

Abstract
Background: Since bromodomain-containing protein 4 (BRD4) facilitates the transcription of genes important for neoplastic cells in a cancer-type specific manner, BRD4-regulated molecules may also include therapeutic targets for mantle cell lymphoma (MCL), a treatment-refractory subtype of malignant lymphoma. Materials and Methods: In order to uncover direct BRD4-regulated targets in MCL, we performed integrated analysis using the pathway database and the results of both gene-expression profiling and chromatin immunoprecipitation with parallel sequencing for BRD4. Results: Treatment with BRD4 inhibitor I-BET151 exerted a dose-dependent inhibitory effect on cell proliferation in MCL cell lines. BRD4 was found to directly regulate series of genes involved in the B-cell receptor (BCR) signaling pathway, including B-cell linker (BLNK), paired box 5 (PAX5), and IKAROS family zinc finger 3 (IKZF3), and several oncogenes, such as MYB. Indeed, the combinatory inhibition of BCR pathway and IKZF showed an additive antitumor effect. Conclusion: Concomitant targeting multiple BRD4-regulated molecules may constitute a rational therapeutic strategy for MCL.
Keywords: Mantle cell lymphoma, BRD4, ChIP-Seq, superenhancer, drug resistance
The advent of immunochemotherapy consisting of cytotoxic agents and monoclonal antibodies, such as the antibody to CD20 rituximab, has markedly improved treatment outcomes for the majority of patients with B-cell lymphomas (BCLs). However, several hard-to-treat disease subtypes still exist for which the treatment outcome needs to be improved (1,2). Mantle cell lymphoma (MCL), originating from CD5-positive naïve B-lymphocytes, is such a treatment-refractory disease subtype which comprises approximately 3% of all non-Hodgkin lymphomas (NHLs). In addition to the disease-specific chromosomal translocation t(11;14)(q13;q32) leading to cyclin D1 (CCND1) overexpression in approximately 90% of patients with MCL, there are additional genetic alterations. Abnormal overexpression of sex-determining region Y-box 11 (SOX11), a transcription factor, and cross talk with aberrant activation of several cell signaling pathways, including B-cell receptor (BCR), nuclear factor-kappa B (NF-ĸB), mitogen-activated protein kinase (MAPK), and NOTCH, coordinately promote MCL development and progression (3,4). Despite the involvement of those known molecular pathways and abnormalities, a promising therapeutic molecular target for MCL has not been defined. The sole inhibition of CCND1 is insufficient for the eradication of tumor cells in experimental MCL models, and the therapeutic significance of SOX11 inhibition has not been found to be relevant to MCL. Although therapeutic moieties that potently target the above described pathways, including the proteasome inhibitor bortezomib which potently inactivates the NF-ĸB pathway and the Bruton kinase (BTK) inhibitor ibrutinib which theoretically shuts down BCR signaling, MCL is rarely cured due to primary and acquired resistance. Therefore, the identification of a rational and targetable therapeutic molecule is needed for the development of a more effective treatment strategy against MCL.
The dysregulation of epigenetic enzymatic ‘writers’, including DNA methyltransferase and ‘erasers’, including histone deacetylases (HDACs), have been the subject of intensive research for various cancer types. The therapeutic targeting of the writers and erasers using compounds, such as either hypomethylating agents or HDAC inhibitors, has been used to treat various types of cancer. One of the epigenetic modification mechanisms involving bromodomain and extra-terminal domain (BET) proteins acts on acetylated histones and facilitates the transcription of target genes as epigenetic ‘readers’. Bromodomain-containing 4 (BRD4), a BET protein, recruits the positive transcription factor b (P-TEFb) complex to genomic lesions in chromatin and, thereby, simultaneously activates RNA polymerase II (Pol II) at specific promoter sites of the target genes. Importantly, recent studies have highlighted the pivotal functional involvement of BRD4 in cancer initiation and progression in cancer by regulating the expression of key molecules responsible for oncogenesis in a disease-type specific manner (5). For example, BRD4 directly governs the expressions of fusion partner genes involved in immunoglobulin heavy chain gene (IgH) translocations in myeloma cells. BRD4 also regulates B-cell lymphoma 2 (BCL2) and cell division protein kinase 6 (CDK6) in acute myeloid leukemia with mixed-lineage leukemia 1 (MLL) gene rearrangement, as well as the interleukin-7 receptor gene in acute lymphoblastic leukemia and the Fos like (FOSL) oncogene in lung adenocarcinoma (6-9). Through these mechanisms, BRD4 regulates the expression and of a number of its downstream direct-target genes. Therefore, BRD4 is an attractive therapeutic target molecule in the context of the molecular pathophysiology of cancers and the dissection of genes targeted by BRD4 might lead to the discovery of disease type-specific and therapeutically targetable key molecules involved in disease development and progression. Based upon this concept, our goal was to identify therapeutic targets for MCL through BRD4 screening, i.e., through the broad screening of BRD4-regulated molecules in MCL by genome-wide approaches which would then identify functionally and therapeutically relevant molecules for MCL.
