

Abstract
Whereas complete loss of Rp function is generally lethal, most heterozygous Rp mutants grow more slowly and are subject to competitive loss from mosaics tissues that also contain wild type cells. The rpS12 gene has a special role in the cell competition of other Ribosomal Protein (Rp) mutant cells in Drosophila. Elimination by cell competition is promoted by higher RpS12 levels and prevented by a specific rpS12 mis-sense mutation, identifying RpS12 as a key effector of cell competition due to mutations in other Rp genes. Here we show that RpS12 is also required for other aspects of Rp mutant phenotypes, including hundreds of gene expression changes that occur in ‘Minute’ Rp heterozygous wing imaginal discs, overall translation rate, and the overall rate of organismal development, all through the bZip protein Xrp1 that is one of the RpS12-regulated genes. Our findings outline the regulatory response to mutations affecting essential Rp genes that controls overall translation, growth, and cell competition, and which may contribute to cancer and other diseases.
Author summary
Ribosomes, the cellular machines that synthesizes new proteins, are themselves made of RNA and proteins. Mutations in Ribosomal protein (Rp) genes affect translation but they are also involved in human diseases and in cancer for reasons that are not yet clear. In the fruitfly Drosophila melanogaster, individual cells carrying mutations in Rp genes can be recognized and eliminated from tissues in a process of ‘cell competition’. Two genes that have been found to be required for cell competition include one specific Rp, RpS12 and the putative transcription factor Xrp1. Previous work has suggested that RpS12 was more specific for cell competition than Xrp1, which was also responsible for other aspects of Rp phenotypes such as changes in gene expression profiles, overall translation, and the rate of organismal development. This paper shows that RpS12 in fact controls Xrp1 levels and all the same aspects of the ‘Minute’ Rp heterozygous phenotype. Xrp1 is required for these RpS12 effects, establishing RpS12 as the most upstream component known of the signaling that occurs in Rp mutants.
Introduction
Around 80 of the many proteins that interact with eukaryotic rRNA during ribosome biogenesis are stably associated with the mature ribosomal Large or Small Subunits [1]. Most of these ribosomal proteins (Rp) are essential to the cell, with roles in ribosome biogenesis and/or in the function of the mature ribosomes. A number of Rp also have additional functions, sometimes at extra-ribosomal locations [2, 3]. The full spectrum of Rp function is far from understood, as illustrated by the recurrent observation of Rp mutations in cancers. Frequent Rp point mutations occur in T-cell Acute Lymphoblastic Leukemia (T-ALL), Chronic Lymphocytic Leukemia (CLL), and colon cancer. Diamond Blackfan Anemia patients, at least 60% of whom carry germline mutations in Rp genes, exhibit higher rates of many cancers, as do patients with 5q syndrome, associated with somatically-acquired deficiency of the RpS14 gene region [4–7]. It seems paradoxical that positive regulators of translation also behave like tumor suppressors.
To help understand the roles played by Rp, we have been studying Rp mutations in the fruitfly Drosophila melanogaster. As in other eukaryotes, homozygous mutation of most Rp loci is lethal in Drosophila. Heterozygotes for most Rp mutations have dominant phenotypes due to haploinsufficiency. In Drosophila, 66 of the 79 Rp are encoded by genes with a shared dominant ‘Minute’ phenotype, characterized by small adult macrochaetae (sensory bristles) and delayed development, and by other defects [8, 9]. These mutations also exhibit cell competition in mosaics, whereupon Rp+/- cells can be eliminated in the presence of nearby wild type cells, which trigger their apoptosis [10–12]. In this paper we use the symbol ‘Rp’ to represent loci encoding ribosomal proteins that have the typical haploinsufficient phenotypes, but ‘rp’ for the small number of loci that are not haploinsufficient.
Genetic screens have identified mutations preventing cell competition, such as mutations in the putative transcription factor Xrp1 [13–15]. Whereas Xrp1 null mutations have no visible phenotype in otherwise wild type flies, Xrp1 expression is up-regulated cell-autonomously in Rp+/- cells, and is required for their elimination ie clones of Rp+/- cells are no longer eliminated by competition even in Xrp1 heterozygotes [14, 16]. In the Xrp1-/- background, even Rp-/- cells survive and proliferate for some time [16]. Xrp1 is also known as a transcriptional target of p53 that is induced following irradiation, as part of a DNA-binding complex that binds to transposable P elements, and as a suppressor of a Drosophila model of Amyotrophic Lateral Sclerosis [17–20].
Xrp1 contributes to other aspects of the ‘Minute’ phenotype besides cell competition. Xrp1 is also responsible for the reduced growth rate of Rp+/- cells, and for their reduced rate of translation, which remarkably therefore depends at least in part on a regulatory response to Rp mutations, and not on any direct effect of Rp+/- genotypes on ribosome activity [16]. Xrp1 is also responsible for some of the developmental delay typical of Rp+/- genotypes, so that a more normal rate of development is restored when Xrp1 is also mutated [16, 21]. Xrp1 mutations have only a small effect on the size of bristles, however [16]. Rp+/- wing imaginal discs show a transcriptional signature distinct from the wild type [22], and more than 80% of these differences depend on Xrp1, corroborating the important role of Xrp1 in the Minute phenotype [16].
A second gene required for elimination of Rp+/- cells by cell competition encodes the ribosomal protein RpS12. RpS12 is an essential ribosomal protein, whose null mutations are lethal as is typical for Rp genes, but is among the minority of Drosophila genes encoding Rp whose mutations are recessive, so that rpS12+/- flies lack any visible ‘Minute’ phenotype [8, 23]. A homozygously viable mis-sense allele, rpS12D97, prevents cell competition of cells that carry mutations in other Rp genes [15, 23]. Multiple results indicate that rpS12D97 represents a loss-of-function mutation affecting a specific function of normal RpS12 in promoting cell competition that is genetically distinct from the essential role of RpS12 in cell growth and survival. For example, clones of cells over-expressing wild-type RpS12 in imaginal discs from ‘Minute’ Rp+/- genotypes are eliminated during growth; by contrast, clones over-expressing the RpS12D97 protein survive normally, indicating that normal RpS12 protein has a function that is deleterious for Rp+/- cells and that RpS12D97 lacks this function [23]. Similarly, cells carrying higher copy numbers of the rpS12+ gene are eliminated from ‘Minute’ genotypes heterozygous for mutations in other Rp genes, but cells carrying extra copies of the rpS12D97 allele are unaffected, again showing that rpS12D97 does not encode this aspect of wild type rpS12+ function. These and other studies indicate that in addition to an essential function, presumably in translation, the wild type RpS12 protein has another function in cell competition. The cell competition function is genetically separable because it is specifically affected by the Gly97Asp substitution [23].
Unlike Xrp1, the rpS12D97 mutation appeared not to rescue the developmental delay of Rp mutants (RpL36+/- and RpS18+/- were tested) [23]. Thus, the rpS12D97 mutation appeared more specific for the process of cell competition than mutations in Xrp1 that affect multiple aspects of the Minute phenotype. This raised the possibility that whereas many genes that affect global aspects of the Minute phenotype would be affected in Xrp1 mutants, the subset whose expression depended on both Xrp1 and RpS12 might identify genes more specifically important in cell competition. It should be noted, however, that the elevated Xrp1 expression in Rp+/- cells was found to be RpS12-dependent, which is surprising if Xrp1 is upstream or parallel to RpS12 [24].
In order to resolve the relationship between Xrp1 and RpS12, and potentially to identify a subset of the Minute cell transcriptional signature that correlated specifically with cell competition rather than other aspects of the Minute phenotype, we compared the transcriptomes of wing discs from wild type Xrp1, and rpS12D97 genotypes and their combinations with Rp+/- genotypes. These studies demonstrated clearly that rpS12 acts upstream of Xrp1 to control the Rp+/- gene expression signature. Consistent with this, we also found that rpS12 is required for the Xrp1-dependent reduction in overall translation of Rp+/-cells, and show through genetic epistasis that rpS12 acts upstream of Xrp1 in the regulation of imaginal disc cell translation and growth. We present new evidence that, although not previously appreciated, rpS12 can affect the developmental delay of Rp+/-genotypes. The reason that the rpS12D97 mutation does not rescue Rp+/-developmental delay as dramatically as Xrp1 mutations may be that rpS12D97 simultaneously retards development through a mechanism independent of Xrp1. Consistent with the notion that rpS12D97 has additional effects, we show that rpS12D97 diminishes longevity and can affect adult organ size.
Our findings provide clear molecular and genetic evidence that RpS12 plays a central and early role in generating multiple aspects of the Rp+/- phenotype in Drosophila imaginal discs by activating Xrp1 expression and activity.
Results
RpS12 regulates the Xrp1-dependent gene expression program of Rp+/-mutant cells
Rp+/- wing discs exhibit an altered pattern of transcription that is thought to encode some of their properties [22]. Our studies identified 253 mRNA’s that were altered in the same direction in both RpS3+/- and RpS17+/- genotype [24]. To identify the RpS12-dependent changes, we now report mRNA-Seq analysis from rpS12D97/D97 RpS3+/- wing imaginal discs, as well as rpS12D97/D97 and Xrp1+/- wing imaginal discs. Strikingly, 201 of the 253 genes (79%) changing expression in Minute Rp+/- genotypes did so in response to rpS12 activity (Fig 1A). Xrp1 also regulates most of the differential mRNA expression in Minute Rp+/- wing discs [24]. Of the 201 RpS12-regulated genes, differential expression of 189 (94%) also depended on Xrp1, strongly suggesting that RpS12 and Xrp1 affected a common pathway (Fig 1B).
Fig 1. RpS12-dependent gene expression in Rp+/- wing discs.

