

Abstract
Mitomycin C (MC), an anti-cancer drug, and its analog, decarbamoylmitomycin C (DMC), are DNA-alkylating agents. MC is currently used in the clinics and its cytotoxicity is mainly due to its ability to form Interstrand Crosslinks (ICLs) which impede DNA replication and, thereby, block cancer cells proliferation. However, both MC and DMC are also able to generate monoadducts with DNA. In particular, we recently discovered that DMC, like MC, can form deoxyadenosine (dA) monoadducts with DNA. The biological role played by these monoadducts is worthy of investigation. To probe the role of these adducts and to detect them in enzymatic digests of DNA extracted from culture cells treated by both drugs, we need access to reference compounds i.e. MC and DMC dA-mononucleoside adducts. Previous biomimetic methods used to generate MC and DMC mononucleoside adducts are cumbersome and very low yielding. Here, we describe the diastereospecific chemical synthesis of both C-1 epimers of MC and DMC deoxyadenosine adducts. The key step of the synthesis involves an aromatic substitution reaction between a 6-fluoropurine 2’-deoxyribonucleoside and appropriately protected stereoisomeric triaminomitosenes to form protected-MC-dA adducts with either an S or R stereochemical configuration at the adenine-mitosene linkage. Fluoride-based deprotection methods generated the final four reference compounds: the two stereoisomeric MC-dA adducts and the two stereoisomeric DMC-dA adducts. The MC and DMC-dA adducts synthesized here will serve as standards for the detection and identification of such adducts formed in the DNA of culture cells treated with both drugs.
Keywords: Mitomycin C, deoxyadenosine, nucleophilic aromatic substitution, stereoisomers, DNA-adducts
Graphical Abstract
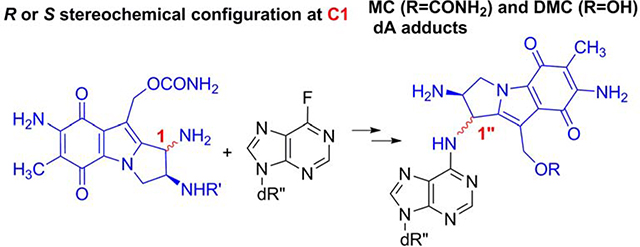
1. Introduction
Mitomycin C (MC, Fig. 1) [1–3] is a well known antineoplastic drug used to treat, among others, bladder and stomach cancers [4–7]. Its application in ophtalmology has also increased in recent years because of its modulatory effects on wound healing [8, 9]. Decarbamoylmitomycin C, (DMC, Fig. 1), is an analog of MC lacking the carbamoyl group on O10. Both MC and DMC are inert in their native state but are able to alkylate DNA upon reductive activation to generate monoadducts and Interstrand Crosslinks (ICLs), thereby inhibiting DNA synthesis [10].
Fig. 1.

(1-column fitting image, color should not be used): Mitomycin C (MC) and Decarbamoylmitomycin C (DMC).
The major monoadducts generated by MC and DMC result from covalent linkage between the exocyclic amino group of deoxyguanosine (dG) and the 1-position of the MC/DMC aziridine moiety (Fig. 1 and 2). These monoadducts can be converted to ICLs upon further activation of the 10-position (10-carbamate in the case of MC, and 10-hydroxyl in the case of DMC) and this results in the formation of ICLs between the exocyclic amino groups of deoxyguanosine residues located on opposing DNA strands. The major adducts identified from cancer cells treated with MC and DMC adopt an opposite stereochemical configuration at the dG-mitosene bond. For MC, the stereochemical configuration at C1” of the major adducts is always R (e.g. 1a, 2a and 3a, Fig. 2); in contrast, for DMC, it is always S (e.g. 2b and 3b, Fig. 2) [4]. In addition, it was recently discovered that ICL 3a (MC major ICL) is only formed at CpG sequences and ICL 3b (DMC major ICL) is only generated at GpC steps [11, 12]. This implies that, for mitomycins, ICLs’ formation is diastereospecific and diastereodivergent. During the course of this work, it was also established that DMC is able to form two stereoisomeric deoxyadenosine (dA) adducts with DNA under certain conditions (Fig. 2, compounds 5a and 5b) and that single stranded DNA seems more prone to dA alkylation than duplex DNA, in contrast to what happens with dG alkylation [13]. This result indicates that unfolded single-stranded structures of nucleic acids could be targets for dA alkylation by MC and DMC in cells [13]. Such substrates would then join the growing list of cellular components targeted by Mitomycins such as rRNA [14] and Thioredoxin Reductase [15].
Fig. 2.

(2-column fitting image, color should be used): Major DNA adducts generated by MC and DMC in cells or purified DNA.
The current consensus is that ICLs 3a and 3b (Fig. 2) are the major lesions responsible for the cytotoxicity of Mitomycins because ICLs impede DNA replication and, thus, are highly harmful to dividing cells. It has been estimated that the presence of approximately 40 unrepaired ICLs can kill mammalian cells [16]. However, the biological role played by other DNA adducts generated by both drugs is worthy of investigation. It is particularly important to assess whether or not MC or DMC monoadducts may generate secondary tumors (i.e. if they are mutagenic) and if they play a role in mitomycins’ cytotoxicity. Relevant to this pursuit, the bio-activity of monoadduct 1a (Fig. 2) has been investigated and 1a was found: 1) to be cytotoxic in Escherichia coli [17]; 2) to be mutagenic and to block replication in human embryonic kidney (HEK) 293T cells [18].
