

Abstract
The complement cascade is an innate immune response to remove pathogens in the peripheral immune system but also plays an important role in microglia-mediated synaptic refinement during brain development. Complement C3 is elevated in Alzheimer’s disease (AD), colocalizing with neuritic plaques, and appears to contribute to Aβ clearance by microglia; however, it is not known whether C3 plays a role in plaque-related synaptic and neuronal health. Previously, we reported that C3-deficient C57BL/6 mice were protected against age-and region-specific hippocampal synapse loss and cognitive decline during normal aging. Furthermore, blocking complement and downstream iC3b/CR3 signaling rescues synapses from Aβ-induced loss in early stages in young Alzheimer’s mice before plaques accumulate. Here, we assessed the effects of C3-deficiency in aged, plaque-rich APPswe/PS1dE9 Tg mice (APP/PS1;C3 KO) on cognition, Aβ plaque deposition and plaque-related neuropathology at later AD stages. We found that 16-month-old APP/PS1;C3 KO mice performed significantly better on a learning and memory task than APP/PS1 mice, despite having more cerebral Aβ plaques. Aged APP/PS1:C3 KO mice also had more ramified-looking microglial and fewer microglia and astrocytes localized within the center of hippocampal Aβ plaques compared to APP/PS1 mice. Several pro-inflammatory cytokines in brain were reduced in APP/PS1;C3 KO mouse brain, consistent with an altered microglial phenotype. C3-deficiency also protected APP/PS1 mice against age-dependent synapse and neuronal loss, even in the context of increased Aβ plaques. Taken together, our studies suggest that complement C3 or downstream complement activation fragments play an important role in plaque pathology, glial response to plaques and neuronal dysfunction in APP/PS1 mouse brains.
One Sentence Summary:
C3-deficiency protects against hippocampal neurodegeneration and cognitive decline in aged AD mice despite the presence of abundant Aβ plaques.
Introduction
The complement cascade, of which C3 is the central molecule, plays a pivotal role in the immune system. Upon activation, classical complement initiating protein C1q binds to and coats dead cells, debris or pathogens, triggering a protease cascade leading to the activation of complement protein C3, which opsonizes material for elimination by triggering phagocytosis of macrophages or leading to the formation of the membrane attack complex, causing cell lysis (1). In addition, complement has been shown to play a role in microglia-mediated synapse elimination in the developing visual system to refine neuronal connections (2–4). We recently demonstrated that C3-deficiency spared age-dependent synaptic and neuronal loss in a region-specific manner, and protected against cognitive impairment in normal aging of C57BL/6 mice (5). Synapse loss occurs early in Alzheimer’s Disease (AD) and correlates with cognitive decline (6, 7). Complement cascade components, including C3 are significantly upregulated and associated with amyloid plaques, typically containing dystrophic neurites, in human AD (8, 9); however, the role of C3 in plaque-related synapse loss and neurodegeneration in AD is not known. Recently, we reported that in early stage AD in mice, pre-plaque Aβ oligomer-induced hippocampal synapse loss was rescued both by genetic deletion of complement and pharmacological treatment with an anti-C1qa blocking antibody (10); however, it is not known if blocking the complement cascade also protects against cognitive impairment and neurodegeneration at later stages in plaque-rich, aged AD mice.
Several complement components including C1q, activated C3 (C3b, C3c, C3d), and C4 (8, 9, 11, 12), may be produced by glia surrounding plaques in AD brain, as Aβ peptides upregulate C3 production by microglia and astrocytes in vitro (13, 14) and C3 expression and/or deposition in AD mouse brain in vivo (10, 15–18). Complement can recruit and activate microglia around fibrillar Aβ deposits (19) and mediates, in part, the uptake and clearance of Aβ by the interaction of complement component iC3b with its receptor CR3 (CD11b/CD18) on the surface of microglia (20). We previously reported that aged C3-deficient J20 hAPP transgenic mice had a higher plaque load, slightly reduced neuron number, and altered macrophage polarization compared to age-matched J20 mice (21). Others reported that C3 inhibition by expression of soluble complement receptor-related protein y (sCrry) in J20 hAPP mice resulted in increased Aβ accumulation, neuronal degeneration and reduced microglia activation (22). In contrast, C1q-deficiency resulted in protection of synapses and neuropathology with decreased glial activation surrounding plaques in two other AD amyloidosis models, Tg2576 and APP/PS1 mice (23). Inhibition of C5a, a C3 downstream activation fragment, using a C5aR antagonist resulted in rescue of a hippocampal-dependent memory task (24). However, none of these previous reports addressed whether complement C3 plays a role in cognitive health. As such, the consequences of C3 in the aging AD brain remain unclear, in regard to both their roles in plaque-related synaptic and neuronal health.
Here, we generated APP/PS1;C3 KO to ask whether C3 plays a role in plaque-related neuropathological changes and cognitive decline. We demonstrate that C3-deficiency in APP/PS1 mice protects against age-related plaque-related synapse and neuron loss, decreases glial reactivity, and spares cognitive decline, despite an increased plaque burden.
Results
Sparing of cognitive decline in plaque-burdened aged APP/PS1;C3 KO mice
Aged APP/PS1 mice (>15 months of age) have been shown to exhibit hyperactivity in the Open Field and impaired learning and spatial memory in the Radial Arm/Morris Water Maze compared with non-transgenic mice (25). To determine whether C3-deficiency affects age-related cognitive changes in mice, we assessed learning and memory using the Water T-Maze (WTM) test which measures spatial learning and memory during acquisition and cognitive flexibility during reversal learning (26), in 16-month-old WT, APP/PS1, APP/PS1;C3 KO and C3 KO mice. Although APP/PS1 mice showed significant impairment in finding the platform on acquisition Day 2 compared to WT mice (p < 0.05; Fig. 1A), there were no significant differences in the percent of mice that reached criterion (≥80% correct choices on each individual day) between genotypes on Day 6, indicating that all groups eventually learned the task (Fig. 1A). Acquisition in APP/PS1;C3 KO mice was between that of WT mice and APP/PS1 mice, indicating a non-significant trend that APP/PS1;C3 KO mice learned the location of the hidden platform more quickly than APP/PS1 mice. During the reversal trials, in which the platform location is switched, APP/PS1 mice demonstrated cognitive impairment by performing significantly worse than WT mice on Days 3–5 (Fig. 1A). Reversal learning in APP/PS1;C3 KO mice was similar to that of WT mice and C3 KO mice, and was significantly better than that of APP/PS1 mice on Days 4 and 5 (Fig. 1A), suggesting that aged APP/PS1;C3 KO mice had enhanced spatial learning and cognitive flexibility compared to APP/PS1 mice.
Fig. 1.

APP/PS1;C3 KO mice showed a significant improvement in cognitive flexibility (reversal) compared to APP/PS1 mice at 16 months of age. A. Percent of mice that reached criterion (≥ 80% correct choices on each individual day) in WTM test. Compared to WT mice, APP/PS1 mice were impaired in acquisition (Days 2) and reversal learning and memory (Days 4 and 5) (*p < 0.05; ** p < 0.01). APP/PS1;C3 KO mice performed significantly better than APP/PS1 mice (## p < 0.01), but similar to WT and C3 KO mice, in the reversal test on Days 4 and 5, suggesting better flexibility in APP/PS1;C3 KO mice compared to APP/PS1 mice. B. In total, fewer APP/PS1 mice reached the reversal criterion (≥80% correct choices over two consecutive days) (*p < 0.05), while the percent of WT, C3 KO and APP/PS1;C3 KO mice that reached criterion in the reversal test was significantly higher compared to APP/PS1 mice (***p < 0.001), indicating that C3-deficiency in APP/PS1 mice had both age-dependent and AD-related effects (WT, n=13; APP/PS1, n=11; APP/PS1;C3 KO, n=10; C3 KO, n = 11). Tests were assessed using one-way ANOVA followed by Fisher’s PLSD post hoc test.
The percent of mice that reached reversal criterion across all days (≥80% correct choices on two or more consecutive days) was significantly higher in WT, C3 KO and APP/PS1;C3 KO mice compared to APP/PS1 mice (Fig. 1B). Importantly, fewer APP/PS1 mice reached reversal criterion compared to APP/PS1;C3 KO mice, whereas C3 KO, APP/PS1;C3 KO and WT mice achieved equivalent reversal criterion, suggesting that C3-deficiency in the APP/PS1 mice protected against both age-and AD-related cognitive impairment.
Consistent with previous reports (25), we found that 16-month-old APP/PS1 mice traveled a significantly greater distance in the Open Field (OF) compared to WT mice (Fig. S1A). Overall, C3-deficiency in APP/PS1 mice had no significant effect on OF distance traveled (Fig. S1A). We also conducted the Elevated Plus Maze (EPM) test to examine anxiolytic-like behavior in 16-month-old mice and found that APP/PS1;C3 KO mice showed anxiolytic-like behavior compared to APP/PS1 mice (Fig. S1 B, C) that was not due to differences in locomotor activity (Fig. S1 D). In summary, our behavioral studies demonstrate that cognitive decline was at least partially spared in APP/PS1;C3 KO mice.