A) 201 of 253 mRNAs altered in Rp+/- wing discs were rpS12-dependent. B) rpS12- and Xrp1-dependent Rp+/- wing disc genes were largely overlapping. Shown in parentheses are the numbers of genes with predicted Xrp1 binding motifs (See also S3 Table). C. Heat map of fold changes in mRNA levels between wing discs from wild type and from indicated genotypes. Upregulation in Rp+/- genotypes was overwhelmingly dependent on both rpS12 and Xrp1. Genes shown here include all those corresponding to the enriched GO terms mature ribosome assembly (R), sulfur compound metabolic process (S), glutathione metabolic process (G), telomere maintenance (T), and DNA repair (D). The individual fold changes are shown in the S1 Table and S2 Table. Panels D-F show GstD1-LacZ expression in wing discs. Panels G-I show Upd3.3-LacZ labeling of wing discs. Genotypes: wild type (D,G); M(3)i55 ubi-GFP FRT80B/+ (E,H); M(3)i55 ubi-GFP FRT80B/FRT82 Xrp1m2-73 (F,I).
We found previously that Rp+/--dependent transcripts were enriched for GO terms glutathione metabolic process, telomere maintenance, DNA recombination, and iron-sulfur cluster assembly [24]. A more recent GO term database also revealed enrichment for ‘mature ribosome assembly’, reflecting elevated transcript levels for the eIF6, CG8549 and CG33158 genes. Our RpS12- and Xrp1-dependent transcript datasets were both enriched for the same GO terms. The genes with the enriched GO terms were overwhelmingly co-regulated by RpS12 and Xrp1 (Fig 1C; S1 Table).
Most apparent differences between our results and gene expression changes in RpS3+/- wing imaginal discs reported by another group [22] only reflect different statistical cutoffs (see methods). By our criteria, the differentially-expressed genes reported previously [22] were significantly enriched for the GO terms oxidation-reduction process, glutathione metabolic process, telomere maintenance, and sensory perception of sweet taste, very similar to our own results.
Despite hypotheses that Rp+/- cells might be less efficient at capturing Dpp [25], or experience elevated Wg signaling [26], representative target genes for Dpp and Wg signals were not affected in RpS3+/- and RpS17+/- wing discs. There were also no gene expression changes of signature genes for Ras, Hh, or Salvador-Warts-Hippo signaling (S2 Table). Socs36E, a target of Jak/Stat signaling was significantly elevated, however, as was a target of JNK signaling (MMP1) and one Notch target gene (E(spl)-m3) was decreased, although other Notch targets were unaffected (Fig 1C, S2 Table). We confirmed that Rp+/- cells exhibit an oxidative stress response using the GstD1-LacZ reporter line, whose expression was elevated in RpS17+/- wing discs in an Xrp1-dependent manner (Fig 1D–1F), and that a Upd-3 reporter also elevated in an Xrp1-dependent manner, which would be expected to signal through Jak/Stat (Fig 1G–1I). We had reported previously that expression of the JNK reporter puc-LacZ in RpS18+/- cells was dependent on Xrp1 [24]. We also found that the loci whose expression was affected in Minute Rp+/- genotypes were enriched for Xrp1 binding motifs. Xrp1 binding sites were among the most highly enriched binding sites in these genes, identified near 77 of these 253 genes (30%)(Fig 1B; S3 Table). ChIP experiments would be required to confirm that these genes were direct Xrp1 targets, however.
We examined transcript levels of the rpS12 and Xrp1 genes themselves to see how they might be related. rpS12 transcript levels were increased in rpS12D97/D97 wing imaginal discs compared to wild type, and reduced in both Minute Rp+/- genotypes (Fig 1C, S1 Table). Because rpS12 transcript levels were elevated in rpS12D97/D97 RpS3+/- wing imaginal discs, we conclude that reduction in RpS3+/- wing discs depended on normal RpS12 function (Fig 1C, S1 Table). Because the rpS12D97 allele affects the coding sequence, this suggests that RpS12 protein might regulate expression of its own mRNA, either directly or indirectly, which could also explain elevated rpS12 mRNA levels in the rpS12D97/D97 genotype. By contrast, rpS12 mRNA levels were not different between RpS3+/- wing discs and RpS3+/- Xrp1+/- wing discs, so Xrp1 was not responsible for altered rpS12 expression in this Minute Rp+/- genotype (Fig 1C, S1 Table).
As reported previously, Xrp1 transcription is elevated in Minute Rp+/- genotypes (Fig 1C, S1 Table) [14, 22, 24]. This increase in Xrp1 transcripts was entirely dependent on rpS12 function (Fig 1C, S1 Table), which is consistent with the reported RpS12-dependence of Xrp1 protein and Xrp1-LacZ enhancer trap expression in RpS17+/- or RpS18+/- wing discs [24]. Xrp1 mRNA levels also depended on Xrp1 function, implying an autoregulatory loop of Xrp1 expression in Rp+/- cells (Fig 1C, S1 Table).
Taken together, these results strongly suggest that RpS12 is an upstream regulator of the Rp+/- phenotype. They indicate that RpS12 activity elevates Xrp1 expression in Minute Rp+/- genotypes, which in turn is directly or indirectly responsible for most of the transcriptional changes seen in Minute Rp+/- wing discs.
RpS12 affects translation and growth through Xrp1
Genetic mosaic experiments were performed to determine whether RpS12 regulates Xrp1 expression cell-autonomously. Clones of RpS3+/- rpS12D97/D97 cells were induced in RpS3+/- rpS12D97/+ wing discs. The RpS3+/- rpS12D97/D97 cells showed lower levels of Xrp1 protein expression, showing that RpS12 regulated Xrp1 levels cell-autonomously (Fig 2A and 2B).
Fig 2. Growth and translation in Rp+/- wing discs.

All panels show wing pouch regions of third-instar wing imaginal discs. Genotypes are as indicated below each figure; the font colors correspond to the labeling of the corresponding genotype (the genotype corresponding to the most brightly labeled cells is shown in white on a black background). A,B). Xrp1 protein levels were lower in RpS3+/- rpS12D97/D97 clones than in RpS3+/- rpS12D97/+ cells. Small RpS3+/- rpS12+/+ clones were detected rarely (eg arrow) C) RpS3+/- rpS12+/+ clones are rarely detected in RpS3+/- rpS12D97/+ wing discs, unlike the reciprocal RpS3+/- rpS12D97/D97 clones. D) In contrast to panel C, in the Xrp1+/- background RpS3+/- rpS12+/+ clones were recovered similarly to the reciprocal RpS3+/- rpS12D97/D97 clones. E,F). Overall translation was reduced in RpS17+/- cells, as was described previously [16]. G,H). Overall translation was not affected in RpS17+/- cells in rpS12D97/D97 wing discs. I,J) Translation in RpS18+/- cells is reduced compared to wt cells, as was described previously [16]. K,L). Further data related to this figure is shown in the S1 Fig. Genotypes: A-C) y w hsF; rpS12D97 FRT80B/P{arm-LacZ} FRT80B M(3)95A armLacZ. D) y w hsF; rpS12D97 FRT80B/P{arm-LacZ} FRT80B Xrp1m2-73 M(3)95A. E,F) y w hsF; RpS174 P{ubi-GFP} FRT80B/FRT80B. G,H) y w hsF; rpS12D97 RpS174 P{ubi-GFP} FRT80B/rpS12D97 FRT80B. I,J) y w hsF; FRT42D P{arm-LacZ} M(2)56i/FRT42D. K,L) y w hsF; FRT42D P{arm-LacZ} M(2)56i/FRT42D; rpS12D97 FRT80B/rpS12D97 FRT80B.
To confirm that Xrp1 functions downstream of RpS12, as suggested by the expression data, we tested whether Xrp1 was required for RpS12 to influence cell competition. In the experiment described above and shown in Fig 2A and 2B, the RpS3+/- cells that have two copies of the wild type rpS12 allele (RpS3+/- rpS12+/+) are frequently lost in the RpS3+/- rpS12D97/+ background because their higher copy number of the rpS12+ locus targets them for elimination by cell competition (Fig 2A and 2C) [23]. This elimination depended on Xrp1, because when Xrp1 was mutated the RpS3+/- rpS12+/+ Xrp1+/- cells survived in the RpS3+/- rpS12D97/+ Xrp1+/- wing discs (Fig 2D). Therefore, RpS12 needs Xrp1 to affect RpS3+/- clone survival.
If RpS12 is the upstream regulator of Xrp1 expression, it should be required for the whole Xrp1-dependent response. Xrp1 is required for the slow growth of Minute Rp+/- genotypes at the cellular level, and regulates their global translation rate. Minute Rp+/- genotypes exhibit slower overall translation rate than wild type cells, and this depends on Xrp1 because normal translation is largely restored in Rp+/- Xrp1+/- cells [24]. If RpS12 acts upstream of Xrp1, we would expect that in rpS12D97 mutants there would also be no difference in translation rate between wild type and Rp+/- cells. As predicted, whereas RpS17+/- and RpS18+/- cells exhibited reduced translation rate compared to wild type cells in the presence of wild type rpS12, both RpS17+/- and RpS18+/- cells showed translation indistinguishable from RpS17+/+ and RpS18+/+ cells in the rpS12D97/D97 genotype(Fig 2E–2L; S1 Fig).
To address this question in another way, rpS12D97/D97 RpS3+/- and rpS12+/+ RpS3+/- clones were examined in rpS12D97/+ RpS3+/- wing discs. As noted above, rpS12+/+ RpS3+/- clones were infrequent and always small, and they generally exhibited reduced translation (S2 Fig panels A,B). The rpS12D97/D97 RpS3+/- cells had higher rates of translation that could often be distinguished from the rpS12D97/+ RpS3+/- background (S2 Fig panels A,B). These rpS12-dependent differences were abolished in the Xrp1+/- background (S2 Fig panel C). We also found that translation in RpS17+/- rpS12D97/+ was quite similar to RpS17+/+(S2 Fig panels D-F).
Taken together, these experiments showed that rpS12 requires Xrp1 to affect the competitiveness of Rp+/-cells, and that the reduction in translation that is mediated by Xrp1 is also downstream of RpS12. These findings strongly support the conclusion derived from gene expression studies: RpS12 appears to be the upstream regulator that signals to Xrp1 in Rp+/-genotypes, and accordingly it is required for Xrp1-dependent effects on translation and growth as well as for cell competition.
