

Abstract
Amyloidosis is a group of disorders characterised by the accumulation of extracellular deposits of insoluble protein aggregates. Clinical management depends on the accurate identification of the amyloid precursor and underlying cause. We describe a rare case of apolipoprotein A-IV cardiac amyloidosis, the diagnosis of which required mass spectrometry-based proteomic analysis.
LEARNING POINTS
To be able to perform the differential diagnosis of cardiac amyloidosis subtypes using imaging methods, analytical results and tissue analysis.
To recognise the added value of mass spectrometry (MS)-based proteomic analysis.
Keywords: Amyloidosis, cardiomyopathy, heart failure, imaging, nuclear medicine, apolipoprotein
CASE DESCRIPTION
A 74-year-old man was evaluated at the Cardiology Clinic in the setting of a syncope episode and atypical thoracic pain.
He had a past history of prostate adenocarcinoma, bilateral carpal tunnel syndrome surgery and a serum creatinine of 1.7 mg/dl. He had no previous hypertension or family history of amyloidosis. Signs of orthostatic hypotension, autonomic neuropathy, purpura, bruising, periorbital ecchymosis or macroglossia were absent.
Initial evaluation disclosed elevated plasma levels of brain natriuretic peptide (2,394 pg/ml), elevated high-sensitivity troponin I readings (154.7 ng/l) and an enlarged mediastinal shadow on thoracic radiography. An EKG showed sinus rhythm and criteria of LV hypertrophy. No conduction abnormalities or significant arrhythmias were detected on 24-hour Holter monitoring. Transthoracic echocardiography confirmed concentric LV hypertrophy, hypokinesis of inferior and posterior mid-basal segments and an LV ejection fraction of 52%.
Due to the presence of thoracic pain and LV segmental wall-motion abnormalities, a cardiac catheterisation was performed and excluded coronary disease. Cardiac magnetic resonance imaging was compatible with cardiac amyloidosis that was further supported by the identification of amyloid deposition on abdominal fat aspirate. Total body scintigraphy showed no cardiac 99mTc-DPD uptake (Fig. 1). Myocardial biopsy showed amyloid deposition at muscle fibre level (Fig. 2). Immunohistochemistry evaluation was inconclusive for kappa and lambda light chains and was negative for serum amyloid A, transthyretin and fibrinogen.
Figure 1.
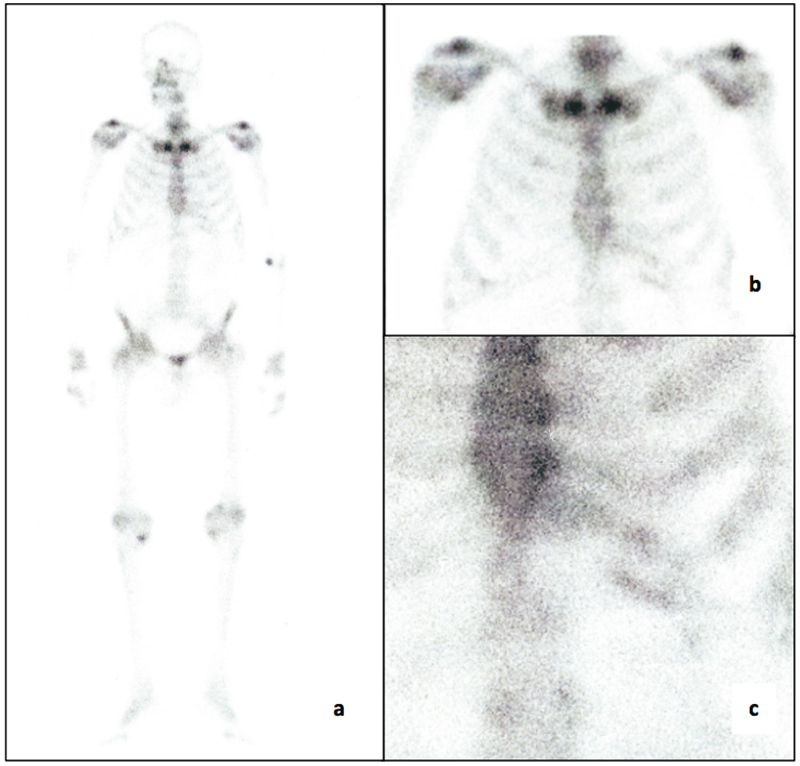
Total body scintigraphic scans at 3 hours (late) after the intravenous injection of 99mTc-DPD: (a) whole-body scan: anterior view, (b) zoom of thoracic view, (c) no uptake of the tracer was evident at the myocardial level.