Elevated cerebral Aβ plaque deposition and insoluble Aβ levels in APP/PS1;C3 KO mice
To determine whether memory improvement in APP/PS1;C3 KO mice was due to decreased Aβ levels, we examined the Aβ plaque load of 4-month-old and 16-month-old APP/PS1;C3 KO mice and APP/PS1 mice. No significant differences in Aβ plaque burden were observed between 4-month-old APP/PS1 and APP/PS1;C3 KO mice at the earliest stages of plaque deposition (Fig. S2). However, by 16 months of age, Aβ plaque deposition was elevated in both the pre-frontal cortex (PFC) and hippocampus (HC) in APP/PS1;C3 KO mice compared to age-matched APP/PS1 mice (Fig. 2A,B; S3). In PFC, plaque deposition was significantly increased by 167.37% for Aβ1-x (3D6; Fig. 2A). In HC, plaque deposition was significantly increased by 63.4% for Aβx-42, 64.1% for Aβx-40, and 65.4% for Aβ1-x (Fig. 2B). Furthermore, we quantified the area of immunoreactivity (IR) for different sizes of Aβx-42-labeled plaques. Larger plaques (>50 μm) were dramatically increased by 127.0% (p < 0.01), whereas small (<20 μm) and medium (20–50 μm) plaques were modestly but significantly increased by 28.7% and by 56.0%, respectively, in the HC of APP/PS1;C3 KO mice compared to APP/PS1 mice (Fig. 2C, D). Thioflavin S-positive staining of fibrillar amyloid deposits was significantly elevated in the HC of APP/PS1;C3 KO mice (Fig. 2C,E).
Fig. 2.

Increased Aβ plaque deposition and Aβ levels by ELISA in 16 month-old APP/PS1;C3 KO mice. A,B. Quantification of Aβx-42, Aβx-40 and Aβ1-x (3D6) IR revealed increased plaque burden in cortex (A) and hippocampus (B) of 16-month-old APP/PS1;C3 KO mice compared to APP/PS1 mice. (* p < 0.05, ** p < 0.01 vs. APP/PS1 mice; n= 9 mice; 6 equidistant planes 150 μm apart; independent unpaired t-test per Aβ species). C. Aβx-42 IR and Thioflavin-S-positive plaque load was higher in the hippocampus of C3-deficient APP/PS1 mice vs. APP/PS1 mice. White circles indicate large plaques (> 50 μm); arrows indicate medium size plaques (> 20 but < 50 μm ). Scale bar = 50 μm. D. Quantification of small, medium and large hippocampal plaques confirmed an increased plaque load in APP/PS1;C3 KO mice, especially for large plaques (* p < 0.05, ** p < 0.01; n = 10; independent unpaired t-tests per plaque size category). E. Quantification of Thio-S in hippocampus confirmed an increase in fibrillar plaques in APP/PS1; C3 KO mice (* p < 0.05 vs APP/PS1 mice; n=6; 3 equidistant planes 300 μm apart; unpaired t-test). F. Increased insoluble cerebral Aβx-40 and Aβx-38 levels were found in APP/PS1;C3 KO mice compared with APP/PS1 (* p < 0.05; n=8) (independent unpaired t-tests per Aβ species).
Aβ ELISAs of hemibrain homogenates revealed that guanidine-soluble (T-per insoluble) Aβx-40 and Aβx-38 levels were significantly higher in APP/PS1;C3 KO mice compared to APP/PS1 mice (Fig. 2F). These data are in agreement with the corresponding increase of Aβx-40 and Aβ1-x plaque loads. These results indicate that the absence of a key complement factor, C3 and/or its downstream activation products in APP/PS1 Tg mice resulted in an age-dependent increase in the accumulation of cerebral Aβ plaques and insoluble Aβ in aged APP/PS1 mice.
Altered plaque-associated gliosis in aged APP/PS1;C3 KO mice
Microglia and astrocytes are frequently associated with plaques in AD brain. Therefore, we examined colocalization of these cells with plaques in mouse brain by immunostaining for Iba-1 and CD68 (microglia/macrophages) and GFAP (astrocytes). Immunoreactivities (IR) to Iba-1 and GFAP were similar between APP/PS1;C3 KO and APP/PS1 mice at P30 and 4 months of age (Fig. S4). However, 16-month-old APP/PS1 Tg mice displayed microglia/macrophage with enlarged cell bodies and thicker, shorter processes with less branching, consistent with the morphological changes associated with reactive microgliosis, compared to Iba-1 IR and CD68 IR cells in APP/PS1;C3 KO mice (Fig. 3A; Fig S5A,B). In addition, GFAP-IR astrocytes were clustered in HC of APP/PS1 mice but more widely distributed and less clustered around plaques in APP/PS1;C3 KO mice (Fig. 3A). Quantitative image analysis revealed that Iba-1-IR (% area) was significantly reduced by 28.8% in hippocampal CA3 and slightly reduced by 19.3% (non-significant trend) in CA1 in APP/PS1;C3 KO mice, but no significant differences were seen in DG (Fig. 3B). Additionally, CD68 IR, indicative of lysosomal phagosomes, was significantly reduced in hippocampal CA3, CA1 and DG in APP/PS1;C3 KO mice (Fig. 3C). GFAP IR of astrocytes in CA3, CA1 and DG was also significantly reduced in APP/PS1;C3 KO mice (Fig. 3D). Further, immunofluorescent intensities of Iba-1 IR and CD68 IR in Aβ plaques of similar size was significantly reduced in hippocampal CA3 of 16-month-old APP/PS1;C3 KO mice compared to APP/PS1 mice (Fig. 3E,F). While glial cell morphology was different, stereological analysis revealed that the number of Iba-1-positive and GFAP-positive cells in hippocampal CA3, CA1 and DG areas was not significantly different between aged APP/PS1;C3 KO mice and APP/PS1 mice (Fig. 3G,H).
Fig. 3.
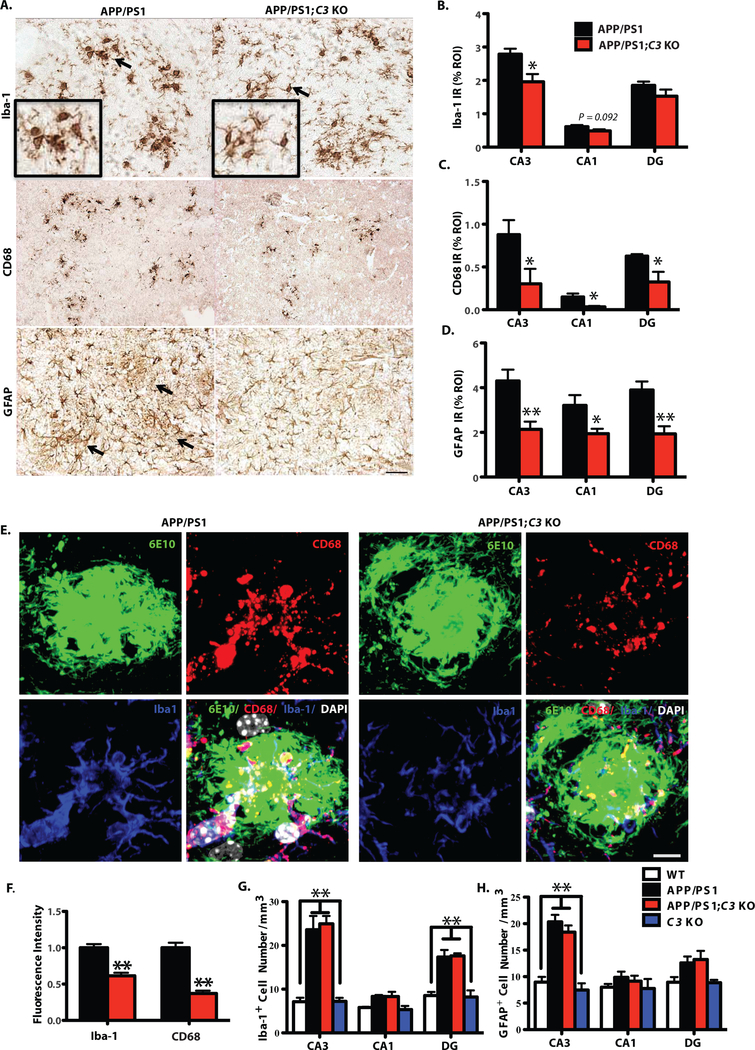
Morphological changes associated with glial activation were reduced in 16-month-old APP/PS1;C3 KO mice. A. Iba-1 IR and CD68 IR show less microglia and macrophage activation (i.e. smaller cells with thinner processes) and less GFAP IR astrocytic clustering in hippocampal CA3 in 16-month-old APP/PS1;C3 KO mice compared to APP/PS1 mice. Scale bar = 50 μm. B-D. Quantification of Iba-1, CD68 and GFAP IR in hippocampal CA3, CA1 and DG shows reduced glial IR in APP/PS1;C3 KO mice compared to APP/PS1 mice (* p < 0.05, ** p < 0.01, n=6–8; 3 equidistant planes, 300μm apart; independent unpaired t-tests per region). E. High-resolution confocal images of Aβ plaques (6E10), microglia/macrophages (Iba-1) and phagocytic cells (CD68) in hippocampal CA3 indicate reduced phagocytosis in APP/PS1;C3 KO mice compared to APP/PS1 mice. Scale bar: 10 = μm. F. Iba-1+ and CD68+ immunofluorescent intensities were significantly lower in APP/PS1;C3 KO mice compared to APP/PS1 mice (** p < 0.01, n=5, independent unpaired t-tests per marker). G.H. Stereological counts of Iba-1 IR (G) and GFAP IR (H) cells (DAB staining) were performed in hippocampal CA3, CA1 and DG. The number of Iba-1 IR microglia/macrophage cells was increased in CA3 and DG in APP/PS1 and APP/PS1;C3 KO mice vs. WT and C3 KO mice (G), whereas the number of GFAP IR astrocytes was increased only in CA3 (H); however, no differences were observed in glial cell numbers between APP/PS1 and APP/PS1;C3 KO mice (** p < 0.01; n=6–8; 3 equidistant planes, 300 μm apart; one-way ANOVA with Bonferroni post-hoc test per region).