RpS12 also makes Xrp1-independent contributions to the rate of development
If RpS12 is the upstream regulator of Xrp1, it is surprising that rpS12 would not affect Rp+/- developmental delay, since Xrp1 is responsible for much of this delay [23, 24]. RpS12 regulates Xrp1 expression, and through Xrp1 gene expression, translation and cell competition in Rp+/- genotypes. Therefore, rpS12 mutations should affect the Rp+/- developmental delay like Xrp1 mutations do.
To revisit the effects of RpS12 on developmental rate, we looked at the effects of increased rpS12 gene dose on Minute Rp+/- genotypes. Compared to wild type controls, RpS3+/- females emerged 24h later, on average, and RpS3+/- males 28h later, on average (Fig 3A and 3B). An additional copy of the rpS12+ locus, encoded on a transgene, further delayed adult emergence by another 24h in rpS12+/+/+ RpS3+/- females, on average, and by another 20h in males (Fig 3A and 3B). No RpS3+/- adults carrying two extra copies of the rpS12+ locus were recovered, although the rpS12+ transgene is homozygously viable by itself. Therefore, although they had no effects in Rp+/+ genotypes, extra copies of rpS12+ exacerbated the Rp+/- developmental delay, showing that RpS12 can indeed regulate developmental rate.
Fig 3. Contribution of RpS12 to rate of development.

All panels show the cumulative percentage of adult flies emerged according to time after egg laying in hours. A,B) A genomic transgene including the rpS12+ locus had little effect on wild type development but significantly retarded development of RpS3+/- flies. C,D) Heterozygosity for rpS12 had little effect on wild type development but modestly accelerated development of RpS18+/- flies. E,F) the genotype rpS12D97/EP3025 modestly delayed development of otherwise wild type flies. G,H) the genotype rpS12D97/D97 modestly delayed development of otherwise wild type flies and this delay was suppressed by a genomic transgene including the rpS12+ locus. I,J) An experiment in which rpS12D97/D97 and rpS12D97/+ genotypes had little effect on the development of RpS3+/- flies. K,L) An experiment in which rpS12D97/D97 and rpS12D97/+ genotypes suppressed the developmental delay of RpS3+/- flies, to a similar extent to RpS3+/- rpS12D97/D97 Xrp1+/- flies. In Fig 3K and 3L, the development of RpS3+/- rpS12D97/D97 flies and RpS3+/- rpS12D97/D97 Xrp1+/- flies are significantly faster than RpS3+/- flies (p<0.00001 in all cases, Mann Whitney procedure). The detailed genotypes used and numerical data corresponding to these graphs is tabulated in the S4 Table.
We then re-examined the effect of rpS12 loss of function in developmental delay. Previously, we reported that RpS18+/- rpS12D97/- flies develop at the same rate as RpS18+/- rpS12D97/+ flies and RpS18+/- rpS12+/+ flies [23]. We noticed, however, that in the same experiments there was a small suppression of developmental delay of RpS18+/- flies that were also rpS12+/- (Fig 3C and 3D). In addition, we previously observed a small developmental delay of rpS12D97/- flies in comparison to wild type [23]. Because this effect was at the limit of what was observable (only 4-5h), and its significance might be questioned, we also examined other rpS12 genotypes. We confirmed a small developmental delay in rpS12D97/EP3025, a different rpS12D97/- genotype from the rpS12D97/s2783 used previously, and in rpS12D97/D97 (Fig 3E–3H). Finally, we found that the developmental delay of rpS12D97/D97 flies was at least partially suppressed by an rpS12+ genomic transgene, suggesting that the mutant rpS12 genotype was at least partly responsible (Fig 3G and 3H). Taken together, these results suggest that the rpS12D97 mutation delays development of otherwise wild type flies, although to a milder degree than the dominant effect of ‘Minute’ mutations at other Rp loci.
Further evidence for a detrimental rpS12D97 phenotype came from studies of longevity and organ size. To compare the longevity of rpS12D97 flies with a control in the same genetic background, we backcrossed the rpS12D97 mutation into a w11-18 background for 6 generations, then established homozygous viable rpS12D97 and wild type lines from single sibling flies from the last generation. In this background the median lifespan of rpS12D97 mutant females was 65 days, compared to 72 days in control females (Fig 4A; S3 Fig). The median lifespan of rpS12D97 mutant males was 58 days, compared to 67 days in controls (Fig 4B). The median lifespan of rpS12D97 mutant These differences were statistically significant. We also noticed that rpS12D97 homozygous adults and adults from a rpS12D97/- genotype often had smaller wings, reduced by up to 8% of total area (Fig 4C and 4D).
Fig 4. Contributions of RpS12 to longevity and organ size.

A) Survival curve of female rpS12G97D flies compared to w11-18 (wild type) controls. Shown is the mean are 95% confidence limits from 3 replicates of 120 flies each. B) Survival curve of male rpS12G97D flies compared to w11-18 (wild type) controls. The difference is significant at p<0.0001 Log-rank (Mantel-Cox) test. See the S3 Fig for individual replicates and the S5 Table for raw data. C) Adult wing size was significantly smaller for the rpS12G97D females compared to w11-18 (p = 0.0165, two-tailed Welch’s t-test) and for rpS12G97D/- females compared to FRT80B (p = 0.000006, two-tailed Welch’s t-test). Wing size was also smaller for rpS12G97D/- males compared to FRT80B (p = 0.00014, two-tailed t-test) but not for rpS12G97D males compared to w11-18 (p = 0.126, two-tailed t-test). Raw data are tabulated in the S7 Table. D) Wings from rpS12G97D/- and FRT80B females overlaid to illustrate the difference. The rpS12G97D/- wing is smaller.
If the rpS12D97 mutation affects non-Minute flies, this suggests how rpS12D97 Rp+/- flies might develop more slowly than Xrp1+/- Rp+/- flies. The rpS12D97 mutation may both suppress Rp+/- developmental delay by preventing Xrp1 expression, at the same time as delaying development independently by another mechanism. Results obtained with RpS3+/- could be consistent with this hypothesis. In multiple experiments, rpS12D97/D97 RpS3+/- flies, and sometimes rpS12D97/+ RpS3+/- flies, were slightly rescued compared to RpS3+/- flies, but always to a lesser degree than observed with Xrp1 mutations (Fig 3I–3L). Similar effects of rpS12 and Xrp1 mutations were seen on the delayed pupariation of RpS3+/- larvae (S3 Fig).
Xrp1-independent regulation of gene expression by RpS12
How the rpS12D97 mutation affects development in Rp+/+ genotypes might be revealed by gene expression studies. DESeq2 identified 258 genes expressed significantly differently in rpS12D97/D97 wing discs compared to wild type, 126 downregulated and 132 upregulated. These genes were significantly enriched for the GO terms related to sensory perception of sweet taste, detection of chemical stimulus involved in the sensory perception of taste, and detection of stimulus involved in sensory perception. Only 35 genes (14%) of these genes were also altered in Rp+/- wing discs, showing that the rpS12D97 mutation had few effects in common with dominant Rp mutations (Fig 5A).
Fig 5. Cross-genotype co-expression analysis.

A. Transcripts that differed significantly between control and rpS12D97/D97 wing discs showed little overlap with those affected in RpS3+/- and RpS17+/- wing discs. B) Transcripts dependent on RpS12 in both Rp+/- and Rp+/+ wing discs showed little overlap with those affected in both RpS3+/- and RpS17+/- wing discs. C) Transcripts that differed significantly between control and Xrp1+/- wing discs showed little overlap with those affected in RpS3+/- and RpS17+/- wing discs. D) Transcripts dependent on Xrp1 in both Rp+/- and Rp+/+ wing discs showed little overlap with those affected in both RpS3+/- and RpS17+/- wing discs. E) RpS12- and Xrp1-dependent transcripts showed little overlap. F) Hierarchical clustering of wing disc samples by their similarities (as Pearson correlation coefficients) in gene expression profiles distinguished samples by genotypes. The Xrp1+/- replicates grouped with wild type controls. G) Co-expressed and similarly-regulated genes (module #2) whose expression was altered in RpS3+/- and RpS17+/- wing discs in an RpS12-dependent, Xrp1-dependent manner. H) Co-expressed and similarly-regulated genes (module #8) whose expression was altered in RpS3+/- and RpS17+/- wing discs in an RpS12-independent, Xrp1-independent manner. I) Co-expressed and similarly-regulated genes (module #6) whose expression was altered in an RpS12-dependent manner regardless of RpS3 genotype. In G-I, the columns in the heatmaps are genes and the rows are samples (3 replicates each as indicated); color scale indicates relative expression across samples. J) Genes in the Module 6 that were downregulated in rpS12D97 genotypes and present in the GO term ‘secretion’. Their fold changes (from DESeq2) are shown here. Bold values were statistically significant (Padj < 0.05). Neither Module 6 genes that were upregulated in rpS12D97 genotypes, not Module 6 as a whole, was enriched for any particular GO terms.
In previous studies it was helpful to focus on Rp+/- -affected genes whose mRNAs were altered in both RpS17+/- and RpS3+/- [24]. We applied a similar cross-comparison to identify RpS12-regulated genes by identifying those whose mRNA levels differed in both rpS12D97/D97 wing discs compared to wild type and in rpS12D97/D97 RpS3+/- wing discs compared to RpS3+/-. The 248 genes identified through this cross-comparison were similar to those described above, and only 23 (9%) were also altered in Rp+/- wing discs (Fig 5B).
DESeq2 identified 77 genes expressed differentially in Xrp1+/- wing discs compared to wild type, 49 downregulated and 38 upregulated. Alternatively, expression of 84 genes differed in both Xrp1m2-73/+ wing discs compared to wild type and in Xrp1m2-73/+ RpS3+/- wing discs compared to RpS3+/-. These Xrp1-regulated genes were not enriched for any biological process GO terms, and showed only 21% and 14% overlap, respectively, with genes altered in Rp+/- wing discs, indicating that Xrp1 largely failed to regulate the same genes in wild type and in Rp+/- wing discs (Fig 5C and 5D).
