

Abstract
The enzyme geranylgeranyl diphosphate synthase (GGDPS) is a potential therapeutic target for multiple myeloma. Malignant plasma cells produce and secrete large amounts of monoclonal protein, and inhibition of GGDPS results in disruption of protein geranylgeranylation which in turn impairs intracellular protein trafficking. Our previous work has demonstrated that some isoprenoid triazole bisphosphonates are potent and selective inhibitors of GGDPS. To explore the possibility of selective delivery of such compounds to plasma cells, new analogues with an ω-hydroxy group have been synthesized and examined for their enzymatic and cellular activity. These studies demonstrate that incorporation of the ω-hydroxy group minimally impairs GGDPS inhibitory activity. Furthermore conjugation of one of the novel ω-hydroxy GGDPS inhibitors to hyaluronic acid resulted in enhanced cellular activity. These results will allow future studies to focus on the in vivo biodistribution of HA-conjugated GGDPS inhibitors.
Keywords: GGDP synthase, inhibition, isoprenoid biosynthesis, triazole, bisphosphonate
Graphical Abstract
Geranylgeranyl diphosphate (1, GGDP, Figure 1) represents an important branch point in isoprenoid biosynthetic pathways. This intermediate is used in plants to afford a tremendous variety of cyclic diterpenoids,1 and in mammals it is used primarily for post-translational modification of proteins.2 Among the proteins modified by reaction with GGDP are those in the Ras superfamily of small GTPases such as the Rho proteins which play roles in cancer cell migration and metastasis,3 and the Rab proteins which are essential for intracellular trafficking processes.4
Figure 1.

Geranylgeranyl diphosphate and known inhibitors of geranylgeranyl diphosphate synthase.
Isoprenoid biosynthesis in humans already is targeted by at least two families of blockbuster drugs, the statins and the nitrogenous bisphosphonates. Statins target the enzyme 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR) which is the rate-limiting step in the mevalonate pathway to higher isoprenoids, and these drugs are widely used as cholesterol-lowering agents and to prevent cardiovascular disease.5 The nitrogenous bisphosphonates target the later enzyme farnesyl diphosphate synthase (FDPS), and are widely used to treat osteoporosis and other diseases of the bone.6 In vitro, both classes of drugs can exert anti-cancer activities as a consequence of disruption of protein prenylation, particularly geranylgeranylation.7 These drugs are not ideal in the setting of systemic anti-cancer therapy however, because standard doses of statins do not alter protein prenylation8–9 and the nitrogenous bisphosphonates in clinical use do not have sufficient systemic exposure.10
For some time there has been interest in identification of compounds that directly inhibit the enzyme geranylgeranyl diphosphate synthase (GGDPS)11–12 to disrupt geranylgeranylation more specifically.11, 13 We and others have focused on the development of GGDPS inhibitors as anti-myeloma agents because disruption of Rab geranylgeranylation results in impairment of monoclonal protein trafficking within myeloma cells, leading to ER stress and cell death.12, 14–15 In this context, we have reported the preparation and biological activity of a number of compounds with significant activity against this enzyme. The first was digeranyl bisphosphonate (2, DGBP),16 which showed an IC50 of ~260 nM for GGDPS and good selectivity over the related enzyme farnesyl diphosphate synthase,17 but cellular activity only at high concentrations (~10 μM). Crystallographic studies indicated that DGBP’s V-shaped structure occupied the enzyme’s active site, with the phosphonates complexed to magnesium ions and the two isoprenoid side chains occupying the FPP and GGPP channels.18 More recently we have focused on monoalkyl compounds assembled through click chemistry and incorporating a triazole ring system. These efforts first yielded the inhibitor 3, which shows enzyme activity at 2.2 μM and cellular activity ~1 μM.19 Surprisingly, as a mixture of olefin isomers the homologue 4 showed an IC50 of 45 nM against GGDPS, high specificity for GGDPS over FDPS, and cellular activity at concentrations as low as 30 nM in multiple myeloma cells.20–21 After preparation of the individual isomers 5 and 6, and methylation of the alpha position, bioassays revealed these compounds had IC50 values of 125 and 86 nM respectively, and cellular activity at 20 and 25 nM levels.21
