cell Tissue Res 356, 15–27 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Szabo A, Kovacs A, Frecska E, Rajnavolgyi E, Psychedelic N,N-dimethyltryptamine and 5-methoxy-N,N-dimethyltryptamine modulate innate and adaptive inflammatory responses through the sigma-1 receptor of human monocyte-derived dendritic cells. PLoS One 9, e106533 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cobos EJ, Entrena JM, Nieto FR, Cendan CM, Del Pozo E, Pharmacology and therapeutic potential of sigma(1) receptor ligands. Current neuropharmacology 6, 344–366 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pozzobon T, Facchinello N, Bossi F, Capitani N, Benagiano M, Di Benedetto G, Zennaro C, West N, Codolo G, Bernardini M, Baldari CT, D'Elios MM, Pellegrini L, Argenton F, de Bernard M, Treponema pallidum (syphilis) antigen TpF1 induces angiogenesis through the activation of the IL-8 pathway. Scientific reports 6, 18785 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roux KJ, Kim DI, Raida M, Burke B, A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. J Cell Biol 196, 801–810 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cross BC, Bond PJ, Sadowski PG, Jha BK, Zak J, Goodman JM, Silverman RH, Neubert TA, Baxendale IR, Ron D, Harding HP, The molecular basis for selective inhibition of unconventional mRNA splicing by an IRE1-binding small molecule. Proc Natl Acad Sci U S A 109, E869–878 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Korennykh AV, Egea PF, Korostelev AA, Finer-Moore J, Zhang C, Shokat KM, Stroud RM, Walter P, The unfolded protein response signals through high-order assembly of Ire1. Nature 457, 687–693 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Copeland S, Warren HS, Lowry SF, Calvano SE, Remick D, Inflammation, I. the Host Response to Injury, Acute inflammatory response to endotoxin in mice and humans. Clinical and diagnostic laboratory immunology 12, 60–67 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oda S, Hirasawa H, Shiga H, Nakanishi K, Matsuda K, Nakamua M, Sequential measurement of IL-6 blood levels in patients with systemic inflammatory response syndrome (SIRS)/sepsis. Cytokine 29, 169–175 (2005). [DOI] [PubMed] [Google Scholar]
- 23.Oslowski CM, Urano F, Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods Enzymol 490, 71–92 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lewis AJ, Seymour CW, Rosengart MR, Current Murine Models of Sepsis. Surg Infect (Larchmt) 17, 385–393 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Remick DG, Bolgos GR, Siddiqui J, Shin J, Nemzek JA, Six at six: interleukin-6 measured 6 h after the initiation of sepsis predicts mortality over 3 days. Shock 17, 463–467 (2002). [DOI] [PubMed] [Google Scholar]
- 26.Shrum B, Anantha RV, Xu SX, Donnelly M, Haeryfar SM, McCormick JK, Mele T, A robust scoring system to evaluate sepsis severity in an animal model. BMC research notes 7, 233 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Papandreou I, Denko NC, Olson M, Van Melckebeke H, Lust S, Tam A, Solow-Cordero DE, Bouley DM, Offner F, Niwa M, Koong AC, Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood 117, 1311–1314 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ghareghani M, Zibara K, Sadeghi H, Dokoohaki S, Sadeghi H, Aryanpour R, Ghanbari A, Fluvoxamine stimulates oligodendrogenesis of cultured neural stem cells and attenuates inflammation and demyelination in an animal model of multiple sclerosis. Sci Rep 7, 4923 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Foster MT Jr., Ceftriaxone in treatment of serious infections. Septicemia. Hosp Pract (Off Ed) 26 Suppl 5, 43–47; discussion 62–43 (1991). [DOI] [PubMed] [Google Scholar]
- 30.Janssens S, Pulendran B, Lambrecht BN, Emerging functions of the unfolded protein response in immunity. Nat Immunol 15, 910–919 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levy MM, Fink MP, Marshall JC, Abraham E, Angus D, Cook D, Cohen J, Opal SM, Vincent JL, Ramsay G, Sccm/Esicm/Accp/Ats/Sis, 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Crit Care Med 31, 1250–1256 (2003). [DOI] [PubMed] [Google Scholar]