Next, glial cell association within and surrounding Aβ plaques was examined to further elucidate a possible mechanism underlying the C3-deficiency-mediated increase in hippocampal Aβ plaque burden in APP/PS1;C3 KO mice. We quantified the immunofluorescent intensity and the number of microglia/macrophage (Iba-1) and astrocytes (GFAP) surrounding Aβ plaques (6E10) of similar size in hippocampal CA3 of 16-month-old APP/PS1;C3 KO mice and APP/PS1 mice using high resolution Z-stack imaging by confocal microscopy (Fig. 4A–L). Orthogonal analysis revealed that Iba1 IR cells and Aβ plaques co-localized (Fig. S5A). 3-D reconstructed images were analyzed for intensity and cell number in the proximal area (within the center of large plaques) and distal area (surrounding large plaques). We observed more Iba-1 IR and GFAP IR cells in the center of plaques in APP/PS1 mice whereas Iba-1 IR and GFAP IR cells tended to surround the plaques in APP/PS1;C3 KO mice (Fig. 4A,G) and were more widely distributed. Upon quantification, we found that the intensity of Iba-1 IR cells in APP/PS1;C3 KO mice was significantly reduced proximal to plaques and significantly increased distal to plaques compared to APP/PS1 mice (Fig. 4C,D). Similarly, the intensity of GFAP IR astrocytes in APP/PS1;C3 KO mice was significantly reduced in the proximal area of plaques and significantly increased distal to plaques compared to APP/PS1 mice (Fig. 4I,J). Although the number of glial cells located within plaques (proximal) was significantly decreased (Fig. 4E,K) while those surrounding plaques (distal) were increased (Fig. 4F,I) in APP/PS1;C3 KO mice, there was no difference in the total number of glial cells in hippocampal CA3 in APP/PS1;C3 KO vs. APP/PS1 mice (Fig. 3G,H). Thus, even though the APP/PS1;C3 KO mice had enhanced plaque deposition, they also had reduced immunofluorescent intensity and localization of glia within the center of plaques, suggesting that C3-deficiency alters the glial response to plaques.
Fig. 4.

Plaque-associated microglia and astrocytes and brain cytokine levels were altered in APP/PS1;C3 KO mice compared to APP/PS1 mice. A,B,G,H: High-resolution confocal images of Iba-1 (red)/6E10 (green)/DAPI (blue) or GFAP (red)/6E10 (green)/DAPI (blue) in APP/PS1 and APP/PS1;C3 KO mice. The inner ring indicates the proximal region of a plaque (i.e. the center) while the outer ring indicates the distal region. Scale bar = 10 μm C,D,I,J: Iba-1+ and GFAP+ intensities were lower in the Aβ plaque proximal area (C, I) and higher in the Aβ plaque distal area (D, J) in APP/PS1;C3 KO mice compared to APP/PS1 mice (*p < 0.05, **p < 0.01, n=6, unpaired t-test). E,F,K,L: The number of Iba-1+ and GFAP+ cells was reduced in the proximal plaque area in APP/PS1;C3 KO mice compared to APP/PS1 mice (E,K) (*p < 0.05) and increased in the distal plaque area (F,L) (*p < 0.05, n=6, unpaired t-test). M. Cytokine ELISA of brain homogenates revealed significant reductions in TNF-α, IFN-γ, and IL-12 and an increased IL-10/IL-12 ratio in 16 month-old APP/PS1;C3 KO mice compared to APP/PS1 mice (* p < 0.05; n=8; independent unpaired t-test per marker followed by Bonferroni correction for multiple comparisons).
In a previous study, we found that CD45 IR was increased in aged J20; C3 KO mice compared to J20 hAPP Tg mice. However, the Iba1 IR and CD68 Western blot levels were not significantly different between J20; C3 KO and J20 mice (15). Here, we performed immunostaining of Iba-1 and CD68 on brains from 18-month-old APP/PS1, APP/PS1; C3 KO, J20, and J20; C3 KO mice. Interestingly, in aged J20 mice, both Iba1 IR and CD68 IR within Aβ plaques were significantly lower than those in aged APP/PS1 mice (Fig. S6A–D). We observed a consistent decrease in Iba1 IR and CD68 IR within Aβ plaques in APP/PS1; C3 KO mice compared to APP/PS1 mice (Fig. S6E,G), but no difference between J20 and J20; C3 KO mice (Fig. S6F,G), consistent with our previous study (16).
Altered cytokines in aged APP/PS1;C3 KO mouse brain
ELISA of T-per soluble brain homogenates revealed that pro-inflammatory cytokines including Tumor Necrosis Factor alpha (TNF-α), Interferon gamma (IFN-γ) and Interleukin 12 (IL-12p70), which have been reported to be elevated in AD brain (27), were significantly reduced in 16-month-old APP/PS1;C3 KO mice compared to APP/PS1 mice (Fig. 4M). The level of Interleukin-10 (IL-10) did not differ between APP/PS1 mice and APP/PS1;C3 KO mice, while the ratio of IL-10/IL-12 was significantly increased in APP/PS1;C3 KO mice compared to APP/PS1 mice (Fig. 4M). Thus, C3-deficiency resulted in the reduction of several pro-inflammatory cytokines in aged AD mice.
Partial protection against age-related hippocampal synaptic degeneration in APP/PS1;C3 KO mice
Decreased synaptic number, mRNA and protein as well as impaired synaptic morphology have been reported in hippocampus and cortex of APP/PS1 mice between 7 and 18 months of age (28–30). To determine the effects of C3 on hippocampal synapses in aged APP/PS1 mice, we performed high resolution confocal puncta analysis of pre-and post-synaptic markers in hippocampal CA3, CA1 and DG in 16-month-old mice and compared WT vs. C3 KO, WT vs. APP/PS1, APP/PS1 vs. APP/PS1;C3 KO and C3 KO vs. APP/PS1;C3 KO mice (Fig. 5). Consistent with our previous results (5), C3 KO mice had significantly more synaptic puncta than WT mice. In addition, C3 KO mice had more pre-synaptic Vglut2 puncta and more post-synaptic GluR1 puncta in than APP/PS1 and APP/PS1;C3 KO mice, although co-localized synaptic puncta were not significantly different between C3 KO and APP/PS1;C3 KO mice. We found that the densities of post-synaptic puncta (GluR1) and co-localized puncta (Vglut2 and GluR1) were significantly reduced in hippocampal CA3 in 16 month-old APP/PS1 mice compared to WT mice (Fig. 5A–C). Interestingly, the density of synaptic puncta was significantly increased in CA3 of APP/PS1;C3 KO mice compared to APP/PS1 mice and normalized to WT levels, suggesting protection against age-related synapse loss (Fig. 5H). Western blotting of pre-synaptic markers, Synapsin-1 (SYN-1) and Synaptophysin (SYP), and post-synaptic markers, GluR1, PSD95 and Homer1, in hippocampal synaptosomes of 16-month-old WT, C3 KO, APP/PS1, and APP/PS1;C3 KO mice revealed that C3 KO mice had elevated levels of GluR1, PSD95 and Homer1 compared to all other groups and APP/PS1 mice had reduced GluR1, PSD95, Homer1, SYN-1 and SYP compared to WT mice (Fig. 5D,E,H). APP/PS1;C3 KO mice had significantly increased levels of all five synaptic proteins compared to APP/PS1 mice and were similar to the levels detected in WT mice (Fig. 5D,E). In addition, microtubule-associated protein 2 (MAP2) IR, a marker for neuronal cell body and dendrites, was significantly reduced within fibrillar Aβ plaques (Thio S positive) and less compacted Aβ plaques (6E10 positive) in 16-month-old APP/PS1 mice, but was much less reduced in similar sized plaques in CA3 of age-matched APP/PS1;C3 KO mice (Fig. S7).
Fig. 5.

