

Abstract
Electrostatic interactions via ion pairs are vital for biological macromolecules. Regarding the free energy of each ion pair as a function of the interionic distance, continuum electrostatic models predict a single energy minimum corresponding to the contact ion-pair (CIP) state, whereas atomically detailed theoretical hydration studies predict multiple energy minima corresponding to the CIP and solvent-separated ion-pair (SIP) states. Through a statistical analysis of high-resolution crystal structures, we present experimental evidence of SIP as a metastable state. The histogram of interionic distances between protein side-chain NH3+ and DNA phosphate groups clearly shows two major peaks corresponding to the CIP and SIP states. The statistical data are consistent with the probability distribution of the CIP-SIP equilibria previously obtained with molecular dynamics simulations. Spatial distributions of NH3+ ions and water molecules around phosphates reveal preferential sites for CIP and SIP formations and show how the ions compete with water molecules.
Keywords: Crystal structures, DNA, free energy, hydration, proteins, statistics
Graphical Abstract
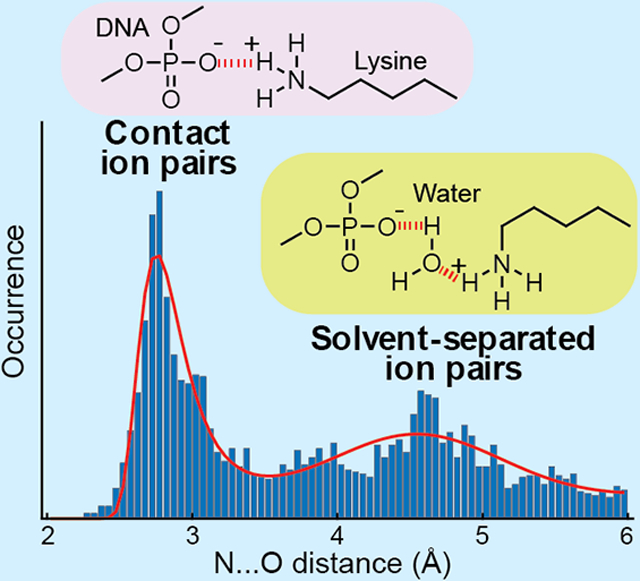
Short-distance electrostatic interactions via ion pairs of positively charged and negatively charged moieties are crucial to the functions of biological macromolecules.1–4 For a deeper understanding of functional mechanisms, the physicochemical properties of macromolecular ion pairs should be delineated. Modeling of the energetics of ion pairs in solution accurately requires an investment in simulations5–8 or many-body theory9. Although continuum electrostatic models successfully explain long-range electrostatic interactions of biological macromolecules,10–11 these models do not capture an important short-ranged structural feature of ion pairs: multiple free energy minima. The simple continuum models and related implicit solvation methods predict only a single minimum in the potential of mean force (PMF) as a function of the interionic distance.12 This minimum corresponds to the contact ion-pair (CIP) state in which the cation and the anion are in direct contact. In contrast, all-atom simulations and other theoretical approaches taking ionic hydration into account predict PMF often involving two major energy minima.6,9,12–16 One of the minima corresponds to the CIP state, and the other to the solvent-separated ion-pair (SIP) state in which the ions are intervened by water molecules (Figure 1A). Equilibria between CIP and SIP states of small ions have been confirmed by experiments.17–26 Nonetheless, experimental confirmation of CIP-SIP equilibria in biological macromolecular systems remains elusive.
Figure 1.

CIP and SIP states of Lys side-chain NH3+–DNA phosphate ion pairs. (A) Concepts of CIP and SIP. Gray circles represent solvent molecules. (B) CIP-SIP model transitions observed in molecular dynamics simulations for protein-DNA complexes.27 (C) Histogram of the dNO distances for 3038 Lys NH3+–DNA phosphate ion pairs found in high-resolution (< 2.0 Å) crystal structures. The solid red line shows a best-fit curve obtained with the sum of a Gaussian distribution function for the SIP peak and a Boltzmann distribution function based on Lennard-Jones and Coulomb potentials (see the Supporting Information). (D) Probability distribution of dNO distance of Lys side-chain NH3+–DNA phosphate ion pairs in 0.6-μs MD trajectories for the Antp homeodomain-DNA complex and Egr-1 zinc-finger–DNA complex (adopted from Chen et al.27). (E, F) Potentials of mean force (PMFs) as a function of the dNO distance protein side-chain NH3+–DNA phosphate ion pairs. Panel E shows PMFs calculated from structure statistical data shown in Figure 1C. Panel F shows PMF calculated from the data shown in Figure 1D (adopted from Chen et al.27). Note that the data profiles in Panels C and E resemble those in Panels D and F, respectively.
