

SUMMARY
Bacteriophages are abundant within the human gastrointestinal tract, yet their interactions with gut bacteria remain poorly understood, particularly with respect to CRISPR-Cas immunity. Here, we show that the Type I-C system in the prevalent gut Actinobacterium Eggerthella lenta is transcribed and sufficient for specific targeting of foreign and chromosomal DNA. Comparative analyses of E. lenta CRISPR-Cas systems across [meta]genomes revealed 2 distinct clades according to cas sequence similarity and spacer content. We assembled a human virome database (HuVirDB), encompassing 1,831 samples enriched for viral DNA, to identify protospacers. This revealed matches for a majority of spacers, a marked increase over other databases, and uncovered “hyper-targeted” phage sequences containing multiple protospacers targeted by several E. lenta strains. Finally, we determined the positional mismatch tolerance of observed spacer/protospacer pairs. This work emphasizes the utility of merging computational and experimental approaches for determining the function and targets of CRISPR-Cas systems.
Graphical Abstract
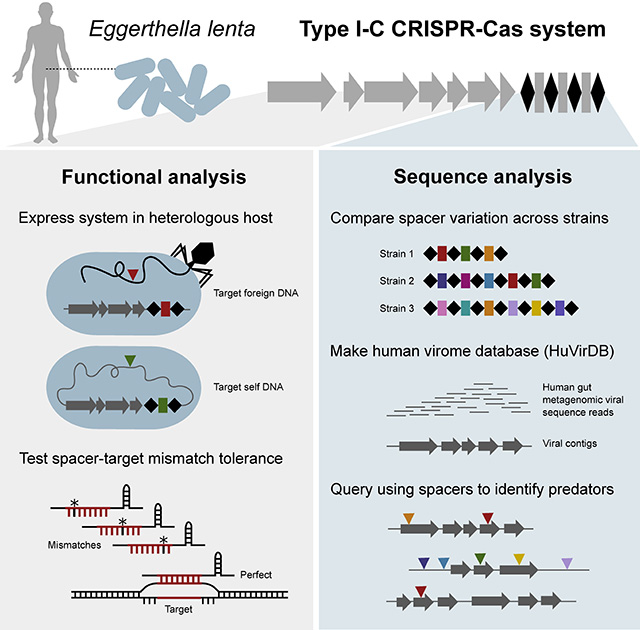
INTRODUCTION
CRISPR-Cas are adaptive immune systems, comprised of RNA-guided nucleases, that protect prokaryotes against infection from parasitic genetic elements by cleaving foreign DNA (Barrangou and Horvath, 2017; Barrangou et al., 2007). A variety of these systems (spanning the mechanistically distinct Types I-VI) have been identified in bacterial and archaeal genomes (Koonin et al., 2017) and function by storing the memory of past exposure to foreign elements as ~30 nt spacers in a CRISPR (clustered regularly interspaced short palindromic repeats) array between direct repeat sequences (Levy et al., 2015; McGinn and Marraffini, 2018). This memory element is subsequently processed, generating RNA guides (crRNA), which are packaged into complexes with Cas (CRISPR-associated) proteins (Brouns et al., 2008) to surveil the cell and mediate the recognition and cleavage of complementary sequences (Garneau et al., 2010). The outcome of these interactions is a limitation of horizontal gene transfer and prevention of phage replication (Bikard et al., 2012).
Most identified spacers cannot be assigned a target, suggesting a ubiquity of unobserved phage and mobile element diversity (Shmakov et al., 2017), especially within the human gut microbiome. Moreover, the relationship between environmental fitness in the gut and CRISPR-Cas remains to be determined given that they defend against phage, but also limit horizontal gene transfer encoding beneficial traits (Barrangou et al., 2007; Bikard et al., 2012; Palmer and Gilmore, 2010).
To date, CRISPR-Cas research in human-associated bacteria has focused on computational analyses (Tajkarimi and Wexler, 2017; Zhang et al., 2014). These studies can both over- and under-estimate CRISPR-Cas prevalence (Zhang and Ye, 2017), motivating the need for experimental demonstration of CRISPR-Cas expression, array processing, and target cleavage. Here, we leverage the use of robust genetic tools in an evolutionary distant bacterium, Pseudomonas aeruginosa, to express cas genes and crRNA, utilizing a generalizable strategy for studying CRISPR-Cas in genetically intractable gut bacteria. We focus on Eggerthella lenta due to: (i) its high prevalence in the human gut (81.6%) (Koppel et al., 2018); (ii) broad impact on the metabolism of drugs (Haiser et al., 2013; Koppel et al., 2018), dietary bioactives (Bess et al., 2018), and endogenous compounds (Harris et al., 2018; Rekdal et al., 2019); and (iii) links to infectious (Chan and Mercer, 2008) and chronic (Qin et al., 2012) disease.
Our work highlights the presence and functionality of a prevalent CRISPR-Cas system in an understudied bacterium and host habitat. In order to identify targets of this immune system, we constructed a specialized database that allowed us to uncover putative phages repeatedly targeted by diverse E. lenta strains. These results serve as a strong foundation for the discovery and mechanistic dissection of phage-bacterial interactions within the gut.
RESULTS
The E. lenta CRISPR-Cas system is transcriptionally active
Analysis of the E. lenta DSM 2243 genome revealed a putative CRISPR-Cas system in the Type I-C subgroup (Figure 1A). Given evidence for Type I-E cas transcriptional-repression during in vitro growth (Pul et al., 2010), we examined transcriptional data from E. lenta DSM 2243 in mid-exponential phase and detected expression of all cas genes (Figures 1A and 1B). We observed heterogeneity across the locus, ranging from cas3 (5.6±0.6 RPKM±SD) to cas5 (181.0±32.2) (Figure 1B), both higher than intragenic expression (0.0747±0.006). The depth of mapped reads (Figure 1A) and predicted transcriptional start sites (Figure S1A) both suggested that this locus produces at least 2 distinct transcripts. We experimentally confirmed this by performing a nested PCR of cDNA using primer pairs that span the junction of each gene pair (Figure S1A). These results, shown in Figure S1B, are consistent with the presence of a monocistronic cas3 transcript and at least one additional polycistronic transcript encompassing the genes from cas5 to cas2.
Figure 1. E. lenta DSM 2243 has a transcriptionally and catalytically active CRISPR-Cas system.

(A) Base coverage of RNA-seq reads to the CRISPR-Cas locus in DSM 2243 indicates active transcription. (B) Expression levels of cas genes during exponential growth measured in reads per kilobase per million mapped reads (RPKM). (C) Transcription from the CRISPR array generates a pre-CRISPR RNA which is processed by the Cas enzymes to form CRISPR RNAs (crRNAs) that direct targeting and cleavage of foreign DNA. (D) Northern blot demonstrates the presence of short RNA species (crRNA) in a CRISPR-positive strain (DSM 2243) but not in a CRISPR-negative strain (Valencia). Growth phase is indicated above the blot: M=mid- exponential (24 hours) and L=late-exponential (37 hours). (E) Growth kinetics (n=3) and (F) cas expression levels demonstrate the stability of cas3 and cas5 across growth phases (n=3, *** P<0.001 two-way ANOVA).
CRISPR array transcription generates a precursor transcript (pre-crRNA) (Figure 1C) whose expression was supported by RNA-sequencing (Figure 1A). Consistent with prior reports, the 5’ end of the array, where new spacers are acquired, was more highly transcribed (Rollie et al., 2015). We sought to test if the pre-crRNA is processed into the short active CRISPR RNA species (crRNA), which are essential for the formation of the interference complex that recruits the endonuclease Cas3 to cleave targets (Figure 1C). Through Northern blot analysis, we detected mature crRNAs, between 50 and 80 nt, during both mid- and late-exponential growth (Figure 1D) which are generated by Cas5 (Hochstrasser et al., 2016). No bands were observed using a control E. lenta strain lacking a CRISPR-Cas system (Figure 1D).