C3-deficiency resulted in partial preservation of synapse density and synaptic protein levels in APP/PS1 mice in spite of an increased plaque load. Comparisons were made between WT vs. C3 KO, WT vs. APP/PS1, APP/PS1 vs. APP/PS1;C3 KO, and C3 KO vs. APP/PS1;C3 KO (H). A. Synaptic puncta of pre-and post-synaptic markers Vglut2 and GluR1, respectively, and their colocalizaton in hippocampal CA3 were analyzed by high resolution confocal microscopy in 16-month-old mice. Scale bar = 5 μm. B. C3 KO mice had increased Vglut1 and GluR1 synaptic densities compared to WT, APP/PS1 and APP/PS1;C3 KO mice. APP/PS1 mice had significantly fewer GluR1 synaptic densities than WT mice while APP/PS1;C3 KO mice had significantly more GluR1 densities than APP/PS1 mice and were not significantly different than WT mice (* p < 0.05, ** p < 0.01; n = 6–8; 3 equidistant planes, 300 μm apart; one-way ANOVA and Bonferroni post-hoc test per marker). C. Colocalization of pre-and post-synaptic puncta reveals increased puncta in C3 KO vs. WT and APP/PS1 but not APP/PS1;C3 KO mice, reduced puncta in APP/PS1 mice vs. WT mice, and a rescue of synaptic puncta in APP/PS1;C3 KO mice compared to APP/PS1 mice, suggesting partial protection against synapse loss by C3-deletion (one-way ANOVA and Bonferroni post-hoc test). D,E. Western blotting of synaptic proteins in hippocampal synaptosomes isolated from aged mice indicates increased post-synaptic proteins GluR1, PSD95 and Homer 1 in C3 KO vs WT, APP/PS1 and APP/PS1;C3 KO mice. APP/PS1 mice had significantly lower post-synaptic GluR1, PSD95 and Homer1 and pre-synaptic SYN-1 and SYP compared to WT mice. APP/PS1;C3 KO mice had significantly more GluR1, PSD95, Homer1, SYN-1 and SYP than APP/PS1 mice, and were not significantly different than WT mice, suggesting a sparing of synaptic loss and normalization to WT levels in aged C3-deficient APP/PS1 mice (* p < 0.05; ** p < 0.01; n = 6; one-way ANOVA and Bonferroni post-hoc test per marker). F,G. Western blotting and quantification of TrkB, mBDNF, CREB, p-CREB in hippocampal homogenates of 16 month-old mice. C3 KO mice had increased mBDNF and p-CREB levels compared to WT, APP/PS1 and APP/PS1;C3 KO mice. APP/PS1 mice had significant reductions in all 4 markers compared to WT mice. APP/PS1;C3 KO mice had significantly higher mBDNF, CREB and pCREB than APP/PS1 mice, and all 4 markers were normalized to WT mouse levels (* p < 0.05, ** p < 0.01; n = 6; one-way ANOVA and Bonferroni post-hoc test per marker), suggesting a partial rescue of age-and/or AD-related lowering of BDNF pathway proteins. H. Table summarizing the effects of C3-deficiency on synaptic and BDNF-related proteins.
Furthermore, mature brain-derived neurotrophic factor (m-BDNF) and its upstream (TrkB) and downstream (CREB, p-CREB) signaling partners, key mediators of synaptic plasticity and memory, were examined by Western blot of brain homogenates and quantified (Fig. 5F,G,H). C3 KO mice had higher mBDNF and pCREB levels than WT, APP/PS1 and APP/PS1;C3 KO mice. APP/PS1 mice had reduced TrkB, m-BDNF, CREB and p-CREB compared to WT mice. APP/PS1;C3 KO mice had significantly higher m-BDNF, CREB and p-CREB levels compared to APP/PS1 mice but lower m-BDNF and pCREB than C3 KO mice. No significant differences were seen in CREB levels between C3 KO and APP/PS1;C3 KO mice. This result correlates well with our cognitive and synaptic puncta data and suggests a pro-cognitive health phenotype in the APP/PS1;C3 KO mice. Thus, while 16-month-old APP/PS1 mice demonstrated hippocampal CA3 synapse loss, APP/PS1;C3 KO mice were at least partially spared synapse loss, which was associated with an increase in BDNF signaling pathway proteins. We also examined the levels of human APP and Presenilin 1 in APP/PS1 and APP/PS1; C3 KO mice and found no significant differences in either protein between the two genotypes (Fig. S8).
Absence of age-dependent hippocampal CA3 neuron loss in APP/PS1:C3 KO mice
As previously reported (5), there was a small (28%) but statistically significant age-dependent reduction in the number of NeuN+ neurons in hippocampal CA3 from P30 to 16 months of age in WT mice that was rescued in C3 KO mice (p < 0.05; Fig 6F). A stronger (40%) reduction in CA3 neurons was seen from P30 to 16 months of age in APP/PS1 mice (p < 0.01; Fig. 6F). Importantly, APP/PS1;C3 KO and C3 KO mice showed no significant age-or plaque-related loss of hippocampal CA3 neurons (Fig. 6F). Instead, 16-month-old APP/PS1;C3 KO mice had significantly more hippocampal CA3 neurons compared to age-matched APP/PS1 mice (Fig. 6A,B). No differences in neuron numbers were found in CA1, DG and PFC in APP/PS1;C3 KO mice compared to APP/PS1 mice (Fig. 6A,C–E). In addition, CA3 neurons were disorganized in 16-month-old APP/PS1 mice but not in APP/PS1;C3 KO mice (Fig. 6A). These data suggest that hippocampal CA3 neurons are more vulnerable to aging than neurons in other brain regions in both WT and APP/PS1 mice, and APP/PS1;C3 KO mice are protected against this age-related neuronal vulnerability even in the presence of abundant plaques.
Fig. 6.

C3-deficiency resulted in partial sparing of neuron loss in hippocampal CA3 in 16-month-old APP/PS1 mice. A. NeuN IR in hippocampal CA3, CA1 and DG and PFC in APP/PS1 and APP/PS1;C3 KO mice. Scale bar: 50 μm. B. APP/PS1;C3 KO mice had more neurons in hippocampal CA3 compared to APP/PS1 mice (*p < 0.05, unpaired t-test). C-E. No significant differences were observed in neuron numbers in CA1, DG and PFC between APP/PS1 and APP/PS1;C3 KO mice. F. Age-dependent neuron loss was observed in WT and APP/PS1 mice from P30 to 16 months of age, and in APP/PS1 mice from 4 months to 16 months of age, but not in C3 KO or APP/PS1;C3 KO mice. (* p < 0.05, ** p < 0.01; n=6–8; 6 equidistant planes 150 μm apart, two-way ANOVA and Bonferroni post-hoc test).
Discussion
Complement C3 is an innate immune system molecule that is important for removing pathogens and eliminating synapses during brain development and aging (2–5). Complement activation is elevated in human AD brain, especially in the presence of fibrillar amyloid plaques containing clusters of reactive microglia (8, 9, 11, 12), and in brain of AD amyloidosis mouse models (10, 17, 31). To address the role of complement C3 in age-dependent, plaque-related neuropathology, we generated APP/PS1;C3 KO mice and analyzed the role of C3 in brain biochemistry, neuropathology and behavior in 16-month-old mice. We demonstrate that C3-deficiency protected against cognitive impairment, loss of hippocampal CA3 synapses and neurons, and glial alterations, even in the context of increased Aβ plaque deposition and, normalized APP/PS1 mice to WT mouse levels. Our results suggest that C3-deficiency protects against both age-and AD-related synapse loss and cognitive decline in aged APP/PS1 mice, despite extensive plaque deposition, by altering the glial response to plaques. These data indicate that plaque load is less critical than the reaction of glia to the plaques, and implicate complement activation as a possible link between fibrillar amyloid and accompanying microglial clustering in late stage AD pathogenesis in mice, similar to that observed in human AD brain.
Progressive cognitive impairment in water maze tests is observed in aged APPswe/PS1dE9 mice (an AD-like mouse model of amyloidosis) and is correlated with progressive Aβ deposition, neuroinflammation and degeneration of synapses and neurons (25, 32). Here, we found that APP/PS1;C3 KO mice were significantly protected against cognitive deficits, especially during WTM reversal, a hippocampal-dependent task (20). This result is in agreement with previous reports, including our own, demonstrating that C3 KO mice are protected against synapse and neuron loss and spared cognitive impairment during normal aging (5, 33). While many of our results in the current study point to age-dependent protection by C3-deficiency in aged APP/PS1 mice, we also demonstrate some AD-related protection by C3-deficiency as there were no significant differences between APP/PS1;C3 KO, WT and C3 KO mice in the percent of mice that reached the reversal criterion in the WTM; similar AD-related protection was also seen for hippocampal SYP and CREB levels. In addition, inhibition of complement C5aR, downstream of C3 activation, with an antagonist has been reported to reduce Aβ pathology and improve cognitive function in Tg2576 AD mice (24), raising the possibility that the protective cognitive effects we observed in aged C3-deficient APP/PS1 mice might be due to the lack of downstream C5a/C5aR signaling. Synaptic degeneration is correlated to cognitive decline in AD (6, 7), therefore our observations of cognitive sparing in aged APP/PS1;C3 KO mice may be due the preservation of hippocampal synapses and neurons, even in the presence of increased plaque load. Whether the protective effects of C3-deficiency overpower the toxic effects of Aβ or cause sequestration of Aβ into plaques remains to be determined. Neuroinflammation (e.g., glial clustering and activation within plaques) was reduced in APP/PS1;C3 KO mice, suggesting that C3 may play a critical role in driving neuroinflammation, which facilitates synaptic decline.
In the brain environment undergoing injury or disease, chronic activation of microglia may contribute to synaptic degeneration and memory decline (34). Elimination of microglia and inhibition of microglial proliferation by inhibiting colony-stimulating factor 1 receptor (CSF1R) have been reported to prevent spine and neuronal loss, and improve memory performance in AD mouse models, without significant modulation of Aβ pathology (35, 36). In addition, we recently demonstrated that complement and microglia mediate synapse loss in AD mouse models at early AD stages prior to plaque deposition (10), whereas our data here demonstrate that complement plays an additional role in the glial response to amyloid deposition and synaptic health at later AD stages as well. Furthermore, early C1q-mediated microglial activation and neurodegenerative changes in retina observed in a mouse model of retinal ischemia and reperfusion was abrogated in C1qa KO mice (37). Similarly, the loss of retinal ganglion cell synapses and dendrites (which precede axon and soma loss) in a genetic mouse model (DBA/2J) and an inducible rat model (microbead) of glaucoma were rescued by genetic deletion of C1qa in the mouse model and C1 inhibition in the rat model (38). Taken together, these findings suggest that inhibition of complement and its interaction with microglia may serve as a target to prevent the progression of synapse and neuron loss in Alzheimer’s disease.