Recently, using nuclear magnetic resonance (NMR) spectroscopy and all-atom molecular dynamics (MD) simulations, Chen et al. studied the CIP-SIP equilibria for the ion pairs of protein side-chain NH3+ and DNA phosphate groups in protein-DNA complexes (Figure 1B).27 MD trajectories showed dynamic transitions between CIP and SIP states on a ps-ns timescale. NMR data of hydrogen-bond scalar couplings clearly indicate the presence of CIPs. NMR order parameters for Lys NH3+ groups and 13Cγ−15Nζ scalar couplings reflecting χ4 dihedral rotations were consistent with those from MD trajectories. These experimental data implicitly support the dynamic CIP-SIP equilibria.
In this paper, we present more direct experimental evidence of SIPs as metastable states for ion pairs of Lys side-chain NH3+ and DNA phosphate groups. This evidence was obtained through a statistical analysis of 3038 ion pairs of Lys side-chain NH3+-DNA phosphate groups in high-resolution (< 2.0 Å) crystal structures of protein-DNA complexes available in the Protein Data Bank (PDB). Comparison of spatial distributions of Lys NH3+ ions and hydration water molecules around DNA phosphate also allowed us to gain further insight into the CIP-SIP equilibria.
We analyzed ion pairs of protein Lys side-chain NH3+ and DNA phosphate groups in PDB crystal structures of protein-DNA complexes that satisfy the following four conditions: 1) the resolution is < 2.0 Å; 2) a crystallographic R-factor is < 0.29; and 3) DNA contains more than 5 nucleotide residues in each strand; 4) a single, unique conformation without missing atoms. 928 structures available at PDB satisfied these conditions. The complete list of the PDB codes of these structures is shown in Table S1 in the Supporting Information (SI). We analyzed structure statistics for these ion pairs. For each Lys Nζ atom in crystal structures of protein-DNA complexes that were solved at a resolution < 2.0 Å, we first identified the closest DNA phosphate group and then examined whether the following conditions are satisfied: 1) the distance dNO between Lys Nζ and closest phosphate oxygen atoms is < 6.0 Å; and 2) the Cα atom of the same residue is within 9.51 Å from the phosphate group. The latter condition was used to exclude any Lys side chains that cannot form CIP no matter what side-chain conformers are adopted. This exclusion is important for us to examine the energy landscape for ion pairs that can undergo CIP-SIP equilibrium. 3038 Lys side-chain NH3+–DNA phosphate ion pairs satisfied these conditions.
Figure 1C shows the histogram of the dNO distances between Lys Nζ and closest oxygen atoms of Lys NH3+–DNA phosphate ion pairs in the high-resolution crystal structures of protein-DNA complexes. Two major peaks are clearly seen in this histogram: one at 2.8 Å and the other at 4.6 Å. The peak at 2.8 Å corresponds to CIPs and the peak at 4.6 Å corresponds to SIPs. The difference in distance between these peaks (i.e., 1.8 Å) is smaller than the apparent diameter of the solvent molecule (2.8 Å) and allows the expected tetrahedrally biased water packing in SIPs (see Figure 1A). Importantly, the statistical distribution of the dNO distance shown in Figure 1C is remarkably similar to the probability distributions of the dNO distances observed in MD trajectories. Figure 1D shows the dNO probability distribution for each Lys NH3+–DNA phosphate ion pair in 0.6-μs all-atom MD simulations with CHARMM27 force-field parameters and TIP3P water for the Antp homeodomain–DNA complex and the Egr-1 zinc-finger–DNA complex.27 The majority of the high-resolution crystal structures analyzed for Figure 1C were solved using diffraction data at a temperature around 100 K (see Figure S1 in SI). The CIP-SIP ensemble of each ion pair in crystals at the cryo-temperature should be similar to the ensemble immediately before conformational fluctuations cease at a temperature around 200 K during flash cooling of crystals.28 The resemblance between the data in Figures 1C and 1D suggests that the statistical distribution of dNO distances for numerous crystal structures reflects a general trend in the free energy landscape of Lys NH3+–DNA phosphate ion pairs, while specific details should depend on the microenvironment surrounding each ion pair.