Materials and Methods
Cell lines and reagents. This study used four human MCL-derived cell lines, JVM-2, MINO and Z138, and KPUM-YY1 which was recently established from a patient with MCL with a t(11;14)(q13;q32) translocation for the IGH/CCND1 fusion gene in our laboratory (10). Cells were maintained in RPMI-1640 containing 10% fetal calf serum, 2 mM L-glutamate, and penicillin/streptomycin. I-BET151, an inhibitor of BRD4, was purchased from TOCRIS Bioscience (Bristol, UK). Lenalidomide and ibrutinib were purchased from Selleck (Houston, TX, USA).
Cell viability and proliferation. Cell growth was analyzed by a modified MTT assay for all four MCL cell lines with triplicated samples. Cells were treated for 72 h with following drug concentrations: I-BET151: 0-10 μM, lenalidomide: 0 - 20 μM, and ibrutinib: 0 - 2 μM in combination with either 0, 1, or 10 μM lenalidomide. Cell numbers were counted using the Cell Counting Kit-8 (Dohjindo Molecular Technologies, Kumamoto, Japan). The MTT assay results were analyzed by Calcusyn software (version 2.0; Biosoft, UK). For apoptosis assay, JVM-2 and MINO cells treated with or without I-BET151 at the 50% inhibitory concentration (IC50) of each cell line for 24 and 48 h were counterstained with both annexin V-fluorescein isothiocyanate (FITC) and propidium iodide (PI) followed by flow cytometric analyses. For analysis of cell cycle distribution by the DNA content, four MCL cell lines were cultured with or without I-BET151 at the IC50 of each cell line for 24 h. Cells were fixed with ice-cold 70% ethanol, stained with PI, and then analyzed by flow cytometry. The flow cytometric results were analyzed by FLOWJO software (version 10.2; Tomy Digital Biology, Tokyo, Japan).
Quantitative reverse transcription-PCR (RQ-PCR). For RQ-PCR analysis, all four MCL cell lines were treated with I-BET151 at the 80% inhibitory concentration (IC80) of each cell line for 3 and 6 h. Total RNA was extracted from the cultured cells using a mirVana miRNA Isolation Kit (Thermo Fisher Scientific, Waltham, MA, USA) and reverse-transcribed by QuantiTect Reverse Transcription Kit (Qiagen, Hilden, Germany). Real-time PCR was performed with triplicate samples as technical replicates using StepOnePlus Real-Time PCR System (Thermo Fisher Scientific). The custom primers (Hokkaido System Science, Hokkaido, Japan) used were as follows: BTK: forward (fwd): 5’-CTT TCT GAA GCG ATC CCA AC-3’, reverse (rev): 5’-ATC CAC CGC TTC CTT AGT TC-3’, CCND1: fwd: 5’-CAA ATG TGT GCA GAA GGA GG-3’, rev: 5’-GGG CGG ATT GGA AAT GAA CT-3’, early B-cell factor 1 (EBF1): fwd: 5’-TTC TCC TTC TCA CCA GCC AA-3’, rev: 5’-TGC ACT TGA AGA TCC CTC TT-3’, IKAROS family zinc finger 1 (IKZF1): fwd: 5’-GGA CGC ACT CCG TCA TTA AA-3’, rev: 5’-GTT GGT CAG GTA GAT GAG AC-3’, MYC: fwd: 5’-GAA ACT TTG CCC ATA GCA G-3’, rev: 5’-AAC TCT GGT TCA CCA TGT C-3’, PAX5: fwd: 5’-AGG ACA TGG AGG AGT GAA TC-3’, rev: 5’-TTG ATG GAA CTG ACG CTA GG-3’, and spleen tyrosine kinase (SYK): fwd: 5’-AGG CAG AAG ATT ACC TGG TC-3’, rev: 5’-CTT CTG ACT GAT GAT GGC CT-3’. Each mRNA level was normalized to that of β-actin using Taqman β-actin control reagents and compared the relative expression level with that of the untreated sample using ΔΔCt method.