Complement C3 plays a pivotal role in plaque deposition and clearance (20); however it is not known whether C3 mediates the glial response in plaque-enriched brain. Here we show that C3-deficiency in aged APP/PS1 mice decreased microglial CD68 activity, maintained microglial branching and increased plaque load, while preserving cognition and hippocampal CA3 synapses and neurons. Both C3-deficiency and inhibition of C3 convertase by overexpression of soluble complement receptor-related protein y (sCrry) in J20 hAPP Tg mice led to an age-dependent increase in plaque burden (21, 22). In agreement with these studies in J20 mice, we found that aged APP/PS1;C3 KO mice had enhanced plaque deposition and increased insoluble Aβ in brain homogenates. Microglial uptake and clearance of Aβ (i.e. phagocytosis) may be mediated by the interaction of iC3b with its receptor, CR3 (CD11b/CD18) on the surface of microglia (20, 39). Previous studies have reported the reduction of microglial markers F4/80, I-A/I-E and/or CD68 in C1q-deficient Tg2576 APP mice (23), J20 hAPP/sCrry mice (22) and J20 hAPP;C3 KO mice (21). Here, we also found that microglia/macrophage and astrocyte markers were reduced in aged APP/PS1;C3 KO mice compared to APP/PS1 mice, even though there was no difference in glial cell number. Moreover, we found fewer microglia and astrocytes located within plaques, suggesting that C3-deficiency alters the glial response to plaques in aged, plaque-rich APP/PS1;C3 KO mice. Thus, these findings suggest that the increased fibrillar Aβ plaque load in the aged APP/PS1;C3 KO mice is due to a muted glial response and reduced Aβ phagocytosis.
Senile plaques have been proposed to act as a potential reservoir of soluble oligomeric Aβ which when non-sequestered, colocalizes with the postsynaptic density, is associated with dendritic spine collapse, and is the major synaptotoxic Aβ species in the AD brain (40). Therefore, in theory, it is possible that while C3-deficiency reduced microglia-mediated phagocytosis of Aβ and increased plaque deposition in APP/PS1 mice, it also may have resulted in more sequestration of Aβ oligomers into plaques. This might reduce the availability of Aβ oligomers to bind to synapses, thereby blocking microglia activation-induced synapse loss in the hippocampus in aged APP/PS1;C3 KO mice. This possibility would be consistent with our previous findings that genetic deletion or antibody-blocking of C1qa protected hippocampal synapses from Aβ oligomer-induced damage (8). While purely speculative at this stage, future studies are underway to further clarify this possibility.
Complement C3 may contribute to region-specific and age-dependent synapse and neuron loss in AD mice via C3-dependent chronic microglial activation. Complement is involved in synaptic elimination in brain development and aging. Upon activation, complement mediates synapse elimination in the developing visual system by complement-tagging of synapses in a microglia-mediated phagocytosis manner (2, 3). Complement C3 contributes to age-dependent and region-specific synapse and neuron loss during normal aging (5). Synapse loss is an early and important change in AD brain (41), and hippocampal synapse loss is observed in APP/PS1 mice (28, 29). While both plaque-associated and plaque-independent hippocampal synaptic degeneration has been observed in aged APP/PS1 mice (42), we found reduced synaptic degeneration despite increased plaque load in aged APP/PS1;C3 KO mice compared to APP/PS1 mice, indicating that the presence of plaques alone did not induce neurodegenerative changes. Since microglia and astrocytes, both of which express complement, are highly phagocytic and participate in synapse pruning (10, 43), our findings that C3-deficiency protected against hippocampal synaptic decline in two mouse models (C3 KO and APP/PS1;C3 KO) suggests that complement C3 or its downstream activation fragments, such as C3a, C5a and C5b-9, play an important role in synapse loss and neurodegeneration. Other reports support this hypothesis. For example, a recent report suggests that complement C3a secretion by astrocytes and binding to C3aR on microglia modulates Aβ levels in vitro and amyloid plaques in vivo in AD mouse models (44). The same group previously demonstrated that Aβ-induced NFκB-mediated release of C3a from astrocytes bound C3aR on neurons and induced neurodegeneration and cognitive impairment while treatment with a C3aR antagonist rescued these effects (14). Others have reported that low-dose treatment with the same C3aR antagonist in an ischemia/reperfusion stroke model in C57BL/6 mice resulted in enhanced neurogenesis, histologic and functional neuroprotection, and an anti-inflammatory effect (e.g., the absence of T lymphocytes expressing C3aR migrating into the ischemic region) 7 days post-injury (45). However, several caveats of the C3aR antagonist including potential off-target and agonistic activity have been reported (46), leaving the role C3a/C3aR signaling in neurodegeneration yet to be fully understood.
Previously, we reported an age-dependent loss of CA3 neurons in 16 month-old C57BL/6 mice (5). Age-dependent neuron loss in APPswe/PS1dE9 mice has been found in striatum (47), but not cortex (48) or hippocampal CA1 (30). Here, we report for the first time, a significant age-dependent loss of hippocampal CA3 neurons in the same APP/PS1 model, which was spared in the absence of complement C3. The preservation of neurons was selective for hippocampal CA3 as no neuron loss was observed in CA1, DG and PFC in aged APP/PS1;C3 KO mice. It is possible that the selective vulnerability of CA3 neurons during aging in APP/PS1 mice is due to early CA3 synapse loss preceding late neuron loss induced by C3-mediated synaptic elimination, similar to what we found in aged WT mice (5). CA3 neurons receive input from other CA3 neurons through recurrent connections as well as medial septum and the diagonal band of Broca, both of which show degenerative changes during aging (49, 50). Interestingly, C1qa-deficient Tg2576 hAPP mice were shown to have increased iC3b deposition in brain, reduced gliosis and neuron loss in several brain regions, and partial protection of SYN IR and MAP2 IR synapses and dendrites, respectively, in the hippocampal CA3 region, suggesting both selective regional vulnerability of synapses and differential effects of various complement pathways on synapses in brain (23).
It has been shown that microglia engulf presynaptic inputs via the C3/CR3-dependent microglia phagocytic signaling pathway in the developing early postnatal brain (3). Recently, Hong et al. reported similar Aβ oligomer-induced, complement-mediated and microglia-dependent synaptic loss in young, pre-plaque AD mice that was rescued by the genetic deletion of C1q or C3, or by treatment with a C1qa blocking antibody (8). This study strongly implicates a role for complement in oligomeric Aβ-induced synaptic pathology at early AD stages prior to plaque deposition. Here, we demonstrate a role for complement C3 signaling in Aβ fibril-induced clustering and activation of microglia within plaques at a much later disease stage in aged APP/PS1 mice. Aβ-induced complement activation is strongly determined by the degree of Aβ fibrillarity (51, 52). In humans, early, non-fibrillar diffuse plaques are associated with little or no microglial clustering (53) and accumulate little or no complement proteins (8, 9). Complement has been shown to promote the nucleation phase of Aβ aggregation (54), suggesting that once activated, complement may perpetuate AD pathogenesis. Indeed, at later stages of AD pathogenesis in humans, various complement proteins and clusters of activated microglia have been found to colocalize with fibrillar amyloid plaques, often containing dystrophic neurites (8, 9). Our current study supports the role of complement in the link between Aβ fibrillarity and clustering of activated microglia, and shows that lifelong C3-deficiency suppresses the glial response to plaques, reduces the production of some pro-inflammatory cytokines, rescues hippocampal synapses and neurons, and spares cognition in aged APP/PS1 mice in spite of increased plaque deposition.
Whether microglia and complement work together to mediate synapse loss in plaque-rich AD brain is unclear but this hypothesis is supported by our data in APP/PS1 mice. Others have suggested that C3-dependent microglia overactivation is responsible for neuroinflammation which accelerates pathogenesis in neurodegenerative diseases (55). For instance, lipopolysaccharide (LPS)-induced loss of dopaminergic neurons was inhibited by C3-deficiency (56). In experimental autoimmune encephalomyelitis (EAE), a mouse model of human multiple sclerosis, C3-overactivation in Crry-deficient mice increased microglial overactivation and exacerbated neurodegeneration (57). Mice deficient for complement receptor 2 (Cr2 KO), which lack the CR1 and CR2 receptors that bind C3 activation fragments, had reduced secondary brain damage after closed head injury including decreased neuron death, astrocytosis and microglial activation, and improved neurological outcome (58). In addition, genetic deletion of C3 (but not C1q) protected against synapse loss and promoted faster recovery one week following axotomy of spinal cord motorneurons by sciatic nerve transection (59). Furthermore, mice deficient for BDNF in microglia have deficits in learning and hippocampal synapse formation indicating that microglial BDNF signaling plays an important role in hippocampal function (60). Taken together, these results provide evidence for a deleterious effect of complement activation and its downstream consequences in brain in the context of aging, trauma and neurodegenerative diseases.
