

Abstract
Atypical hemolytic uremic syndrome (aHUS) is a complex complement-mediated disease that progresses to end-stage renal failure (ESRF) in 50% of cases. Dysregulation of the alternative pathway (AP) of the complement cascade manifests as microangiopathic anaemia and thrombocytopenia. Multiple genes in the AP have been implicated in disease pathogenesis. Here, we report the clinical presentation of an affected patient that was inconsistent with genotype–phenotype data for carriers of CD46 mutations. Tests of AP function in this patient suggested additional genetic factors, and in-depth studies revealed a de novo heterozygous deletion that creates a novel CFH/CFHR1 fusion protein.
Keywords: aHUS, complement, deletion, kidney transplantation
Background
Atypical hemolytic uremic syndrome (aHUS) is a rare renal disease caused by mutations in several different complement genes. Included in this list are CFH, CFI, C3, THBD, CFB and CD46. Phenotype–genotype studies have shown that the initial clinical course of aHUS, as well as the likelihood of progression to end-stage renal failure (ESRF) and prognosis for graft survival following renal transplantation, is strongly correlated with the mutated gene.
Complement factor H (CFH), the primary regulator of the alternative pathway (AP), is most frequently mutated, typically carrying missense mutations in the last two short consensus repeats (SCRs) of this 20 SCR protein [1–3]. However, in addition to single base-pair mutations, non-homologous allelic recombination (NHAR) between CFH and the two immediate down-stream genes, complement factor H-related 1 and 3 (CFHR), also causes aHUS. The reported NHAR event creates a CFH/CFHR1 fusion protein that includes the first 18 SCRs of CFH and the last two SCRs of CFHR1 [4]. aHUS patients with CFH mutations do poorly—60–70% progress to ESRF, and 70–90% lose their allografts following transplantation [3].
Case report
A patient presented with a HUS-05a at 11 months of age with pallor, lethargy and oliguria following several days of fever and irritability. He was anaemic (haemoglobin, 7.4 gm/dL; hematocrit, 19.1%) and had schistocytes by peripheral smear. Pertinent findings included the following: platelet count, 141 K/mm3 (high); LDH, 5938 U/L (high); BUN, 65 mg/dL (high); and serum creatinine, 1.5 mg/dL (high). Blood and protein were noted on urinalysis, stool was negative for Escherichiacoli strain 0157:H7, and serum C3, C4 and CH50 levels were normal (109 mg/dL, 18.6 mg/dL and 43 U/mL, respectively).
Diagnosed with aHUS, the patient received red blood cell transfusions, but within 3 days, marked thrombocytopenia (30 K/mm3) necessitated q12h plasma infusions (10 mL/kg). In this protocol, the platelet count (271 K/mm3) and renal function (BUN, 24 mg/dL; creatinine, 0.6 mg/dL) improved, but when infusions were discontinued, thrombocytopenia and hemolysis recurred. He was therefore maintained on an every-third-day infusion regimen and remained asymptomatic until age 15 months when a central line infection triggered a fever and generalized seizures. Two months later, aHUS again flared following an infection. Both episodes responded to more frequent infusions.
At age 21 months, an attempt to increase the interval between infusions to 10 days was unsuccessful, and within 2 weeks, without signs of illness, a relapse occurred. Over the next several years, the patient was maintained on infusions 1–3 times per week. During this time, a partial genetic work-up showed that CFH was normal, factor H antibodies were absent and factor H serum levels were normal.
At age 6, a severe relapse of aHUS was triggered by Serratiamarcescens sepsis. Fluid overload and respiratory failure necessitated emergent intubation, mechanical ventilation and acute hemodialysis. At age 6.5 years, long-term dialysis was initiated, and further genetic studies were requested. The earlier genetic findings were confirmed, and in addition, a mutation was identified in CD46.