Figure 2.
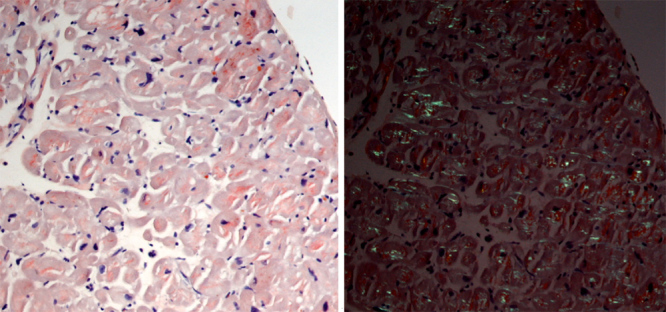
Myocardial biopsy. Congo red staining showed reddish amyloid material in the myocardium (left) with apple-green birefringence under polarised light (right), ×200.
Serum and urine electrophoresis and immunofixation studies were negative for monoclonal proteins and the free light chain (kappa and lambda) ratio was normal.
A bone marrow biopsy was also carried out, excluding plasma cell dyscrasia and revealing an absence of Congo red positivity. During a 4-year follow-up, the patient’s serum creatinine gradually rose to 2.1 mg/dl, and a 24-hour urine collection disclosed a proteinuria of 0.51 g, with unremarkable urinary sediment and renal ultrasound. The patient was submitted to a renal biopsy that showed amyloid deposition restricted to the medulla with no involvement of glomeruli (Fig. 3), which raised the suspicion of systemic amyloidosis related to apolipoprotein deposition. MS-based proteomic analysis, performed to determine the proteome profile of the cardiac amyloid, identified and quantified more than 800 proteins and disclosed large amounts of AApoAIV amyloid deposits, one of the most abundant proteins detected in the biopsy (Fig. 4).
Figure 3.
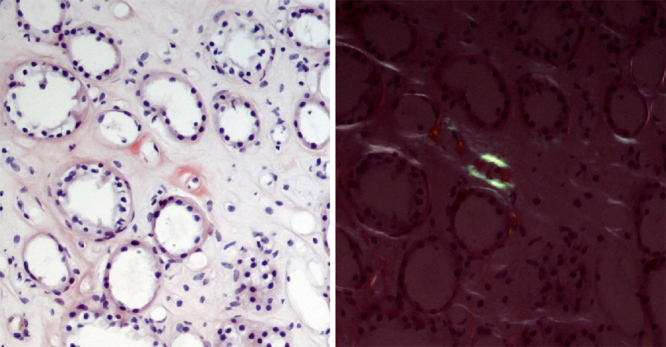
Renal biopsy. Congo red staining showed reddish amyloid material in the renal medulla (left) with apple-green birefringence under polarised light (right), ×200.
Figure 4.

The table shows the list of the most abundant proteins identified in the tested sample (myocardial biopsy). The ApoA4 protein is highlighted with 90% sequence coverage. The graphic indicates the relative abundance of peptide (y-axis) in relation to peptide retention time (x-axis).
Two years later, the patient developed progressive fatigue, exertional dyspnoea and leg oedema. The EKG showed sinus rhythm and frequent ventricular extrasystoles. Cardiac ultrasound revealed severe LV dysfunction with an ejection fraction of 28%, a restrictive mitral inflow pattern and an E/e′ ratio of 19.5 compatible with high LV filling pressures (Fig. 5). Due to the severity of LV dysfunction, it was decided to implant a cardioverter-defibrillator for primary prevention of sudden cardiac death. The patient was treated with furosemide, low doses of beta-blockers and an angiotensin II antagonist.
Figure 5.

Transthoracic echocardiogram. (a) Longitudinal axis and short axis showing increased thickness of LV wall, (b) restrictive transmitral filling pattern, (c) longitudinal strain imaging showing a basal to apical gradient.