Based on this idea, we estimated the free energy landscape in terms of the one-dimensional potential of mean force (PMF) using the structure statistical data of dNO distances of Lys NH3+–DNA phosphate ion pairs (Figure 1E). We did this analysis in two different manners. In the first approach, we conducted fitting to the dNO histogram using the sum of a Gaussian distribution function for the SIP state and a Boltzmann distribution function based on the Lennard-Jones and Coulomb potentials for the CIP state. The best-fit curve through this calculation is shown in red in Figure 1C. A PMF was obtained from the log of the obtained probability distribution function. In the second approach, we calculated another PMF directly from the histogram without assuming any physical models. Despite the statistical use of numerous different systems’ ion pairs, the obtained PMFs appear to be similar to the PMFs previously obtained from MD trajectories for ion pairs undergoing dynamic CIP-SIP transitions in the Antp homeodomain-DNA complex and the Egr-1 zinc-finger-DNA complex (Figure 1F) showing the strong influence of local chemistry. Thus, we argue that the statistical data shown in Figure 1C represent explicit experimental evidence of SIPs as metastable states in electrostatically dominated interionic interactions.
To gain more insight into the CIP and SIP states, we also analyzed the three-dimensional distribution of Lys NH3+ ions around DNA phosphate. Figure 2A shows a probability density map of Lys Nζ atoms around DNA for CIPs with dNO < 3.2 Å. The vast majority (98.6%) of CIPs were formed at the OP1 or OP2 atoms, whereas CIPs formed at O5’ or O3’ atoms were rare (1.4%), which seems to be largely due to steric exclusion near O5’ and O3’ atoms. Although one may expect spherical distribution around the OP1 or OP2 atoms, the actual spatial distribution of Lys Nζ atoms in the CIP state appears more like a combination of a horseshoe-shaped distribution around the OP1 atom and a lobe-shaped distribution around the OP2 atom (see Figure 2A). Figures 2B and 2C show the spatial distribution of Lys NH3+ ions with the dNO distance ranging between 3.2 and 6.0 Å. Compared to the Lys Nζ distribution in CIPs, the three-dimensional distribution of Lys Nζ atoms in SIPs appears to be more spherical and more widespread, probably reflecting a flatter effective energy surface for the SIP state. Nevertheless, four high-density zones were found: three near the OP1 atom and one near the OP2 atom. The density maxima of three of the four zones were located within 4.6 ± 0.2 Å from the OP1 or OP2 atom. These zones largely account for the second peak at dNO = 4.6 Å representing SIPs in the statistical distribution of the dNO distance shown in Figure 1C.
Figure 2.

Spatial distributions of Lys NH3+ ions and water molecules that make direct contact with a DNA phosphate in high-resolution crystal structures. To simplify the view, Nζ atoms whose closest oxygen is OP1 and those whose closest oxygen atom is OP2 are separately shown in Panels A-C and E. (A) The spatial distribution of Lys Nζ atoms around DNA phosphates in the CIP states. The probability density map shows regions enclosing 90% of observed CIP Lys Nζ atoms. (B) The spatial distribution of Lys Nζ atoms around DNA phosphate with the dNO distance being between 3.2 and 6.0 Å. (C) Two-dimensional projections of Nζ atoms on the plane of P-OP1 (or P-OP2) and P-OP2 (or P-OP1) vectors. Data points of CIPs (dNO < 3.2 Å) are shown in red. (D) The spatial distribution of hydration water molecules around DNA phosphates. The hydration sites W11, W12, W13, W21, W22, and W23 (shown in blue) were identified as defined by Schneider and Berman.29 The probability density map shows regions enclosing 90% of observed water molecules. (E) Positions of high-density clusters of SIP Nζ atoms with respect to the hydration clouds. The probability density map (shown in cyan) shows regions enclosing 20% of Lys Nζ atoms with the dNO distance being between 3.2 and 6.0 Å. (F) Some representative SIPs with two water molecules shared by a Lys NH3+ ion and a DNA phosphate in crystal structures (PDB 4U0Y, left; and 5VHV, right). The structures and the probability density maps were drawn using the Chimera software.30
To better understand the spatial distribution of Lys Nζ atoms around DNA phosphate, we also analyzed the spatial distribution of water molecules around DNA phosphate. The hydration of DNA phosphates was previously studied with simulation.31–32 An extensive study of the experimental data for the time was made by Schneider and Berman in 1998:29 the number of high-resolution (< 2.0 Å) crystal structures of DNA duplexes or DNA-protein complexes was far fewer at that time (220 structures in the year 1998 vs. 1152 in the year 2019). We updated the hydration statistics for a larger current dataset, analyzing 10305 water molecules around DNA phosphates that do not contact proteins in the same set of 928 crystal structures of protein-DNA complexes (details are described in SI). Figure 2D shows a probability density map of hydration water molecules around DNA phosphate. We were able to confirm the 6 major hydration sites around DNA phosphate, which Schneider and Berman referred to as the sites W11, W12, and W13 around the OP1 atom and as the sites W21, W22, and W23 around the OP2 atom, which are shown as blue balls in Figure 2D.29 These sites exhibit the highest probability densities. However, hydration water molecules are distributed broadly around these sites, and the actual probability distribution (referred to as the ‘hydration clouds’ hereafter) appear like a combination of two doughnut-shaped distributions around the OP1 and OP2 atoms, as shown in Figure 2D.