Only 25 RpS12-regulated genes (10%) showed similar regulation by Xrp1 indicating that, in the absence of Minute Rp mutations, rpS12 and Xrp1 mutations affected largely distinct pathways.
Because the rpS12-dependent genes were not obviously enriched for growth genes (in principle genes affecting taste might affect food intake but we would not expect their expression in wing imaginal discs to be responsible), we analyzed gene expression in another way to search for genes and pathways that may be affected in a more subtle way. We performed unsigned Weighted Gene Co-expression Network Analysis (WGCNA) to identify sets of genes with similar co-expression patterns, referred as gene modules. Importantly, WGCNA can identify changes in gene expression that may not reach the statistical threshold for detecting differential expression of individual genes and allow integrated analysis of samples across multiple genotypes. Clustering analysis of samples by their gene expression similarities showed that samples of different genotype clustered separately, except that Xrp1+/- samples were intermingled with the wild type control, suggesting that in non-Minute genotypes Xrp1 has only a minimal effect on mRNA abundance (Fig 5F). WGCNA analysis of the 21 samples identified 20 gene modules, containing various numbers of genes with significantly positively or negatively correlated expression patterns. Of these, Module #2 contained genes elevated in both RpS3+/- and RpS17+/-, in an RpS12/Xrp1-dependent manner (Fig 5G). Interestingly, Module #2 contained many of the RpS12/Xrp1-regulated genes already identified as differential expression in Minute Rp mutants. Module #8 contained genes elevated in both RpS3+/- and RpS17+/-, in an RpS12/Xrp1-independent manner (Fig 5H). Module #8 was enriched for GO terms associated with cytoplasmic translation, indicating that Minute Rp mutants may also affect translation independently of RpS12/Xrp1. Module #6 contained RpS12-regulated genes (altered in rpS12D97/D97 RpS3+/- compared to RpS3+/- and in rpS12D97/D97 compared to wild type) (Fig 5I). The Module #6 genes that were down-regulated in rpS12D97 genotypes were enriched for genes involved in protein secretion, although with the exception of pastrel (pst) the effects on gene expression were minor (Fig 5J). We did not detect any module of genes clearly regulated by Xrp1 alone.
Although these experiments did not identify how the rpS12D97 mutation might affect growth independently of Xrp1, they confirmed that individually, Rp+/-, rpS12D97 and Xrp1+/- mutants had little in common, suggesting that the RpS12-Xrp1 regulatory axis is not active in non-Minute wing discs. Indeed, gene expression in Xrp1+/- wing discs was indistinguishable from the controls by WGCNA analysis (Fig 5F).
Regulation of Rp gene transcripts
The GO term ‘cytoplasmic translation’ was enriched among transcripts that were affected by Minute Rp mutants independently of RpS12 and Xrp1 (Module 8: Fig 5H). This reflects presence of many Rp gene transcripts in this module. We therefore looked at the transcript levels of all Rp genes in wing discs (Fig 6). Rp transcripts are highly expressed (>36% of total transcripts in wild type wing discs). Both RpS3+/- and RpS17+/- led to a general reduction in mRNA levels of Rp genes (median fold change -13%). In addition, RpS9, RpS12, Rack1 and RpS27A genes stood out as being more strongly reduced (Fig 6A). This picture changed very little in RpS3+/- Xrp1+/- wing discs, indicating that these changes in Rp expression were independent of Xrp1(Fig 6B). In RpS3+/- rpS12D97/D97 wing discs, however, only RpS9 and Rack1 were still significantly lower than the general distribution of Rp transcripts (Fig 6C). Thus, reduction in RpS27A mRNA levels in RpS3+/- discs was a specific function of rpS12. The rpS12 mRNA itself was significantly elevated in RpS3+/- rpS12D97/D97 wing discs(Fig 6C). This could reflect a role of RpS12 in suppressing its own expression, as revealed when rpS12D97/D97 wing discs were compared to controls. In this case all other Rp mRNA levels were at control levels (median fold change 1%), but RpS12 levels were elevated by 55% (Fig 6D).
Fig 6. Ribosomal protein transcripts.

All panels show fold changes of Rp mRNA levels from mRNA-seq analysis of mutant wing imaginal discs in comparison to wild type controls. Loci are arranged from most reduced to most increased in each case. Transcripts of duplicated Rp genes that are expressed in at low levels because their paralog dominates in wing discs are not included. RpS9, rpS12, RACK1, RpS27A transcripts highlighted. A) Rp genes show an overall reduction in transcript levels in RpS3+/- and RpS17+/- wing discs, with RpS9, rpS12, Rack1, and RpS27A affected to a greater extent. Fold changes are the mean of RpS3+/- and RpS17+/- values except for RpS3 and RpS17 themselves; transcript levels of these genes is 50% reduced in their own mutants so only the value for the other mutant is shown here. B) Rp mRNAs in RpS3+/- Xrp1+/- wing discs resembled those in RpS3+/- wing discs. RpS3 mRNA levels are not included. C) RpS9 and Rack1 transcript levels remained reduced in rpS12D97/D97 RpS3+/- but rpS12 and RpS27A were restored to wild type or higher levels. RpS3 mRNA levels are not included. D) The only Rp whose transcript levels were affected in rpS12D97/D97 wing discs was rpS12 itself.
These findings identify a hierarchy of Rp transcript regulation. Both RpS3+/- and RpS17+/- genotypes modesty reduce mRNA levels of all Rp genes in an RpS12-Xrp1-independent manner (median fold change 13–20% in the various genotypes). The reductions of RpS9 and Rack1 levels stand out as still more extreme (-39% and -37% fold change, respectively). RpS27A and rpS12 are also reduced, but in an RpS12-dependent manner that does not depend on Xrp1. These findings illustrate that not all aspects of the Rp+/- phenotype depend on rpS12 or Xrp1. Since it is Xrp1 that is responsible for much of the reduction in bulk translation rate that occurs in ‘Minute’ Rp+/- genotypes [24], how much these modest, Xrp1-independent changes in Rp transcripts affect steady-state ribosome number or overall translation is uncertain.
Evidence for an RpS12-independent contribution of Xrp1 to the rate of development
To help understand how RpS12 and Xrp1 function together, developmental delay was assessed in the triple mutant combination rpS12D97/D97 RpS3+/- Xrp1+/-. If the hypothesis that RpS12 acts through Xrp1 is correct, we would expect that Xrp1 would make no contribution to the developmental delay of rpS12D97/D97 RpS3+/- flies, in which Xrp1 expression is not elevated, so that rpS12D97/D97 RpS3+/- Xrp1+/- and rpS12D97/D97 RpS3+/- would develop similarly. In the experiment described in a previous section, where rpS12D97/D97 RpS3+/-partially suppressed the developmental delay of RpS3+/-, a similar or greater suppression was seen with rpS12D97/D97 RpS3+/- Xrp1+/- (Fig 3K and 3L). We do not always, however, see suppression of developmental delay by rpS12D97 [23]. In an independent experiment where rpS12D97/D97 RpS3+/- and rpS12D97/D97 RpS17+/- were not rescued in comparison to RpS3+/- and RpS17+/-, developmental delay was partially suppressed in rpS12D97/D97 RpS3+/- Xrp1+/- and suppressed in rpS12D97/D97 RpS17+/- Xrp1+/- to the same degree as in RpS17+/- Xrp1+/- (Fig 7A–7D). The variable phenotype of rpS12D97 Rp+/- genotypes is unexpected. We do not think this reflects inaccuracy in the measurement method. The rescue observed in Fig 3K and 3L was highly significant statistically and also observed when timing of pupariation was measured (S3 Fig). The various panels of Fig 3 and Fig 7 together contained 34 internal controls of independently replicated measurements (eg of multiple wild type genotypes) that illustrate very high reproducibility for other genotypes. It is possible that rpS12D97 Rp+/- genotypes behave differently because their rate of development arises as a balance of multiple positive and negative processes.
Fig 7. Effects of Xrp1 and of tetracycline on rate of development.

All panels show the cumulative percentage of adult flies emerged according to time after egg laying in hours. A,B) An experiment in which rpS12D97/D97 and rpS12D97/+ genotypes did not accelerate the development of RpS3+/- flies, and in which RpS3+/- rpS12D97/D97 Xrp1+/- flies developed more slowly than RpS3+/- Xrp1+/- flies. C,D) An experiment in which rpS12D97/D97 and rpS12D97/+ genotypes did not accelerate the development of RpS17+/- flies, but in which RpS17+/- rpS12D97/D97 Xrp1+/- flies developed as rapidly as RpS17+/- Xrp1+/- flies. E,F) Tetracyclin feeding modestly retarded development of wild type flies. G,H) Tetracyclin feeding modestly retarded development of RpS3+/- and RpS17+/- flies. I,J) After tetracycline feeding, rpS12D97/D97 and rpS12D97/+ genotypes did not accelerate the development of RpS3+/- flies, but in which RpS3+/- rpS12D97/D97 Xrp1+/- flies developed as rapidly as RpS3+/- Xrp1+/- flies. K,L) After tetracycline feeding, rpS12D97/D97 and rpS12D97/+ genotypes did not accelerate the development of RpS3+/- flies, but in which RpS3+/- rpS12D97/D97 Xrp1+/- flies developed as rapidly as RpS3+/- Xrp1+/- flies. The genotypes used and numerical data corresponding to these graphs is tabulated in the S8 Table.