Consistent with these results, the cas genes were also stably transcribed throughout exponential growth (Figures 1E and 1F). The relative expression level between cas5 and cas3 was also stable over time; cas5 was expressed at 17.3±1.2 fold higher levels than cas3 (Pgene<0.001, two-way ANOVA, Figure 1F). This transcriptional control of cas3 has been proposed to keep low but sufficient levels of the protein in order to provide immunity while avoiding off-target nuclease activity (Majsec et al., 2016). Together, these results indicate that the Type I-C CRISPR-Cas system of E. lenta DSM 2243 is transcriptionally active and that mature crRNAs are generated during in vitro growth.
The E. lenta CRISPR-Cas system is sufficient to target phage and chromosomal DNA
To definitively demonstrate targeting by the E. lenta CRISPR-Cas system, and to circumvent the lack of genetic tools available, we designed a heterologous expression system in P. aeruginosa PA01, which lacks an endogenous system. The resulting strain (PA01 tn7::lentaIC) expresses the minimal machinery from the E. lenta system required for targeting and cleavage (cas5, cas8c, cas7, and cas3) (Figure 2A). To complete the interference complex, we constructed a plasmid expressing a minimal CRISPR array (Figure 2A). To target sequences of interest, we used the Type I-C canonical protospacer adjacent motif (PAM), responsible for identifying non-self DNA, sequence (TTC).
Figure 2. Heterologous expression in P. aeruginosa demonstrates the ability to target phage and chromosomal DNA.

(A) P. aeruginosa strain (PA01 tn7::lentaIC) constructed to inducibly express the minimal cas genes required for interference and a plasmid containing a minimal CRISPR array. (B) Expression of gJBD30 (phage-targeting) causes a 120-fold reduction in the number of plaque forming units (PFUs) when compared to a non-targeting (NT) control (n=4, *P=0.0286, Mann-Whitney U). (C) Expression of gPhzM (chromosome-targeting) decreases the colony forming units (CFUs) by 13,450-fold (n=5, **P=0.0079, Mann-Whitney U). (D) Distribution of spacer lengths found in the E. lenta isolate genomes, metagenomes, and merged datasets. (E) Variable length crRNAs decrease the number of PFUs with the exception of a 40 nt crRNA (n=4).
We tested the system’s ability to target foreign DNA by providing a 34-nt spacer targeting the phage JBD30 (gJBD30). When challenged with JBD30, there was a 120-fold reduction in plaque formation compared to a non-targeting (NT) control (P=0.0286, Mann-Whitney U-test; Figure 2B). Phage targeting was also evident from plaque morphology: individual plaques became smaller and less opaque, indicative of inhibited lytic activity (Figure 2B). Next, to determine if the system was capable of targeting self DNA, we designed a 34-nt spacer to target the region upstream of the pyocyanin pigment biosynthetic gene (phzM) in the host genome. Expression of this crRNA (gPhzM) resulted in a >10,000-fold reduction in colony formation compared to the NT control (P=0.0079, Mann-Whitney U, Figure 2C). Together, these results demonstrate that the E. lenta Type I-C CRISPR-Cas effector complex is sufficient for the specific recognition and cleavage of foreign and self-DNA.
We designed both spacers, gJBD30 and gPhzM, to be 34 nt long; however, spacers within E. lenta isolates naturally vary from 32 to 38 nt with 74.2% of the spacers being 33 or 34 nt (Figure 2D). This spacer length variation has been observed in soil bacteria with a Type I-C system (Lee et al., 2018). To determine the effect of spacer length on targeting efficiency, we designed multiple spacers varying from 30–40 nt against a single JBD30 protospacer. We observed similar plaquing efficiencies for all spacer lengths with the exception of the 40 nt spacer (Figure 2E), demonstrating that all of the naturally occurring spacer lengths are efficient at phage targeting.
The presence of CRISPR-Cas systems varies within the E. lenta species
Multiple studies have emphasized strain-level variation in metabolism (Koppel et al., 2018), immune response (Belkaid and Hand, 2014), and pathogenesis (Britton and Young, 2014). To assess if CRISPR-Cas presence in E. lenta is similarly strain-specific, we expanded our analysis to include a collection of human-associated E. lenta strains (Bisanz et al., 2018). These genomes have a mean size of 3.53 Mb, a minimum contig length covering 50% of the genome (N50) of 431,316bp, and Ncontigs=59. Of the 24 E. lenta genomes analyzed, 15 had a Type I-C CRISPR-Cas system (Figure 3A) and no other complete CRISPR-Cas system types were observed. For CRISPR-Cas-encoding strains, the genomic context was conserved and phylogenetic analysis based on cas alignment revealed 2 distinct clades: A and B (Figure 3A). The number of spacers per CRISPR array ranged from 10 to 64 (median 52; Figure 3A) with a total of 210 unique spacers across the 15 E. lenta genomes.
Figure 3. Strain-level variation in the E. lenta Type I-C CRISPR-Cas system.

(A) 15 sequenced isolate genomes contain a Type I-C system that clusters into two distinct clades based on an alignment of the cas genes. The global nucleotide identity to the DSM 2243 ortholog is shown. Diamonds indicate the number of direct repeats and the exact number of spacers in each array is displayed. (B) CRISPR spacer conservation between strains. Spacers are indicated as colored diamonds with identical spacers linked by a grey line.
Strains C592 and 28B were annotated as having a 5’ truncated cas3 (Figure 3A). We observed and confirmed a single base insertion in the 28B cas3 sequence that caused a premature stop codon, leading to an internal ATG sequence being identified as the cas3 translational start (Figure S2A). Prediction of functional domains revealed that the insertion separated the helicase and endonuclease domains into 2 separate coding sequences (Figures S2B and S2C). More work is necessary to determine if this leads to inactivation or if these open reading frames are still able to generate functional polypeptides carrying out their respective activities, as shown in other systems (Makarova et al., 2011; Plagens et al., 2012).
The spacers found on the 3’ end of the array were more conserved even across cas clades (Figure 3B). In most instances, exemplified by strains DSM 11767 and DSM 15644, unique spacers are found near the 5’ end of the array, consistent with acquisition of spacers over time. Spacers interrupting stretches of highly correlated spacer order could be due to loss via recombination or low-frequency spacer acquisition in the middle rather than the start of the array (Deveau et al., 2008).
To enrich our sampling of E. lenta CRISPR-diversity, we leveraged metagenomic data and the nature of the CRISPR direct repeat. An alignment of the direct repeat sequences from our reference genomes revealed a highly conserved 33 nt motif (Figure 4A). This appears to be unique to E. lenta, as it is absent from the CRISPR-Cas systems of other members of the Coriobacteriia with the nearest homologous direct repeat observed in Bifidobacterium thermophilum RBl67 (5 mismatches) (Grissa et al., 2007). Due to the low abundance of E. lenta within the human gut microbiota (Bisanz et al., 2018), we utilized a select set of 96 gut metagenomes that we previously found to have high E. lenta genome coverage (Koppel et al., 2018) to identify spacers by retrieving and assembling reads containing the direct repeat and then extracting spacers flanked by repeats containing no more than 3 mismatches from our consensus motif (Figure S3).
Figure 4. Direct repeat and spacer conservation across reference genomes and metagenomic datasets.

(A) The 33 nt E. lenta direct repeat was found to be highly conserved in all 15 CRISPR-positive isolate genomes. (B) Analysis of shared spacer content between strains provides evidence of a clade-specific pattern of conservation. Spacers were numbered 1–493 ordered by frequency of occurrence. (C) Venn diagram of shared spacer content between isolate genome clades and metagenomic data. (D) Evaluation of self (targeting of genome by spacer encoded within genome) and inter-strain targeting. Alignment of spacers to the E. lenta “super genome”: a 7-Mbp non-redundant sequence representing the aggregate genomes of this bacterial species. Red triangles indicate spacer matches within putative prophage and mobile elements.