Seemingly contradictory results of complement have been observed among different AD mouse models. C3 inhibition enhanced plaque deposition and promoted neurodegeneration in J20 hAPP mice either by over-expression of sCrry (22) or genetic deletion of C3 (21). The addition of a mutant human PS1 transgene is one possible explanation for difference in C3-deficiency between J20 mice in the previous studies and APP/PS1 mice in this study. Previous reports demonstrated upregulation of C1q in hippocampus and GFAP and Cathepsin S in cortex and hippocampus in conditional PS1/2 double knockout mice, and increased expression of complement cascade components C1q, C3aR and C4b in hippocampi of PS1/2 double knockout mice (61, 62). Thus, it is possible that the presence of the mutant human PS1 gene in the APP/PS1 mice may explain, in part, the differential effect of C3-deficiency on neurodegeneration compared to J20 hAPP mice. However, we found no significant difference in APP or PS1 levels in the APP/PS1 vs. APP/PS1;C3 KO mice used here. Other studies demonstrated that life-long C1q-deficiency rescued synaptophysin and MAP-2 in hippocampal CA3 in aged Tg2576 APP and APP/PS1 mice (23), similar to our findings in 16-month-old APP/PS1;C3 KO mice, although an additional study suggests that C1q does not appear to play a role in synapse loss during aging but instead maintains brain functionality (31). Here, we observed less reactive microgliosis and reduced astrocytosis in aged APP/PS1;C3 KO mice, compared to no change in Iba-1 and CD68 markers in J20;C3 KO mice, similar to our previous study. In addition, our current findings showing hippocampal region-specific and glia/synapse marker-specific effects described in APP/PS1;C3 KO mice may have been missed in our previous J20;C3 KO study that used fewer markers and examined the entire hippocampus, not individual sub-regions. Taken together, the differences in baseline glial response to plaques and effects of C3-deletion on microglia in aged J20 vs. aged APP/PS1 mice may explain the differences in neurodegeneration that we have observed in the two models.
Lastly, we acknowledge the limitations of our study, including the overexpression of human mutant APP and PS1 in mice that likely generate other APP fragments that could impact behavior and, the absence of tau pathology which, similar to synaptic loss, is better correlated with cognitive decline in human AD. Future studies in APP knockin mice and/or mice that have both amyloid and tau pathologies (e.g., 3xTg-AD mice), would be useful to confirm the protective effects of C3-deficiency in aged APP/PS1 mice in this study and might better reflect human AD.
In summary, we found that C3-deficiency in APP/PS1 mice confers neuroprotection against age-and AD-related neurodegeneration and cognitive decline despite enhancing plaque burden. Our results suggest that C3 may play a detrimental role in synaptic and neuronal function in plaque-rich, aged brain and suggest that inhibition of C3 signaling may be a potential therapeutic target for AD treatment. Our future studies using inducible C3 conditional KO mice will determine whether depleting C3 after brain development or after the onset of AD pathology will protect synapses and spare cognitive decline. Positive results from these studies may lead to new therapies targeting complement.
Materials and Methods
Study Design
The objective of our study was to determine whether complement C3 plays a role in late stage AD pathogenesis and cognitive decline. We crossbred an AD-like mouse model with C3 KO mice and aged them to 16 months. The APP/PS1;C3 KO mice were compared with APP/PS1, WT and C3 KO mice in terms of behavior, neuropathological and biochemical endpoints. Operators were blinded to mouse genotype for all outcome measures.
Mice
C57BL/6J mice, APPswe/PS1dE9 mice and homozygous C3-deficient breeder mice (C3−/−; line B6.129S4-C3tm1Crr/J) (63) were obtained from The Jackson Laboratory. C3 KO mice were crossed with APP/PS1 mice to generate APP/PS1 (heterozygotes);C3 KO (heterozygotes) mice, which were backcrossed with C3 KO mice to generate APP/PS1 (heterozygotes);C3 KO (homozygotes). These mice were then bred with C3 KO mice to generate APP/PS1;C3 KO mice and littermate C3 KO mice. However, a separate cohort of APP/PS1 mice was bred with C57BL/6J mice to generate WT littermates and heterozygotes APP/PS1 mice. Mice were genotyped by PCR using the following primers: APP/PS1: 5’-GACTGACCACTC GACCAGCTT-3’ and 5’-CTTGTAAGTTGGATTCTCATAT-3’; C3: 5’-ATCTTGAGTGCACCAAGCC-3’ and 5’-GGTTGCAGCAGTCTATGAAGG-3’ (C3 WT); 5’-CTTGGGTGGAGAGGCTATTC-3’ and 5’-AGGTGAGATGACAGGAGATC-3’ (C3 mutant). Mice were aged to postnatal day 30, 4 mo, or 16 mo of age. Only males were used for this study to reduce gender-specific variability in the behavioral tests. At the end of the study, mice were anesthetized, blood collected, and the brain perfused with saline prior to harvest.
All animal protocols were approved by the Harvard Medical Area Standing Committee on Animals and studies were performed in accordance with all state and federal regulations. The Harvard Medical School animal management program is accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care, International (AAALAC), and meets all National Institutes of Health standards as demonstrated by an approved Assurance of Compliance (A3431–01) filed at the Office of Laboratory Animal Welfare (OLAW).
Water T-maze.
The water T-maze (WTM) behavioral paradigm assesses spatial learning and memory by training mice to use the spatial cues in a room to navigate to a hidden platform to escape water. Testing was performed as previously reported (5). Note: the 16 month-old WT and C3 KO mice used for behavioral testing in this large study were the same mice that we reported in (5) even though they were tested concurrently with the APP/PS1 and APP/PS1;C3 KO mice in the present study. In the current study, all analyses were performed on these same mice.
Immunohistochemistry.
Hemibrains were fixed in 4% PFA for 24 h and immunohistochemistry was performed as described previously (5). Fixed, frozen sections were incubated with anti-Aβx-42 (1:200, Covance), 3D6 (Aβ1–5) (1:1000, Elan), 6E10 (1:1000, Covance), MAP-2 (1:500, Millipore) and NeuN (1:200, Serotec) mouse monoclonal antibodies, or Aβ40 (1:200, Covance), Iba-1 (1:200, Wako) and GFAP (1:1000, DAKO) rabbit polyclonal antibodies, or CD68 (1:250, Serotec) rat polyclonal antibody overnight at 4°C. After washing with TBS, sections were incubated with biotinylated secondary antibodies and developed using Vector ELITE ABC kits (Vector Laboratories) and 3,3-diaminobenzidine (Sigma-Aldrich) or immunofluorescent-labeled secondary antibodies and cover-slipped with mounting media (Vector). Thioflavin S staining of amyloid fibrils was performed as previously described (21).
Aβ load analysis.
Images of Aβ immunoreactivity were captured in a single session under a Nikon Eclipse E400 microscope. Quantification of Aβ plaque load and analysis of Aβ plaque size was measured within a region of interest (ROI) using the Bioquant image analysis system (Nashville, TN).
Three of six equidistant planes of 10 μm thick sections from each mouse were analyzed. The operator was blinded to mouse genotype and age when analyzing.
Aβ and cytokine ELISAs.
MSD Aβ Triplex ELISA was performed on Tper-insoluble, guanidine hydrochloride-extracted brain homogenates from mouse hemibrain, including cortex and hippocampus. The V-Plex Pro-inflammatory Cytokine ELISA kit (Meso Scale Discovery, Cat K15048) was used for detecting cytokine levels by MSD plate reader QuickPlex SQ 120 (Meso Scale Discovery). Brain homogenization and ELISAs were performed as previously described (5).
Confocal analysis of glia morphology and association with Aβ-plaques.
Immunofluorescence for Aβ plaques (6E10), microglia/macrophages (Iba-1) and, astrocytes (GFAP) was detected by confocal microscopy (Zeiss, LSM 710; Carl Zeiss) using methodology described previously (64). Images were collected using the same exposure settings. Confocal Z-stack images (optical slices of 0.2 μm) of plaques and surrounding glia were collected using a 63x objective. Three images were acquired from 3 equidistant planes 500 μm apart per mouse. Immunofluorescent intensity analysis and 3D reconstruction of Z-stack images were performed with confocal image analysis software, Zein black (Carl Zeiss). Glia counts were performed using stereological methods after collecting images.
Synaptic puncta staining and analysis was performed as previous described (5, 65).
Confocal imaging was performed using a ZEISS LSM710 confocal microscope and a 63x oil objective. Images were acquired using a 1 Airy Unit (AU) pinhole, while holding constant the gain and offset parameters for all sections and mice per experiment.
Preparation of synaptosome fractions.
Synaptosome fractions were prepared as described previously (66).
Western blotting of synapse markers and BDNF signaling proteins.
Brain hippocampal and cortical tissues were homogenized and Western blotting performed as previously described (5) using rabbit polyclonal antibodies Synapsin-1 (SYN-1, 1:200; Millipore), BDNF (Abcam, 1:200), pCREB and CREB (Chemicon, 1000), and mouse monoclonal antibodies PSD95 (1:200; Millipore) and GAPDH (1:200; Millipore). Blots were scanned using a LiCor Odyssey Infrared Imaging System. Intensity of bands was measured by LiCor Odyssey software.
Stereological quantification of neuron and glia counts.