The identification and quantification of DNA adducts in culture cells treated by MC or DMC is performed at the nucleoside level following DNA digestion with phosphodiesterases and phosphatases, and it requires the availability of authentic standards of the adducts being identified [10–12, 19]. Therefore, in order to detect the formation of MC-dA and DMC-dA monoadducts in treated cells and investigate their role in the biological effects of mitomycins, we need to synthesize MC and DMC dA-mononucleosides so they can be used as reference compounds. One approach for the synthesis of such adducts involves the direct reaction of dA with reduced MC or DMC and we have successfully isolated adducts 5a and 5b using this method in the past [13]. This direct method provides the adducts in a single step, but their isolation from the reaction mixtures is cumbersome, expensive and very low yielding (0.8% yield). An alternative approach to obtain N-substituted-purine mononucleoside derivatives involves chemical synthesis, using a coupling reaction between an electrophilic nucleoside derivative and the corresponding amine. Our laboratory has successfully used this synthetic approach to obtain deoxyguanosine adducts of mitomycins such as N2-dG adduct 1a [20] (Fig. 2) as well as the N2-dG adduct of the major Mitomycin C metabolite, 2,7-diaminomitosene [21]. Here, we present a diastereospecific chemical synthesis of the four N6 deoxyadenosine-adducts formed by mitomycin C (4a and 4b) and decarbamoylmitomycin C (5a and 5b) (Fig. 2). The key step of the synthesis involves a nucleophilic aromatic substitution reaction between a ((2-trimethylsilyl)ethoxycarbonyl)-protected aminomitosene with either an R or S stereochemical configuration at C1 and a C-6 fluoropurine 2’-deoxyribonucleoside. The final deprotected MC and DMC dA adducts (4a, 4b, 5a and 5b, Fig. 2) were obtained after treatment with fluoride-containing reagents. These reference compounds will allow the detection and quantitation of such adducts in cellular environments using techniques similar to those used previously for the detection of quantitation of other MC/DMC adducts [10–12, 19]. Two features of the chemical synthesis of these adducts are discussed in detail: (a) the efficiencies of 2’-deoxyadenosine adduct syntheses by fluorine displacement using a C-6 fluoropurine 2’-deoxyribonucleoside and the influence of the stereochemical configurations of the mitosenes on the nucleophilic substitution reaction rate and (b) the influence of the stereochemical configurations of the dA adducts on the deprotection steps.
2. Materials and methods
2.1. General Information
1H NMR and 13C NMR spectra were recorded using a JEOL ECX 300 (300 MHz), a Varian Inova 500 (500 MHz) or a Bruker AVANCE 500 (500 MHz) spectrometer. Spectra were recorded at 298 K and the residual solvent peak was used as the internal reference. Chemical shifts are reported in parts per million and coupling constants are in hertz (Hz). The conventional numbering system is used for the mitosene moiety and the purine carbons are numbered 1–6 also as per convention. The sugar carbons are numbered 1’−5’ beginning at the anomeric carbon and proceeding via the carbon chain to the primary carbinol center. We will refer to mitosene derivatives with an R configuration at carbon 1 as “α” or trans, while those with an S configuration at carbon 1 are termed cis or “β”.
Reagents were obtained from commercial sources and were used without further purification. All reactions were carried out under an atmosphere of Argon unless otherwise stated. Thin layer chromatographic analyses were carried out on 250 mm silica gel plates containing a fluorescent indicator. Column chromatographic purifications were performed using 200–300 mesh silica gel. Triaminomitosene precursors 6a and 6b and 6-fluoropurine 2’-deoxyriboside were synthesized according to previously described procedures with slight modifications, as detailed in sections 2.2 to 2.4. HRMS spectra were recorded by direct infusion using Bruker’s micrOTOF-II ESI instrument at Notre Dame University Mass Spectrometry Facility.
Circular Dichroism (CD) and Ultraviolet and Visible (UV-Vis) spectra were recorded on a Jasco J-1500 (serial number A0045PM539) spectrophotometer. The experiment parameters are the following: band width, 1 nm; data pitch, 0.1 nm; scanning speed, 100 nm/min. CD Spectra were recorded in the interval 650 nm – 200 nm. The CD/UV-Vis spectrum of methanol (blank) was subtracted from each of the recorded spectra also in methanol. Kaleidagraph software (version 4.1.3) was used to generate the graphs presented in the manuscript.
2.2. Synthesis of 9-(2-Deoxy-β-D-erythro-pentofuranosyl)purin-6-yl-trimethylammonium chloride
This synthesis was adapted from previous work [22]. The following changes were made: The starting material, 6-chloro-9-(2-Deoxy-β-D-erythro-pentofuranosyl)purine, was dried under high vacuum while being slightly heated for 30 minutes to eliminate all moisture. The salt was stored under high vacuum (lyophilizer) until the next step. The reaction was performed under dry Argon.
2.3. Synthesis of 6-fluoro-9-(2-Deoxy-β-D-erythro-pentofuranosyl)purine
This synthesis was adapted from previous work [22]. The following changes were made: KF was dried under high vacuum while being slightly heated for 30 minutes to eliminate all moisture.
2.4. Synthesis of amine precursors 6a and 6b from mitomycin C (scheme 1)
Scheme 1.

(2-column fitting image, color should not be used): Synthesis of triaminomitosenes 6a and 6b. R=Ph for 6a R=Me for 6b; Teoc=2-(trimethylsilyl)ethoxycarbonyl; Teoc-ONp=2-(trimethylsilyl)-ethyl-4-nitrophenyl carbonate.
This synthesis was adapted from our previous work [19, 20]. The following changes were made: the liquid-liquid extraction of the azide intermediate was performed using dichloromethane rather than ethyl acetate to improve recovery.
2.5. Synthesis of 7a
6-fluoro-9-(2-Deoxy-β-D-erythro-pentofuranosyl)purine (22 mg, 0.086 mmol) was dissolved in dry DMSO (95 μL). Compound 6a (20 mg, 0.043 mmol) and diisopropylethylamine (10 μL, 7.42 mg, 0.96 mmol) were added to the reaction mixture which was incubated at 45 °C for 2 days. The resulting mixture was diluted with water (1 mL) and lyophilized. The desired product was isolated by preparative thin layer chromatography (SiO2: NH4OH, 1.5%; hexane, 5%; CH3OH, 15%; CH2Cl2, 78.5% v/v) to give 24 mg (81 % yield) of 7a (Rf = 0.39). 1H NMR (CD3OD, 300 MHz): δ 8.24 (s, Har, 1H), 6.41 (s, dd, J = 8.4, 6.4 Hz, H1’, 1H), 5.93 (s, br, H1”, 1H), 4.99 (s, H10”, 2H), 4.76 (m, overlapping signals for H2” and H3’, 2H), 4.56 (dd, J = 5.5, 2.5 Hz, H3”a, 1H), 4.05 (m, overlapping signals for CH2-O, H4’ and H3”b, 4H), 3.83 and 3.72 (doublets of ABq, J = 3.3 and 12.3 Hz, H5’a and H5’b, 2H), 2.80 (app quint, J = 7.9 Hz, H2’a, 1H), 2.38 (ddd, J = 13.4, 6.0, 2.6 Hz, H2’b, 1H), 1.78 (s, CH3, 3H), 0.91 (t, J = 8.4 Hz, CH2-Si, 2H), −0.01 (s, Si(CH3)3, 9H). 13C NMR (CD3OD, 300 MHz): δ 178.4, 177.6, 158.0, 157.2, 154.4, 151.2, 148.4, 147.8, 140.1, 138.3, 128.9, 122.0, 120.1, 112.8, 105.1, 88.6, 85.8, 71.8, 62.8, 62.3, 60.8, 56.9, 53.3, 50.6, 40.2, 17.1, 6.8, −2.9. HRMS m/z calcd for C30H40N9O9Si [M + H]+: 698.2718, found: 698.2681.