Intriguingly, the spatial distribution of Lys Nζ atoms in direct contact with phosphate resembles the hydration clouds and overlap with five hydration sites W11, W12, W13, W21, and W23 (Figure 2D). This resemblance suggests that a Lys NH3+ ion and water molecules dynamically compete for these sites around a DNA phosphate. CIP formation must involve displacement of hydration water molecules, which is usually considered to increase entropy due to the freedom gained by the released water molecules.9,19 When Figures 2A and 2D are compared, it becomes obvious that the Lys Nζ distribution for CIPs exhibits a significantly lower density at the W22 site. This suggests that the hydration water molecule at the W22 site is more difficult to displace with a Lys NH3+ ion. The stability of the hydration water at the W22 site is also inferred by the highest probability density among the six sites (Figure S3 in SI).
As shown in Figure 2E, the high-density zones for Lys NH3+ ions to form a SIP with a DNA phosphate appear to be located at positions suited to form hydrogen bonds with water molecules in the hydration clouds. Although our analysis identified SIPs based on the dNO distance rather than based on the presence of water molecules between Lys Nζ and phosphate atoms in crystal structures, many high-resolution crystal structures actually show one or two water molecules intervening Lys Nζ and phosphate oxygen atoms. Two examples are shown in Figure 2F. Considering the hydration clouds and the wide distribution of Lys Nζ atoms (Figure 2B–E), it seems that a SIP can be formed between DNA phosphate and Lys side chains in various conformations, which could be entropically favorable. Comparison of spatial distributions of Lys NH3+ ions and hydration water molecules around DNA phosphates provide unique insight into the CIP-SIP equilibrium.
It seems that SIPs as metastable states are general for other types of ion pairs as well. Using the same set of crystal structures, we conducted a statistical analysis of the shortest N…O distances (dNO,min) for ion pairs of Arg guanidinium and DNA phosphate groups. It is not straightforward to analyze their PMF because simultaneous formation of a CIP with one phosphate and a SIP with another is quite common for Arg side chains. Nonetheless, the obtained histograms clearly show a peak of SIPs, suggesting their metastable nature (Figure S4). The statistical analysis of the shortest O…O distances (dOO,min) of like ion pairs of Asp/Glu carboxylate and DNA phosphate groups shows the major peak of SIPs (Figure S5), implying that solvation can counteract the electrostatic penalty of bringing two like-charge ions together. SIPs seem to exist as metastable states for protein-protein ion pairs as well. In 1997, Nussinov and coworkers conducted a survey of ion pairs of protein-protein interfaces of 555 PDB structures (resolution < 3.0 Å) and obtained data on distribution of the distance between opposite charges.33 They found a strong peak at 2.75 Å corresponding to the CIP states. The distance distribution data (Fig. 13b in Ref.33) clearly show another peak at 4.75 Å, although the authors did not mention anything about this second peak in the paper. Based on our current study, we interpret that this peak at 4.75 Å represents another piece of evidence of SIPs as metastable states of ion pairs.
In conclusion, the structure statistics of ion pairs in high-resolution crystal structures provides explicit experimental evidence of SIPs as metastable states in electrostatic interactions in biological macromolecules. The statistical analysis also provides unique insight into how NH3+ ions compete with hydration water molecules in the CIP-SIP equilibria. The experimental confirmation of SIPs as metastable states for protein-nucleic acids systems may illuminate a problem of continuum electrostatic models and relevant implicit solvation models, which do not predict the CIP-SIP equilibria.12 In this regard, our current work may encourage development of improved models of biopolymers that may be able to capture the CIP-SIP equilibria (e.g., the semi-explicit assembly solvation model34). Such improvement would facilitate protein engineering and drug development in the future.