DISCUSSION
Cardiac amyloidosis is a myocardial disease frequently manifested as hypertrophic or restrictive cardiomyopathy after the sixth decade of life.
Clinical suspicion may derive from abnormalities detected on an EKG (disproportion between left ventricle wall thickness and QRS voltages, pseudonecrosis pattern, atrioventricular block), an echocardiogram (“speckled” appearance, reduced myocardial deformation sparing the apex, thickening of the free wall of the right ventricle, atrioventricular valves or interatrial septum, pericardial effusions) or from cardiac magnetic resonance (subendocardial distribution of late gadolinium enhancement, T1 signal abnormalities, marked extracellular volume expansion).
Amyloidogenic proteins frequently affecting the heart are ATTR and AL, but 11 different proteins have already been documented in cardiac amyloidosis [1].
ATTR could be diagnosed using bone-tracer scintigraphy[2]. Diagnosis of AL amyloidosis requires the identification of plasma cell dyscrasia by serum and urine immunofixation, quantification of serum free light chains and tissue biopsy confirmation of amyloid protein deposits.
AApoAIV amyloidosis is a rare systemic disease[3]. ApoAIV is a 46 kDa protein, predominantly synthesised by the enterocytes in the small intestine, which has an important role in absorption, transportation and metabolism of lipids[4]. It has anti-oxidant, anti-inflammatory and atheroprotective functions and plasma levels increase with age[5]. The mechanisms that promote the development of AApoAIV amyloidosis are largely unknown. Heart involvement in those patients presents with clinical and histopathological features indistinct from other amyloidosis types, mainly hypertrophic cardiomyopathy, with or without LVOT obstruction[6]. It is more prevalent in men, and commonly diagnosed after 60 years of age. Involvement of the microvasculature is also a prominent feature in cardiac AApoAIV amyloidosis[6]. In our patient, we cannot exclude the possibility that the presence of LV segmental wall-motion abnormalities on the first echocardiogram could be secondary to obstructive microvascular amyloidosis, although this involvement was not apparent in the analysed tissue fragment.
Myocardial uptake was negative in 99mTc-DPD scintigraphy, a result similar to that found in ApoAII-related disease but dissimilar to the ApoAI amyloidosis reported case[7,8]. As far as we know, this is the first report of the use of 99mTc-DPD scintigraphy in a patient with amyloidotic cardiomyopathy due to AApoAIV amyloidosis. The reason underlying different uptake patterns on 99mTc-DPD scintigraphy among several amyloidosis subtypes remains unclear.
Renal disease in AApoAIV amyloidosis presents with slow deteriorating kidney function over years, which may be accelerated in the presence of hypertension or diabetes[3]. Unlike most forms of amyloidosis, urinalysis is unremarkable, proteinuria is absent or minimal and the amyloid deposits are restricted to the renal medulla, features also noted in our patient.
Tissue biopsy is crucial for the accurate diagnosis of the underlying amyloid protein. Immunohistochemistry is the current standard approach for amyloid protein identification. However, the accuracy of diagnosis depends on the antibody panel directed against known amyloidogenic proteins, a lack of commercially available amyloid-specific antibodies, antibody binding to circulating proteins present in the tissue specimen, background staining, correlation between immunohistochemistry staining and Congo red-positive areas and the experience and expertise of the operator[9]. On the other hand, ApoAIV is among the signature chaperone proteins found in different amyloid deposits[3] and immunohistochemistry may not be useful to specifically diagnose AApoAIV amyloidosis. As MS provides a semiquantitative estimate of the amount of protein deposits, ApoAIV protein would be considered the primary amyloid protein if no other known amyloidogenic proteins are present or if ApoAIV is present in a significantly higher quantity[10]. An MS-based proteomic analysis may be a determinant of the diagnosis, and is now the gold standard for fibril typing. It can also identify mutant genotypes.
At the present time, there are no specific treatment options available for AApoAIV amyloidosis, although the prognosis may be more favourable than for other subtypes of cardiac amyloidosis[6].