This analysis increased the total number of E. lenta-derived spacers (210 to 493; 2.3-fold). Consistent with our reference genomes, spacer length varied from 32–38 nt in metagenomes with 69.4% being 33 and 34 nt. When both isolate and metagenomes are combined and dereplicated it is apparent that both datasets display a similar length distribution (Figure 2D). No assembled arrays were detected in a control set of 96 randomly selected metagenomes that contain E. lenta below the limit of detection (Nayfach et al., 2015). We next looked at shared spacers across reference genomes and metagenomes observing correspondence between spacer content and cas clade (Figure 4B). Metagenome-assembled CRISPR arrays were interwoven between clades suggesting strains of E. lenta representing both clades can be found within the human gastrointestinal tract. The correspondence between spacer content clade was correlated with strain phylogeny (Figure S4), consistent with the idea that these sequences at least partially reflect evolutionary history. We also detected evidence for horizontal gene transfer: strain AB8n2 phylogenetically clusters with strains from Clade B but contains a Clade A system. While a common set of 47 spacers was observed across clades A, B, and metagenomes, each had a unique set with considerably higher diversity in the metagenomic data (57.4% of unique spacers; Figure 4C).
To determine the extent to which CRISPR-targeting occurs within the E. lenta pangenome, spacers were compared to a non-redundant representation of the E. lenta species genome. We found 60 putative protospacers present in 18 strains targeting a limited number of loci (Figure 4D). Of these protospacers, 8 occur within the genome encoding the spacer, which may suggest self-targeting (the remaining 52 were inter-strain and intra-species). Closer inspection of the protospacers revealed that 6 occur in a putative prophage observed in two of these strains (Bisanz et al., 2018), one in a suspected integrated plasmid, and another in a region adjacent to a tetracycline resistance gene (Table S1). Neither perfect alignments nor a flanking sequence indicative of a PAM were observed, suggesting that the E. lenta system does not actively target these sites.
Protospacer identification reveals undescribed E. lenta phages
Most spacers found in sequenced prokaryotic genomes lack a predictable target, emphasizing that many mobile genetic elements and phages remain unknown (Shmakov et al., 2017). To identify potential parasitic elements targeted by CRISPR, we queried 3 publicly available databases for matches to previously characterized plasmids or viruses; however, no significant matches were found. The NCBI non-redundant database allowed us to assign 1.6% of the spacers to chromosomal genes of cryptic function and origin (Figure 5A). These results are consistent with the vast viral diversity within humans and its limited representation in established databases.
Figure 5. Discovering E. lenta predators based on protospacer enrichment.

(A) Comparison of protospacer matches within HuVirDB (249/493) versus other publicly accessible databases, including isolated and sequenced plasmids and phage from Refseq. (B) To facilitate phage discovery, public virome sequencing data was collected and assembled for our Human Virome Database (HuVirDB). (C) The 5’ flanking sequence was enriched for the canonical Type I-C protospacer adjacent motif (PAM) TTC. (D) Detailed annotation of a representative phage (ELM P1), identified based on a high frequency of matching E. lenta spacers.
To enable a more comprehensive platform for the identification of protospacers, we built a custom Human Virome Database (HuVirDB) that integrates data from 18 publicly available virome studies representing 1,831 samples from 730 humans from 9 countries (Figure 5B, Tables S2 and S3). We assembled 19.4 Gbp of sequence from 1,783 samples recovering 3,386 putative protospacers representing 249/493 (50.5%) of spacers. These protospacers were observed across 218 human samples, 161 individuals, and 14 studies representing a broad geographical distribution (Table S4). Furthermore, we used this data to determine the PAM sequence through motif analysis of the protospacer-adjacent regions. This revealed the canonical 5’ Type I-C PAM “TTC” (Figure 5C) with no strong conservation in the 3’ region. In recovering protospacers, HuVirDB outperformed the NCBI environmental non-redundant database (NCBI env nt), which is 6.4-fold larger (124.1 Gbp) but resulted in half the matches (25.0%) with higher computational overhead and without easily accessible metadata (Figure 4A). We similarly contrasted our recovery of protospacers against the Integrated Microbial Genome/Virus 2.0 (IMG VR) (Paez-Espino et al., 2019) finding >2-fold increased protospacer identification with HuVirDB for E. lenta.
To examine the utility of this approach for the study of other gut bacterial species, we extracted spacers from the Human Microbiome Project (HMP) reference genomes, Pathosystems Resource Integration Center (PATRIC) genomes of human gastrointestinal origin, and a subset thereof belonging to 28 strains of Akkermansia muciniphila. Similar to E. lenta, A. muciniphila showed improved protospacer identification in HuVirDB compared against all other databases (Figure S5A). However, the overall HMP and PATRIC datasets had higher protospacer identification with IMG VR and the two NCBI databases, likely due to the presence of data from pathogens and bacteria from other body habitats in these other databases. Consistent with these observations, network analysis of CRISPR-array containing genomes linked through common targets revealed the presence of strong clade specificity of CRISPR targeting (Figure S5B). These results emphasize the value of having complementary databases for protospacer identification depending on the specific bacterial host of interest.
To examine E. lenta target diversity, we clustered the protospacer-containing scaffolds at 80% global nucleotide identity into 13 non-singleton phage genomes (Figure 6A). Analysis of a representative sequence for each of these families revealed as many as 96 distinct protospacers, suggesting that E. lenta has repeatedly been exposed to these “hyper-targeted” phage (Figure 6A). A representative phage, referred to as Eggerthella lenta metagenomic phage 1 (ELM P1), with a genome size of 19,474 bp, contains 37 distinct protospacers (Figure 5D). This phage possesses genes homologous to Actinomyces phage AV-1 and Bacillus phage phi29, which are small dsDNA phages of the podophage families with genome size in the 17–22 kbp range (Delisle et al., 2006; Meijer et al., 2001). The protospacer sequences were concentrated within discrete portions of phage genomes, which may indicate bias towards the sequence injected earliest (Modell et al., 2017) and/or primed acquisition (Fineran et al., 2014; Kunne et al., 2016). In almost all of the targeted phage sequences we found annotated genes that suggest the presence of tail, collar, and head proteins (Figure 6A, Table S5).
Figure 6. E. lenta metagenomic (ELM) phage genomes.

(A) Genomes are presented with annotated genes colored by high-level function and protospacer locations indicated by dashes. Protospacers were allowed to have up to 4 mismatches to the spacer sequence. The number of unique [meta]genomes targeting the seed phage and the number of unique spacers is shown. (B) Clustering for taxonomic annotation of ELM phages with prokaryotic viral genomes from Viral RefSeq v.85 based on gene-sharing (Bin Jang et al., 2019) demonstrates a unique clade formed by 7 ELM phage. Only non-singleton clusters are depicted. (C) A phylogenetic tree of the ELM phages based on genome-wide BLAST distances (Meier-Kolthoff and Göker, 2017). Numbers on tree represent pseudo-bootstrap support values from 100 replications.
To better understand the taxonomy and phylogeny of these ELM phages, we began by clustering with previously described phages with approved taxonomies by the International Committee for the Taxonomy of Viruses (ICTV) (Bin Jang et al., 2019). We found that a subset (7/13) ELM phages formed a subcluster which could not be assigned even a family-level taxonomy while the remaining phages were singletons (Figure 6B). We next built a phylogenetic tree which grouped 6/7 of these related ELM phages into a single cluster (Figure 6C), supporting their close taxonomic and phylogenetic relationship. The remaining ELM phages were clustered into at least two additional groups. Of note, ELM P1 grouped together with the other 6 phage by taxonomy but was in a distinct cluster based on phylogeny, potentially due to the mosaic nature of phage genomes. These results suggest that the metagenomic E. lenta phages we have observed represent a previously undescribed branch of phage diversity targeting gut Actinobacteria.