2.6. Synthesis of 7 b
Compound 6 b (15 mg, 0.032 mmol); 6-fluoro-9-(2-Deoxy-β-D-erythro-pentofuranosyl)purine (16 mg, 0.064 mmol) and diisopropylamine (10 μL, 7.42 mg, 0.96 mmol) were stirred in 95 μL of DMSO. The reaction mixture was incubated at 45 °C for 4 days. The resulting mixture was diluted with water (1 mL) and lyophilized. The final compound was purified by preparative thin layer chromatography (SiO2: NH4OH, 1.5%; hexane, 5%; CH3OH, 15%; CH2Cl2, 78.5% v/v) yielding 17 mg (75 % yield) of 7b (Rf = 0.42). 1H NMR (CD3OD, 300 MHz): δ 8.26 (s, Har, 1H), 8.22 (s, Har, 1H), 6.40 (t, J = 6.4 Hz, H1’, 1H), 6.02 (s, br, H1”, 1H), 5.09 (s, H10”, 2H), 5.07 (app q, J = 7.2 Hz, H2”, 1H), 4.56 (m, overlapping signals for H3’ and H3”a, 2H), 4.14 (dd, J = 12.7, 7.2 Hz, H3”b, 1H), 4.05 (app q, J = 2.4 Hz, H4’, 1H), 3.96 (app q, J = 8.1 Hz, CH2-O, 2H), 3.82 and 3.73 (doublets of ABq, J = 3.1 and 12.4 Hz, H5’a and H5’b, 2H), 2.79 (app quint, J = 8.4 Hz, H2’a, 1H), 2.37 (ddd, J = 13.4, 5.8, 2.4 Hz, H2’b, 1H), 1.76 (s, CH3, 3H), 0.74 (t, J=8.0 Hz, CH2-Si, 2H), −0.1 (s, Si(CH3)3, 9H). 13C NMR (CD3OD, 300 MHz): δ 178.9, 177.0, 157.0, 156.2, 154.4, 152.5, 149.2, 147.6, 140.3, 139.8, 128.9, 121.9, 120.3, 112.8, 105.2, 88.6, 84.6, 71.5, 62.5, 62.4, 56.9, 56.6, 55.6, 50.8, 19.0, 17.6, 8.9, −1.1. HRMS m/z calcd for C30H40N9O9Si [M + H]+: 698.2718, found: 698.2688.
2.7. Synthesis of 4a
TBAF (tetra-n-butylammonium fluoride, 1 M in THF, 36 μL, 0.036 mmol) was added to compound 7a (5 mg, 0.0072 mmol). An additional 40 μL of THF was added. The reaction mixture was incubated at 45 °C for 15 h. and the solvent was evaporated by Argon flow. Compound 4a was isolated by preparative thin layer chromatography (SiO2: NH4OH, 3%; CH3OH, 15%; CH3Cl, 82% v/v; Rf =0.10). The residue was triturated with hexane and filtered to remove TBAF, giving 4a (2.71 mg, 68%) as a pale pink solid. 1H NMR (C5D5N with 20% v/v D2O, 300 MHz) δ 8.62 (s, Har, 1H), 6.91 (t, J = 6.6 Hz, H1’, 1H), 5.97 (s, br, H1”, 1H), 5.67 and 5.52 (ABq, J = 12.4 Hz, H10”a and H10”b, 2H), 5.17 (m, br, H3’, 1H), 5.00–4.94 (m, br, H3”a, 1H), 4.66 (app q, J = 3 Hz, H4’, 1H), 4.47–4.40 (m, br, H2”, 1H), 4.42 (dd, J = 16, 4.9 Hz, H3”b, 1H), 4.22 and 4.15 (doublets of ABq, J = 3.3 and 12.3 Hz, H5’a and H5’b, 2H), 3.11 (app quin, J = 6.2 Hz, H2’a, 1H), 2.88 (ddd, J = 12.1, 4.5, 2.3 Hz, H2’b, 1H), 2.17 (s, CH3, 3H). 13C NMR (C5D5N with 20% v/v D2O, 300 MHz): δ 180.6, 179.1, 159.7, 153.9, 141.5, 140.7, 130.4, 123.6, 122.2, 115.1, 107.2, 90.3, 87.3, 77.3, 73.2, 64.5, 64.2, 59.1, 55.3, 42.1, 9.8. Note: There are three unobserved resonances hidden under the pyridine signals. HRMS m/z calcd for C24H28N9O7 [M + H]+: 554.2112, found: 554.2114.
2.8. Synthesis of 4b
TAS-F (trisulfonium difluorotrimethylsilicate, 0.15 mmol, 42.5 mg) was added to 8 mg of 7b (0.011 mmol) in 400 μL of DMF. The reaction mixture was incubated at 45 °C for 5 h. followed by dilution with 1 mL of water and lyophilization. 4b was isolated by preparative thin layer chromatography (SiO2: NH4OH, 3%; CH3OH, 15%; CH3Cl, 82% v/v; Rf =0.12) to give 5 mg (82 % yield) of 4b. 1H NMR (C5D5N with 20% v/v D2O, 300 MHz) δ 8.71 (s, Har, 1H), 6.84 (t, J = 6.8 Hz, H1’, 1H), 6.20 (s, br, H1”, 1H), 5.47 (s, H10”, 2H), 5.12 (app t, J = 2.6 Hz, H3’,1H), 4.90 (dd, J = 12.6, 7.2 Hz, H3”a, 1H), 4.75 (app q, J = 6.9 Hz, H4’, 1H), 4.65 (app d, J = 2.6 Hz, H2”, 1H), 4.49 (dd, J = 12.6, 6.9 Hz, H3”b, 1H), 4.26 and 4.20 (doublets of ABq, J = 4.4 and 12.4 Hz, H5’a and H5’b, 2H), 3.08 (m, H2’a, 1H), 2.93 (m, H2’b, 1H), 2.14 (s, CH3, 3H). 13C NMR (C5D5N with 20% v/v D2O, 300 MHz): δ 180.1, 179.7, 160.3, 154.3, 141.0, 140.8, 130.8, 123.3, 121.8, 115.3, 107.6, 89.9, 87.2, 77.2, 73.4, 63.9, 59.7, 58.5, 53.9, 41.9, 9.8. Note: There are three unobserved resonances hidden under the pyridine signals. HRMS m/z calcd for C24H28N9O7 [M + H]+: 554.2112, found: 554.2095.