Western blotting. To assess the molecular expression effects by I-BET151 at protein level, all four cell lines were treated with I-BET151 at either IC50s or IC80s for 12 or 24 h. For lenalidomide, four cell lines were treated with either 1μM or 10μM lenalidomide for 48 h. Cells were lysed with lysis buffer, incubated on ice for 1 h, centrifuged at 13,400 g for 10 min at 4˚C and supernatants were obtained as protein extracts. Each 30 μg of extracted protein samples were denatured at 100˚C for 5 min, applied into Novex WedgeWell 12% Tris-Glycine Gels (Thermo Fisher Scientific) and separated by SDS-PAGE at 110 V for 1.5 h. Protein samples were electroblotted onto a Hybond-PDVF membrane (Amersham Biosciences, Uppsala, Sweden) at 25 V for 2.5 h. The membranes were saturated with 5% (wt/vol) non-fat dry milk in phosphate-buffered saline (PBS) containing 0.1% (vol/vol) Tween 20 (Sigma-Aldrich, Saint Louis, MO, USA) at room temperature (RT) for 1 h. The blocked membranes were incubated with primary antibodies against CCND1, PAX5 (Becton Dickinson, San Diego, CA, USA), MYC, interferon regulatory factor 4 (IRF4) (Santa Cruz Biotechnology, Dallas, TX, USA), BTK, SYK, IKZF1 (Cell Signaling Technology, Beverly, MA, USA), or β-ACTIN (Sigma-Aldrich) at 4˚C overnight. Antibodies were detected by horseradish peroxidase-conjugated secondary antibodies and enhanced chemiluminescence (ECL) Prime (Amersham Biosciences) and qualitatively assessed.
Microarray analysis and signal pathway analysis. JVM-2 and Z138 cells were cultured with dimethyl sulfoxide (DMSO) or I-BET151 at each IC80 for 6 h. Total RNA was extracted from the cultured cells using a mirVana miRNA Isolation Kit. The gene-expression prolife (GEP) analysis was determined by Affymetrix Gene Chip arrays and GeneChip Scanner 3000 (Affymetrix, Santa Clara, CA, USA). Data were analyzed using Affymetrix GeneChip operating software ver. 1.0. Genes showing at least a 1.5-fold difference in expression levels from control were considered to be positive. Signal pathway analysis was determined with the Ingenuity pathway analysis (IPA) software (Ingenuity Systems, Mountain View, CA, USA).