CONCLUSIONS
The use of MS-based proteomic analysis allowed the precise diagnosis and typing of AApoAIV amyloidosis in this patient. This is a rare type of systemic amyloidosis. It involves the heart, with indistinct clinical and histopathological features, and the kidneys with distinct presentation characterised by slowly progressive renal dysfunction, no significant proteinuria and amyloid deposition restricted to the renal medulla. A high degree of suspicion is critical to diagnose rare diseases with unknown prevalence and risk factors. In this case, cardiac biopsy and MS-based proteomic analysis were crucial for the accurate diagnosis of the underlying amyloidogenic protein and for further treatment strategy establishment. Given the possibility of codeposition of different amyloid precursors, tissue demonstration of amyloid deposition is necessary for diagnostic confirmation of the disease.
Acknowledgements
The Authors acknowledge Rede Nacional de Espectrometria de Massa, ROTEIRO/0028/2013, ref. LISBOA-01-0145-FEDER-022125, supported by COMPETE and North Portugal Regional Operational Programme (Norte2020), under the PORTUGAL 2020 Partnership Agreement, through the European Regional Development Fund (ERDF). This work is partially funded by FCT - Fundação para a Ciência e a Tecnologia [UID/BIM/04293/2013].
Footnotes
Conflicts of Interests: The Authors declare that there are no competing interest
REFERENCES
- 1.Benson MD, Buxbaum JN, Eisenberg DS, Merlini G, Saraiva MJM, Sekijima Y, et al. Amyloid nomenclature 2018: recommendations by the International Society of Amyloidosis (ISA) nomenclature committee. Amyloid. 2018;25:215–219. doi: 10.1080/13506129.2018.1549825. [DOI] [PubMed] [Google Scholar]
- 2.Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, Dispenzieri A, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133:2404–2412. doi: 10.1161/CIRCULATIONAHA.116.021612. [DOI] [PubMed] [Google Scholar]
- 3.Dasari S, Amin MS, Kurtin PJ, Vrana JA, Theis JD, Grogg KL, et al. Clinical, biopsy, and mass spectrometry characteristics of renal apolipoprotein A-IV amyloidosis. Kidney Int. 2016;90:658–664. doi: 10.1016/j.kint.2016.04.003. [DOI] [PubMed] [Google Scholar]
- 4.Wang F, Kohan AB, Lo CM, Liu M, Howles P, Tso P. Apolipoprotein A-IV: a protein intimately involved in metabolism. J Lipid Res. 2015;56:1403–1418. doi: 10.1194/jlr.R052753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sun Z, Larson IA, Ordovas JM, Barnard JR, Schaefer EJ. Effects of age, gender, and lifestyle factors on plasma apolipoprotein A-IV concentrations. Atherosclerosis. 2000;151:381–388. doi: 10.1016/s0021-9150(99)00395-0. [DOI] [PubMed] [Google Scholar]
- 6.Bois MC, Dasari S, Mills JR, Theis J, Highsmith WE, Vrana JA, et al. Apolipoprotein A-IV-associated cardiac amyloidosis. J Am Coll Cardiol. 2017;69:2248–2249. doi: 10.1016/j.jacc.2017.02.047. [DOI] [PubMed] [Google Scholar]
- 7.Longhi S, Bonfiglioli R, Obici L, Gagliardi C, Milandri A, Lorenzini M, et al. Etiology of amyloidosis determines myocardial 99mTc-DPD uptake in amyloidotic cardiomyopathy. Clin Nucl Med. 2015;40:446–447. doi: 10.1097/RLU.0000000000000741. [DOI] [PubMed] [Google Scholar]
- 8.Quarta CC, Obici L, Guidalotti PL, Pieroni M, Longhi S, Perlini S, et al. High 99mTc-DPD myocardial uptake in a patient with apolipoprotein AI-related amyloidotic cardiomyopathy. Amyloid. 2013;20:48–51. doi: 10.3109/13506129.2012.746938. [DOI] [PubMed] [Google Scholar]
- 9.Picken MM. Amyloidosis-where are we now and where are we heading? Arch Pathol Lab Med. 2010;134:545–551. doi: 10.5858/134.4.545. [DOI] [PubMed] [Google Scholar]
- 10.Vrana JA, Theis JD, Dasari S, Mereuta OM, Dispenzieri A, Zeldenrust SR, et al. Clinical diagnosis and typing of systemic amyloidosis in subcutaneous fat aspirates by mass spectrometry-based proteomics. Haematologica. 2014;99:1239–1247. doi: 10.3324/haematol.2013.102764. [DOI] [PMC free article] [PubMed] [Google Scholar]