2.9. Synthesis of 5a
TAS-F (trisulfonium difluorotrimethylsilicate, 0.12 mmol, 33 mg) in 40 μL of DMF was added to compound 7a (17 mg, 0.024 mmol). The reaction mixture was incubated at 40 °C for 5 h, diluted in 1 mL of water and lyophilized. Adduct 5a was isolated by preparative thin layer chromatography (SiO2: NH4OH, 3%; CH3OH, 15%; CH3Cl, 82% v/v) to give 7.84 mg (63% yield) of 5a (Rf =0.12). 1H NMR (C5D5N with 20% v/v D2O, 500 MHz): δ 8.64 (s, Ar-H2, 1H), 8.58 (s, Ar-H8, 1H), 6.89 (t, J = 6.8 Hz, H1’, 1H), 5.91 (s, br, H1”, 1H), 5.18 (m, br, H3’, 1H), 5.17 and 5.14 (ABq, J = 14.5 Hz, H10”a and H10”b, 2H), 4.99–4.92 (m, br, H3”a, 1H), 4.67–4.64 (m, br, H4’, 1H), 4.54–4.45 (m, br, H2”, 1H), 4.46 (dd, J = 12.8, 3.6 Hz, H3”b, 1H), 4.23 and 4.17 (doublets of ABq, J = 3.2 and 12.4 Hz, H5’a and H5’b, 2H), 3.05 (app quint, J = 6.2 Hz, H2’a, 1H), 2.91 (ddd, J = 13.5, 6.1, 2.9 Hz, H2’b, 1H), 2.15 (s, CH3, 3H). 13C NMR (C5D5N with 20% v/v D2O, 500 MHz): δ 179.5, 177.9, 154.7, 152.6, 146.8, 140.2, 137.9, 129.3, 122.1, 120.5, 106.0, 89.0, 85.8, 72.0, 63.1, 62.7, 55.7, 52.3, 53.7, 40.8, 8.5. Note: There are two unobserved resonances hidden under the pyridine signals. HRMS m/z calcd for C23H27N8O6 [M + H]+: 511.2048, found: 511.2046.
2.10. Synthesis of 5b
TBAF (tetra-n-butylammonium fluoride, 1 M in THF, 57 μL, 0.057 mmol) was added to compound 7b (8 mg, 0.011 mmol). An additional 64 μL of THF was added. The reaction mixture was incubated at 40 °C for 4 h and the solvent was evaporated by argon bubbling. The products were first isolated by preparative thin layer chromatography (SiO2: NH4OH, 3%; CH3OH, 15%; CH3Cl, 82% v/v). The residue was then triturated with hexane and filtered to remove TBAF, giving 5b (6.5 mg, 63%) as a pale pink solid (Rf =0.27). 1H NMR (C5D5N with 20% v/v D2O and 0.05% v/v TMS, 500 MHz): δ 8.66 (s, Ar-H2, 1H), 8.60 (s, Ar-H8, 1H), 6.88 (t, J = 6.8 Hz, H1’, 1H), 6.07 (s, br, H1”, 1H), 5.21 and 5.15 (ABq, J = 13.9 Hz, H10”a and H10”b, 2H), 5.16 (s, br, H3’, 1H), 4.88 (dd, J = 12.6, 7.2 Hz, H3”a, 1H), 4.70 (app q, J = 7.2 Hz, H2”, 1H), 4.64 (app q, J = 3.2 Hz, H4’, 1H), 4.47 (dd, J = 12.6, 7.4 Hz, H3”b, 1H), 4.21 and 4.15 (doublets of ABq, J = 3.2 and 12.2 Hz, H5’a and H5’b, 2H), 3.08 (app quint, J = 6.2 Hz, H2’a, 1H), 2.91 (ddd, J = 13.3, 5.8, 2.9 Hz, H2’b, 1H), 2.16 (s, CH3, 3H). 13C NMR (C5D5N with 20% v/v D2O, 300 MHz): δ 179.6, 178.0, 154.7, 152.6, 148.1, 140.3, 138.3, 129.4, 121.6, 120.5, 106.1, 89.1, 85.7, 72.0, 62.7, 57.8, 56.3, 52.3, 50.6, 40.9, 8.6. Note: There are two unobserved resonances hidden under the pyridine signals. HRMS m/z calcd for C23H27N8O6 [M + H]+: 511.2048, found: 511.2039.
2.11. Oligonucleotide Alkylation by DMC
Self-complimentary oligonucleotide 5’-TATATATATATA (10 A260 unit scale; Tm: 57°C) was dissolved in 10 mM potassium phosphate buffer (pH 5.8, 355 μL) and annealed by heating (90°C, 10 min) followed by slow cooling to 0°C. The reaction mixture was put under ice and deaerated via argon bubbling (30 min) while kept at 0°C. Excess Na2S2O4 (3.24 μmol in 20 μL of potassium phosphate buffer, 10 mM, pH 5.8) from a freshly prepared anaerobic solution was then added to the mixture quickly and immediately followed by addition of DMC (1.30 μmol). The reaction mixture was allowed to stir with argon bubbling for 1 hr and then opened to air, followed by gentle mixing until a consistent purple color was obtained. The mixture was then allowed to stir for 20 min under air and chromatographed on a 2.5*56 cm Sephadex G-25 column using 20 mM NH4HCO3 as eluent. Oligonucleotide containing fractions were lyophilized.