- 32.Davenport EE, Burnham KL, Radhakrishnan J, Humburg P, Hutton P, Mills TC, Rautanen A, Gordon AC, Garrard C, Hill AV, Hinds CJ, Knight JC, Genomic landscape of the individual host response and outcomes in sepsis: a prospective cohort study. Lancet Respir Med 4, 259–271 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hu SB, Zider A, Deng JC, When host defense goes awry: Modeling sepsis-induced immunosuppression. Drug Discov Today Dis Models 9, e33–e38 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Salminen A, Kauppinen A, Suuronen T, Kaarniranta K, Ojala J, ER stress in Alzheimer's disease: a novel neuronal trigger for inflammation and Alzheimer’s pathology. Journal of neuroinflammation 6, 41 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marrazzo A, Caraci F, Salinaro ET, Su TP, Copani A, Ronsisvalle G, Neuroprotective effects of sigma-1 receptor agonists against beta-amyloid-induced toxicity. Neuroreport 16, 1223–1226 (2005). [DOI] [PubMed] [Google Scholar]
- 36.Kikuchi H, Almer G, Yamashita S, Guegan C, Nagai M, Xu Z, Sosunov AA, McKhann GM 2nd, Przedborski S, Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc Natl Acad Sci U S A 103, 6025–6030 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hayashi T, Su TP, Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 131, 596–610 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Fontanilla D, Johannessen M, Hajipour AR, Cozzi NV, Jackson MB, Ruoho AE, The hallucinogen N,N-dimethyltryptamine (DMT) is an endogenous sigma-1 receptor regulator. Science 323, 934–937 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shajib MS, Khan WI, The role of serotonin and its receptors in activation of immune responses and inflammation. Acta Physiol (Oxf) 213, 561–574 (2014). [DOI] [PubMed] [Google Scholar]
- 40.Ha Y, Saul A, Tawfik A, Williams C, Bollinger K, Smith R, Tachikawa M, Zorrilla E, Ganapathy V, Smith SB, Late-onset inner retinal dysfunction in mice lacking sigma receptor 1 (sigmaR1). Invest Ophthalmol Vis Sci 52, 7749–7760 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sabino V, Cottone P, Parylak SL, Steardo L, Zorrilla EP, Sigma-1 receptor knockout mice display a depressive-like phenotype. Behav Brain Res 198, 472–476 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seluanov A, Vaidya A, Gorbunova V, Establishing primary adult fibroblast cultures from rodents. Journal of visualized experiments : JoVE, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang X, Goncalves R, Mosser DM, The isolation and characterization of murine macrophages. Curr Protoc Immunol Chapter 14, Unit 14 11 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gaultier A, Arandjelovic S, Niessen S, Overton CD, Linton MF, Fazio S, Campana WM, Cravatt BF 3rd, Gonias SL, Regulation of tumor necrosis factor receptor-1 and the IKK-NF-kappaB pathway by LDL receptor-related protein explains the antiinflammatory activity of this receptor. Blood 111, 5316–5325 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Girard CA, Wunderlich FT, Shimomura K, Collins S, Kaizik S, Proks P, Abdulkader F, Clark A, Ball V, Zubcevic L, Bentley L, Clark R, Church C, Hugill A, Galvanovskis J, Cox R, Rorsman P, Bruning JC, Ashcroft FM, Expression of an activating mutation in the gene encoding the KATP channel subunit Kir6.2 in mouse pancreatic beta cells recapitulates neonatal diabetes. J Clin Invest 119, 80–90 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kumar Y, Valdivia RH, Actin and intermediate filaments stabilize the Chlamydia trachomatis vacuole by forming dynamic structural scaffolds. Cell Host Microbe 4, 159–169 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F, Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3, RESEARCH0034 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Seki SM, Stevenson M, Rosen AM, Arandjelovic S, Gemta L, Bullock TNJ, Gaultier A, Lineage-Specific Metabolic Properties and Vulnerabilities of T Cells in the Demyelinating Central Nervous System. J Immunol 198, 4607–4617 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thurm CW, Halsey JF, Measurement of cytokine production using whole blood. Curr Protoc Immunol Chapter 7, Unit 7 18B (2005). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
fig. S1. S1R deletion influences a subset of LPS-induced processes without causing global perturbation.
fig. S2. Canonical TLR4 signaling in macrophages is unperturbed by S1R deletion.
fig. S3. Gating strategy and quantification of immunophenotyping on blood.
fig. S4. Gating strategy and quantification of immunophenotyping on peritoneal contents.
fig. S5. Gating strategy and quantification of immunophenotyping on spleen and lymph node.
fig. S6. S1R controls inflammation in primary fibroblasts
fig. S7. STF affects cytokine production in vitro and in vivo.
fig. S8. Anti-inflammatory effect of fluvoxamine does not globally suppress cytokine production in human tissue.
fig. S9. Proposed mechanism of action of S1R during LPS mediated inflammatory response.
Data file S1: Primary data (Excel file)