Chromatin immunoprecipitation with parallel sequencing (ChIP-Seq). ChIP was performed using the SimpleChIP Plus Enzymatic Chromatin IP Kit (Magnetic Beads; Cell Signaling Technology) according to the manufacturer’s protocol. Four million JVM-2 cells were treated at a cell density of 2.0×105 cells/ml with either 10 μM I-BET151 or DMSO for 6 h and crosslinked with 1.7% formaldehyde in PBS for 5 min at RT. The cross-linking reaction was stopped by the addition of 1x glycine solution (provided with the kit) for 5 min at RT followed by two washes with PBS. Cross-linked cells were lysed with buffer A supplemented with a dithiothreitol and protease inhibitor cocktail, and the nuclear fraction was collected and treated with micrococcal nuclease in buffer B containing dithiothreitol at 37˚C for 20 min. After sonication in 1x ChIP buffer supplemented with a protease inhibitor cocktail, lysates were clarified by centrifugation at 9,300 g for 10 min at 4˚C. Chromatin samples were diluted into five-fold in ChIP buffer containing a protease inhibitor cocktail, and incubated with either anti-BRD4 antibody (E2A7X, Cell Signaling Technology) or anti-IgG antibody (background control) overnight at 4˚C. The antibody-bound complexes were captured by incubation with protein G magnetic beads for 2 h at 4˚C and washed in low and high salt ChIP buffer. DNA-protein complexes were eluted with elution buffer at 65˚C for 30 min, and the DNA-protein cross-links were reversed by adding NaCl and Proteinase K followed by incubation for 2 h at 65˚C. The immunoprecipitated DNA was purified using a spin column. ChIP-Seq libraries for sequencing were prepared using the TruSeq ChIP Sample Prep Kit (Illumina, San Diego, CA, USA). The libraries were subjected to parallel sequencing with a HiSeq2500 sequencer (Illumina) using the single-end 50 bp sequencing length protocol. Next generation sequencing (NGS) raw data were converted into FASTQ files using CASAVA software (version 1.8.2), and each data set was aligned to the human genome reference (UCSC hg19) using the Burrows-Wheeler Aligner (version 0.7.12) (11). ChIP-Seq peak calling was performed using the MACS2 program (version 2.0.1) with the default parameters but -q value 0.05, with the input data for subtraction (12). Peak comparison with the control and I-BET151 samples was performed with Diffbind, an R package, using a DeSeq2 algorithm, where the false-discovery rate of 0.1 was considered significant (13). Super-enhancers were identified from the set of peaks detected in DMSO-treated JVM-2 cells with the super-enhancers software ROSE (14). ChIP-Seq peaks were annotated with an R package of ChIPpeakAnno, and promoters were defined as BRD4-enriched regions within 1 kb from the transcription start site (15). The target gene of those super-enhancers was assigned to the nearest gene from the center of the enhancer to each transcription start site. Interaction analysis using the Reactome Pathway Database (version 2014) was performed and visualized with ReactomeFIViz, a Cytoscape plug-in, where the functional interaction of genes down-regulated by BRD4 according to GEP analysis is displayed as a network, and genes that are regulated are colored based on the ChIP-Seq analysis result (16,17).
Results
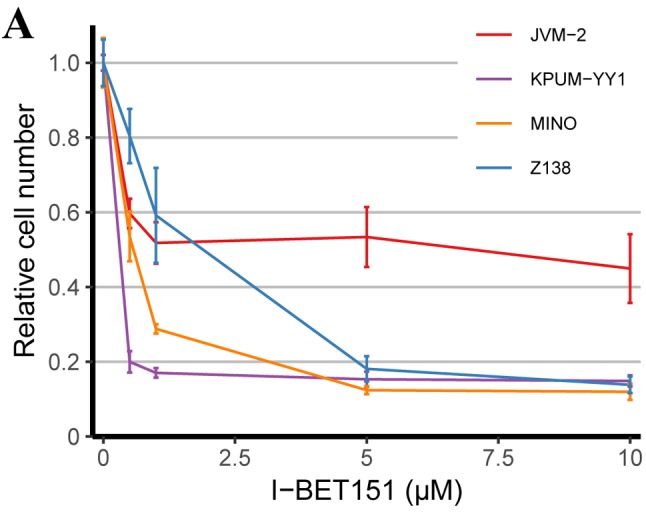
BRD4 inhibitor I-BET151 inhibits the growth and survival of MCL-derived cell lines via G1/S cell cycle arrest and apoptosis, respectively. After treatment with I-BET151 for 72 h, all four MCL-derived cell lines showed a dose-dependent inhibition of cell proliferation with the IC50s of 3.6 μM, 3.5 μM, 2.6 μM and 3.0 μM for JVM-2, KPUM-YY1, MINO, and Z138 cells, respectively (Figure 1A). The underlying mechanisms for the anti-proliferative effect on the MCL cell lines included both G1/S cell-cycle arrest (Figure 1B) and apoptosis (Figure 1C).