2.12. Enzymatic Digestion of Alkylated Oligonucleotides
1 A260 unit of oligonucleotide and 2 units of nuclease P1 were incubated at 37°C for 2 hr in 0.8 mL of 20 mM ammonium acetate (pH 5.5); 100 mM MgCl2 (20μL) was added, and the pH was adjusted to 8.2 by addition of 20 μL of 200 mM NaOH. SVD (2 units) and AP (2 units) were added and incubation was continued at 37°C for 2.5 h.
2.13. Analysis of DNA Adducts after Enzymatic Digestion
The digestion mixture was directly analyzed by HPLC using an Agilent 1200 HPLC system and a XBridge C-18 reverse phase column (5 μm, 0.46*25 cm). The elution system was 6–18% acetonitrile in 30 mM potassium phosphate (pH 5.4), in 60 min, 1 mL/min flow rate.
3. Results and Discussion
3.1. Synthesis of protected 2’-deoxyadenosine adducts 7a and 7b. Efficiency of nucleophilic aromatic substitutions using 6-fluoro-9-(2-Deoxy-β-D-erythro-pentofuranosyl)purine and aminomitosenes
The reaction of 2-fluoropurine or 6-fluoropurine nucleosides with amines has been used to synthesize N-substituted dG or dA derivatives [19–21, 23, 24]. We have previously used the coupling reaction between a 2-fluoropurine derivative and aminomitosenes to synthesize mitosene-deoxyguanosine adducts (MC-dG and DMC-dG adducts 1a and 2b, Fig. 2) [19, 20]. Our approach is analogous to the method developed by Harris and Rizzo to generate adducts of exocyclic amines of nucleosides [25]. In this work, a similar strategy was employed for the synthesis of MC and DMC deoxyadenosine (dA) adducts 4a, 4b, 5a and 5b. The synthesis commenced with the preparation of the isomeric protected triamino mitosenes 6a (1,2 trans) and 6b (1,2 cis) (Scheme 1) which can be obtained from mitomycin C according to a route that has been developed in our laboratory [19, 20].
These precursors 6a and 6b (Scheme 1) were then coupled to a 6-fluoropurine deoxyribonucleoside (Scheme 2, left column) to yield 7a and 7b. The coupling reaction showed diastereomer-dependent reactivity. The fluoride displacement reaction by the 1-R triaminomitosene 6a (trans isomer) to produce 7a was faster and higher yielding than in the case of the 1-S triaminomitosene 6b (cis isomer) to give 7b (Table 1). The same diastereomer-dependent reactivity was also observed in our previous work leading to dG adducts 9a and 9b (Scheme 2, right column, and Table 1) [19, 20]. This effect is probably the result of the differences in steric hindrance caused by the protected 1-amino group of 6a and 6b in the coupling reaction: For the 1-S triaminomitosene (cis isomer) the electrophile must approach from the same side of the bulky substituent at C2, making the reaction slower than in the case of the 1-R triaminomitosene (trans isomer), where the electrophile reacts from the less hindered opposite side.
Scheme 2.

(2-column fitting image, color should be used): Synthesis of 7a, 7b, 9a, 9b via nucleophilic aromatic substitution. R = deoxyribose, R’ = 2-(trimethylsilyl)ethoxycarbonyl (teoc), R” = 2-p-nitrophenylethyl.
Table 1:
Comparison between nucleophilic aromatic substitutions using 6-fluoro-9-(2-Deoxy-β-D-erythro-pentofuranosyl)purine (for the synthesis of dA adducts 7a and 7b) or 2-fluoro-O6-(2-p-nitrophenylethyl)-2’-deoxyinosine (for the synthesis of dG adducts 9a [20] and 9b [19])
Nucleophilic aromatic substitutions between 6a/6b and 6-fluoropurines were faster and proceeded in better yields than similar reactions with 2-fluopurines (Table 1). This effect can be attributed to functional group dependence. The 2-fluoropurine derivatives contain an electron-donating substituent at C6, while the 6-fluoropurine ring has no substituents other than deoxyribose and fluoride. Differences in reactivity between the two fluoropurines have been well known in the chemical carcinogenesis field. 6-fluoropurine reacts via a classic SNAr whereas 2-fluoropurine likely reacts by an extended conjugate addition followed by elimination. This could be the underlying cause for the observed differences [26].
3.2. Efficiency of deprotection methods. Influence of the stereochemical configuration of the adenine-mitosene bond
We previously found that the 2-(trimethylsilyl)ethoxycarbonyl (teoc) group was a convenient protecting group for the 2-amino group on mitosene derivatives, since its fluoride-mediated removal is compatible with the labile nature of mitosene-nucleoside and mitosene-oligonucletide adducts [19–21]. For the deprotection of the teoc group in 7a and 7b, we initially used tetrabutylammoniumfluoride (Scheme 3) as the source of fluoride anions. Treatment of 7a with excess of TBAF led to efficient teoc deprotection to give the α adduct 4a. However, the reaction of the β isomer 7b under the same conditions, resulted in the removal of both the teoc protecting group and the carbamoyl group at C10 to yield the β adduct 5b (Scheme 3 and Table 2). Additionally, the rate of removal of both groups (teoc and carbamoyl) in 7b was faster (4h) than the removal of the teoc group in 7a (15 h) (Table 2). This difference in reactivity toward TBAF between the 2 isomers is not totally unexpected since side reactions have been commonly observed during deprotection with TBAF [27].
Scheme 3.

(2-column fitting image, color should be used): Synthesis of 4a, 4b, 5a, 5b. Fluoride mediated deprotection of the 2-(trimethylsilyl)ethoxycarbonyl (teoc) group with or without concomitant decarbamoylation.
Table 2:
Selective conditions for the deprotection of the 2-(trimethylsilyl)ethoxycarbonyl (teoc) group with or without concomitant decarbamoylation.
| Starting Material | Reagent | Equivalent | Time | Products (yield) |
|---|---|---|---|---|
| 7a 0.6M | TAS-F or TBAF, 3 M | 5 | 5 h | 5a (63%) |
| 7a 0.095M | TBAF, 0.47 M | 5 | 15 h | 4a (68%) |
| 7b 0.091 M | TBAF, 0.47 M | 5.2 | 4 h | 5b (63%) |
| 7b 0.028 M | TAS-F, 0.37 M | 14 | 24 h | 4b (82%) |
Therefore, in order to obtain the β (R) MC-dA adduct 4b, we decided to use a milder reagent for fluoride mediated teoc deprotection. TAS-F (tris(dimethylamino)sulfonium difluorotrimethylsilicate) is a good alternative to TBAF and has the added advantage to prevent undesirable side reactions which may occur when TBAF is used [27]. Indeed, in the case of 7b, treatment with TAS-F afforded the desired β-MC-dA adduct 4b without decarbamoylation. The last adduct of the series, the α-DMC-dA adduct 5a, was obtained using either TAS-F or TBAF as the source of fluoride ion, but the reaction required a much higher concentration of reactants. Under these conditions both the carbamoyl and the teoc groups were removed from 7a in one step to afford 5a.