The Type I-C CRISPR-Cas system can accommodate common protospacer mismatches
Further examination revealed that only 40.2% of protospacers were a perfect match to the spacer before dereplication into seed sequences (Table S4). Partial matches could indicate accumulated mutations that allow the phage to evade CRISPR-mediated immunity (Semenova et al., 2011) or that this system can accommodate mismatches, as has been demonstrated in other CRISPR-Cas system types (Pyenson et al., 2017). In order to distinguish between these two alternatives, we examined mismatch frequency as a function of the spacer/protospacer nucleotide position. The most commonly observed mismatched positions occur, in order of frequency, at nucleotides 18, 1, 33, and 9 (Figure 7A). We designed a series of spacers against JBD30 containing point mutations and examined their efficiency (Figures 7B and 7C).
Figure 7. Most mismatches between the spacer and protospacer sequences still provide immunity against phage.

(A) The occurrence of mismatches throughout the length of spacer was calculated using HuVirDB. (B) Plaque assays revealed two phenotypes: opaque plaques (efficient targeting) and clearer plaques (poor targeting). Controls include: a perfect match positive control and a non-targeting (NT) negative control. A representative mismatch (A31G) is shown. (C) Plaquing efficiency [log-estimation in tested gRNA / log-estimation in NT control] reveals that mutations in the seed sequence of the crRNA allow the phage to escape CRISPR-Cas immunity (n = 4). The dashed line at 10−5 denotes the average value for the perfect match control gRNA. *P<0.05 Kruskal-Wallis with uncorrected Dunnett’s (compared to targeting control).
Mutations in the 5’ spacer region, called the seed sequence, have been shown to provide phage an opportunity for escaping CRISPR immunity (Semenova et al., 2011). In accordance with this, we observed a high efficiency of plaquing for crRNAs with a mutation at the 3rd position or insertion at the 2nd position (P=0.021, Kruskal-Wallis with Dunnett’s post test; Figure 7C). Interestingly, we noted that a mutation in the 31st nucleotide also allowed the phage to evade CRISPR interference (P=0.021; Figures 7B and 7C). In contrast, single, double, and triple mutations in the middle of the spacer were tolerated, thus still providing immunity. Together, these results demonstrate that most naturally-occuring mismatches still allow for efficient targeting of the invading sequence.
DISCUSSION
Here, we report the characterization of an active Type I-C CRISPR-Cas system in a prevalent member of the human gut microbiota, revealing undescribed hyper-targeted phages that infect gut Actinobacteria, which have eluded isolation despite their prevalence. By combining a systematic meta-analysis of virome datasets, metagenomics, and comparative genomics, we were able to uncover putative targets for >50% of E. lenta spacers. These results support the critical role of CRISPR-Cas systems in adaptive immunity to bacteriophage, while also raising the question as to whether or not the remaining spacers target bacteriophage that remain to be discovered, mobile genetic elements, and/or as-of-yet unknown novel targets. These spacer- protospacer matches provide more definitive evidence for the host range of phages identified in virome datasets, as exemplified by the discovery of hyper-targeted phage that appear to have been repeatedly encountered and targeted by geographically diverse E. lenta CRISPR-Cas systems. The identified hyper-targeted phage are likely major determinants of E. lenta fitness and their isolation or synthetic reconstitution would provide a major step forward in understanding the biology of this neglected bacterial species and determining whether or not the presence of multiple spacers within a single array is necessary for robust immunity.
Despite common mismatches detectable in gut viromes, we found that the E. lenta CRISPR-Cas system could tolerate single and even double or triple mutations within the middle of the spacer, as described in other types of systems (Pyenson et al., 2017). This suggests that phage may have a limited ability to escape targeting by mutation, requiring a mismatch in the first few nucleotides of the spacer or the PAM motif (both of which we detected in our computational analysis). Surprisingly, we also found a significant impact of point mutations in nucleotide 31, more work is necessary to determine why this particular nucleotide matters, either through disrupting complex formation, target binding, and/or nuclease activity.
Our results emphasize the critical importance of providing experimental support for CRISPR-Cas system function. In addition to the previously described false-positives driven by incomplete systems (Zhang and Ye, 2017) and other types of genomic repeats (Zhang and Ye, 2017) in other environments, we found that of the 24 E. lenta genomes analyzed, 9 lacked the entire Type I-C system. The 15 strains that encoded a complete system could be binned into two distinct clades based on cas gene homology and spacer content, emphasizing the strain- level variation of these systems. Given this strain level heterogeneity, our results emphasize the challenges in predicting bacterial interactions with phages based only on species abundance and the need for continued progress towards the functional characterization and mechanistic dissections of these systems within their natural host bacteria and physiological context.
These results also emphasize the utility of combining the computational and functional dissection of CRISPR-Cas systems in bacterial reference genome and metagenomic datasets to gain insights into the bacterial and viral components of the human microbiome. The approaches we have used do not require genetic tools in the target microorganism, enabling mechanistic insights into the vast majority of human-associated bacteria which remain genetically intractable (Burstein et al., 2016). More broadly, our development of HuVirDB provides a useful resource with rich metadata, enabling the study of predator-prey relationships across the human microbiome. While our current studies have focused on the E. lenta Type I-C system, this database could be readily queried for matches to spacers from other human gut bacteria of interest. To facilitate the rapid adoption of this tool in the microbiome and CRISPR- Cas community we have made all of the data publicly accessible and have integrated it into a widely-used graphical tool for spacer matching (Biswas et al., 2013).
Finally, our work provides fundamental biological insights into endogenous CRISPR-Cas systems found within the human gut microbiome, an essential prerequisite for efforts to reprogram these systems to more precisely impact the structure and function of complex host-associated microbial communities. Continued progress in this area will require the development of approaches for gene delivery within the gastrointestinal tract, robust methods to engineer bacteriophage or other vectors, and the identification of bacterial targets with readily quantifiable impacts on host pathophysiology.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Peter Turnbaugh (Peter.Turnbaugh@ucsf.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial and phage culture
All strains used for these studies are listed in the Key Resources Table. Pseudomonas aeruginosa phage JBD30 was propagated on P. aeruginosa PA01 (Stover et al., 2000) and stored in SM buffer at 4°C. The titer of the phage was determined by performing serial dilutions and mixing 10 μl of phage with 150 μl of P. aeruginosa PA01 (grown overnight) in 0.7% LB agar and incubating overnight at 30°C. Routine culturing of E. lenta was done under anaerobic conditions (Coy Lab Products) using BHI++ media (BHI with 1% arginine, 0.05% L-cysteine-HCl, 1 μg/mL vitamin K, 5 μg/mL hemin, and 0.0001% w/v resazurin (Bisanz et al., 2018). Routine culturing of P. aeruginosa was done aerobically with rotation in LB media. For P. aeruginosa PA01 tn7::lentalC with plasmid pJB3 carrying the crRNA construct, the cells were grown in LB supplemented with 50 μg/ml of gentamicin.
METHOD DETAILS
RNA extraction
RNA extractions were performed as described previously (Bess et al., 2018). Briefly, a 24-hour broth culture of Eggerthella lenta DSM 2243 was subcultured at 1% v/v in BHI++ and allowed to grow for 24 hours (until mid-exponential, OD600 of ~0.3) in an anaerobic chamber (Coy Laboratory Products). The cells were spun down at max speed, 4°C, for 10 minutes. Cells were resuspended in TRI Reagent (Sigma) and lysed by a bead beater (BioSpec Products). Chloroform was added 1:5 to the mixture, incubated at room temperature for 10 minutes, and then spun down at 16,000 × g for 15 minutes. The top phase was placed in a clean tube and mixed 1:1 with 100% ethanol. The RNA extraction was then done using Purelink™ RNA Mini Kit (Invitrogen) according to the manufacturer’s protocol, with the addition of an in-column DNase treatment. A second DNase treatment was done after eluting using TURBO-DNase (Ambion).