We considered the possibility that microbiome differences might affect our results. Cultures of Rp+/- genotypes can grow poorly and may be more prone to microbial growth, which can be suppressed by supplementing fly food with antibiotics. Interestingly, tetracycline measurably delayed development of all genotypes, including wild type controls (eg Fig 7E–7H). Whether this reflects a positive contribution of the microbiome to fly growth, or a deleterious effect of tetracycline [27, 28] is uncertain. In the presence of antibiotic, rpS12D97/D97 RpS3+/-and rpS12D97/D97 RpS17+/-developed no faster than RpS3+/-or RpS17+/-, but rpS12D97/D97 RpS3+/- Xrp1+/-, RpS3+/- Xrp1+/-, rpS12D97/D97 RpS17+/- Xrp1+/- and RpS17+/- Xrp1+/- all showed a similar acceleration of development compared to RpS3+/-and RpS17+/-(Fig 7I–7L). Therefore, although the results are complicated by variability of rpS12D97 Rp+/- genotypes, Xrp1 mutation frequently accelerated the development of rpS12D97/D97 RpS3+/- flies, despite the fact that Xrp1 expression was not elevated in rpS12D97/D97 RpS3+/- wing discs.
Discussion
Our findings provide compelling evidence that RpS12 contributes to the ‘Minute’ Rp+/- phenotype through Xrp1. In contrast to initial impressions that RpS12 might play a more specific role in cell competition than Xrp1, because rpS12 mutations seemed to suppress less of the ‘Minute’ Rp+/- phenotype, it is in fact RpS12 that is the more upstream of the two cell competition genes (Fig 8).
Fig 8. Model for regulatory effects of Rp mutations.
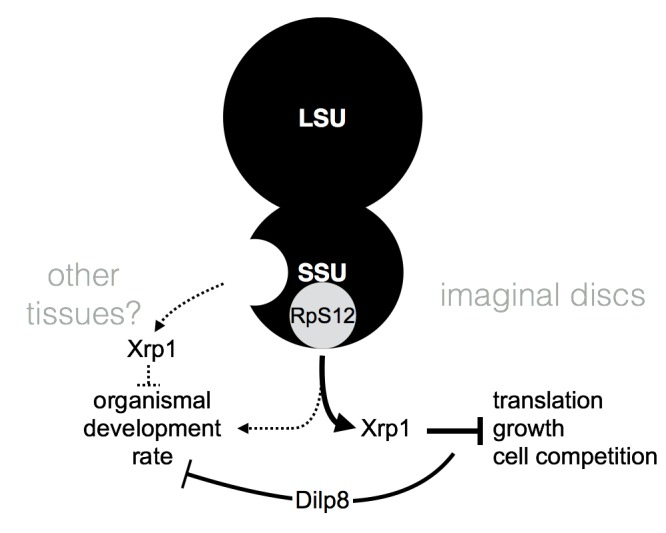
When one of the many haploinsufficient Rp is limiting, an RpS12-dependent signal is activated that elevates Xrp1 expression, inhibiting imaginal disc cell translation and growth and making Rp+/- cells less competitive. Analyses of imaginal disc gene expression and genetic epistasis studies provide compelling evidence that RpS12 acts through Xrp1 in imaginal discs. Defects in imaginal disc growth delay overall organism development, which may depend on Xrp1-dependent signaling by Dilp8 [21]. Other data suggest that RpS12 also contributes positively to the rate of development, independently of Minute Rp mutations, and that Minute Rp mutations and Xrp1 may also slow organism development independently of RpS12.
The RpS12-dependent and Xrp1-dependent components of the Rp+/- gene expression signature overlap almost completely, indicating that they affect the same pathway (Fig 1A–1C). These RNA-Seq results also suggest that RpS12 acts upstream of Xrp1. They show that Xrp1 expression is RpS12-dependent; conversely, Xrp1 makes no contribution to RpS12 expression in Rp+/- genotypes. There are also transcripts, such as RpS27A, that are altered in Rp+/- wing discs in a way that depends on RpS12 but not on Xrp1 (Fig 1C, Fig 6).
Previously we had noted that RpS12 is required for the elevated Xrp1 expression seen in Rp+/- cells [16]. Here we confirm this regulation is cell-autonomous, and importantly we show, through genetic epistasis, that Xrp1 is indeed required downstream of RpS12 in cell competition, as the regulation of Xrp1 expression levels would suggest (Fig 2A–2D). In addition we showed that, as would be expected if RpS12 acts through Xrp1, the rpS12D97 mutation also prevents the reduction in overall translation rate that Xrp1 expression causes in ‘Minute’ Rp+/- wing discs (Fig 2E–2L).
These molecular and genetic findings demonstrate clearly that RpS12 not only affects Xrp1 expression but that through Xrp1 it is required for almost the entire gene expression program of Rp+/- wing discs as well as their growth and global translation rate. The close correspondence between the Xrp1 and the rpS12D97 mutations argues that Xrp1 is the main target of RpS12 in Rp+/- wing discs. Because RpS12 is a ribosomal protein, it may be the signal, or part of the signal, that communicates a failure of ribosome assembly to the cell (Fig 8) [23]. While the signaling mechanism is not yet known, genetic studies indicate that the cell-competition mutation rpS12D97 affects a different aspect of RpS12 function from the essential function that reflects the role of RpS12 in general translation (which is also not yet understood) [23], and here we report accordingly that the gene expression changes caused by the rpS12D97 mutations are largely unrelated to those caused by mutations in most other Rp genes (Fig 5). Many ribosomal proteins, all of which are very highly expressed proteins, have a minor pool that participates in extra-ribosomal activities [2], but it also cannot be excluded that RpS12 might make two distinct contributions to ribosome function, only one of which is affected by rpS12D97 [23].
The role of RpS12 as an upstream regulator of Xrp1 was surprising at first, because rpS12D97 had not seemed to suppress the developmental delay of ‘Minute’ Rp+/- genotypes as Xrp1 mutations did [23]. If Xrp1 expression causes the developmental delay, and RpS12 is required for elevated Xrp1 expression, then RpS12 should be required for the developmental delay. Several new lines of evidence suggest that this is the case. First, we found that extra copies of RpS12 enhanced the developmental delay of RpS3+/-, suggesting that wild type RpS12 does promote developmental delay (Fig 3A and 3B). Secondly, we now find multiple examples where rpS12 mutations do suppress the developmental delay of Minutes, albeit always to a lesser extent than Xrp1 mutations did (Fig 3C, 3D, 3I–3L).
A potential explanation why the rpS12D97 mutation has less effect on the developmental delay of Rp+/- genotypes than Xrp1, and sometimes no effect at all, could be that rpS12D97 has other deleterious effects on development, so that in rpS12D97/D97 Rp+/- genotypes the negative effects of the rpS12D97 mutation partially or completely cancel out the effect of reduced Xrp1 expression. Several lines of evidence are now consistent with this hypothesis. First, a modest but consistent delay of development is observed in rpS12D97/D97 and rpS12D97/- genotypes in the absence of other Rp mutations (Fig 3E–3H). Secondly, rpS12D97/D97 and rpS12D97/- exhibit reduced longevity and reduced adult wing size, consistent with a deleterious effect even in the absence of other Rp mutations (Fig 4). We did not, however, identify any candidate growth pathway from gene expression studies of rpS12D97/D97 wing discs, which seems to share little in common with the effects of mutations at haploinsufficient loci such as RpS3 and RpS17 (Fig 5A, 5B, 5F and 5J). Given that rpS12D97/D97 mutant clones grow at the same rate in wing discs as wild type controls [23], the possibility exists that the rpS12D97mutation might affect developmental growth rate through another tissue, in which case these effects may not be not reflected in wing disc mRNA sequencing results.
A recent study has also implicated RpS12 and Xrp1 in the systemic delay of organismal development that results from damaging the wing disc by Rp knockdown. The wing disc damage induces organismal delay through RpS12- and Xrp1-dependent regulation of Dilp8, a further example where RpS12 signals through Xrp1 [21]. Rp+/- genotypes, although probably less severe than Rp knockdown, also elevate dilp8 transcription (Fig 1C and [16]), so there could also be a systemic component to the ‘Minute’ Rp+/- developmental delay. If the overall developmental rate of ‘Minute’ Rp+/- flies is determined by interactions between multiple tissues, not just the rate of imaginal disc growth alone, this would be consistent with some of the effects of RpS12 and Xrp1 could occur in other tissues.
Because rpS12D97/D97 flies are unable to eliminate Rp+/- cells by cell competition, defects in these flies could also reflect positive roles for cell competition in healthy aging. This has been suggested based on reduced longevity of and accumulation of developmental defects in azot mutants, encoding another gene implicated in cell competition [29]. It is notable, however, that loss of Xrp1 does not affect longevity [18], and we do not see developmental defects in either rpS12D97/D97 or Xrp1+/- flies, suggesting that these aspects of the azot phenotype are not due to failure to eliminate Rp+/- cells, although they could reflect failure to eliminate other kinds of cells.
Xrp1 appears to slow growth cell-autonomously by reducing overall translation [24]. In Rp+/- genotypes, Xrp1 is responsible for regulating three regulators of cytoplasmic ribosome assembly, eIF6, CG8549 and CG33158 (Fig 1A). CG8549 encodes the Drosophila homolog of the Shwachman-Diamond syndrome protein. In humans and in yeast these genes provide a quality control mechanism at the final step in LSU assembly that functionally activates the 60S subunit [30, 31], although eIF6 also promotes growth through other mechanisms [32]. These genes are up-regulated by Xrp1 in Rp+/- wing discs, however, which should not make them limiting for ribosome biogenesis or growth (Fig 1A). The Rp+/- genotypes also show a modest (~15%) reduction in mRNA levels of most Rp genes, but this was Xrp1-independent, suggesting that this is not the mechanism reducing translation in Rp+/- genotypes, although it could contribute to Xrp1-independent growth reduction in Rp+/- genotypes(Fig 6).
The RpS12-Xrp1 axis adds to the notion that many effects of Rp mutations are due to regulatory changes, not direct effects of a hypothetical reduction in ribosome numbers [16]. A regulatory pathway of nucleolar stress is also thought to contribute to Diamond-Blackfan Anemia, the human disorder associated with heterozygous mutations in Rp genes, and might contribute to the properties of human tumors with Rp mutations. In mammalian cells where ribosome assembly is reduced, unassembled RpL5 and RpL11 stabilize p53 [33, 34]. It is not yet clear how much p53 contributes to the phenotype, for example it is not yet established whether p53 is responsible for reducing translation, and it is debated whether p53 is responsible for anemia [35–39]. How mammalian RpS12 might be involved in Diambond Blackfan Anemia or cancer has not been described, although a possible association has been reported between RpS12 deletion and Diffuse Large B Cell Lymphoma [7, 40].