3.3. Compound Characterization
All compounds synthesized were characterized by UV, CD and NMR spectroscopy. HRMS spectra confirmed the structures of diastereoisomeric pairs: 7a/b; 5a/b and 4a/b. The UV spectra of all compounds show the presence of a deoxyadenosine chromophore (λmax 267 nm) and a 7-aminomitosene chromophore (λmax 313 nm). All spectra are identical to the spectra of previously isolated and characterized MC and DMC-N6-deoxyadenosine adducts [13, 28].
Adducts 7a, 7b, 4a, 4b, 5a and 5b were characterized via NMR spectroscopy. The protected adducts 7a and 7b are soluble in common organic solvents such as DMSO-d6 and Methanol-d4 with good line resolution. However, spectra for adducts 5a, 5b, 4a and 4b could only be obtained using a mixture of D2O and pyridine-d6. Differences in chemical shifts of protons from the mitosene moiety between the 6 adducts are relatively small. In terms of the magnitude of the difference, the protons most affected are H1” and H2” on the tetrahydropyrole ring. The H1” and H2” protons on the R (α, trans) dA adducts 7a, 4a and 5a are more shielded compared to protons on the S (β, cis) isomers 7b, 4b and 5b with |δΔ|=0.09–0.27 ppm for H1” and |δΔ|=0.22–0.31 ppm for H2”. Regarding protons from the glycosidic moiety, the R (α, trans) and S (β, cis) isomers from each pair (7a/7b; 4a/4b; 5a/5b) exhibit signals at similar frequencies. In contrast, there is a marked difference in chemical shifts between the glycosidic protons from the teoc protected pair (7a/7b) and those from the deprotected pairs (5a/5b; 4a/4b). Protons from the protected dA adduct pair, 7a and 7b, are more shielded than their analogues from both the MC-dA adducts 4a/4b and DMC-dA adducts 5a/5b by: |δΔ|=0.59–0.70 ppm for H4’; |δΔ|=0.38–0.44 ppm for H5’a; |δΔ|=0.42–0.48 ppm for H5’b; |δΔ|=0.40–0.51 ppm for H1’; |δΔ|=0.36–0.62 ppm for H3’; |δΔ|=0.25–0.32 ppm for H2’a; |δΔ|=0.50–0.57 ppm for H2’b. The chemical shifts of the deoxyribose ring protons, especially those at C2’ are predicted to be affected by the magnitude of the glycosidic torsion angle that results from ring current and the local magnetic anisotropy of their bases [29, 30]. Therefore, one possible explanation for the difference in chemical shifts between the glycosidic protons on teoc protected 7a/7b and those on teoc deprotected 5a/5b; 4a/4b is that the teoc group has a direct effect on the glycosidic torsion angle.
Circular dichroism is a reliable method to assign the absolute configuration of mitosene adducts at C1” [31, 32]. The band at around 530 nm, generated by the weak and broad absorption of the mitosene chromophore (which has an extinction coefficient value of 800 at 225 nm) is diagnostic of the β or α configurations of C1” and is independent of the chemical nature of substituents at C1” and of the chemical nature of substituents on other stereogenic centers nearby (such as C2”) [31, 32]. Mitosene derivatives with an α configuration at C1” display a negative cotton effect (CE) centered around 530 nm whereas those with a β configuration display a positive CE. Both the mitosene and the adenosine chromophores present absorptions in the 200–400 nm region and it has been previously hypothesized that coupled CD signals are present in this region [31]. Fig. 3 shows the CD and UV spectra of MC-protected-dA adducts 7a/7b and Fig. 4 of MC-dA adducts 4a/4b and DMC-dA adducts 5a/5b. Cis and trans dA-adduct pairs bear a nearly mirror image relationship and all CD spectra are consistent with previously published data of mitosenes and mitosene-nucleoside adducts [13, 28, 31]. These spectra confirm the stereochemical configuration at C1” of all adducts synthesized in this work.
Fig. 3:

(2-column fitting image, no color should be used) (a) CD spectra of MC-protected-dA adducts 7b (dotted line) and 7a (full line); (b) enlargement of the area showing the cotton effect around 530 nm; (c) UV spectrum of 7b; (d) UV spectrum of 7a.
Fig. 4:

(2-column fitting image, no color should be used) (a) CD spectra of MC-dA adducts 4b (dotted line) and 4a (full line); (b) enlargement of the area showing the cotton effect around 530 nm; (c) UV spectrum of 4b; (d) UV spectrum of 4a; (e) CD spectra of DMC-dA adducts 5b (dotted line) and 5a (full line); (f) enlargement of the area showing the cotton effect around 530 nm; (g) UV spectrum of 5b; (h) UV spectrum of 5a.
3.4. Use of adducts 5a and 5b as reference compounds for the detection of DMC-dA adducts formed in DNA
DMC-dA adducts 5a and 5b, synthesized according to the new route described above, were used as reference compounds to determine the presence of dA adducts formed during the alkylation of duplex oligonucleotide (TATATATATATA)2 with DMC under bifunctional reductive acitvation conditions. The reaction is described in the experimental section and in previous work [13]. The alkylated oligonucleotide was digested to the nucleoside level using AP (alkaline phosphatase), SVD (snake venom diesterase) and nuclease P1. HPLC analysis of the enzymatic digest showed the presence of two DMC-nucleoside adducts that eluted with retention times of 23 and 36 minutes (Fig. 5c). In order to demonstrate that the adducts detected in the digest are the same compounds as the synthesized reference compounds 5a and 5b, we performed 2 co-injections with the digest of the alkylated oligonucleotide: one sample spiked with pure 5a (Fig 5d ) and another one spiked with pure 5b (Fig 5e). HPLC chromatograms of the spiked samples clearly show that the two adducts formed between duplex (TATATATATATA)2 and DMC are identical to compounds 5a and 5b.