RT-qPCR
Reverse transcription was carried out using 500 ng of total RNA and the iScript™ Reverse Transcription SuperMix according to the manufacturer’s protocol (Bio-Rad). The qPCR assays were performed using SYBR Select Master Mix for CFX (Applied Biosystems) run in a CFX384 Real-Time System (BioRad) using 10μl reactions according to the manufacturer’s recommendations. 200 nM primers were used to quantify gene expression as listed in the key resources table. All primers used are listed in Table S6.
Northern Blot
The Northern blot was carried out as previously described (Bondy-Denomy et al., 2013). Briefly, the probe was generated by amplifying a fragment spanning the first four spacers of the DSM 2243 CRISPR array, cleaning the PCR product (QIAGEN PCR Purification Kit) and labeling 300 ng of the clean product with Alpha-32P dCTP using Klenow polymerase (NEB M0210L). 5 μg of total RNA from E. lenta DSM 2243 (grown to mid-exponential) were used to run (per lane) in a denaturing gel. The RNA was transferred to a positively charged nylon membrane (Roche) using the semi-dry setting in a Trans-Blot Turbo (Bio-Rad) and crosslinked with 10 mJ UV burst over 30 seconds (Stratagene). The membrane was blocked with pre-hybridization buffer, consisting of 50% formamide, 5x Denhardts solution, 6x SSC, and 100 μg/ml of salmon sperm DNA, at 42°C for 1 hour. Probing was done at 42°C for 16–18 hours using the probe labeled with >4×105 cpm of dCTP. Afterwards, the blot was washed with wash solution 1 (2xSSC and 1% SDS) twice for 10 minutes at 18°C, two 30 minutes washes at 65°C, and wash solution 2 (0.2x SSC and 0.1%SDS) for 10 minutes at 18.C. The blot was developed using a phosphoimager.
RNA-Sequencing Analysis
The RNA-sequencing of E. lenta DSM 2243 was described elsewhere (Bess et al., 2018) and reads are available under Sequence Read Archive Project SRP140684. Briefly, RNA was extracted from triplicate mid-exponential cultures as described above and rRNA depletion (Illumina Ribo-Zero) was used for subsequent library construction (NEBNext Ultra RNA). Sequencing was conducted via Illumina HiSeq 2500 with single ended 51 bp chemistry. Using Bowtie2 (Langmead and Salzberg, 2012), the reads were mapped to the reference DSM 2243 assembly (GCA_000024265.1) with the following parameters: --end-to-end --sensitive --trim5 5 --trim3 5. Next counts per feature were determined using htseq-count (Anders et al., 2015) and normalized using the reads per million per kilobase (RPKM) method. Sequencing coverage over the entire CRISPR-Cas locus was visualized using Gviz (Hahne and Ivanek, 2016). The calculation of background expression levels was done by averaging the reads of intergenic regions (leaving out ±200 bp from coding sequences).
Construction of the Pseudomonas aeruginosa strain carrying the Eggerthella lenta cas genes
Chromosomal insertion of cas5–8-7–3 genes into P. aeruginosa was done as previously described, with insertion at the Tn7 location via a helper transposase vector (Choi and Schweizer, 2006). The cas3 gene was cloned downstream of cas7 to mitigate toxicity due to overexpression. Gentamicin resistant strains were selected and the insertion location confirmed via PCR. The gentamicin marker was then flipped out via FLP recombinase, generating a gentamicin sensitive strain with stably integrated and IPTG-inducible Cas proteins. To introduce crRNAs, the pHERD30T vector (Qiu et al., 2008) was used, a high copy gentamicin resistance, arabinose inducible shuttle vector. An “entry” array was designed containing a repeat-pseudospacer-repeat organization (pJB3). The pseudospacer possessed two Bsal sites to enable the cloning of annealed oligonucleotides as described previously (Marino et al., 2018).
crRNA cloning
The vector pJB3 was digested using the enzyme Bsal (NEB) and the fragment was gel extracted (Invitrogen Gel Extraction Kit). The primers (IDT & Sigma), carrying the point mutations of interest, were annealed and phosphorylated in a single reaction with 10x T4 Ligation Buffer (NEB) and T4 Polynucleotide Kinase (NEB) by incubating at 37°C for 2 hours, 95°C for 5 minutes, and ramp down to 20°C at 5°C/minute. Afterwards, they were diluted 1:500 in water and 1 μl was used to ligate to 60 ng of digested pJB3. The ligation was carried out overnight and stopped by incubating at 65°C for 20 minutes. 2 μl of the ligation were used to transform into NEB 5-alpha competent E. coli following the manufacturer’s protocol. The cells were grown in LB agar supplemented with 30 μg/ml of gentamicin. Cloning was verified by Sanger sequencing and plasmids were used to transform Pseudomonas aeruginosa tn7::lentaIC.
Transformation of Pseudomonas aeruginosa
A seed culture of the P. aeruginosa strain was subcultured 1:100 in fresh LB media and allowed to grow for 18 hours. 2 ml of the culture were spun down and washed twice with 300 mM sucrose and then re-suspended in 225 μl of 300 mM sucrose. 100 μl of the washed cells and 10–100 ng of plasmid DNA were used per transformation reaction. The cells were electroporated in a 0.2 mm cuvette using 25 μF, 200 ohm, and 2.5 kV. After the pulse, 800 μl of LB were added to the cells and then incubated at 37°C with shaking for 45 minutes. 100 μl of the reaction were used to spread in an LB agar plate supplemented with 50 μg/ml of gentamicin.
Phage plaque assays
Bacterial lawns were made by mixing 150 μl of an overnight culture of host bacteria with 4 ml of 0.7% LB agar with 10 mM MgSO4, 50 μg/ml of gentamicin and the inducers of expression (0.5 mM IPTG and 0.1% arabinose). Phage dilutions were made by diluting the phage in SM Buffer and 3 μl of each dilution were used to spot on the bacterial lawn. The plates were incubated at 30°C for 16–18 hours, after which the PFUs were quantified.
Metagenomic CRISPR spacer arrays
As previously identified (Koppel et al., 2018), paired end sequences from 96 E. lenta-enriched metagenomes were retrieved from the NCBI Sequence Read Archive. Reads were filtered using Trimmomatic (Bolger et al., 2014) to remove potential adapters and trimmed using the default sliding window filter. Next reads containing the consensus direct repeat were identified using vsearch (Rognes et al., 2016) with the following parameters: --usearch_global --id 0.87 --maxgaps 1 --maxsubs 4 --mincols 32 --maxaccepts 0 -- maxrejects 0 --strand both. Assembly was then carried out with SPAdes 3.7.0 (Bankevich et al., 2012). 96 E. lenta-deficient metagenomes, as determined from Metaquery (Nayfach et al., 2015), were also included and used as negative controls. CRISPR-array assemblies could not be generated from any of the E. lenta-deficient metagenomes. Spacers were extracted from these assemblies as below.