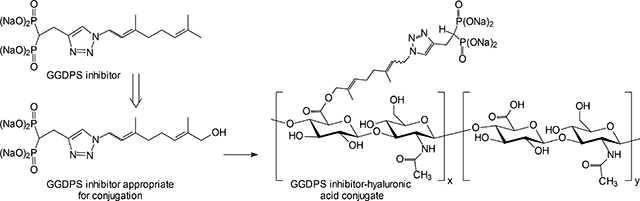
The potency of these GGDPS inhibitors in both enzyme and cell assays is relevant from a therapeutic perspective. Preclinical studies of compounds 4–6 have shown systemic distribution, prolonged half-lives and metabolic stability,22–23 all of which are important drug-like features. The dose-limiting toxicity for these compounds has been hepatic in nature22–23 and it would be desirable to enhance their anti-myeloma activity by optimizing drug delivery to the target organs of interest and thereby minimize off-target effects. A prodrug approach might help in this regard,24–27 but it may be preferable to utilize GGDPS inhibitors that can be conjugated to other agents. Hyaluronic acid (HA), a non-sulfated glycosaminoglycan, can be readily optimized for delivery of varied cargoes including small molecule chemotherapeutic agents,28–30 and has thus far found clinical use in ophthalmology, rheumatology and wound healing applications.31–32 Furthermore, work done with fluorescent dyes has revealed that conjugation of the dye to HA can lead to improved tumor uptake relative to surrounding tissue and alter biodistribution profiles.33 HA is the native ligand for CD44 and multiple studies have demonstrated that HA can target both to CD44-overexpressed solid tumors34–36 and to cells of hematological malignancies37 including myeloma cells.38 As an initial foray into this area, we describe the synthesis and biological activity of two ω-hydroxy triazole bisphosphonates. Furthermore, we demonstrate that the ω-OH modification allows linkage to HA via ester formation and report the cellular activity of the first HA-GGDPS inhibitor conjugate.
As an initial test of this strategy, the first target chosen was the triazole bisphosphonate 15 (Scheme I) which was viewed as reasonably accessible. The synthesis of this compound started with selenium dioxide catalyzed allylic oxidation of commercially available geranyl acetate (7).39 While oxidation provided a mixture of the desired alcohol and the corresponding aldehyde, treatment with NaBH4 to reduce that aldehyde increased the yield of the desired alcohol 8 to an acceptable level. After protection of the free alcohol 8 as the TBS ether 9, base catalyzed hydrolysis of the acetate afforded compound 10.40 Conversion to the bromide 1141 was accomplished in near-quantitative yield by reaction with PBr3. Reaction of the bromide 11 with sodium azide proceeded cleanly, but gave the allylic azide as a mixture of E and Z isomers due to a well known [3,3] sigmatropic rearrangement.42 The azide then was allowed to react with the acetylene 1343 to afford the TBS protected triazole, which was immediately carried to acidic hydrolysis to afford the desired alcohol 14. Standard McKenna hydrolysis44 of the tetraethyl ester 14 by treatment with TMSBr followed by NaOH provided the tetra-sodium salt 15. Based on the 1H NMR spectrum of this product, the E/Z ratio was found to be ~2:1 in favor of the E isomer.
Scheme 1.

Synthesis of compound 15, the ω-hydroxy analogue of compound 3.
The second synthetic target in this series was the ω-hydroxy analogue of compound 6, which required a longer synthesis but is one of the compounds with better cellular activity. This compound could be envisioned as arising from homonerol through a sequence parallel to that used to obtain compound 15. However, our previous route to homonerol employed an 8-step sequence45 and while it gave isomerically pure material, to avoid an even longer sequence an alternative route to homonerol was developed. For this route, nerol (16) first was oxidized to neral (17) under either of two reaction conditions. A TEMPO oxidation46 gave the desired aldehyde in just four hours while an MnO2 oxidation required several days but gave the same aldehyde in nearly quantitative yield. Wittig olefination of neral produced the isomerically pure triene 18 in high yield. Regioselective hydroboration-oxidation of the terminal double bond in the triene 18 gave homonerol (19) in modest yield,47 but the brevity of this reaction sequence made that acceptable.