Methods
928 high-resolution (< 2.0 Å) crystal structures that satisfy the aforementioned criteria were retrieved from the Research Collaboratory for Structural Bioinformatics (RCSB) PDB server.35 The statistical analysis of the ion pairs in high-resolution crystal structures was conducted using scripts of MATLAB software (MathWorks, Inc.). Other details of calculations are described in the SI.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by Grants R35-GM130326 (to J.I.) and R01-GM037657 (to B.M.P.) from the National Institutes of Health, Grant H-0013 from the Robert A. Welch foundation (to B.M.P.), and Grant CHE-1608866 (to J.I.) from the National Science Foundation. We thank Drs. Mark White and Hashim Al-Hashimi for useful discussion.
Footnotes
Supporting Information.
The following file is available free of charge.
A PDF containing methods for statistical analysis of dNO distances, spatial distribution of Lys Nζ atoms, and hydration around DNA phosphates; histograms of crystallographic resolutions and temperatures (Figure S1); histograms of dNO distances obtained with different criteria (Figure S2); and relative probability density of water molecules at hydration sites (Figure S3); histograms of dNO,min distances for ion pairs of Arg guanidinium and DNA phosphate groups (Figure S4); a histogram of dOO,min distances for like ion pairs of Asp/Glu carboxylate and DNA phosphate groups (Figure S5).
The authors declare no competing financial interests.
REFERENCES
- 1.Brooks CL; Karplus M; Pettitt BM Proteins: A theoretical perspective of dynamics, structure, and thermodynamics. Adv. Chem. Phys 2006, 71, 1–259. [Google Scholar]
- 2.Iwahara J; Esadze A; Zandarashvili L Physicochemical properties of ion pairs of biological macromolecules. Biomolecules 2015, 5, 2435–2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kumar S; Nussinov R Close-range electrostatic interactions in proteins. Chembiochem 2002, 3, 604–617. [DOI] [PubMed] [Google Scholar]
- 4.Privalov PL; Dragan AI; Crane-Robinson C Interpreting protein/DNA interactions: distinguishing specific from non-specific and electrostatic from non-electrostatic components. Nucleic Acids Res. 2011, 39, 2483–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Berkowitz M; Karim OA; McCammon JA; Rossky PJ Sodium chloride ion pair interaction in water: computer simulation. Chem. Phys. Lett 1984, 105, 577–580. [Google Scholar]
- 6.Fennell CJ; Bizjak A; Vlachy V; Dill KA Ion pairing in molecular simulations of aqueous alkali halide solutions. J. Phys. Chem. B 2009, 113, 6782–6791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Patey GN; Carnie SL Theoretical results for aqueous electrolytes. Ion-ion potentials of mean force and the solute-dependent dielectric constant. J. Chem. Phys 1983, 78, 5183–5190. [Google Scholar]
- 8.Patey GN; Valleau JP A Monte Carlo method for obtaining the interionic potential of mean force in ionic solution. J. Chem. Phys 1975, 63, 2334–2339. [Google Scholar]
- 9.Pettitt BM; Rossky PJ Alkali-Halides in Water - Ion Solvent Correlations and Ion Ion Potentials of Mean Force at Infinite Dilution. J. Chem. Phys 1986, 84, 5836–5844. [Google Scholar]
- 10.Baker NA; Sept D; Joseph S; Holst MJ; McCammon JA Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U. S. A 2001, 98, 10037–10041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Honig B; Nicholls A Classical electrostatics in biology and chemistry. Science 1995, 268, 1144–1149. [DOI] [PubMed] [Google Scholar]
- 12.Masunov A; Lazaridis T Potentials of mean force between ionizable amino acid side chains in water. J. Am. Chem. Soc 2003, 125, 1722–1730. [DOI] [PubMed] [Google Scholar]
- 13.Debiec KT; Gronenborn AM; Chong LT Evaluating the strength of salt bridges: a comparison of current biomolecular force fields. J. Phys. Chem. B 2014, 118, 6561–6569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Resat H; Mezei M; McCammon JA Use of the grand canonical ensemble in potential of mean force calculations. J. Phys. Chem 1996, 100, 1426–1433. [Google Scholar]