Comparative genomics and spacer identification
The collection and sequencing of the E. lenta genomes is described elsewhere (Bisanz et al., 2018). Annotation of the cas genes of E. lenta strain 28B was done using CRISPRCasFinder (Couvin et al., 2018) (Figure S3C). The presence of CRISPR arrays and their direct repeats in genome assemblies was first determined using the MINced 0.2.0 (github.com/ctSkennerton/minced). The consensus direct repeat sequence was determined via the MSA package (Bodenhofer et al., 2015) and ggseqlogo (Wagih, 2017). Arrays were then recalled from both isolate and meta-genomes by extracting regions flanked by the consensus 5’-GTCACTCCCCGCATGGGGAGTGCGGGTTGAAAT-3’ allowing for up to 3 mismatches from the consensus. The uniqueness of the E. lenta direct repeat was determined through comparison against our Coriobacteriia collection and through the use of the CRISPRdb (Grissa et al., 2007). The locus diagram was prepared through identification of orthologous gene clusters containing the DSM 2243 cas genes and extracting their genomic coordinates from Genbank transfer format files (Bisanz et al., 2018). Relative base position was determined by recentering coordinates on the 5’ translational start site of cas5. Nucleotide identity was determined by a Needleman-Wunsch global alignment of nucleotide sequence with percent ID calculated as 100*(identical positions) / (aligned positions + internal gap positions). The cas gene phylogenetic tree was created by concatenating the individual alignments of the cas genes as before, and building a tree with FastTree (Price et al., 2009). The super genome alignment was created using the Progressive Mauve algorithm (Darling et al., 2010) and plotting hits on this set of super-coordinates.
HuVirDB
We queried the NCBI Sequence Read Archive for studies of human-associated phage communities with shotgun sequencing data available. 18 studies were identified with sufficient metadata for inclusion (Table S2). Where possible, relevant per-sample metadata was preserved as identified in the SRA or in the original publication. A total of 1831 samples were collected for assembly and matching runs determined using the SRAdb package (Zhu et al., 2013), however 49 low-coverage samples (2.7%) failed assembly and were not pursued further. Trimmomatic was used to remove possible adapter contamination with sliding window filtering, then metaSPAdes (or SPAdes when SE reads) was used for assembly. When 454 sequencing was applied, error correction was bypassed using the --only-assembler flag. Resulting contigs were identified by their default identifier concatenated to their SRA sample accession and merged to form a single large database. Assembly statistics were generated using QUAST with a minimum contig size of 200 (Gurevich et al., 2013). To identify E. lenta protospacers, persample databases were queried through BLASTN (-task blastn-short) reporting 100 alignments with no more than 4 misaligned bases (qlen-nident<=4) allowed and filtered to ensure that the flanking sequences (Samtools 1.9) (Li et al., 2009) did not contain either the E. lenta direct repeat, or other repetitive sequence that could indicate the hit was a component of a contaminating CRISPR array. Phages of interest were annotated using a combination of RASTtk (Brettin et al., 2015) and PHASTER (Arndt et al., 2016) and visualized using gggenes (github.com/wilkox/gggenes). Based on these annotations, the genes were manually grouped into distinct functional categories: structural, packaging, replication, infection, other, and not annotated. E. lenta-targeted contigs were dereplicated based on a all-versus-all global nucleotide alignment strategy with 80% identity (measured as identities over the length of the shorter sequence) used as the clustering threshold. The largest phage assembly within the cluster served as the seed sequence, and if a fragment could be assigned with equal confidence to multiple seeds, one was randomly selected. Seed phage sequences are available at github.com/jbisanz/HuVirDB. Taxonomic clustering of ELM phages was carried out using VConTACT2 v0.9.9 against the ProkaryoticViralRefSeq85-ICTV database and visualized in R using ggnet v0.1.0. To generate the phylogenetic tree of ELM phages, all pairwise comparisons of the nucleotide sequences were conducted using the Genome-BLAST Distance Phylogeny (GBDP) method (Meier-Kolthoff et al., 2013) under settings recommended for prokaryotic viruses (Meier-Kolthoff and Goker, 2017). The resulting intergenomic distances were used to infer a balanced minimum evolution tree with branch support via FASTME including SPR postprocessing (Lefort et al., 2015) for formula D0. Branch support was inferred from 100 pseudo-bootstrap replicates each. Trees were rooted at the midpoint and visualized with FigTree v1.4.3. Taxon boundaries were estimated with the OPTSIL program (Göker et al., 2009), the recommended clustering thresholds (Meier-Kolthoff and Göker, 2017) and an F value (fraction of links required for cluster fusion) of 0.5 (Meier-Kolthoff et al., 2014).
To contrast databases, HMP reference genomes and PATRIC reference genomes had CRISPR-arrays extracted as above using MINced which were then merged with the E. lenta spacers previously identified. These were queried against BLAST databases as above including a concatenated HuVirDB, NCBI environmental non-redundant (env nt), IMG VR (January 2018 release), NCBI non-redundant nucleotide (nt), PATRIC phage, and Refseq viral and plasmid databases. Akkermansia muciniphila spacers were identified by being encoded in a genome annotated as A. muciniphila according to PATRIC metadata.
QUANTIFICATION AND STATISTICAL ANALYSIS
Where applicable, statistical analysis was carried out using either Prism 7 (Graphpad Software) or R 3.5.0 using Mann-Whitney U-tests or Kruskal-Wallis one-way analysis of variance with Dunnett’s multiple comparison post-hoc test. Phage plaque counts were estimated to the nearest 10-fold dilution with representative images of plaque morphology provided.
DATA AND CODE AVAILABILITY
HuVirDB metadata and related information is available at github.com/jbisanz/HuVirDB and the database itself for download at opengut.ucsf.edu/HuVirDB-1.0.fasta.gz. HuVirDB has been made available in CRISPRTarget as an available database (http://crispr.otago.ac.nz/CRISPRTarget/crispr_analysis.html) for general queries. Genome assemblies are available under the following BioProjects: PRJNA412637, PRJNA384908, PRJNA21093, PRJNA46413, PRJNA40023, PRJNA59527. HMP reference genomes were retrieved from NCBI using BioProject PRJNA28331 (retrieved 23 August 2018). PATRIC reference genomes were retrieved from NCBI on 23 August 2018 by assembly accession as identified in the PATRIC genome catalog (patricbrc.org/view/Taxonomy/2#view_tab=genomes) after filtering for host_name containing human or sapiens, and an isolation_source containing stool, faecal, faeces, fecal, feces, gastrointestinal, gut, intestine, rectal, or rectum.
Supplementary Material
Table S1. Putative protospacers within Eggerthella lenta genomes related to Figure 4.
Table S2. Studies included in HuVirDB related to Figure 5.
Table S3. HuVirDB sample metadata related to Figure 5.
Table S4. Putative protospacer sequences in HuVirDB related to Figure 5.
Table S5. E. lenta metagenomic phage genome annotations related to Figure 6.
Table S6. Oligonucleotide sequences used in the study related to STAR Methods.
Highlights.
Eggerthella lenta, a human gut Actinobacterium, encodes a functional CRISPR-Cas system
Strain-level variations exist in system presence, cas gene sequence, and spacer content
HuVirDB is a generalizable human virome database to search for CRISPR targets
Hyper-targeted phage that harbor multiple protospacers were discovered
Soto-Perez, Bisanz et al. focus on a Type I-C CRISPR-Cas system encoded by Eggerthella lenta, a prevalent human gut Actinobacterium implicated in metabolism and pathogenesis. Through computational and experimental approaches, they determine the system’s activity and strain-level variation, while also generating a human virome database to identify common phage predators.
ACKNOWLEDGEMENTS
We are grateful to Stephen Nayfach for providing the E. lenta enriched-metagenomes, Elizabeth Bess for providing RNA sequencing data, Chris Brown and Peter Fineran for including HuVirDB as a database for CRISPRTarget, and all of the members of the Turnbaugh and Bondy-Denomy labs for helpful discussion. This work was supported by the National Institutes of Health (R01HL122593), Searle Scholars Program (SSP-2016-1352), and the UCSF Program for Breakthrough Biomedical Research (partially funded by the Sandler Foundation). PJT holds an Investigators in the Pathogenesis of Infectious Disease Award from the Burroughs Wellcome Fund, is a Chan Zuckerberg Biohub investigator, and a Nadia’s Gift Foundation Innovator supported, in part, by the Damon Runyon Cancer Research Foundation (DRR-42-16). Fellowship support was provided by the Canadian Institutes of Health Research (KNL), the Natural Sciences and Engineering Research Council (JEB), and the UCSF Discovery Fellows (PSP). JBD was supported by an NIH Office of the Director Early Independence Award (DP5-OD021344).