Once homonerol (19) was in hand, multiple attempts to accomplish a SeO2 oxidation went unrewarded. Therefore, the reaction sequence employed in Scheme 1 was reorganized so that the selenium dioxide oxidation could be pursued at a later stage. Treatment of homonerol (19) with methanesulfonyl chloride and subsequent reaction of the mesylate with sodium bromide gave homoneryl bromide (20) in good yield.21 At this stage of the sequence, the selenium dioxide mediated allylic oxidation48 was modestly successful, and furnished the desired alcohol 21 in ~25% yield. While this yield might still be improved, once the alcohol 21 was in hand replacement of bromide with azide was performed and the product immediately was carried to the next step. For this click reaction, the alkyne 23 was synthesized by a known procedure from tetraethyl vinyl bisphosphonate (22) through a two-step process.21 The click reaction then was conducted under standard conditions to afford the ester 24. McKenna hydrolysis of the phosphonate esters provided the desired salt 25.
Once the two new triazoles 15 and 25 were available, they were tested for their ability to disrupt protein geranylgeranylation in cell assays and to inhibit GGDPS in enzyme assays. The activity of the new ω-hydroxy compounds was compared directly to their respective parent compounds 3 and 6. As shown in Figure 2, the disruption of protein geranylgeranylation was evaluated using two methods: 1) immunoblot analysis for unmodified Rap1a (a representative substrate for geranylgeranyl transferase (GGTase) I; and, 2) ELISA for intracellular lambda light chain levels as a marker for disruption of Rab geranylgeranylation.14 Lovastatin was included as a positive control.14 Both compounds 15 and 25 induced concentration-dependent effects in these assays, consistent with GGDPS inhibitory activity. In both cases, the addition of the ω-OH group did modestly diminish cellular potency, with compound 15 approximately 10-fold less potent than 3 and compound 25 approximately 10-fold less potent than 6. Enzyme assays utilizing recombinant GGDPS and FDPS confirmed the specificity of these new compounds as GGDPS inhibitors (Table 1).
Figure 2. Comparison of the effects of the novel ω-hydroxyl triazole bisphosphonates to the parent analogues on protein geranylgeranylation.

RPMI-8226 cells were incubated for 48 hours in the presence or absence of lovastatin (Lov, 10 μM) or varying concentrations of the test compounds. A) Immunoblot analysis of uRap1a (antibody detects only unmodified protein) and β-tubulin (as a loading control). B) Intracellular lambda light chain concentrations were determined via ELISA. Data are expressed as a percentage of control (mean ± SD, n=3). The * denotes p < 0.05 per unpaired two-tailed t-test.
Table 1.
Summary of the bioassay results of the novel ω-OH-triazole bisphosphonates.
| Compound | GGDPS IC50 (μM) | FDPS IC50 (μM) | Fold-selectivity for GGDPS compared to FDPS | Cellular LEC1 (μM) |
|---|---|---|---|---|
| 15 | 11.6 + 2.6 | 91.2 + 25.0 | 8 | 10 |
| 25 | 0.67 + 0.30 | 81.4 + 23.5 | 121 | 0.25 |
Cellular LEC (lowest effective concentration) is defined as the lowest concentration for which an unmodified Rap1a band is visible in the immunoblot and a statistically significant increase in intracellular lambda light chain is observed in the ELISA.
Because it was more readily available, we then joined the alcohol 15 to HA (26, 10K) via NHS/EDC conjugation chemistry49–52 to prepare the drug conjugate 27. Conjugation was confirmed by the presence of the aromatic signal from the triazole moiety and acetyl hydrogens from HA in the 1H-NMR spectrum as well as by the presence of a phosphonate resonance in the 31P NMR spectrum.
Next, we compared the cellular activity of the parent ω-OH compound 15 to the HA conjugate 27. As shown in Figure 3, the HA-conjugate 27 induced greater accumulation of unmodified Rap1a than the free drug 15 in two different human myeloma cell lines. This enhanced cellular potency is likely a consequence of improved cellular uptake due to CD44 cell surface expression. Presumably cellular esterases53–54 then hydrolyze the ω-OH-GGDPS inhibitor from the polymer, releasing free drug in the cell.
Figure 3. Comparison of the effects of the ω-hydroxyl triazole bisphosphonate 15 and the HA-conjugate 27 on protein geranylgeranylation.