Figure 1. The antitumor effect of bromodomain-containing 4 (BRD4) inhibitor I-BET151 on mantle cell lymphoma (MCL)-derived cell lines. A: Treatment with I-BET151 for 72 h inhibited the proliferation of four MCL cell lines in a dose-dependent manner. Results are expressed as the mean of triplicates. B: I-BET151 treatment at the 50% inhibitory concentration (IC50) for the indicated periods induced G1/S cell-cycle arrest and an increase in the sub-G1 population in MCL-derived cell lines. The DNA content was examined by propidium iodide (PI) staining of formalinfixed cells using flow cytometric analysis. The x-axis represents DNA content, and the y-axis represents the relative cell numbers. Data shown are representative results for JVM-2, KPUM-YY1, MINO, and Z138 cells. Green: G1 phase; yellow: S phase; light blue: G2/M phase; white: sub-G1 population, a hallmark of apoptosis induction. C: I-BET151 treatment induced apoptosis in MCL-derived cell lines. Cells were treated with I-BET151 at the IC50s for each cell line for 24 h and were subjected to double staining with annexin-V (AV) and PI. Data shown are representative of results for JVM-2 cells and MINO cells. x-Axis: AV positivity; y-axis: PI positivity. Fraction I: AV−/PI− (viable cells), fraction II: AV+/PI−(cells undergoing early apoptosis), fraction III: AV+/PI+ (cells undergoing late apoptosis), and fraction IV: AV−/PI+ (necrotic cells) populations.

Global transcription in MCL-derived cell lines is affected by BRD4 inhibition with I-BET151. Next, we analyzed the global transcriptional changes in MCL-derived cell lines induced by I-BET151 treatment to investigate the molecular sequelae of BRD4 inhibition. We selected two cell lines, JVM-2 and Z138, for this analysis because their sensitivities to I-BET151 were intermediate among the four cell lines. After 6-hour treatment with I-BET151 at the IC80 concentration for each cell line, GEP analyses revealed that 609 were commonly up-regulated in both cell lines by more than 1.5-fold, and 665 genes down-regulated by less than 0.67-fold (Figure 2A).
Figure 2. Molecular expression changes induced by bromodomain-containing 4 (BRD4) inhibitor I-BET151 in mantle cell lymphoma (MCL)-derived cell lines. A: Venn diagrams showing the numbers of genes up-regulated by more than 1.5-fold or down-regulated by less than 0.67-fold, respectively, in JVM-2 and Z138 cells treated with I-BET151. B: Average fold change in relative expression of 665 commonly down-regulated genes in JVM-2 and Z138 cells after I-BET151 treatment as identified by microarray analysis are illustrated with the annotation of genes associated with B-cell biology. C: Quantitative reverse transcription-polymerase chain reaction identified genes associated with B-cell biology and MYC. Cells were treated with their respective 80% inhibitory concentrations (IC80s) with I-BET151 for the indicated periods. Significantly different at *p<0.05 and **p<0.01 compared with untreated cells (t-test). D: Western blot analyses. Cells were treated with their respective 50% inhibitory concentrations (IC50s) or IC80s for I-BET151 for the indicated periods. Non-specific bands are indicated by arrowheads. Ctrl: Untreated cells.