Mutations in CD46 are generally associated with a favourable course post-transplantation. At age 8, arrangements for surgery were made, including native kidney embolization to control hypertension. Post-embolization, seizures and respiratory distress necessitated intubation. The planned transplantation was halted and a genetic re-evaluation was requested.
Materials and methods
Genomic analysis
Genomic DNA was extracted from peripheral blood, and mutation screening of CFH, CFHR5, CFI, CD46, CFB, C3 and THBD was completed [3].
Functional assays
Fluid-phase AP activity assay.
Fluid-phase AP activity was measured with the Wieslab complement AP assay (Euro-Diagnostica, Westport, CT, USA).
Cell surface AP activity.
Cell surface AP activity was measured using a sheep erythrocyte hemolytic assay [5].
Autoantibody assays
CFH autoantibodies.
Autoantibodies to CFH were analysed by ELISA [6].
Copy number variation
The possibility of copy number variation (CNV) was investigated by multiplex ligation-dependent probe amplification (MLPA) across the CFH–CFHR5 genomic region [7].
Breakpoint mapping
Genomic DNA was amplified with primers spanning the deletion using Takara Long Range polymerase (Fischer Scientific, Pittsburgh, PA, USA).
Western blot
Diluted patient and control sera were run on an SDS–PAGE gel and blotted with a polyclonal sheep anti-human CFH antibody (Abcam, Cat # ab8842-1) (dilution 1:1000) using a standard protocol.
Results
Genetic analysis
A heterozygous missense mutation c.1058C > T (p.A353V) was found in CD46 (MCP). This variant is a rare SNP (rs35366573) previously observed in aHUS patients [8]. It was not detected in 176 ethnically matched healthy controls.
Functional assays
Fluid-phase AP activity was normal. The hemolytic assays showed complete hemolysis (cell surface AP dysregulation).
Autoantibody assays
CFH autoantibodies were absent.
Copy number variation
MLPA amplification from exon 22 (encoding SCR20) of CFH to exon 5 (encoding SCR4) of CFHR1 revealed one copy of this genomic interval (Figure 1A). Flanking probes detected two copies of CFH up to and including exon 21 (encoding SCR19) and two copies of CFHR1 beginning at exon 6 (encoding SCR5) and extending distally. No other deletions of the CFHR region were detected. Our screen of other aHUS patients identified one additional patient with this rearrangement (2/150, 1.5%). It was not seen in 720 control chromosomes.
Fig. 1.
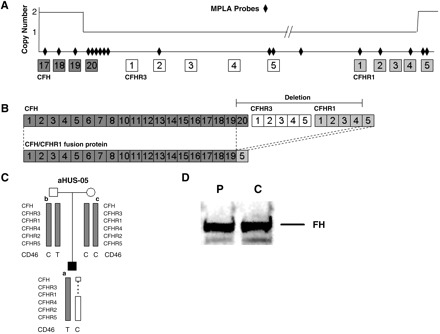
A novel CFH/CFHR1 fusion protein is created in this patient by NAHR. (A) MLPA determination of copy number over the CFH–CFHR region identified a deletion beginning before CFH SCR 20 (encoded by CFH exon 22) and ending after CFHR1 SCR4 (encoded by CFHR1 exon 5). MLPA probes are represented by diamonds; boxes show SCRs and lines between the boxes represent either introns or intergenic sequence (not to scale). (B) The resulting fusion protein comprised SCRs 1–19 of CFH and SCR 5 of CFHR1. (C) Nuclear family showing the segregation of the CD46 c.1058C > T, p.A353V and the de novo NAHR event. (D) Western blotting of CFH using a polyclonal CFH antibody against sera from the patient (P) and a control (C) resolves bands of equal intensity and does not identify additional CFH bands in the patient.