RPMI-8226 (R) or MM.1S (M) cells were incubated for 48 hours in the presence or absence of lovastatin (Lov, 10 μM) or varying concentrations of the test compounds (for the HA-conjugate 27, concentration is based on the percent weight of the inhibitor in the HA polymer conjugate). Immunoblot analysis of uRap1a (antibody detects only unmodified protein) and β-tubulin (as a loading control). These blots are representative of three independent experiments.
In conclusion, after introduction of the ω-hydroxyl group to geranyl acetate (7), and its immediate protection as a TBS ether, the synthetic sequence to compound 15 closely parallels our earlier synthesis of compound 3. In contrast, the preparation of compound 25 employs a different synthetic sequence for preparation of homonerol (19), a sequence that is just three steps long rather than the eight used previously. As a result, this intermediate was available in sufficient quantity that the disappointing yield for the selenium dioxide oxidation to afford compound 21 could be tolerated. The remaining steps in the sequence then run parallel to our earlier synthesis of compound 6.
While our prior studies have included substantial structure-function analysis of the triazole bisphosphonate class of GGDPS inhibitors, we have not previously evaluated the impact of modification of the terminal component of the isoprenoid chain. In the studies presented here we demonstrate that the addition of an ω-hydroxyl group to two of our previously reported inhibitors results in retention of GGDPS inhibitory activity. While the potency of these new analogues is diminished relative to the parent compounds, the hydroxy group affords the ability to conjugate these inhibitors to other agents such as HA with the goal of modifying drug biodistribution patterns and enhancing the therapeutic index. Our preliminary studies with the conjugate 27 demonstrate enhanced cellular activity of the HA-GGDPS inhibitor conjugate compared to the free drug. Future studies now can examine the biodistribution and toxicity profile of the HA-GGDPS inhibitor conjugates in vivo, as well as explore alternative modifications at the ω-position to create additional “linkable” inhibitors.
Supplementary Material
Scheme 2.

Synthesis of compound 25, the ω-hydroxy analogue of compound 6.
Scheme 3.

Generation of HA-GGDPS inhibitor polymer conjugate 27.
Acknowledgements
We thank Jack Neal for his assistance with preparation of some of the synthetic intermediates. This project was supported in part by the Roy J. Carver Charitable Trust (01-224 to DFW) and the National Institutes of Health (R01CA-172070 to SAH, P20 GM103480 and P30 CA036727). DB was supported through a University of Nebraska Medical Center Program of Excellence Assistantship, and WMP was supported by fellowships from the PhRMA Foundation and Blue Waters Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Davis EM; Croteau R, Cyclization enzymes in the biosynthesis of monoterpenes, sesquiterpenes, and diterpenes In Biosynthesis: Aromatic Polyketides, Isoprenoids, Alkaloids, Leeper FJ; Vederas JC, Eds. 2000; Vol. 209, pp 53–95. [Google Scholar]
- 2.Wiemer AJ; Wiemer DF; Hohl RJ, Geranylgeranyl Diphosphate Synthase: An Emerging Therapeutic Target. Clin. Pharmacol. Ther. 2011, 90 (6), 804–812. [DOI] [PubMed] [Google Scholar]
- 3.Lawson CD; Ridley AJ, Rho GTPase signaling complexes in cell migration and invasion. The Journal of cell biology 2018, 217 (2), 447–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grosshans BL; Ortiz D; Novick P, Rabs and their effectors: achieving specificity in membrane traffic. Proceedings of the National Academy of Sciences of the United States of America 2006, 103 (32), 11821–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chou R; Dana T; Blazina I; Daeges M; Jeanne TL, Statins for Prevention of Cardiovascular Disease in Adults: Evidence Report and Systematic Review for the US Preventive Services Task Force. Jama 2016, 316 (19), 2008–2024. [DOI] [PubMed] [Google Scholar]