Fig. 5:

(1-column fitting image, color should be used) HPLC chromatograms: (a) synthesized adduct 5a; (b) synthesized adduct 5b; (c) enzymatic digest of DMC-alkylated (TATATATATATA)2; (d): co-injection between (a) and (c); (e): co-injection between (a) and (c)
4. Conclusion
In conclusion, we have successfully used organic synthesis to obtain and characterize the four deoxyadenosine adducts that MC and DMC generate in their reactions with DNA. The synthesis is diastereoselective and the alkylated mononucleosides were obtained in good yields. Nucleophilic aromatic substitutions between the 6-fluoropurine derivative and triaminomitosenes to yield the protected dA adducts were faster and proceeded in better yields than similar reactions with 2-fluoropurine derivatives leading to protected dG adducts. In the course of this synthesis, we observed differences in reactivity from the diastereomeric dA-adducts in the final deprotection reaction, with TBAF (at low concentration) removing the carbamoyl at C10 only in the case of the cis diastereomer.
Direct alkylation reactions between dA and MC/DMC to generate MC/DMC dA adducts are very low yielding and they did not provide sufficient quantities of the adducts for a full and rigorous characterization. The synthesis presented here allows for an easy access to such conjugates, which is crucial to investigate their role in cellular mechanisms. The newly synthesized reference compounds allowed us to determine the presence of DMC adducts when a T-A rich duplex oligonucleotide was treated by DMC. In the future, the reference compounds synthesized in this work will allow us to detect the presence of such adducts in the enzymatic digest of cellular DNA extracted from cells treated with either MC or DMC.
Highlights.
We synthesized dA adducts generated by MC and DMC in their reactions with DNA.
We used 6-fluoropurine 2’-deoxyribonucleosides in aromatic substitution reactions.
We examined the influence of the stereochemical configuration at C1 on the reaction rate.
We confirmed the stereochemical configuration at C1 by CD spectroscopy.
We compared the efficiencies of dA adducts versus dG adducts formation.
We used the dA adducts synthesized to detect them in DNA.
Acknowledgements
This work was supported by NIH grant 2SC3GM105460-05 to Elise Champeil as well as by the Program for Research Initiatives for Science Majors (PRISM) at John Jay College.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Hata T, Sano Y, Sugawara R, Matsumae A, Kanamorei K, Shima T, Hoshi T, Mitomycin, a New Antibiotic from Streptomyces, J. Antibiotics, Ser. A 9 (1956) 141–146. [PubMed] [Google Scholar]
- [2].Tomasz M, Mitomycin C: small, fast and deadly (but very selective), Chem. Biol 2 (1995) 575–579. [DOI] [PubMed] [Google Scholar]
- [3].Paz MM, Pritsos CA, The Molecular Toxicology of Mitomycin C, in: Fishbein JC (Eds.), Advances in Molecular Toxicology Vol. 6, Elsevier Science Publishers B. V., Amsterdam, 2012, pp. 244–286. [Google Scholar]
- [4].Verweij J, Pinedo H, Cancer Chemotherapy and Biological Modifiers, Annual 11, Pinedo HM, Chabner BA, Longo DL (Eds.), Elsevier Science Publishers B. V., Amsterdam, 1990, p. 67. [Google Scholar]
- [5].Chabner BA, Amrein PC, Druker BJ, Michaelson MD, Mitsiades CS, Goss PE, Ryan DP, Ramachandra S, Richardson PJ, Supko JG, Wilson WH, Antineoplastic Agents, in: Brunton LL, Lazo JS, Parker KL (Eds.), Goodman & Gilman’s The Pharmacological Basis of Therapeutics, McGraw-Hill Publishers, New York, 2005, pp. 1315–1403. [Google Scholar]
- [6].Bradner WT, Mitomycin C: a Clinical Update, Cancer Treat. Rev 27 (2001) 35–50. [DOI] [PubMed] [Google Scholar]
- [7].Bass PD, Gubler DA, Judd TC, Williams RM, Mitomycinoid Alkaloids: Mechanism of Action, Biosynthesis, Total Syntheses, and Synthetic Approaches, Chem. Rev 113 (2013) 6816–6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Cincik H, Güngör A, Cekin E, Saglam O, Yildirim S, Poyrazoglu E, Candan H, Effects of Topical Application of mitomycin-C and 5-fluorouracil on Myringotomy in Rats, Otol. Neurotol 26 (2005) 351–354. [DOI] [PubMed] [Google Scholar]
- [9].Kim TI, Choi SI, Lee HK, Cho YJ, Kim EK, Mitomycin C Induces Apoptosis in Cultured Corneal Fibroblasts Derived from Type II Granular Corneal Dystrophy Corneas. Mol. Vis 14 (2008) 1222–1228. [PMC free article] [PubMed] [Google Scholar]
- [10].Palom Y, Suresh Kumar G, Tang LQ, Paz MM, Musser SM, S Rockwell M Tomasz, Relative Toxicities of DNA Cross-links and Monoadducts: New Insights from Studies of Decarbamoyl mitomycin C and Mitomycin C, Chem. Res. Toxicol 15 (2002) 1398–1406. [DOI] [PubMed] [Google Scholar]
- [11].Aguilar W, Paz MM, Vargas A, Cheng S-Y, Clement C, Champeil E, Sequence-Dependent Diastereospecific and Diastereodivergent Crosslinking of DNA by Decarbamoylmitomycin C, Chem. Eur. J 24 (2018) 6030–6035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Aguilar W, Paz MM, Vargas A, Zheng M, Cheng S-Y, Champeil E, Interdependent Sequence-Selectivity and Diastereoselectivity in the Alkylation of DNA by Decarbamoylmitomycin C, Chem. Eur. J 24 (2018) 13278–13289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zacarias O, Aguilar W, Paz MM, Tsukanov S, Zheng M, Cheng S-Y, Pradhan P, Champeil E, Isolation and Rationale for the Formation of Isomeric Decarbamoylmitomycin C-N6-deoxyadenosine Adducts in DNA, Chem. Res. Toxicol 31 (2018) 762–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Snodgrass RG, Collier AC, Coon AE, Pritsos CA, Mitomycin C Inhibits Ribosomal RNA: A Novel Cytotoxic Mechanism for Bioreductive Drugs, J. Biol. Chem 285 (2010) 19068–19075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Paz MM, Zhang X, Lu J, Holmgren A, A New Mechanism of Action for the Anticancer Drug Mitomycin C: Mechanism-Based Inhibition of Thioredoxin Reductase, Chem. Res. Toxicol 25 (2012) 1502–1511. [DOI] [PubMed] [Google Scholar]
