

Abstract
Objective
To describe the case of an African patient who was diagnosed with cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL).
Methods
Case report and literature review.
Results
We present a 39-year-old Gabonese man who developed progressive gait difficulty at the age of 32, followed by insidious tetraparesis, urinary sphincter disturbance, spastic dysarthria, cognitive dysfunction, and seizures. Brain imaging was performed many years after disease onset and revealed diffuse confluent white matter lesions and lacunar infarcts. He tested negative for acquired white matter disease, but genetic screening detected a genetic variant of HTRA1 gene (G283R), which has not been previously reported.
Conclusions
CARASIL is a disease that usually affects Asian patients. This case report describes a unique case of an African patient diagnosed with CARASIL and a novel genetic mutation in HTRA1 that has not been previously described in the literature.
Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL) is a familial autosomal recessive vascular disorder characterized by nonhypertensive cerebral small vessel disease with early adulthood onset of subcortical infarcts, progressive motor and cognitive impairment, alopecia, and spondylosis.1 It is a single-gene disorder that directly affects the cerebral small blood vessels. The disease is caused by mutation in the HTRA1 gene on chromosome 10q, which encodes a serine protease (HTRA1) that normally represses the expression of transforming growth factor (TGF)-β family.2
CARASIL is a rare disease, with less than 70 cases published in the literature thus far.3,4 Most of the reported cases are from Asia, with only few cases described in Europe.4–7 There are still no known reports from Africa.
Methods
We present a case of CARASIL in an African patient affected by a unique HTRA1 mutation.
Standard protocol approvals, registrations, and patient consents
The study was preapproved by the Research and Ethics Committee of the Neuro Spinal Hospital, Dubai, and written informed consent was obtained from the patient's wife because the patient was not in a position to do so because of cognitive impairment. The written consent also gave authorization to disclose any potentially recognizable information in the report.
Data availability
The authors confirm that all data supporting the findings of this study are available within the article.
Case report
A 39-year-old man from Gabon in Central Africa was referred to our hospital with a 7-year history of slowly progressive bilateral lower limb weakness and gradual gait deterioration. Earlier spine imaging in his home country had revealed a small thoracic syrinx (figure 1), and he had undergone thoracic laminectomy at T3 and T4 levels based on the assumption that his symptoms were related to the syrinx. He had continued to deteriorate despite the surgical intervention and had developed weakness in the upper limbs eventually. Further investigation 1 year later had revealed cervical disc herniation, and he had subsequently undergone cervical disc surgery, which also had not improved his symptoms. In the year before presentation, he had developed cognitive symptoms in addition to progressive dysarthria, urinary incontinence, and epileptic seizures. Both of his parents were Gabonese, and he had 2 siblings, none of whom had any history of neurologic disease at the time of this writing. His 11-year-old daughter is also free of neurologic disease.
Figure 1. Patient's dorsal spine MRI showing cord findings and previous intervention.
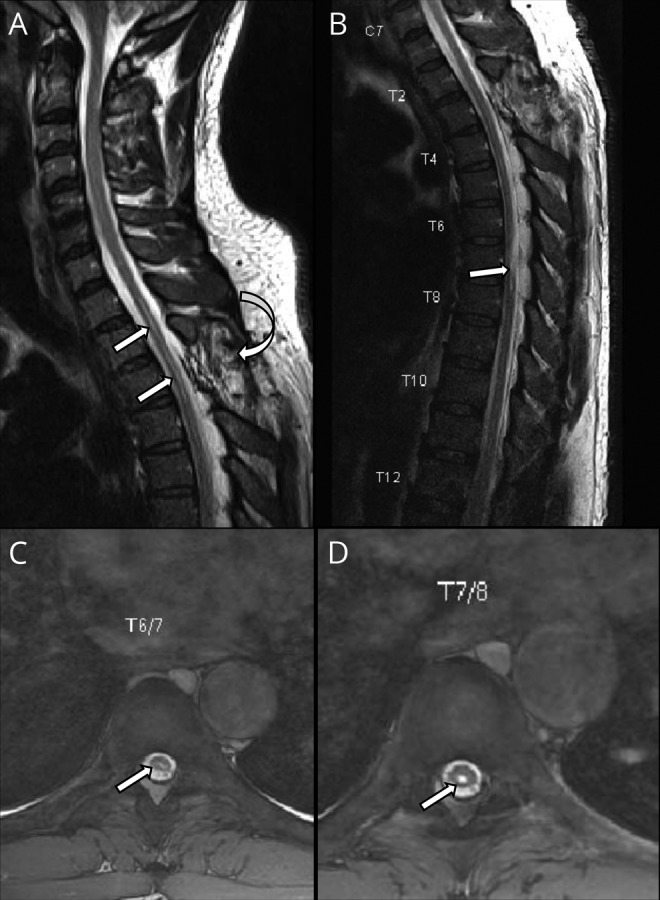
(A) Sagittal T2WI of the dorsal spine showing the site of the previous surgery (curved arrow) and intramedullary syrinx of the upper thoracic cord (arrows). (B) Sagittal and (C and D) axial T2WI of the thoracic spine showing cord affection by intramedullary patch of hyper intensity (arrows).