- 6.Cremers S; Drake MT; Ebetino FH; Bilezikian JP; Russell RGG, Pharmacology of Bisphosphonates. British journal of clinical pharmacology 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thurnher M; Nussbaumer O; Gruenbacher G, Novel aspects of mevalonate pathway inhibitors as antitumor agents. Clinical cancer research : an official journal of the American Association for Cancer Research 2012, 18 (13), 3524–31. [DOI] [PubMed] [Google Scholar]
- 8.Schachter M, Chemical, pharmacokinetic and pharmacodynamic properties of statins: an update. Fundam Clin Pharmacol 2005, 19 (1), 117–25. [DOI] [PubMed] [Google Scholar]
- 9.Pan HY; DeVault AR; Wang-Iverson D; Ivashkiv E; Swanson BN; Sugerman AA, Comparative pharmacokinetics and pharmacodynamics of pravastatin and lovastatin. J Clin Pharmacol 1990, 30 (12), 1128–35. [DOI] [PubMed] [Google Scholar]
- 10.Chen T; Berenson J; Vescio R; Swift R; Gilchick A; Goodin S; LoRusso P; Ma P; Ravera C; Deckert F; Schran H; Seaman J; Skerjanec A, Pharmacokinetics and pharmacodynamics of zoledronic acid in cancer patients with bone metastases. J. Clin. Pharmacol 2002, 42 (11), 1228–36. [DOI] [PubMed] [Google Scholar]
- 11.Haney SL; Wills VS; Wiemer DF; Holstein SA, Recent advances in the development of mammalian geranylgeranyl diphosphate synthase inhibitors. Molecules 2017, 22 (6), 886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lacbay CM; Waller DD; Park J; Palou MG; Vincent F; Huang XF; Ta V; Berghuis AM; Sebag M; Tsantrizos YS, Unraveling the Prenylation-Cancer Paradox in Multiple Myeloma with Novel Geranylgeranyl Pyrophosphate Synthase (GGPPS) Inhibitors. J. Med. Chem 2018, 61 (15), 6904–6917. [DOI] [PubMed] [Google Scholar]
- 13.Agabiti SS; Liang YL; Wiemer AJ, Molecular mechanisms linking geranylgeranyl diphosphate synthase to cell survival and proliferation. Mol. Membr. Biol 2016, 33 (1–2), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holstein SA; Hohl RJ, Isoprenoid biosynthetic pathway inhibition disrupts monoclonal protein secretion and induces the unfolded protein response pathway in multiple myeloma cells. Leukemia research 2011, 35 (4), 551–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dykstra KM; Allen C; Born EJ; Tong H; Holstein SA, Mechanisms for autophagy modulation by isoprenoid biosynthetic pathway inhibitors in multiple myeloma cells. Oncotarget 2015, 6 (39), 41535–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shull LW; Wiemer AJ; Hohl RJ; Wiemer DF, Synthesis and biological activity of isoprenoid bisphosphonates. Bioorganic & medicinal chemistry 2006, 14 (12), 4130–6. [DOI] [PubMed] [Google Scholar]
- 17.Wiemer AJ; Tong H; Swanson KM; Hohl RJ, Digeranyl bisphosphonate inhibits geranylgeranyl pyrophosphate synthase. Biochem. Biophys. Res. Commun 2006. [DOI] [PubMed] [Google Scholar]
- 18.Chen CKM; Hudock MP; Zhang YH; Guo RT; Cao R; No JH; Liang PH; Ko TP; Chang TH; Chang S; Song YC; Axelson J; Kumar A; Wang AHJ; Oldfield E, Inhibition of geranylgeranyl diphosphate synthase by bisphosphonates: A crystallographic and computational investigation. J. Med. Chem 2008, 51 (18), 5594–5607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou X; Ferree SD; Wills VS; Born EJ; Tong H; Wiemer DF; Holstein SA, Geranyl and neryl triazole bisphosphonates as inhibitors of geranylgeranyl diphosphate synthase. Bioorg. Med. Chem 2014, 22 (9), 2791–2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wills VS; Allen C; Holstein SA; Wiemer DF, Potent triazole bisphosphonate inhibitor of geranylgeranyl diphosphate synthase. ACS medicinal chemistry letters 2015, 6 (12), 1195–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matthiesen RA; Varney ML; Xu PC; Rier AS; Wiemer DF; Holstein SA, alpha-Methylation enhances the potency of