Methods
Experimental animals
Species: Drosophila melanogaster. Strains were generally maintained at 25°C on medium containing the following ingredients per 1L: 18g yeast; 22g molasses; 80g malt extract; 9g agar; 65g cornmeal; 2.3g methyl para-benzoic acid; 6.35ml propionic acid unless otherwise indicated. Sex of larvae dissected for most imaginal disc studies was not differentiated.
Fly stocks
Transgenes strains used in this study included: P{arm-LacZ, w+} [41], P{ubi-GFP, w+} [42]; P{rpS12+-8kb, w+} [23]. Mutant alleles used in this study: rpS12D97 [23], Xrp1M2-73 [13], M(2)56F (laboratory of Y. Hiromi), RpS174 [10], RpS3 [43].
Clonal analysis
Genetic mosaics were generated using the FLP/FRT system employing hsFLP and eyFLP transgenic strains [44–46]. For making clones using inducible hsFLP, larvae of non-Minute genotypes were subjected to 1 hour heat shock at 37°C, 60 ± 12 hours after egg laying and dissected 60hr later. For Minute/+ genotypes, heat shock was administered after 84 ± 12 hours of egg laying and dissected 72 hours later. Detailed genotypes employed are listed in figure legends.
Measurement of developmental timing
For measurements of developmental rate, adults were allowed to lay eggs on yeast-glucose media changed at 8h intervals, with or without tetracycline at 20 μg/ml final concentration [47, 48]. Multiple cultures were established in parallel to generate sufficient numbers, and typically maintained for 7–10 days of passage every 8h. Overcrowded or near-barren vials were discarded. Adults were collected every 8h to record emergence time. Whenever possible, genotypes emerging in the same cultures were compared and in other cases, control genotypes that were also present could be compared between crosses to verify comparable conditions (see the S4 Table & S8 Table). In most experiments shown, multiple independent estimates were obtained for the developmental rate of some genotypes within these crosses. These were very similar in every case, verifying the reproducibility of the assays (see Figs 3 & 7). For some experiments, pupariation was also recorded at 8h intervals.
Immunohistochemistry and antibody labeling
For antibody labeling, imaginal discs were dissected from late 3rd instar larvae, fixed and processed for immunohistochemistry as described previously [49]. Primary Antibodies are described in the Key Resources Table. Secondary Antibodies were Cy2-, Cy3- and Cy5- conjugates from Jackson Immunoresearch.
Image acquisition and processing
Confocal images were recorded using Leica SP2, SP5 and SP8 confocal microscopes using 20x and 40x objectives. Images were processed using Image J1.44j and Adobe Photoshop CS5 Extended.
Measurement of translation in vivo
Translation was detected by the Click-iT Plus OPP Alexa Fluor 594 or 488 Protein Synthesis Assay Kits (Thermofisher) as described [50] with modifications as described [24]. OPP (o-propargyl puromycin) is a puromycin analog that is incorporated into nascent polypeptide chains during a 15 min incubation with explanted imaginal discs.
mRNA-Seq
These mRNA-Seq analyses described here were performed in parallel to studies of other genotypes described previously, using the same methods [24]. Precise genotypes for which data are described for the first time were:
w11-18; rpS12D97 FRT80B / rpS12D97 FRT80B
w11-18; rpS12D97 FRT80B / rpS12D97 FRT82B RpS3
w11-18; FRT82B Xrp1m2-73 / +
At least 65,000,000 clean RNA-seq reads past quality controls were obtained from every sample and for every sample at least 89% of them were mapped to the fly genome. Bioconductor DESeq2 [51] was used to identify genes expressed significantly differently between control and rpS12D97/D97 wing discs and between control and Xrp1m2-73/+ wing discs, using the criteria log2(fold change)>1, adjusted p-value (Padj)<0.05. We also exploited cross genotype comparisons to define RpS12-regulated genes as those that were expressed differentially from control in rpS12D97/D97 and also expressed differently from RpS3+/- in rpS12D97/D97 RpS3+/-, with |log2fold change|>0.5 and Padj<0.1 in both cases, and to define Xrp1-regulated genes as those that were expressed differentially from control in Xrp1+/- and expressed differently from RpS3+/- in Xrp1+/- RpS3+/-, with |log2fold change|>0.5 and Padj<0.1 in both cases. GO enrichment analysis was performed using Gene Ontology Consortium tool PANTHER [52–54]. We considered GO terms to be significantly enriched when p<0.05 after Benjamini correction for multiple tests. This correction contributes to some differences in enriched terms in our analyses compared to those from another group [22].
Gene co-expression network
A total of 8,321 expressed genes, with FPKM (Fragments Per Kilobase of transcript per Million mapped read) > 0 in at least one sample, were used for weighted gene correlation network analysis (WGCNA) [55, 56]. We transferred the FPKM values into Transcripts Per Million (TPM) and utilized “normalizeBetweenArrays” function in the “limma” package [57] to normalize the TPM values across samples. We chose the power threshold of 9 and minimal module size > 30, and obtained 20 gene modules, with sizes ranging from 1,299 to 48 genes. Since we used the parameter “unsigned” in computing correlation in order to maximize the gene co-expression network, the expression of a module gene could be either positively or negatively correlated to the module eigengene (the first principal component of each gene module).
Quantification and statistical analyses
Statistical comparisons between individual mutant and control experiments were by Student’s t-test or by the Mann-Whitney procedure, as indicated in Figure Legends. Significance of differential gene expression derived from mRNA-Seq results was the adjusted P value determined by DESEQ2.
Supporting information
Fold changes (determined by DESeq2) in mRNA levels between wing discs from wild type and from indicated genotypes. Significant differences (Padj<0.05) indicated in bold. Genes shown here include all those corresponding to the enriched GO terms mature ribosome assembly (R), sulfur compound metabolic process (S), glutathione metabolic process (G), telomere maintenance (T), and DNA repair (D). These data correspond to the heatmap in Fig 1C.
(PDF)
Fold changes (determined by DESeq2) in mRNA levels between wing discs from wild type and from indicated genotypes. Significant differences (Padj<0.05) indicated in bold.
(PDF)
(PDF)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Comparing translation rates measured by OPP incorporation between different regions of wing discs is complicated to some extent by the dynamic patterns of translation that occur the in wild type [16], perhaps reflecting the dynamic and patchy activity of TORC1 that is revealed by RpS6 phosphorylation patterns[58]. Changes due to mutations in Rp genes are superimposed upon this variable background and are best assessed by directly observing how translation changes cell-autonomously along sharp clone boundaries. In support of the conclusion that RpS17 mutations reduce translation in an RpS12-dependent manner (see Fig 2E–2H), we present additional examples of cell autonomous differences in translation rate between RpS17+/- cells, labeled with GFP, and unlabeled RpS17+/+ clones (panels A-G). Translation, shown by OPP incorporation in panels A’-G’, is consistently lower in RpS17+/- regions. In contrast to these rpS12+/+examples, clones of RpS17+/+ rpS12D97/D97cells in RpS17+/- rpS12D97/D97wing discs (panels H-L) show no difference in translation rate (panels H’-L’).
(TIF)
Panels A-F show wing discs containing clones of indicated genotypes. Corresponding translation rate (OPP incorporation) is shown in panels A’-F”, and the overlay of translation and genotype in panels A”-F”. A,B,C indicated that rpS12 had cell-autonomous, Xrp1-dependent effects on translation in RpS3+/- cells. (A-A” and B-B”) RpS3+/- rpS12+/+ clones often show lower translation than the RpS3+/- rpS12D97/+ background (eg orange arrows). Translation was often higher in RpS3+/- rpS12D97/D97 clones (eg yellow arrows). (C-C”). In the Xrp1+/- background, translation rates were unaffected by rpS12 genotype. D,E,F show RpS17+/- genotypes. Because rpS12 and RpS17 both map to chromosome 3L, FLP-mediated recombination cannot generate RpS17+/- rpS12D97/D97 and RpS17+/- rpS12+/+ clones in the same disc. These figures show that, unlike RpS17+/- rpS12+/+ cells (see main text Fig 2E and 2F), translation in the RpS17+/- rpS12D97/+ genotype was only sometimes distinguishable from that of RpS17+/+ cells. (D-D”). In some cases, RpS17+/+ rpS12+/+ clones showed translation rates higher than the RpS17+/- rpS12D97/+ background (eg cyan arrows). (E-E”). In most cases, RpS17+/+ rpS12+/+ clones typically showed translation rates similar to the RpS17+/- rpS12D97/+ background (eg cyan arrows). (F-F”) Little or no translation difference was seen between RpS17+/+ rpS12D97/D97 clones and the RpS17+/- rpS12D97/+ background (eg blue arrows). Genotypes used. A,B) y w hsF; rpS1297D FRT80B M(3)95A /rpS1297D FRT80B P{arm-LacZ}. C) y w hsF; rpS1297D FRT80B M(3)95A/rpS1297D FRT80B P{arm-LacZ} Xrp1m2-73. D,E) y w hsF; RpS174 rpS1297D P{ubi-GFP} FRT80B/FRT80B. F) y w hsF; RpS174 P{ubi-GFP} FRT80B/rpS1297D FRT80B.
(TIF)
The timing of pupariation was measured in the same experiment shown in Fig 3K and 3L. Like the effects on adult emergence, rpS12D97/D97 and rpS12D97/+ genotypes suppressed the delay to pupariation on RpS3+/- larvae, to a similar extent to RpS3+/- rpS12D97/D97 Xrp1+/- larvae. The detailed genotypes used and numerical data corresponding to these graphs is tabulated in S5 Table.