On examination, he showed cognitive impairment in the form of deficient attention span, slow mentation, apathy, poor episodic memory and difficulty following verbal instructions. Pupillary examination and fundoscopy were unremarkable. He had spastic dysarthria and spastic tetraparesis, with power of grade 2 of 5 in the lower limbs and grade 4 of 5 in the upper limbs. He was using a wheelchair and had suspended sensory deficit to pinprick from the umbilicus to the shoulders bilaterally. He was on urinary catheter because of urinary incontinence.
Brain MRI performed at our hospital showed confluent, symmetric hyperintense lesions in the periventricular and deep white matter as well as lacunar infarcts, with relative sparing of the U-fibers (figure 2). His dorsal spine MRI showed a linear central intramedullary hyperintensity representing a small syrinx (figure 1). In the differential diagnosis, we excluded the common acquired causes of white matter disease, including sporadic vascular diseases, vasculitis, MS, and other immunologic and neuroinflammatory disorders. Notably, vasculitis screening, CSF oligoclonal bands, antiaquaporin 4 antibody, antimyelin oligodendrocyte antibody, serum and CSF angiotensin converting enzyme assay, CSF venereal disease research laboratory test, and HIV test were all negative. The patient also did not have any of the known traditional vascular risk factors such as hypertension and diabetes mellitus.
Figure 2. Patient's brain MRI showing hemispheric and brainstem findings.
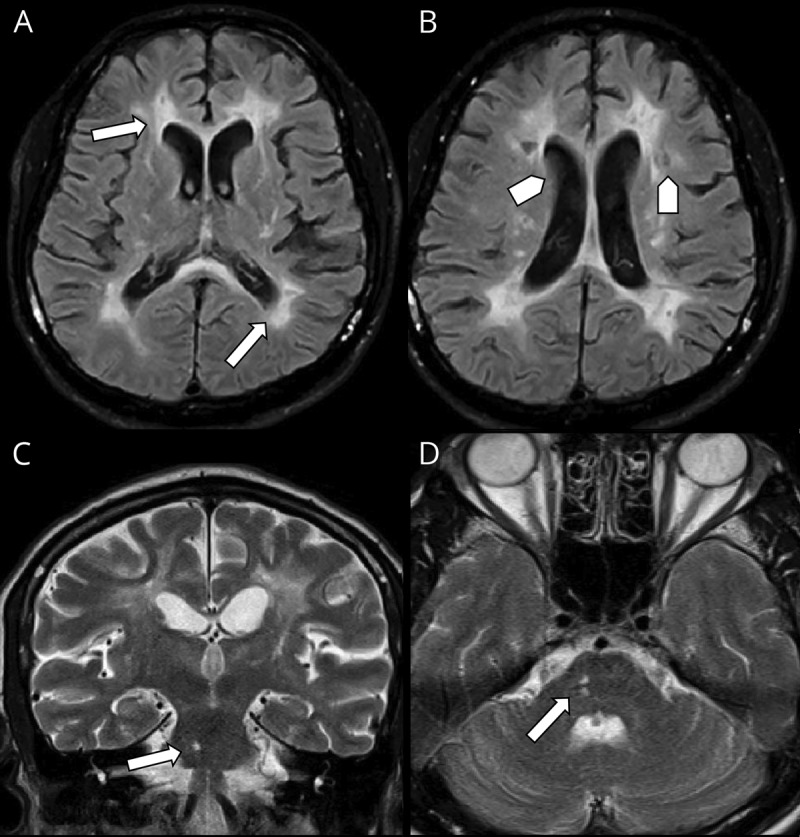
Pattern of periventricular white matter involvement in different pulse sequences in axial FLAIR (A and B), note lacunar infarcts (arrow head) is seen in B. (C) Coronal T2WI and (D) Axial T2WI showing affection of the right aspect of pons (arrows) creeping to the middle cerebral peduncle. FLAIR = fluid-attenuated inversion recovery.
Genetic testing for an expanded panel of genes associated with leukodystrophies and other white matter diseases was carried out and showed a heterozygous mutation in the HTRA1 gene, HTRA1 G283R, in which glycine was replaced by arginine at position 283 in the amino acid sequence of the HTRA1 protein (Gly283Arg) because of a missense mutation in which adenine was substituted for guanine at codon 847 (GGA > AGA). We could not carry out genetic screening on his parents because his father had died at the time of his presentation. We were thus unable to determine if this mutation was inherited or arose de novo. He continued to deteriorate cognitively and suffered further seizures until his death in April 2019, 8 years after disease onset.
Discussion
CARASIL is a rare genetic vascular disorder which has been linked to a single-gene mutation involving HTRA1.2 This gene was determined in 20092 and is believed to encode a serine protease (HTRA1) that represses the expression of TGF-β family.2 The resultant effect of this mutation is an increased expression of the TGF-β family within the tunica intima and media of cerebral small vessels, which ultimately leads to arteriolosclerosis.2
The disorder is inherited in an autosomal recessive manner, and heterozygotes are not expected to manifest disease. Nevertheless, symptomatic heterozygote carriers have been reported.8,9 This category of patients has less frequent non-neurological features such as alopecia.8,9 Likewise, our patient had no alopecia, thus showing some similarities to the reported heterozygous cases.