Footnotes
DECLARATION OF INTERESTS
PJT is on the scientific advisory board for Kaleido, Seres, SNIPRbiome, uBiome, and WholeBiome and JBD is on the scientific advisory board for SNIPRbiome and Excision Biotherapeutics; there is no direct overlap between the current study and these consulting duties.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arndt D, Grant JR, Marcu A, Sajed T, Pon A, Liang Y, and Wishart DS (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol 19, 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrangou R, and Horvath P (2017). A decade of discovery: CRISPR functions and applications. Nat Microbiol 2, 17092. [DOI] [PubMed] [Google Scholar]
- Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, Romero DA, and Horvath P (2007). CRISPR provides acquired resistance against viruses in prokaryotes. Science 315, 1709–1712. [DOI] [PubMed] [Google Scholar]
- Belkaid Y, and Hand TW (2014). Role of the microbiota in immunity and inflammation. Cell 157, 121–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bess EN, Bisanz JE, Spanogiannopoulos P, Ang QY, Bustion A, Kitamura S, Alba DL, Wolan DW, Koliwad SK, and Turnbaugh PJ (2018). The genetic basis for the cooperative bioactivation of plant lignans by a human gut bacterial consortium. bioRxiv 357640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bikard D, Hatoum-Aslan A, Mucida D, and Marraffini LA (2012). CRISPR interference can prevent natural transformation and virulence acquisition during in vivo bacterial infection. Cell Host Microbe 12, 177–186. [DOI] [PubMed] [Google Scholar]
- Bin Jang H, Bolduc B, Zablocki O, Kuhn JH, Roux S, Adriaenssens EM, Brister JR, Kropinski AM, Krupovic M, Lavigne R, et al. (2019). Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol 37, 632–639. [DOI] [PubMed] [Google Scholar]
- Bisanz JE, Soto-Perez P, Lam KN, Bess EN, Haiser HJ, Allen-Vercoe E, Rekdal VM, Balskus EP, and Turnbaugh PJ (2018). Illuminating the microbiome’s dark matter: a functional genomic toolkit for the study of human gut Actinobacteria. bioRxiv 304840. [Google Scholar]
- Biswas A, Gagnon JN, Brouns SJJ, Fineran PC, and Brown CM (2013). CRISPRTarget: bioinformatic prediction and analysis of crRNA targets. RNA Biol. 10, 817–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodenhofer U, Bonatesta E, Horejs-Kainrath C, and Hochreiter S (2015). msa: an R package for multiple sequence alignment. Bioinformatics 31, 3997–3999. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondy-Denomy J, Pawluk A, Maxwell KL, and Davidson AR (2013). Bacteriophage genes that inactivate the CRISPR/Cas bacterial immune system. Nature 493, 429–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brettin T, Davis JJ, Disz T, Edwards RA, Gerdes S, Olsen GJ, Olson R, Overbeek R, Parrello B, Pusch GD, et al. (2015). RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep 5, 8365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britton RA, and Young VB (2014). Role of the intestinal microbiota in resistance to colonization by Clostridium difficile. Gastroenterology 146, 1547–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouns SJJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJH, Snijders APL, Dickman MJ, Makarova KS, Koonin EV, and van der Oost J (2008). Small CRISPR RNAs guide antiviral defense in prokaryotes. Science 321, 960–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burstein D, Harrington LB, Strutt SC, Probst AJ, Anantharaman K, Thomas BC, Doudna JA, and Banfield JF (2016). New CRISPR-Cas systems from uncultivated microbes. Nature 542, 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan RC, and Mercer J (2008). First Australian description of Eggerthella lenta bacteraemia identified by 16S rRNA gene sequencing. Pathology 40, 409–410. [DOI] [PubMed] [Google Scholar]
- Choi K-H, and Schweizer HP (2006). mini-Tn7 insertion in bacteria with single attTn7 sites: example Pseudomonas aeruginosa. Nat. Protoc 1, 153–161. [DOI] [PubMed] [Google Scholar]
- Couvin D, Bernheim A, Toffano-Nioche C, Touchon M, Michalik J, Néron B, Rocha C, Vergnaud EP, Gautheret G, D., and Pourcel C (2018). CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 46, W246–W251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling AE, Mau B, and Perna NT (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5, e11147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delisle AL, Barcak GJ, and Guo M (2006). Isolation and expression of the lysis genes of Actinomyces naeslundii phage Av-1. Appl. Environ. Microbiol 72, 1110–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deveau H, Barrangou R, Garneau JE, Labonté J, Fremaux C, Boyaval P, Romero DA, Horvath P, and Moineau S (2008). Phage response to CRISPR-encoded resistance in Streptococcus thermophilus. J. Bacteriol 190, 1390–1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fineran PC, Gerritzen MJH, Suarez-Diez M, Künne T, Boekhorst J, van Hijum SAFT, Staals RHJ, and Brouns SJJ (2014). Degenerate target sites mediate rapid primed CRISPR adaptation. Proc. Natl. Acad. Sci. U. S. A 111, E1629–E1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garneau JE, Dupuis M-È, Villion M, Romero DA, Barrangou R, Boyaval P, Fremaux C, Horvath P, Magadan AH, and Moineau S (2010). The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature 468, 67–71. [DOI] [PubMed] [Google Scholar]
- Goker M, Garcia-Blazquez G, Voglmayr H, Telleria MT, and Martín MP (2009). Molecular taxonomy of phytopathogenic fungi: a case study in Peronospora. PLoS One 4, e6319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grissa I, Vergnaud G, and Pourcel C (2007). The CRISPRdb database and tools to display CRISPRs and to generate dictionaries of spacers and repeats. BMC Bioinformatics 8, 172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich A, Saveliev V, Vyahhi N, and Tesler G (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahne F, and Ivanek R (2016). Visualizing genomic data using Gviz and Bioconductor. Methods Mol. Biol 1418, 335–351. [DOI] [PubMed] [Google Scholar]
- Haiser HJ, Gootenberg DB, Chatman K, Sirasani G, Balskus EP, and Turnbaugh PJ (2013). Predicting and manipulating cardiac drug inactivation by the human gut bacterium Eggerthella lenta. Science 341, 295–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris SC, Devendran S, Mendez-Garcia C, Mythen SM, Wright CL, Fields CJ, Hernandez AG, Cann I, Hylemon PB, and Ridlon JM (2018). Bile acid oxidation by Eggerthella lenta strains C592 and DSM 2243T. Gut Microbes 9, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochstrasser ML, Taylor DW, Kornfeld JE, Nogales E, and Doudna JA (2016). DNA targeting by a minimal CRISPR RNA-guided cascade. Mol. Cell 63, 840–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin EV, Makarova KS, and Zhang F (2017). Diversity, classification and evolution of CRISPR-Cas systems. Curr. Opin. Microbiol 37, 67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppel N, Bisanz JE, Pandelia M-E, Turnbaugh PJ, and Balskus EP (2018). Discovery and characterization of a prevalent human gut bacterial enzyme sufficient for the inactivation of a family of plant toxins. eLife Sciences 7, e33953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunne T, Kieper SN, Bannenberg JW, Vogel AIM, Miellet WR, Klein M, Depken M, Suarez-Diez M, and Brouns SJJ (2016). Cas3-derived target DNA degradation fragments fuel primed CRISPR adaptation. Mol. Cell 63, 852–864. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Zhou Y, Taylor DW, and Sashital DG (2018). Cas4-dependent prespacer processing ensures high-fidelity programming of CRISPR arrays. Mol. Cell 70, 48–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefort V, Desper R, and Gascuel O (2015). FastME 2.0: a comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Biol. Evol 32, 2798–2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy A, Goren MG, Yosef I, Auster O, Manor M, Amitai G, Edgar R, Qimron U, and Sorek R (2015). CRISPR adaptation biases explain preference for acquisition of foreign DNA. Nature 520, 505–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majsec K, Bolt EL, and Ivancic-Bace I (2016). Cas3 is a limiting factor for CRISPR-Cas immunity in Escherichia coli cells lacking H-NS. BMC Microbiol. 16, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Haft DH, Barrangou R, Brouns SJJ, Charpentier E, Horvath P, Moineau S, Mojica FJM, Wolf YI, Yakunin AF, et al. (2011). Evolution and classification of the CRISPR-Cas systems. Nat. Rev. Microbiol 9, 467–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino ND, Zhang JY, Borges AL, Sousa AA, Leon LM, Rauch BJ, Walton RT, Berry JD, Joung JK, Kleinstiver BP, et al. (2018). Discovery of widespread type I and type V CRISPR-Cas inhibitors. Science 362, 240–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinn J, and Marraffini LA (2018). Molecular mechanisms of CRISPR-Cas spacer acquisition. Nat. Rev. Microbiol 17, 7–12. [DOI] [PubMed] [Google Scholar]