- [16].Lawley PD, Phillips DH, DNA Adducts from Chemotherapeutic Agents, Mutat. Res 355 (1996) 13–40. [DOI] [PubMed] [Google Scholar]
- [17].Ramos LA, Lipman R, Tomasz M, Basu AK, The Major Mitomycin C-DNA Monoadduct is Cytotoxic but not Mutagenic in Escherichia coli, Chem. Res. Toxicol. 11 (1998) 64–69. [DOI] [PubMed] [Google Scholar]
- [18].Bose A, Surugihalli C, Pande P, Champeil E, Basu AK, Comparative Error-free and Error-prone Translesion Synthesis of the N2-2’-deoxyguanosine Adducts Formed by Mitomycin C and its Metabolite, 2,7-diaminomitosene, in Human Cells, Chem. Res. Toxicol 29 (2016), 933–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Champeil E, Cheng S-Y, Huang BT, Conchero-Guisan M, Martinez T, Paz MM, Sapse AM, Synthesis of Mitomycin C and Decarbamoylmitomycin C N2 Deoxyguanosine-adducts, Bioorg.Chem 65 (2016) 90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Champeil E, Paz M, Lukasiewicz E, Kong W, Watson S, Sapse AM, Synthesis of a Major Mitomycin C DNA Adduct via a Triaminomitosene, Bioorg. Med. Chem. Lett 22 (2012) 7198–7200. [DOI] [PubMed] [Google Scholar]
- [21].Champeil E, Paz MM, Ladwa S, Clement C, Zatorski A, Tomasz M, Synthesis of an Oligodeoxyribonucleotide Adduct of Mitomycin C by the Postoligomerization Method via a Triamino mitosene, J. Am. Chem. Soc 130 (2008) 9556–9565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Robbins MJ, Bason GL, Nucleic Acid Related Compounds. 8. Direct Conversion of 2′-Deoxyinosine to 6-Chloropurine 2′-Deoxyriboside and Selected 6-Substituted Deoxynucleosides and Their Evaluation As Substrates of Adenosine Deaminase, Can. J. Chem 51 (1973) 3161–3169. [Google Scholar]
- [23].Lee H, Hinz M, Stezowski JJ, Harvey RG, Syntheses of Polycyclic Aromatic Hydrocarbon-Nucleoside and Oligonucleotide Adducts Specifically Alkylated on the Amino Functions of Deoxyguanosine and Deoxyadenosine, Tetrahedron Lett. 31 (1990) 6773–6776. [Google Scholar]
- [24].Lakshman MK, Keeler JC, Ngassa FN, Hilmer JH, Pradhan P, Zajc B, Thomasson KA, Highly Diastereoselective Synthesis of Nucleoside Adducts from the Carcinogenic Benzo[a]pyrene Diol Epoxide and a Computational Analysis, J. Am. Chem. Soc 129 (2007), 68–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].(a) Harris CM, Zhou L, Strand EA, Harris TM, New strategy for the synthesis of oligodeoxynucleotides bearing adducts at exocyclic amino sites of purine nucleosides, J. Am. Chem. Soc 113 (1991) 4328–4329 [Google Scholar]; (b) DeCorte BL, Tsarouhtsis D, Kuchimanchi S, Cooper MD, Horton P, Harris CM, Harris TM, Chem. Res. Toxicol 9 (1996) 630–637 [DOI] [PubMed] [Google Scholar]; (c) Christov PP, Brown KL, Kozekov ID, Stone MP, Harris TM, Rizzo CJ, Site-specific synthesis and characterization of oligonucleotides containing an N6-(2-Deoxy-D-erythro-pentofuranosyl)-2,6-diamino-3,4-dihydro-4-oxo-5-N-methylformamidopyrimidine lesion, the ring-opened product from N7-methylation of deoxyguanosine, Chem. Res. Toxicol 21 (2008) 630–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Jones AM, Winship PCM, Caldwell JJ, Collins I, Chemistry of fluorinated purines, in: Nenajdenko V (Eds.), Fluorine in Heterocyclic Chemistry, Vol. 2: 6-Membered Heterocycles, Springer International Publishing, Switzerland, 2014, pp. 717–752. [Google Scholar]
- [27].Roy M, Talyor RE, Tris(dimethlyamino)sulfonium Difluorotrimethylsilicate, First Update, in: Fuchs PL (Eds), Handbook of Reagents for Organic Synthesis, Reagents for Silicon-Mediated Organic Synthesis, John Wiley and Sons Ltd Publishers, Chichester, 2011, pp 741–743. [Google Scholar]
- [28].Palom Y, Lipman R, Musser SM, Tomasz M, A mitomycin-N6-deoxyadenosine adduct isolated from DNA, Chem. Res. Toxicol 11 (1988) 203–210. [DOI] [PubMed] [Google Scholar]
- [29].Wijmenga SS, Kruithof M, Hilbers CW, Analysis of 1H Chemical Shifts in DNA: Assessment of the Reliability of 1H Chemical Shift Calculations for Use in Structure Refinement, J. Biomol. NMR 10 (1997) 337–350. [DOI] [PubMed] [Google Scholar]
- [30].Fonville JM, Swart M, Volkáčová Z, Sychrovský V, Šponer JE, Šponer J, Hilbers CW, Bickelhaupt FM, Wijmenga SS, Chemical Shifts in Nucleic Acids Studied by Density Functional Theory Calculations and Comparison with Experiment, Chem. Eur. J 18 (2012) 12372–12387 [DOI] [PubMed] [Google Scholar]
- [31].Tomasz M, Jung M, Verdine G, Nakanishi K, Circular Dichroism Spectroscopy as a Probe for the Stereochemistry of Aziridine Cleavage Reactions of Mitomycin C. Application to Adducts of Mitomycin with DNA constituents, J. Am. Chem. Soc 106 (1984) 7367–7370. [Google Scholar]
- [32].Fiallo MML, Kozlowski H, Garnier-Suillerit A, Mitomycin Antitumor Compounds Part 1. CD Studies on their Molecular Structure, Eur. J. Pharm. Sci 12 (2001) 487–494. [DOI] [PubMed] [Google Scholar]