Because BRD4 inhibition leads to the down-regulation of its direct target genes and also affects the expressions of downstream target genes, we next performed the Ingenuity Canonical Pathway analyses focusing on the 665 genes down-regulated by I-BET151 treatment in both JVM-2 and Z138 cells. BRD4 inhibition affects BCR signaling, PI3K signaling in B-lymphocytes, and genes associated with B-cell development as the major pathways, suggesting that BRD4 regulates multiple genes crucial for B-cell functions, i.e. development, differentiation, proliferation, and activation (Table I). Furthermore, GEP revealed that multiple genes involved in B-cell master regulation, such as IKZF1, IKZF2, IKZF3, PAX5, and EBF1, and those associated with BCR signaling pathways, such as BLNK, CD22, BTK, SYK, BLK, CD19, and CD79B, were simultaneously down-regulated by BRD4 inhibition (Figure 2B). We also confirmed that I-BET151 treatment caused a reduction in multiple molecules associated with B-cell biology at the transcriptional level and at the protein level in four of the cell lines examined (Figure 2C and D). In addition, MYC expression was also down-regulated by I-BET151 treatment in all of the cell lines examined, while expression of CCND1, which is the hallmark of MCL, was reduced by I-BET151 in only two out of the four cell lines (Figure 2D). Major cyclin-dependent kinase inhibitors, p21WAF/CIP1 and p27KIP1, were also up-regulated following I-BET151 treatment (Figure 2D), suggesting their involvement in G1/S cell-cycle arrest.
Table I. Transcriptional expression changes induced by I-BET151 treatment in JVM-2 and Z138 cells. The top 10 signal pathways suggested by Ingenuity Canonical Pathway analysis involving genes commonly down-regulated by I-BET151 in JVM-2 and Z138 cells.

Identification of BRD4 direct binding partner genes by ChIP-Seq. We next performed ChIP-Seq analysis of I-BET151-untreated and -treated JVM-2 cells to identify the target genes that bound to BRD4 out of all of the genes down-regulated by I-BET151. ChIP-Seq analysis identified 14,267 and 3,091 sites bound to BRD4 in the untreated and I-BET151-treated cells, respectively, including 2,386 sites which commonly bound with BRD4 regardless of I-BET151 treatment (Figure 3A). By precise comparison of the ChIP signal intensities of treated and untreated cells, 2,890 out of 14,267 sites were significantly affected by I-BET151 according to the DeSeq2 algorithm, including those of genes involved in BCR signaling, B-cell development, and oncogene MYB expression (Figure 3B and 3C). Among all of the BRD4-binding regions, 547 were characterized as super-enhancers, including BLNK, IKZF3, and PAX5 (Figure 4).
Figure 3. Results of bromodomain-containing 4 (BRD4) chromatin immunoprecipitation with parallel sequencing (ChIP-Seq) in JVM-2 cells. A: Venn diagram showing the numbers of peaks detected by BRD4 ChIP-Seq when treated with either Control or I-BET151, an inhibitor of BRD4. B: Heatmaps of BRD4 signals between control and I-BET151-treated cells identified 2,890 promoters significantly affected by I-BET151. C: Gene tracks of BRD4 signals near the transcription start site of B-cell linker (BLNK), MYB, and paired box 5 (PAX5) genes in untreated (red) and IBET151- treated (blue) cells. Light blue bar indicates the region of a peak detected by the computational tool.

Figure 4. Super-enhancers identified by bromodomain-containing 4 (BRD4) chromatin immunoprecipitation with parallel sequencing (ChIP-Seq) in JVM-2 cells. A: A total of 547 BRD4-binding regions were characterized as super-enhancers. B: Gene tracks of BRD4 signals at B-cell linker (BLNK), IKAROS family zinc finger 3 (IKZF3), and paired box 5 (PAX5) genes, including super-enhancer regions in untreated cells are shown. Light blue bar indicates a super-enhancer.
We then combined the results of the GEP and ChIP-Seq analyses to determine the genes most likely to be directly regulated by BRD4, and data were integrated with the Reactome Pathway Database. Visualization of these results showed that in the MCL cell lines, BRD4 directly and indirectly regulates a network of genes involved in the BCR pathway and B-cell biology [BLNK, PAX5, EBF1, IKZF3, pre-B-cell leukemia transcription factor 2 (PBX2), IRF5, tumor necrosis factor receptor superfamily member 17 (TNFRSF17), etc.], as well as genes regulated by MYC (IKZF3, etc.) and genes for cell proliferation and survival [MYB, signal transducer and activator of transcription 6 (STAT6), ETS variant 6 (ETV6), etc.] (Figure 5). Unexpectedly, MYC was not directly regulated by BRD4.