(TIF)
A-F) Survival curves of 3 replicates comparing rpS12G97D flies with w11-18 (wild type) controls. 120 flies per sex per genotype per replicate. For A,C,E,F, P<0.0001; For B, p = 0.0012; For D, p = 0.0284 by Log-rank (Mantel-Cox) test. For raw data see the S6 Table.
(TIF)
Acknowledgments
This paper is dedicated to the memory of our colleague Dr. J. Warner. The authors also thank D. Rio for anti-Xrp1 antibody and J. Secombe and C. Khan for comments on the manuscript. Drosophila strains from the Bloomington Drosophila Stock Center were used in this study. Confocal Imaging was performed at the Analytical Imaging Facility, Albert Einstein College of Medicine. This paper includes data from theses partially fulfilling of the requirements for the Degree of Doctor of Philosophy in the Graduate Division of Medical Sciences, Albert Einstein College of Medicine, Yeshiva University.
Data Availability
Primary mRNA-Seq data and DESeq2 analyses are available from the GEO (accession numbers GSE112864 and GSE124924). Other relevant data are within the manuscript and its Supporting Information files.
Funding Statement
NEB received awards GM104213 & GM120451 from the National Institutes of General Medical Sciences (https://www.nigms.nih.gov/), and from the Albert Einstein College of Medicine Human Genetics Program. The Bloomington Drosophila Stock Center was supported by NIH (P40OD018537). The Analytical Imaging Facility, Albert Einstein College of Medicine, was supported by NCI (P30CA013330) and by NIH (SIG 1S10 OD023591). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.de la Cruz J, Karbstein K, Woolford JL, Jr. Functions of ribosomal proteins in assembly of eukaryotic ribosomes in vivo. Annu Rev Biochem. 2015;84:93–129. 10.1146/annurev-biochem-060614-033917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Warner JR, McIntosh KB. How common are extraribosomal functions of ribosomal proteins? Mol Cell. 2009;34(1):3–11. 10.1016/j.molcel.2009.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou X, Liao WJ, Liao JM, Liao P, Lu H. Ribosomal proteins: functions beyond the ribosome. J Mol Cell Biol. 2015;7(2):92–104. 10.1093/jmcb/mjv014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vlachos A, Rosenberg PS, Atsidaftos E, Alter BP, Lipton JM. Incidence of neoplasia in Diamond Blackfan anemia: a report from the Diamond Blackfan Anemia Registry. Blood. 2012;119(16):3815–9. Epub 2012/03/01. 10.1182/blood-2011-08-375972 blood-2011-08-375972 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.De Keersmaecker K, Sulima SO, Dinman JD. Ribosomopathies and the paradox of cellular hypo- to hyperproliferation. Blood. 2015;125(9):1377–82. 10.1182/blood-2014-10-569616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sulima SO, Hofman IJF, De Keersmaecker K, Dinman JD. How Ribosomes Translate Cancer. Cancer Discov. 2017;7(10):1069–87. 10.1158/2159-8290.CD-17-0550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ulirsch JC, Verboon JM, Kazerounian S, Guo MH, Yuan D, Ludwig LS, et al. The Genetic Landscape of Diamond-Blackfan Anemia. Am J Hum Genet. 2018;103(6):930–47. 10.1016/j.ajhg.2018.10.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marygold SJ, Roote J, Reuter G, Lambertsson A, Ashburner M, Millburn GH, et al. The ribosomal protein genes and Minute loci of Drosophila melanogaster. Genome Biol. 2007;8(10):R216 Epub 2007/10/12. 10.1186/gb-2007-8-10-r216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lambertsson A. The Minute genes in Drosophila and their molecular functions. Advances in Genetics. 1998;38:69–134. 10.1016/s0065-2660(08)60142-x [DOI] [PubMed] [Google Scholar]
- 10.Morata G, Ripoll P. Minutes: mutants of Drosophila autonomously affecting cell division rate. Developmental Biology. 1975;42:211–21. 10.1016/0012-1606(75)90330-9 [DOI] [PubMed] [Google Scholar]
- 11.Claveria C, Torres M. Cell Competition: Mechanisms and Physiological Roles. Annu Rev Cell Dev Biol. 2016;32:411–39. 10.1146/annurev-cellbio-111315-125142 . [DOI] [PubMed] [Google Scholar]
- 12.Baker NE. Mechanisms of cell competition emerging from Drosophila studies. Curr Opin Cell Biol. 2017;48:40–6. 10.1016/j.ceb.2017.05.002 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee CH, Rimesso G, Reynolds DM, Cai J, Baker NE. Whole-Genome Sequencing and iPLEX MassARRAY Genotyping Map an EMS-Induced Mutation Affecting Cell Competition in Drosophila melanogaster. G3 (Bethesda). 2016;6(10):3207–17. 10.1534/g3.116.029421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baillon L, Germani F, Rockel C, Hilchenbach J, Basler K. Xrp1 is a transcription factor required for cell competition-driven elimination of loser cells. Sci Rep. 2018;8(1):17712 10.1038/s41598-018-36277-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tyler DM, Li W, Zhuo N, Pellock B, Baker NE. Genes affecting cell competition in Drosophila. Genetics. 2007;175(2):643–57. Epub 2006/11/18. genetics.106.061929 [pii] 10.1534/genetics.106.061929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee CH, Kiparaki M, Blanco J, Folgado V, Ji Z, Kumar A, et al. A Regulatory Response to Ribosomal Protein Mutations Controls Translation, Growth, and Cell Competition. Dev Cell. 2018;46(4):456–69 e4. 10.1016/j.devcel.2018.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Francis MJ, Roche S, Cho MJ, Beall E, Min B, Panganiban RP, et al. Drosophila IRBP bZIP heterodimer binds P-element DNA and affects hybrid dysgenesis. Proc Natl Acad Sci U S A. 2016;113(46):13003–8. 10.1073/pnas.1613508113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mallik M, Catinozzi M, Hug CB, Zhang L, Wagner M, Bussmann J, et al. Xrp1 genetically interacts with the ALS-associated FUS orthologue caz and mediates its toxicity. J Cell Biol. 2018. 10.1083/jcb.201802151 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brodsky MH, Weinert BT, Tsang G, Rong YS, McGinnis NM, Golic KG, et al. Drosophila melanogaster MNK/Chk2 and p53 regulate multiple DNA repair and apoptotic pathways following DNA damage. Mol Cell Biol. 2004;24(3):1219–31. Epub 2004/01/20. 10.1128/MCB.24.3.1219-1231.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Akdemir F, Christich A, Sogame N, Chapo J, Abrams JM. p53 directs focused genomic responses in Drosophila. Oncogene. 2007;26(36):5184–93. Epub 2007/02/22. 10.1038/sj.onc.1210328 . [DOI] [PubMed] [Google Scholar]
- 21.Boulan L, Andersen D, Colombani J, Boone E, Leopold P. Inter-Organ Growth Coordination Is Mediated by the Xrp1-Dilp8 Axis in Drosophila. Dev Cell. 2019;49(5):811–8 e4. 10.1016/j.devcel.2019.03.016 . [DOI] [PubMed] [Google Scholar]
- 22.Kucinski I, Dinan M, Kolahgar G, Piddini E. Chronic activation of JNK JAK/STAT and oxidative stress signalling causes the loser cell status. Nat Commun. 2017;8(1):136 10.1038/s41467-017-00145-y . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kale A, Ji Z, Kiparaki M, Blanco J, Rimesso G, Flibotte S, et al. Ribosomal Protein S12e Has a Distinct Function in Cell Competition. Dev Cell. 2018;44(1):42–55 e4. 10.1016/j.devcel.2017.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lee CH, Kiparaki M, Blanco J, Folgado V, Ji Z, Kumar A, et al. A Regulatory Response to Ribosomal Protein Mutations Controls Translation, Growth, and Cell Competition. Dev Cell. 2018;46:456–69. 10.1016/j.devcel.2018.07.003 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moreno E, Basler K, Morata G. Cells compete for decapentaplegic survival factor to prevent apoptosis in Drosophila wing development. Nature. 2002;416:755–9. 10.1038/416755a [DOI] [PubMed] [Google Scholar]
- 26.Akai N, Igaki T, Ohsawa S. Wingless signaling regulates winner/loser status in Minute cell competition. Genes Cells. 2018;23(3):234–40. 10.1111/gtc.12568 . [DOI] [PubMed] [Google Scholar]
- 27.Moullan N, Mouchiroud L, Wang X, Ryu D, Williams EG, Mottis A, et al. Tetracyclines Disturb Mitochondrial Function across Eukaryotic Models: A Call for Caution in Biomedical Research. Cell Rep. 2015. 10.1016/j.celrep.2015.02.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mortison JD, Schenone M, Myers JA, Zhang Z, Chen L, Ciarlo C, et al. Tetracyclines Modify Translation by Targeting Key Human rRNA Substructures. Cell Chem Biol. 2018;25(12):1506–18 e13. 10.1016/j.chembiol.2018.09.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Merino MM, Rhiner C, Lopez-Gay JM, Buechel D, Hauert B, Moreno E. Elimination of unfit cells maintains tissue health and prolongs lifespan. Cell. 2015;160(3):461–76. Epub 2015/01/21. 10.1016/j.cell.2014.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Warren AJ. Molecular basis of the human ribosomopathy Shwachman-Diamond syndrome. Adv Biol Regul. 2018;67:109–27. 10.1016/j.jbior.2017.09.002 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weis F, Giudice E, Churcher M, Jin L, Hilcenko C, Wong CC, et al. Mechanism of eIF6 release from the nascent 60S ribosomal subunit. Nat Struct Mol Biol. 2015;22(11):914–9. 10.1038/nsmb.3112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miluzio A, Ricciardi S, Manfrini N, Alfieri R, Oliveto S, Brina D, et al. Translational control by mTOR-independent routes: how eIF6 organizes metabolism. Biochem Soc Trans. 2016;44(6):1667–73. 10.1042/BST20160179 . [DOI] [PubMed] [Google Scholar]