isoprenoid triazole bisphosphonates as geranylgeranyl diphosphate synthase inhibitors. Bioorg. Med. Chem 2018, 26 (2), 376–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Haney SL; Chhonker YS; Varney ML; Talmon G; Murry DJ; Holstein SA, Preclinical investigation of a potent geranylgeranyl diphosphate synthase inhibitor. Investigational new drugs 2018, 36 (5), 810–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haney SL; Chhonker YS; Varney ML; Talmon G; Murry DJ; Holstein SA, In Vivo Evaluation of Novel Geranylgeranyl Diphosphate Synthase Inhibitors. Blood 2018, 132 (Suppl 1), 215–215. [Google Scholar]
- 24.Wiemer AJ; Wiemer DF, Prodrugs of Phosphonates and Phosphates: Crossing the Membrane Barrier In Phosphorus Chemistry I: Asymmetric Synthesis and Bioactive Compounds, Montchamp JL, Ed. 2015; Vol. 360, pp 115–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pradere U; Garnier-Amblard EC; Coats SJ; Amblard F; Schinazi RF, Synthesis of Nucleoside Phosphate and Phosphonate Prodrugs. Chem. Rev 2014, 114 (18), 9154–9218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lentini NA; Foust BJ; Hsiao CHC; Wiemer AJ; Wiemer DF, Phosphonamidate Prodrugs of a Butyrophilin Ligand Display Plasma Stability and Potent V gamma 9 V delta 2 T Cell Stimulation. J. Med. Chem 2018, 61 (19), 8658–8669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Foust BJ; Poe MM; Lentini N; Hsiao CHC; Wiemer AJ; Wiemer DF, Mixed Aryl Phosphonate Prodrugs of a Butyrophilin Ligand. Acs Medicinal Chemistry Letters 2017, 8 (9), 914–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Choi KY; Jeon EJ; Yoon HY; Lee BS; Na JH; Min KH; Kim SY; Myung S-J; Lee S; Chen X; Kwon IC; Choi K; Jeong SY; Kim K; Park JH, Theranostic nanoparticles based on PEGylated hyaluronic acid for the diagnosis, therapy and monitoring of colon cancer. Biomaterials 2012, 33 (26), 6186–6193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Choi KY; Yoon HY; Kim J-H; Bae SM; Park R-W; Kang YM; Kim I-S; Kwon IC; Choi K; Jeong SY; Kim K; Park JH, Smart Nanocarrier Based on PEGylated Hyaluronic Acid for Cancer Therapy. ACS Nano 2011, 5 (11), 8591–8599. [DOI] [PubMed] [Google Scholar]
- 30.Huang G; Huang H, Application of hyaluronic acid as carriers in drug delivery. Drug delivery 2018, 25 (1), 766–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kogan G; Soltés L; Stern R; Gemeiner P, Hyaluronic acid: a natural biopolymer with a broad range of biomedical and industrial applications. Biotechnol Lett 2007, 29 (1), 17–25. [DOI] [PubMed] [Google Scholar]
- 32.Gaffney J; Matou-Nasri S; Grau-Olivares M; Slevin M, Therapeutic applications of hyaluronan. Mol. BioSyst 2010, 6 (3), 437–443. [DOI] [PubMed] [Google Scholar]
- 33.Souchek JJ; Wojtynek NE; Payne WM; Holmes MB; Dutta S; Qi B; Datta K; LaGrange CA; Mohs AM, Hyaluronic acid formulation of near infrared fluorophores optimizes surgical imaging in a prostate tumor xenograft. Acta biomaterialia 2018, 75, 323–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ganesh S; Iyer AK; Morrissey DV; Amiji MM, Hyaluronic acid based self-assembling nanosystems for CD44 target mediated siRNA delivery to solid tumors. Biomaterials 2013, 34 (13), 3489–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cohen K; Emmanuel R; Kisin-Finfer E; Shabat D; Peer D, Modulation of drug resistance in ovarian adenocarcinoma using chemotherapy entrapped in hyaluronan-grafted nanoparticle clusters. ACS Nano 2014, 8 (3), 2183–95. [DOI] [PubMed] [Google Scholar]
- 36.Yan H; Song J; Jia X; Zhang Z, Hyaluronic acid-modified didecyldimethylammonium bromide/ d-a-tocopheryl polyethylene glycol succinate mixed micelles for delivery of baohuoside I against non-small cell lung cancer: in vitro and in vivo evaluation. Drug delivery 2017, 24 (1), 30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jin L; Hope KJ; Zhai Q; Smadja-Joffe F; Dick JE, Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nature medicine 2006, 12 (10), 1167–74. [DOI] [PubMed] [Google Scholar]