- 15.Rozanska X; Chipot C Modeling ion-ion interaction in proteins: A molecular dynamics free energy calculation of the guanidinium-acetate association. J. Chem. Phys 2000, 112, 9691–9694. [Google Scholar]
- 16.Thomas AS; Elcock AH Direct observation of salt effects on molecular interactions through explicit-solvent molecular dynamics simulations: differential effects on electrostatic and hydrophobic interactions and comparisons to Poisson-Boltzmann theory. J. Am. Chem. Soc 2006, 128, 7796–7806. [DOI] [PubMed] [Google Scholar]
- 17.Enderby JE; Neilson GW The structure of electrolyte solutions. Rep. Prog. Phys 1981, 44, 593–653. [Google Scholar]
- 18.Lü JM; Rosokha SV; Lindeman SV; Neretin IS; Kochi JK “Separated” versus “contact” ion-pair structures in solution from their crystalline states: dynamic effects on dinitrobenzenide as a mixed-valence anion. J. Am. Chem. Soc 2005, 127, 1797–1809. [DOI] [PubMed] [Google Scholar]
- 19.Marcus Y; Hefter G Ion pairing. Chem. Rev 2006, 106, 4585–4621. [DOI] [PubMed] [Google Scholar]
- 20.Masnovi JM; Kochi JK Direct observation of ion-pair dynamics. J. Am. Chem. Soc 1985, 107, 7880–7893. [Google Scholar]
- 21.Neilson GW; Enderby JE Structure of an aqueous solution of nickel chloride. Proc. Royal Soc. Lond. A 1983, 390, 353–371. [Google Scholar]
- 22.Peters KS; Li BL Picosecond dynamics of contact ion-pairs and solvent-separated ion-pairs in the photosolvolysis of diphenylmethyl chloride. J. Phys. Chem 1994, 98, 401–403. [Google Scholar]
- 23.Simon JD; Peters KS Picosecond dynamics of ion-pairs - the effect of hydrogen-bonding on ion-pair intermediates. J. Am. Chem. Soc 1982, 104, 6542–6547. [Google Scholar]
- 24.Simon JD; Peters KS Direct observation of the special salt effect - picosecond dynamics of ion-pair exchange. J. Am. Chem. Soc 1982, 104, 6142–6144. [Google Scholar]
- 25.Winstein S; Klinedinst PE; Robinson GC Salt effects and ion pairs in solvolysis and related reactions .17. Induced common ion rate depression and mechanism of special salt effect. J. Am. Chem. Soc 1961, 83, 885-&. [Google Scholar]
- 26.Yabe T; Kochi JK Contact ion-pairs - picosecond dynamics of solvent separation, internal return, and special salt effect. J. Am. Chem. Soc 1992, 114, 4491–4500. [Google Scholar]
- 27.Chen CY; Esadze A; Zandarashvili L; Nguyen D; Pettitt BM; Iwahara J Dynamic equilibria of short-range electrostatic interactions at molecular interfaces of protein-DNA complexes. J. Phys. Chem. Lett 2015, 6, 2733–2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weik M; Colletier JP Temperature-dependent macromolecular X-ray crystallography. Acta Crystallogr. D Biol. Crystallogr 2010, 66, 437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schneider B; Patel K; Berman HM Hydration of the phosphate group in double-helical DNA. Biophys. J 1998, 75, 2422–2434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- 31.Beveridge DL; Swaminathan S; Ravishanker G; Withka JM; Srinivasan J; Prevost C; Louise-May S; Langley DR; DiCapua FM; Bolton PH Molecular dynamics simulations on the hydration, structure, and motions of DNA oligomers In Water and Biological Macromolecules, Westhof E, Ed CRC Press: Boca Raton, 1993; pp 165–225. [Google Scholar]
- 32.Feig M; Pettitt BM Modeling high-resolution hydration patterns in correlation with DNA sequence and conformation. J. Mol. Biol 1999, 286, 1075–1095. [DOI] [PubMed] [Google Scholar]
- 33.Xu D; Tsai CJ; Nussinov R Hydrogen bonds and salt bridges across protein-protein interfaces. Protein Eng. 1997, 10, 999–1012. [DOI] [PubMed] [Google Scholar]
- 34.Fennell CJ; Kehoe CW; Dill KA Modeling aqueous solvation with semi-explicit assembly. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 3234–3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berman HM; Westbrook J; Feng Z; Gilliland G; Bhat TN; Weissig H; Shindyalov IN; Bourne PE The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.