- 33.Deisenroth C, Franklin DA, Zhang Y. The Evolution of the Ribosomal Protein-MDM2-p53 Pathway. Cold Spring Harb Perspect Med. 2016;6(12). 10.1101/cshperspect.a026138 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Donati G, Peddigari S, Mercer CA, Thomas G. 5S ribosomal RNA is an essential component of a nascent ribosomal precursor complex that regulates the Hdm2-p53 checkpoint. Cell Rep. 2013;4(1):87–98. 10.1016/j.celrep.2013.05.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Raiser DM, Narla A, Ebert BL. The emerging importance of ribosomal dysfunction in the pathogenesis of hematologic disorders. Leukemia & lymphoma. 2014;55(3):491–500. Epub 2013/07/19. 10.3109/10428194.2013.812786 . [DOI] [PubMed] [Google Scholar]
- 36.Danilova N, Gazda HT. Ribosomopathies: how a common root can cause a tree of pathologies. Dis Model Mech. 2015;8(9):1013–26. 10.1242/dmm.020529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ellis SR. Nucleolar stress in Diamond Blackfan anemia pathophysiology. Biochim Biophys Acta. 2014;1842(6):765–8. 10.1016/j.bbadis.2013.12.013 . [DOI] [PubMed] [Google Scholar]
- 38.Khajuria RK, Munschauer M, Ulirsch JC, Fiorini C, Ludwig LS, McFarland SK, et al. Ribosome Levels Selectively Regulate Translation and Lineage Commitment in Human Hematopoiesis. Cell. 2018;173(1):90–103 e19. 10.1016/j.cell.2018.02.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mills EW, Green R. Ribosomopathies: There's strength in numbers. Science. 2017;358(6363). 10.1126/science.aan2755 . [DOI] [PubMed] [Google Scholar]
- 40.Derenzini E, Agostinelli C, Rossi A, Rossi M, Scellato F, Melle F, et al. Genomic alterations of ribosomal protein genes in diffuse large B cell lymphoma. Br J Haematol. 2019;185(2):330–4. 10.1111/bjh.15442 . [DOI] [PubMed] [Google Scholar]
- 41.Vincent J, Girdham C, O'Farrell P. A cell-autonomous, ubiquitous marker for the analysis of Drosophila genetic mosaics. Developmental Biology. 1994;164:328–31. 10.1006/dbio.1994.1203 [DOI] [PubMed] [Google Scholar]
- 42.Janody F, Lee JD, Jahren N, Hazelett DJ, Benlali A, Miura GI, et al. A mosaic genetic screen reveals distinct roles for trithorax and polycomb group genes in Drosophila eye development. Genetics. 2004;166(1):187–200. 10.1534/genetics.166.1.187 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burke R, Basler K. Dpp receptors are autonomously required for cell proliferation in the entire developing Drosophila wing. Development. 1996;122:2261–9. [DOI] [PubMed] [Google Scholar]
- 44.Golic KG. Site-specific recombination between homologous chromosomes in Drosophila. Science. 1991;252:958–61. 10.1126/science.2035025 [DOI] [PubMed] [Google Scholar]
- 45.Xu T, Rubin GM. Analysis of genetic mosaics in the developing and adult Drosophila tissues. Development. 1993;117:1223–36. [DOI] [PubMed] [Google Scholar]
- 46.Newsome TP, Asling B, Dickson BJ. Analysis of Drosophila photoreceptor axon guidance in eye-specific mosaics. Development. 2000;127:851–60. [DOI] [PubMed] [Google Scholar]
- 47.Sullivan W, Ashburner M, Hawley RS. Drosophila protocols: Cold Spring Harbor Laboratory Press; 2000. [Google Scholar]
- 48.Sultan R, Stampas A, Goldberg MB, Baker NE. Drug resistance of bacteria commensal with Drosophila melanogaster in laboratory cultures. Drosoph Inf Serv. 2001;84(December 2001):176–80. [Google Scholar]
- 49.Baker NE, Li K, Quiquand M, Ruggiero R, Wang LH. Eye development. Methods. 2014;68(1):252–9. 10.1016/j.ymeth.2014.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sanchez CG, Teixeira FK, Czech B, Preall JB, Zamparini AL, Seifert JR, et al. Regulation of Ribosome Biogenesis and Protein Synthesis Controls Germline Stem Cell Differentiation. Cell Stem Cell. 2016;18(2):276–90. 10.1016/j.stem.2015.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550 10.1186/s13059-014-0550-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25(1):25–9. 10.1038/75556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mi H, Huang X, Muruganujan A, Tang H, Mills C, Kang D, et al. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 2017;45(D1):D183–D9. 10.1093/nar/gkw1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.The Gene Ontology C. Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 2017;45(D1):D331–D8. 10.1093/nar/gkw1108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559 10.1186/1471-2105-9-559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang B, Horvath S. A general framework for weighted gene co-expression network analysis. Stat Appl Genet Mol Biol. 2005;4:Article17. 10.2202/1544-6115.1128 . [DOI] [PubMed] [Google Scholar]
- 57.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47 10.1093/nar/gkv007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Romero-Pozuelo J, Demetriades C, Schroeder P, Teleman AA. CycD/Cdk4 and Discontinuities in Dpp Signaling Activate TORC1 in the Drosophila Wing Disc. Dev Cell. 2017;42(4):376–87 e5. 10.1016/j.devcel.2017.07.019 . [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fold changes (determined by DESeq2) in mRNA levels between wing discs from wild type and from indicated genotypes. Significant differences (Padj<0.05) indicated in bold. Genes shown here include all those corresponding to the enriched GO terms mature ribosome assembly (R), sulfur compound metabolic process (S), glutathione metabolic process (G), telomere maintenance (T), and DNA repair (D). These data correspond to the heatmap in Fig 1C.
(PDF)
Fold changes (determined by DESeq2) in mRNA levels between wing discs from wild type and from indicated genotypes. Significant differences (Padj<0.05) indicated in bold.
(PDF)
(PDF)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
(XLSX)
Comparing translation rates measured by OPP incorporation between different regions of wing discs is complicated to some extent by the dynamic patterns of translation that occur the in wild type [16], perhaps reflecting the dynamic and patchy activity of TORC1 that is revealed by RpS6 phosphorylation patterns[58]. Changes due to mutations in Rp genes are superimposed upon this variable background and are best assessed by directly observing how translation changes cell-autonomously along sharp clone boundaries. In support of the conclusion that RpS17 mutations reduce translation in an RpS12-dependent manner (see Fig 2E–2H), we present additional examples of cell autonomous differences in translation rate between RpS17+/- cells, labeled with GFP, and unlabeled RpS17+/+ clones (panels A-G). Translation, shown by OPP incorporation in panels A’-G’, is consistently lower in RpS17+/- regions. In contrast to these rpS12+/+examples, clones of RpS17+/+ rpS12D97/D97cells in RpS17+/- rpS12D97/D97wing discs (panels H-L) show no difference in translation rate (panels H’-L’).
(TIF)
Panels A-F show wing discs containing clones of indicated genotypes. Corresponding translation rate (OPP incorporation) is shown in panels A’-F”, and the overlay of translation and genotype in panels A”-F”. A,B,C indicated that rpS12 had cell-autonomous, Xrp1-dependent effects on translation in RpS3+/- cells. (A-A” and B-B”) RpS3+/- rpS12+/+ clones often show lower translation than the RpS3+/- rpS12D97/+ background (eg orange arrows). Translation was often higher in RpS3+/- rpS12D97/D97 clones (eg yellow arrows). (C-C”). In the Xrp1+/- background, translation rates were unaffected by rpS12 genotype. D,E,F show RpS17+/- genotypes. Because rpS12 and RpS17 both map to chromosome 3L, FLP-mediated recombination cannot generate RpS17+/- rpS12D97/D97 and RpS17+/- rpS12+/+ clones in the same disc. These figures show that, unlike RpS17+/- rpS12+/+ cells (see main text Fig 2E and 2F), translation in the RpS17+/- rpS12D97/+ genotype was only sometimes distinguishable from that of RpS17+/+ cells. (D-D”). In some cases, RpS17+/+ rpS12+/+ clones showed translation rates higher than the RpS17+/- rpS12D97/+ background (eg cyan arrows). (E-E”). In most cases, RpS17+/+ rpS12+/+ clones typically showed translation rates similar to the RpS17+/- rpS12D97/+ background (eg cyan arrows). (F-F”) Little or no translation difference was seen between RpS17+/+ rpS12D97/D97 clones and the RpS17+/- rpS12D97/+ background (eg blue arrows). Genotypes used. A,B) y w hsF; rpS1297D FRT80B M(3)95A /rpS1297D FRT80B P{arm-LacZ}. C) y w hsF; rpS1297D FRT80B M(3)95A/rpS1297D FRT80B P{arm-LacZ} Xrp1m2-73. D,E) y w hsF; RpS174 rpS1297D P{ubi-GFP} FRT80B/FRT80B. F) y w hsF; RpS174 P{ubi-GFP} FRT80B/rpS1297D FRT80B.
(TIF)
The timing of pupariation was measured in the same experiment shown in Fig 3K and 3L. Like the effects on adult emergence, rpS12D97/D97 and rpS12D97/+ genotypes suppressed the delay to pupariation on RpS3+/- larvae, to a similar extent to RpS3+/- rpS12D97/D97 Xrp1+/- larvae. The detailed genotypes used and numerical data corresponding to these graphs is tabulated in S5 Table.
(TIF)
A-F) Survival curves of 3 replicates comparing rpS12G97D flies with w11-18 (wild type) controls. 120 flies per sex per genotype per replicate. For A,C,E,F, P<0.0001; For B, p = 0.0012; For D, p = 0.0284 by Log-rank (Mantel-Cox) test. For raw data see the S6 Table.
(TIF)
Data Availability Statement
Primary mRNA-Seq data and DESeq2 analyses are available from the GEO (accession numbers GSE112864 and GSE124924). Other relevant data are within the manuscript and its Supporting Information files.