The radiologic hallmarks of CARASIL include high-signal-intensity lesions in the periventricular white matter and deep white matter as well as multiple lacunar infarctions in the basal ganglia and thalamus, with a relative sparing of the superficial white matter (U-fibers).10 Many of these features were found in our patient. The diffuse white matter lesions may also extend to the temporal poles or external capsules,10 although no temporal pole lesions were found in our case (figure 2).
About 48 cases of this disorder have been published from Japan and China, slightly more than a dozen cases from Europe and, more recently, 8 cases from Turkey.4–7 Up to the time of writing this report, occurrence of this disease is yet to be reported in Africans.
Hitherto, to our knowledge, the G283R variant has neither been documented as a pathogenic variant nor has it been reported as a benign variant. However, we surmise that this variant is pathogenic, considering that a heterozygous missense substitution at the same position (G283E) has been reported in an individual with cerebral small vessel disease, cognitive impairment, and gait disturbance.9 In that publication, functional analysis suggested that the mutant gene might cause disease through defective trimerization with the wild type.9 Although functional analysis was not performed in our case, we suppose that G283R may possibly have similar molecular characteristics as G283E and may have similar pathogenicity.
Our case presents unique peculiarities in comparison with similar cases described in the literature. First, this disease is yet to be reported in Africans. Second, we report a genetic variant of HTRA1 gene mutation that has not been previously reported in the literature (G283R), and finally, we add to the few reported cases of symptomatic heterozygote forms of HTRA1 mutation.
Glossary
- CARASIL
Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy
Author contributions
O.J. Oluwole was involved in study conceptualization, data acquisition and interpretation, manuscript writing, final editing, and manuscript submission. H. Ibrahim was involved in data acquisition and interpretation, manuscript writing, and final editing. D. Garrozo was involved in manuscript writing and final editing. K Ben Hamouda was involved in final editing of the scientific content. S.I Mostafa Hassan was involved in data acquisition and final editing. A.M. Hegazy was involved in final editing of the scientific content. A.K. Msaddi was involved in the final editing of the scientific content.
Study funding
Genetic testing was funded out of pocket by the authors.
Disclosure
Disclosures available: Neurology.org/NG.
References
- 1.Yanagawa S, Ito N, Arima K, Ikeda S. Cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy. Neurology 2002;58:817–820. [DOI] [PubMed] [Google Scholar]
- 2.Hara K, Shiga A, Fukutake T, et al. . Association of HTRA1 mutations and familial ischemic cerebral small-vessel disease. N Engl J Med 2009;360:1729–1739. [DOI] [PubMed] [Google Scholar]
- 3.Wang XL, Li CF, Guo HW, Cao BZ. A novel mutation in the HTRA1 gene identified in Chinese CARASIL pedigree. CNS Neurosci Ther 2012;18:867–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen Y, He Z, Meng S, Li L, Yang H, Zhang X. A novel mutation of the high-temperature requirement A serine peptidase 1 (HTRA1) gene in a Chinese family with cerebral autosomal recessive arteriopathy with subcortical infarcts and leukoencephalopathy (CARASIL). J Int Med Res 2013;41:1445–1455. [DOI] [PubMed] [Google Scholar]
- 5.Cordeiro IM, Nzwalo H, Sá F, et al. . Shifting the CARASIL paradigm report of a non-Asian family and literature review. Stroke 2015;46:1110–1112. [DOI] [PubMed] [Google Scholar]
- 6.Bianchi S, Di Palma C, Gallus GN, et al. . Two novel HTRA1 mutations in a European CARASIL patient. Neurology 2014;82:898–900. [DOI] [PubMed] [Google Scholar]
- 7.Bayrakli F, Balaban H, Gurelik M, Hizmetli S, Topaktas S. Mutation in the HTRA1 gene in a patient with degenerated spine as a component of CARASIL syndrome. Turk Neurosurg 2014;24:67–69. [DOI] [PubMed] [Google Scholar]
- 8.Verdura E, Hervé D, Scharrer E, et al. . Heterozygous HTRA1 mutations are associated with autosomal dominant cerebral small vessel disease. Brain 2015;138:2347–2358. [DOI] [PubMed] [Google Scholar]
- 9.Nozaki H, Kato T, Nihonmatsu M, et al. . Distinct molecular mechanisms of HTRA1 mutants in manifesting heterozygotes with CARASIL. Neurology 2016;86:1964–1974. [DOI] [PubMed] [Google Scholar]
- 10.Fukutake T, Shimoe Y, Hattori T. Differences in MRI lesion patterns of two hereditary vascular leukoencephalopathy: CADASIL and CARASIL. J Stroke Cerebrovasc Dis 2000;9(suppl 1):263–264. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that all data supporting the findings of this study are available within the article.