Immunohistochemistry for NeuN, Iba-1 and GFAP was performed as described previously (21). Stereological counting of neurons and glia was performed on 10 μm sagittal, immunostained stained sections at each of 6 planes 250μ m apart per mouse using the optical dissector method. Cells were counted in an area of ~300 μm within a 10 μm depth per brain region examined. The number of neurons and glia per section in each brain region was estimated using ~15 optical dissectors and the Bioquant image analysis system according to the principles of Cavalieri (67).
Statistical analysis.
Data are expressed as mean ± SEM. Comparisons were made between WT vs. C3 KO, WT vs. APP/PS1, APP/PS1 vs. APP/PS1;C3 KO and C3 KO vs APP/PS1;C3 KO mice. Significance for all behavioral tests was assessed by one-way ANOVA followed by Fisher’s PLSD using StatView Version 5.0 software. All other data were analyzed by one-way or two-way ANOVAs followed by Bonferroni’s post hoc test or the Student t-test using Prism Version 6.0 (GraphPad) software. A p value of < 0.05 was considered significant.
Supplementary Material
Acknowledgments:
We thank Jeffrey Frost, Jessica Kenison and Yanli Hu (ARCND) and Kate Merry (BCH) for technical assistance. We thank Charles White (ARCND) for statistics advice and Bin Liu (ARCND) for data discussions. We are grateful to Sarah Matousek (ARCND) and Ken Colodner (BCH) for assistance with mouse breeding. We thank Dennis Selkoe (ARCND) for providing the 3D6 antibody for immunohistochemistry and Oleg Butovsky (ARCND) for critical reading of the manuscript.
Funding: This work was funded by Fidelity Biosciences Research Initiative (F-PRIME) (C.A.L. and B.S.), NIH/NIA R21 AG044713 (C.A.L.), a BrightFocus Foundation Fellowship (Q.S.) and an Edward R. and Anne G. Lefler Fellowship (S.H.).
Footnotes
Competing Interests: B.S. serves on the Scientific Advisory Board and is a minor shareholder of Annexon LLC. No other authors have competing interests related to this project.
References and Notes:
- 1.Veerhuis R, Nielsen HM, Tenner AJ, Complement in the brain. Mol Immunol 48, 1592–1603 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SW, Barres BA, The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, Stevens B, Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bialas AR, Stevens B, TGF-beta signaling regulates neuronal C1q expression and developmental synaptic refinement. Nat Neurosci 16, 1773–1782 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 5.Shi Q, Colodner KJ, Matousek SB, Merry K, Hong S, Kenison JE, Frost JL, Le KX, Li S, Dodart JC, Caldarone BJ, Stevens B, Lemere CA, Complement C3-Deficient Mice Fail to Display Age-Related Hippocampal Decline. J Neurosci 35, 13029–13042 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R, Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol 30, 572–580 (1991). [DOI] [PubMed] [Google Scholar]
- 7.DeKosky ST, Scheff SW, Synapse loss in frontal cortex biopsies in Alzheimer’s disease: correlation with cognitive severity. Ann Neurol 27, 457–464 (1990). [DOI] [PubMed] [Google Scholar]
- 8.Stoltzner SE, Grenfell TJ, Mori C, Wisniewski KE, Wisniewski TM, Selkoe DJ, Lemere CA, Temporal accrual of complement proteins in amyloid plaques in Down’s syndrome with Alzheimer’s disease. Am J Pathol 156, 489–499 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eikelenboom P, Hack CE, Kamphorst W, Rozemuller JM, Distribution pattern and functional state of complement proteins and alpha 1-antichymotrypsin in cerebral beta/A4 deposits in Alzheimer’s disease. Res Immunol 143, 617–620 (1992). [DOI] [PubMed] [Google Scholar]
- 10.Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, Lemere CA, Selkoe DJ, Stevens B, Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Veerhuis R, Histological and direct evidence for the role of complement in the neuroinflammation of AD. Curr Alzheimer Res 8, 34–58 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Tenner AJ, Complement in Alzheimer’s disease: opportunities for modulating protective and pathogenic events. Neurobiol Aging 22, 849–861 (2001). [DOI] [PubMed] [Google Scholar]
- 13.Haga S, Ikeda K, Sato M, Ishii T, Synthetic Alzheimer amyloid beta/A4 peptides enhance production of complement C3 component by cultured microglial cells. Brain Res 601, 88–94 (1993). [DOI] [PubMed] [Google Scholar]
- 14.Lian H, Yang L, Cole A, Sun L, Chiang AC, Fowler SW, Shim DJ, Rodriguez-Rivera J, Taglialatela G, Jankowsky JL, Lu HC, Zheng H, NFkappaB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron 85, 101–115 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Matsuoka Y, Picciano M, Malester B, LaFrancois J, Zehr C, Daeschner JM, Olschowka JA, Fonseca MI, O’Banion MK, Tenner AJ, Lemere CA, Duff K, Inflammatory responses to amyloidosis in a transgenic mouse model of Alzheimer’s disease. Am J Pathol 158, 1345–1354 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fan R, DeFilippis K, Van Nostrand WE, Induction of complement proteins in a mouse model for cerebral microvascular A beta deposition. J Neuroinflammation 4, 22 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou J, Fonseca MI, Pisalyaput K, Tenner AJ, Complement C3 and C4 expression in C1q sufficient and deficient mouse models of Alzheimer’s disease. J Neurochem 106, 2080–2092 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reichwald J, Danner S, Wiederhold KH, Staufenbiel M, Expression of complement system components during aging and amyloid deposition in APP transgenic mice. J Neuroinflammation 6, 35 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Eikelenboom P, Veerhuis R, The role of complement and activated microglia in the pathogenesis of Alzheimer’s disease. Neurobiol Aging 17, 673–680 (1996). [DOI] [PubMed] [Google Scholar]
- 20.Fu H, Liu B, Frost JL, Hong S, Jin M, Ostaszewski B, Shankar GM, Costantino IM, Carroll MC, Mayadas TN, Lemere CA, Complement component C3 and complement receptor type 3 contribute to the phagocytosis and clearance of fibrillar Abeta by microglia. Glia 60, 993–1003 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maier M, Peng Y, Jiang L, Seabrook TJ, Carroll MC, Lemere CA, Complement C3 deficiency leads to accelerated amyloid beta plaque deposition and neurodegeneration and modulation of the microglia/macrophage phenotype in amyloid precursor protein transgenic mice. J Neurosci 28, 6333–6341 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wyss-Coray T, Yan F, Lin AH, Lambris JD, Alexander JJ, Quigg RJ, Masliah E, Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer’s mice. Proc Natl Acad Sci U S A 99, 10837–10842 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fonseca MI, Zhou J, Botto M, Tenner AJ, Absence of C1q leads to less neuropathology in transgenic mouse models of Alzheimer’s disease. J Neurosci 24, 6457–6465 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fonseca MI, Ager RR, Chu SH, Yazan O, Sanderson SD, LaFerla FM, Taylor SM, Woodruff TM, Tenner AJ, Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer’s disease. J Immunol 183, 1375–1383 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arendash GW, King DL, Gordon MN, Morgan D, Hatcher JM, Hope CE, Diamond DM, Progressive, age-related behavioral impairments in transgenic mice carrying both mutant amyloid precursor protein and presenilin-1 transgenes. Brain Res 891, 42–53 (2001). [DOI] [PubMed] [Google Scholar]
- 26.Kleinknecht KR, Bedenk BT, Kaltwasser SF, Grunecker B, Yen YC, Czisch M, Wotjak CT, Hippocampus-dependent place learning enables spatial flexibility in C57BL6/N mice. Front Behav Neurosci 6, 87 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O’Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T, Inflammation and Alzheimer’s disease. Neurobiol Aging 21, 383–421 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dickey CA, Loring JF, Montgomery J, Gordon MN, Eastman PS, Morgan D, Selectively reduced expression of synaptic plasticity-related genes in amyloid precursor protein + presenilin-1 transgenic mice. J Neurosci 23, 5219–5226 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alonso-Nanclares L, Merino-Serrais P, Gonzalez S, DeFelipe J, Synaptic changes in the dentate gyrus of APP/PS1 transgenic mice revealed by electron microscopy. J Neuropathol Exp Neurol 72, 386–395 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.West MJ, Bach G, Soderman A, Jensen JL, Synaptic contact number and size in stratum radiatum CA1 of APP/PS1DeltaE9 transgenic mice. Neurobiol Aging 30, 1756–1776 (2009). [DOI] [PubMed] [Google Scholar]
- 31.Stephan AH, Madison DV, Mateos JM, Fraser DA, Lovelett EA, Coutellier L, Kim L, Tsai HH, Huang EJ, Rowitch DH, Berns DS, Tenner AJ, Shamloo M, Barres BA, A dramatic increase of C1q protein in the CNS during normal aging. J Neurosci 33, 13460–13474 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Filali M, Lalonde R, Age-related cognitive decline and nesting behavior in an APPswe/PS1 bigenic model of Alzheimer’s disease. Brain Res 1292, 93–99 (2009). [DOI] [PubMed] [Google Scholar]