- Meier-Kolthoff JP, and Goker M (2017). VICTOR: genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 33, 3396–3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier-Kolthoff JP, Auch AF, Klenk H-P, and Goker M (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14, 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier-Kolthoff JP, Hahnke RL, Petersen J, Scheuner C, Michael V, Fiebig A, Rohde C, Rohde M, Fartmann B, Goodwin LA, et al. (2014). Complete genome sequence of DSM 30083T, the type strain (U5/41T) of Escherichia coli, and a proposal for delineating subspecies in microbial taxonomy. Standards in Genomic Sciences 9, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer WJ, Horcajadas JA, and Salas M (2001). Phi29 family of phages. Microbiol. Mol. Biol. Rev 65, 261–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modell JW, Jiang W, and Marraffini LA (2017). CRISPR-Cas systems exploit viral DNA injection to establish and maintain adaptive immunity. Nature 544, 101–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayfach S, Fischbach MA, and Pollard KS (2015). MetaQuery: a web server for rapid annotation and quantitative analysis of specific genes in the human gut microbiome. Bioinformatics 31, 3368–3370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paez-Espino D, Roux S, Chen I-MA, Palaniappan K, Ratner A, Chu K, Huntemann M, Reddy TBK, Pons JC, Llabres M, et al. (2019). IMG/VR v.2.0: an integrated data management and analysis system for cultivated and environmental viral genomes. Nucleic Acids Res. 47, D678–D686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer KL, and Gilmore MS (2010). Multidrug-resistant enterococci lack CRISPR-cas. MBio 1, e00227–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plagens A, Tjaden B, Hagemann A, Randau L, and Hensel R (2012). Characterization of the CRISPR/Cas subtype IA system of the hyperthermophilic crenarchaeon Thermoproteus tenax. J. Bacteriol 194, 2491–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price MN, Dehal PS, and Arkin AP (2009). FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol 26, 1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pul U, Wurm R, Arslan Z, Geissen R, Hofmann N, and Wagner R (2010). Identification and characterization of E. coli CRISPR-cas promoters and their silencing by H-NS. Mol. Microbiol 75, 1495–1512. [DOI] [PubMed] [Google Scholar]
- Pyenson NC, Gayvert K, Varble A, Elemento O, and Marraffini LA (2017). Broad targeting specificity during bacterial type III CRISPR-Cas immunity constrains viral escape. Cell Host Microbe 22, 343–353.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, et al. (2012). A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490, 55–60. [DOI] [PubMed] [Google Scholar]
- Qiu D, Damron FH, Mima T, Schweizer HP, and Yu HD (2008). PBAD-based shuttle vectors for functional analysis of toxic and highly regulated genes in Pseudomonas and Burkholderia spp. and other bacteria. Appl. Environ. Microbiol 74, 7422–7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rekdal VM, Bess EN, Bisanz JE, Turnbaugh PJ, and Balskus EP (2019). Discovery and inhibition of an interspecies gut bacterial pathway for Levodopa metabolism. Science 364, eaau6323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rognes T, Flouri T, Nichols B, Quince C, and Mahe F (2016). VSEARCH: a versatile open source tool for metagenomics. PeerJ 4, e2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rollie C, Schneider S, Brinkmann AS, Bolt EL, and White MF (2015). Intrinsic sequence specificity of the Cas1 integrase directs new spacer acquisition. Elife 4, e087l6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenova E, Jore MM, Datsenko KA, Semenova A, Westra ER, Wanner B, van der Oost J, Brouns SJJ, and Severinov K (2011). Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RnA is governed by a seed sequence. Proc. Natl. Acad. Sci. U. S. A 108, 10098–10103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmakov SA, Sitnik V, Makarova KS, Wolf YI, Severinov KV, and Koonin EV (2017). The CRISPR spacer space Is dominated by sequences from species-specific mobilomes. MBio 8, e01397–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stover CK, Pham XQ, Erwin AL, Mizoguchi SD, Warrener P, Hickey MJ, Brinkman FS, Hufnagle WO, Kowalik DJ, Lagrou M, et al. (2000). Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406, 959–964. [DOI] [PubMed] [Google Scholar]
- Tajkarimi M, and Wexler HM (2017). CRISPR-Cas systems in Bacteroides fragilis, an important pathobiont in the human gut microbiome. Front. Microbiol 8, 2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagih O (2017). ggseqlogo: a versatile R package for drawing sequence logos. Bioinformatics 33, 3645–3647. [DOI] [PubMed] [Google Scholar]
- Zhang Q, and Ye Y (2017). Not all predicted CRISPR-Cas systems are equal: isolated cas genes and classes of CRISPR like elements. BMC Bioinformatics 18, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Doak TG, and Ye Y (2014). Expanding the catalog of cas genes with metagenomes. Nucleic Acids Res. 42, 2448–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Stephens RM, Meltzer PS, and Davis SR (2013). SRAdb: query and use public next-generation sequencing data from within R. BMC Bioinformatics 14, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Putative protospacers within Eggerthella lenta genomes related to Figure 4.
Table S2. Studies included in HuVirDB related to Figure 5.
Table S3. HuVirDB sample metadata related to Figure 5.
Table S4. Putative protospacer sequences in HuVirDB related to Figure 5.
Table S5. E. lenta metagenomic phage genome annotations related to Figure 6.
Table S6. Oligonucleotide sequences used in the study related to STAR Methods.
Data Availability Statement
HuVirDB metadata and related information is available at github.com/jbisanz/HuVirDB and the database itself for download at opengut.ucsf.edu/HuVirDB-1.0.fasta.gz. HuVirDB has been made available in CRISPRTarget as an available database (http://crispr.otago.ac.nz/CRISPRTarget/crispr_analysis.html) for general queries. Genome assemblies are available under the following BioProjects: PRJNA412637, PRJNA384908, PRJNA21093, PRJNA46413, PRJNA40023, PRJNA59527. HMP reference genomes were retrieved from NCBI using BioProject PRJNA28331 (retrieved 23 August 2018). PATRIC reference genomes were retrieved from NCBI on 23 August 2018 by assembly accession as identified in the PATRIC genome catalog (patricbrc.org/view/Taxonomy/2#view_tab=genomes) after filtering for host_name containing human or sapiens, and an isolation_source containing stool, faecal, faeces, fecal, feces, gastrointestinal, gut, intestine, rectal, or rectum.