- 38.Gu Z; Wang X; Cheng R; Cheng L; Zhong Z, Hyaluronic acid shell and disulfide-crosslinked core micelles for in vivo targeted delivery of bortezomib for the treatment of multiple myeloma. Acta biomaterialia 2018, 80, 288–295. [DOI] [PubMed] [Google Scholar]
- 39.Yu X-J; Zhang H; Xiong F-J; Chen X-X; Chen F-E, An Improved Convergent Strategy for the Synthesis of Oligoprenols. Helv. Chim. Acta 2008, 91 (10), 1967–1974. [Google Scholar]
- 40.Reardon MB; Xu M; Tan Q; Baumgartel PG; Augur DJ; Huo S; Jakobsche CE, Long-Range Reactivity Modulations in Geranyl Chloride Derivatives. J. Org. Chem 2016, 81 (22), 10964–10974. [DOI] [PubMed] [Google Scholar]
- 41.Lee ER; Lakomy I; Bigler P; Scheffold R, Reductive radical cyclizations of bromo acetals and (bromomethyl)silyl ethers of terpenoid alcohols. Helv. Chim. Acta 1991, 74 (1), 146–162. [Google Scholar]
- 42.Padwa A; Sa MM, Intramolecular O-H insertion reaction of azido substituted diazoesters and its relevance to the mechanism of the allylic azide rearrangement. Tetrahedron Lett. 1997, 38 (29), 5087–5090. [Google Scholar]
- 43.Zhou X; Hartman SV; Born EJ; Smits JP; Holstein SA; Wiemer DF, Triazole-based inhibitors of geranylgeranyltransferase II. Bioorg. Med. Chem. Lett 2013, 23 (3), 764–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.McKenna CE; Higa MT; Cheung NH; McKenna MC, Facile Dealkylation of Phosphonic Acid Dialkyl Esters by Bromotrimethylsilane. Tetrahedron Lett. 1977, (2), 155–158. [Google Scholar]
- 45.Matthiesen RA; Wills VS; Metzger JI; Holstein SA; Wiemer DF, Stereoselective Synthesis of Homoneryl and Homogeranyl Triazole Bisphosphonates. J. Org. Chem 2016, 81 (19), 9438–9442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kliman LT; Mlynarski SN; Ferris GE; Morken JP, Catalytic Enantioselective 1,2-Diboration of 1,3-Dienes: Versatile Reagents for Stereoselective Allylation. Angewandte Chemie-International Edition 2012, 51 (2), 521–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huang HJ; Yang WB, Synthesis of moenocinol and its analogs using BT-sulfone in Julia-Kocienski olefination. Tetrahedron Lett. 2007, 48 (8), 1429–1433. [Google Scholar]
- 48.Huang QH; Rawal VH, Total synthesis of (+/−)-bipinnatin J. Organic Letters 2006, 8 (3), 543–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hill TK; Abdulahad A; Kelkar SS; Marini FC; Long TE; Provenzale JM; Mohs AM, Indocyanine Green-Loaded Nanoparticles for Image-Guided Tumor Surgery. Bioconjug Chem 2015, 26 (2), 1416–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hill TK; Davis AL; Wheeler FB; Kelkar SS; Freund EC; Lowther WT; Kridel SJ; Mohs AM, Development of a Self-Assembled Nanoparticle Formulation of Orlistat, Nano-ORL, with Increased Cytotoxicity against Human Tumor Cell Lines. Molecular pharmaceutics 2016, 13 (3), 720–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hill TK; Kelkar SS; Wojtynek NE; Souchek JJ; Payne WM; Stumpf K; Marini FC; Mohs AM, Near Infrared Fluorescent Nanoparticles Derived from Hyaluronic Acid Improve Tumor Contrast for Image-Guided Surgery. Theranostics 2016, 6 (13), 2314–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kelkar SS; Hill TK; Marini FC; Mohs AM, Near infrared fluorescent nanoparticles based on hyaluronic acid: Self-assembly, optical properties, and cell interaction. Acta biomaterialia 2016, 36, 112–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hatfield JM; Wierdl M; Wadkins RM; Potter PM, Modifications of human carboxylesterase for improved prodrug activation. Expert opinion on drug metabolism & toxicology 2008, 4 (9), 1153–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fukami T; Yokoi T, The emerging role of human esterases. Drug Metab Pharmacokinet 2012, 27 (5), 466–77. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.