- 33.Perez-Alcazar M, Daborg J, Stokowska A, Wasling P, Bjorefeldt A, Kalm M, Zetterberg H, Carlstrom KE, Blomgren K, Ekdahl CT, Hanse E, Pekna M, Altered cognitive performance and synaptic function in the hippocampus of mice lacking C3. Exp Neurol 253, 154–164 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Spangenberg EE, Green KN, Inflammation in Alzheimer’s disease: Lessons learned from microglia-depletion models. Brain Behav Immun, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Spangenberg EE, Lee RJ, Najafi AR, Rice RA, Elmore MR, Blurton-Jones M, West BL, Green KN, Eliminating microglia in Alzheimer’s mice prevents neuronal loss without modulating amyloid-beta pathology. Brain 139, 1265–1281 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Olmos-Alonso A, Schetters ST, Sri S, Askew K, Mancuso R, Vargas-Caballero M, Holscher C, Perry VH, Gomez-Nicola D, Pharmacological targeting of CSF1R inhibits microglial proliferation and prevents the progression of Alzheimer’s-like pathology. Brain 139, 891–907 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Silverman SM, Kim BJ, Howell GR, Miller J, John SW, Wordinger RJ, Clark AF, C1q propagates microglial activation and neurodegeneration in the visual axis following retinal ischemia/reperfusion injury. Mol Neurodegener 11, 24 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Williams PA, Tribble JR, Pepper KW, Cross SD, Morgan BP, Morgan JE, John SW, Howell GR, Inhibition of the classical pathway of the complement cascade prevents early dendritic and synaptic degeneration in glaucoma. Mol Neurodegener 11, 26 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choucair-Jaafar N, Laporte V, Levy R, Poindron P, Lombard Y, Gies JP, Complement receptor 3 (CD11b/CD18) is implicated in the elimination of beta-amyloid peptides. Fundam Clin Pharmacol 25, 115–122 (2011). [DOI] [PubMed] [Google Scholar]
- 40.Koffie RM, Meyer-Luehmann M, Hashimoto T, Adams KW, Mielke ML, Garcia-Alloza M, Micheva KD, Smith SJ, Kim ML, Lee VM, Hyman BT, Spires-Jones TL, Oligomeric amyloid beta associates with postsynaptic densities and correlates with excitatory synapse loss near senile plaques. Proc Natl Acad Sci U S A 106, 4012–4017 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Selkoe DJ, Alzheimer’s disease is a synaptic failure. Science 298, 789–791 (2002). [DOI] [PubMed] [Google Scholar]
- 42.Knafo S, Alonso-Nanclares L, Gonzalez-Soriano J, Merino-Serrais P, Fernaud-Espinosa I, Ferrer I, DeFelipe J, Widespread changes in dendritic spines in a model of Alzheimer’s disease. Cereb Cortex 19, 586–592 (2009). [DOI] [PubMed] [Google Scholar]
- 43.Chung WS, Verghese PB, Chakraborty C, Joung J, Hyman BT, Ulrich JD, Holtzman DM, Barres BA, Novel allele-dependent role for APOE in controlling the rate of synapse pruning by astrocytes. Proc Natl Acad Sci U S A 113, 10186–10191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lian H, Litvinchuk A, Chiang AC, Aithmitti N, Jankowsky JL, Zheng H, Astrocyte-Microglia Cross Talk through Complement Activation Modulates Amyloid Pathology in Mouse Models of Alzheimer’s Disease. J Neurosci 36, 577–589 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ducruet AF, Zacharia BE, Sosunov SA, Gigante PR, Yeh ML, Gorski JW, Otten ML, Hwang RY, DeRosa PA, Hickman ZL, Sergot P, Connolly ES Jr., Complement inhibition promotes endogenous neurogenesis and sustained anti-inflammatory neuroprotection following reperfused stroke. PLoS One 7, e38664 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Woodruff TM, Tenner AJ, A Commentary On: “NFkappaB-Activated Astroglial Release of Complement C3 Compromises Neuronal Morphology and Function Associated with Alzheimer’s Disease”. A cautionary note regarding C3aR. Front Immunol 6, 220 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Richner M, Bach G, West MJ, Over expression of amyloid beta-protein reduces the number of neurons in the striatum of APPswe/PS1DeltaE9. Brain Res 1266, 87–92 (2009). [DOI] [PubMed] [Google Scholar]
- 48.Oh ES, Savonenko AV, King JF, Fangmark Tucker SM, Rudow GL, Xu G, Borchelt DR, Troncoso JC, Amyloid precursor protein increases cortical neuron size in transgenic mice. Neurobiol Aging 30, 1238–1244 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baskerville KA, Kent C, Nicolle MM, Gallagher M, McKinney M, Aging causes partial loss of basal forebrain but no loss of pontine reticular cholinergic neurons. Neuroreport 17, 1819–1823 (2006). [DOI] [PubMed] [Google Scholar]
- 50.Li D, Wang J, Yew DT, Lucy Forster E, Yao Z, Age-related alterations of Nestin-immunoreactive neurons in rat basal forebrain with aged memory deficit. Neurochem Int 53, 270–277 (2008). [DOI] [PubMed] [Google Scholar]
- 51.Rogers J, Cooper NR, Webster S, Schultz J, McGeer PL, Styren SD, Civin WH, Brachova L, Bradt B, Ward P, et al. , Complement activation by beta-amyloid in Alzheimer disease. Proc Natl Acad Sci U S A 89, 10016–10020 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Snyder SW, Wang GT, Barrett L, Ladror US, Casuto D, Lee CM, Krafft GA, Holzman RB, Holzman TF, Complement C1q does not bind monomeric beta-amyloid. Exp Neurol 128, 136–142 (1994). [DOI] [PubMed] [Google Scholar]
- 53.Sasaki A, Yamaguchi H, Ogawa A, Sugihara S, Nakazato Y, Microglial activation in early stages of amyloid beta protein deposition. Acta Neuropathol 94, 316–322 (1997). [DOI] [PubMed] [Google Scholar]
- 54.Webster S, Glabe C, Rogers J, Multivalent binding of complement protein C1Q to the amyloid beta-peptide (A beta) promotes the nucleation phase of A beta aggregation. Biochem Biophys Res Commun 217, 869–875 (1995). [DOI] [PubMed] [Google Scholar]
- 55.Perry VH, Holmes C, Microglial priming in neurodegenerative disease. Nat Rev Neurol 10, 217–224 (2014). [DOI] [PubMed] [Google Scholar]
- 56.Bodea LG, Wang Y, Linnartz-Gerlach B, Kopatz J, Sinkkonen L, Musgrove R, Kaoma T, Muller A, Vallar L, Di Monte DA, Balling R, Neumann H, Neurodegeneration by activation of the microglial complement-phagosome pathway. J Neurosci 34, 8546–8556 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ramaglia V, Hughes TR, Donev RM, Ruseva MM, Wu X, Huitinga I, Baas F, Neal JW, Morgan BP, C3-dependent mechanism of microglial priming relevant to multiple sclerosis. Proc Natl Acad Sci U S A 109, 965–970 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Neher MD, Rich MC, Keene CN, Weckbach S, Bolden AL, Losacco JT, Patane J, Flierl MA, Kulik L, Holers VM, Stahel PF, Deficiency of complement receptors CR2/CR1 in Cr2(−)/(−) mice reduces the extent of secondary brain damage after closed head injury. J Neuroinflammation 11, 95 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Berg A, Zelano J, Stephan A, Thams S, Barres BA, Pekny M, Pekna M, Cullheim S, Reduced removal of synaptic terminals from axotomized spinal motoneurons in the absence of complement C3. Exp Neurol 237, 8–17 (2012). [DOI] [PubMed] [Google Scholar]
- 60.Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR 3rd, Lafaille JJ, Hempstead BL, Littman DR, Gan WB, Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155, 1596–1609 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Beglopoulos V, Sun X, Saura CA, Lemere CA, Kim RD, Shen J, Reduced beta-amyloid production and increased inflammatory responses in presenilin conditional knock-out mice. J Biol Chem 279, 46907–46914 (2004). [DOI] [PubMed] [Google Scholar]
- 62.Mirnics K, Norstrom EM, Garbett K, Choi SH, Zhang X, Ebert P, Sisodia SS, Molecular signatures of neurodegeneration in the cortex of PS1/PS2 double knockout mice. Mol Neurodegener 3, 14 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wessels MR, Butko P, Ma M, Warren HB, Lage AL, Carroll MC, Studies of group B streptococcal infection in mice deficient in complement component C3 or C4 demonstrate an essential role for complement in both innate and acquired immunity. Proc Natl Acad Sci U S A 92, 11490–11494 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jordao JF, Thevenot E, Markham-Coultes K, Scarcelli T, Weng YQ, Xhima K, O’Reilly M, Huang Y, McLaurin J, Hynynen K, Aubert I, Amyloid-beta plaque reduction, endogenous antibody delivery and glial activation by brain-targeted, transcranial focused ultrasound. Exp Neurol 248, 16–29 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ippolito DM, Eroglu C, Quantifying synapses: an immunocytochemistry-based assay to quantify synapse number. J Vis Exp, (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jin M, Shepardson N, Yang T, Chen G, Walsh D, Selkoe DJ, Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci U S A 108, 5819–5824 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.West MJ, Gundersen HJ, Unbiased stereological estimation of the number of neurons in the human hippocampus. J Comp Neurol 296, 1–22 (1990). [DOI] [PubMed] [Google Scholar]
- 68.Morrison HW, Filosa JA, A quantitative spatiotemporal analysis of microglia morphology during ischemic stroke and reperfusion. J Neuroinflammation 10, 4 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L, High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci 20, 4050–4058 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.