Breakpoint mapping
The breakpoint of this deletion is between the last SNP of CFH in intron 21 and the first SNP of CFHR1 in intron 5. Between these SNPs are 118 nucleotide bases, which place the exact breakpoint between chr1 bp 84 717 and 84 835 bp. This NAHR event creates a novel hybrid CFH/CFHR1 protein (Figure 1B). Haplotype segregation in the family identified this rearrangement as a de novo NAHR event (Figure 1C). To rule out the possibility of a truncated protein, we performed a western blot with patient and control sera, which showed no size differences in CFH (Figure 1D).
Discussion
The clinical course of this patient was more severe than expected for CD46 mutation carriers [9]. In addition, functional assays using patient serum showed cell surface AP dysregulation, which is not consistent with a CD46 mutation. Extensive MLPA assays demonstrated a novel de novo CFH/CFHR1 hybrid protein. We characterized this rearrangement (Maga–Meyer deletion) and demonstrated that the created fusion protein is comprised of amino acids encoded by CFH exons 1–21 and CFHR1 exon 6. Because CFH exon 21 and CFHR1 exon 5 encode identical proteins, the Venables deletion and this deletion produce identical fusion proteins despite differing NAHR sites [4].
Our finding is noteworthy for two reasons. First, it suggests that NAHR may be a more common cause of aHUS than suspected, especially given the high homology between factor H-related family members (95% at nucleotide level). Second, this finding is of clinical relevance because it predicts a poor outcome for this patient following renal transplantation unless adjuvant eculizumab therapy or combined hepato-renal transplantation is considered [10].
In conclusion, this case highlights the importance of diligent genetic testing and the careful interpretation of genetic data in the context of the clinical presentation in patients with aHUS.
Acknowledgments
We thank the Foundation for Children with Atypical HUS (RJHS) for grant sponsorship.
Conflict of interest statement. None declared.
References
- 1.Caprioli J, Noris M, Brioschi S, et al. Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood. 2006;108:1267–1279. doi: 10.1182/blood-2005-10-007252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361:1676–1687. doi: 10.1056/NEJMra0902814. [DOI] [PubMed] [Google Scholar]
- 3.Maga TK, Nishimura CJ, Weaver AE, et al. Mutations in alternative pathway complement proteins in American patients with atypical hemolytic uremic syndrome. Hum Mutat. 2010;31:E1445–E1460. doi: 10.1002/humu.21256. [DOI] [PubMed] [Google Scholar]
- 4.Venables JP, Strain L, Routledge D, et al. Atypical haemolytic uraemic syndrome associated with a hybrid complement gene. PLoS Med. 2006;3:e431. doi: 10.1371/journal.pmed.0030431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanchez-Corral P, Gonzalez-Rubio C, Rodriguez de Cordoba S, et al. Functional analysis in serum from atypical hemolytic uremic syndrome patients reveals impaired protection of host cells associated with mutations in factor H. Mol Immunol. 2004;41:81–84. doi: 10.1016/j.molimm.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 6.Dragon-Durey MA, Loirat C, Cloarec S, et al. Anti-Factor H autoantibodies associated with atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2005;16:555–563. doi: 10.1681/ASN.2004050380. [DOI] [PubMed] [Google Scholar]
- 7.Schouten JP, McElgunn CJ, Waaijer R, et al. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30:e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fang CJ, Fremeaux-Bacchi V, Liszewski MK, et al. Membrane cofactor protein mutations in atypical hemolytic uremic syndrome (aHUS), fatal Stx-HUS, C3 glomerulonephritis, and the HELLP syndrome. Blood. 2008;111:624–632. doi: 10.1182/blood-2007-04-084533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loirat C, Fremeaux-Bacchi V. Hemolytic uremic syndrome recurrence after renal transplantation. Pediatr Transplant. 2008;12:619–629. doi: 10.1111/j.1399-3046.2008.00910.x. [DOI] [PubMed] [Google Scholar]
- 10.Nurnberger J, Philipp T, Witzke O, et al. Eculizumab for atypical hemolytic-uremic syndrome. N Engl J Med. 2009;360:542–544. doi: 10.1056/NEJMc0808527. [DOI] [PubMed] [Google Scholar]