Figure 5. Integrated analysis of the results of gene-expression profiling (GEP) and chromatin immunoprecipitation with parallel sequencing (ChIPSeq) with the Reactome Pathway Database. Blue circles indicate genes significantly reduced by bromodomain-containing 4 (BRD4) inhibitor IBET151 treatment, red; genes with super-enhancers, pink; genes with super-enhancers where the BRD4 signal was significantly reduced by IBET151 treatment, green; genes indirectly down-regulated by BRD4 according to GEP that were not related to the BRD4 promoter.
Silencing single signaling pathways downstream of BRD4 is not effective therapeutically. Finally, we investigated the degree of involvement of the IKZF family and the BCR signaling pathway on the cell viability of MCL-derived cell lines, since these have been the therapeutic molecular targets in BCLs. Treatment with up to 20 μM lenalidomide, an inhibitor of the IKZF family, resulted in the reduction of IKZF1 protein expression (Figure 6A), however, cell proliferation was only modestly reduced in the KPUM-YY1 and MINO cells, and not significantly reduced in the JVM-2, and Z138 cells (Figure 6B). Cell proliferation was moderately suppressed by up to 2 μM ibrutinib, a BTK inhibitor that blocks BCR signaling, in two out of the four cell lines, KPUM-YY1 and MINO, in a dose-dependent manner, while the effect was modest in the other two cell lines, JVM-2, and Z138 (Figure 6C). When ibrutinib and lenalidomide were combined, an additive effect on cell proliferation in the moderately sensitive cell lines MINO and KPUM-YY1 was observed (Figure 6C).
Figure 6. Silencing signaling pathways in mantle cell lymphoma (MCL)-derived cells. A: Western blot analyses show the changes in protein levels. Cells were treated with the indicated concentrations of lenalidomide (Len) for 24 h. B: MTT analysis shows cell proliferation was only modestly reduced in KPUM-YY1 and MINO cells on treatment with lenalidomide for 72 h. C: MTT analysis shows cell proliferation was moderately suppressed in KPUM-YY1 and MINO cells in a dose-dependent manner by ibrutinib (Ibr) for 72 h with concurrent treatment with lenalidomide. The additive effect of lenalidomide was observed in these two cell lines.
Discussion
Accumulating evidence in studies of carcinogenesis has demonstrated the involvement of super-enhancers, regulatory regions with high levels of acetylated histone H3 lysine 27 as the transcriptional mediator complexes involving BRD4. Super-enhancers are enriched at oncogenic genes and regulate their expression in various types of cancer cells (5,14,18-20). Therefore, the blockade of BRD4 potently down-regulates the expression of super-enhancer-associated oncogenic genes and potentially constitutes an anticancer treatment in a broad range of cancer types. The blockade of BRD4 activity has been proposed as a potential therapeutic for MCL. The BRD4 inhibitor JQ1 showed high anti-proliferative efficacy which was accompanied by the down-regulation of MYC, CDK4/6 and RelA in MCL-derived cell lines (21). We proposed that additional BRD4-regulated targets for MCL likely exist. Supported by recent findings on the promising utility of BRD4-regulated enhancer profiling as a strategy for the identification of disease-specific oncogenic molecules (22), we further identified BRD4 targets which promote MCL development and progression by taking advantage of comprehensive screening of genes down-regulated by I-BET151.
BRD4 ChIP-Seq analysis identified several super-enhancers as BRD4 targets in MCL, and, in combination with the results obtained by GEP, our study revealed that genes associated with the BCR signaling pathway and the IKZF family, presumable upstream of the MYC/IRF4 axis (23,24), are crucially involved in MCL pathophysiology. These results support the current clinical therapeutic strategy for MCL (25). The introduction of targeted agents, such as ibrutinib for BTK, which is a mediator of BCR signaling, bortezomib for the NF-ĸB pathway, which is downstream of the BCR signaling pathway, and lenalidomide for IKZF family proteins, has improved treatment outcomes for MCL (26-28). However, despite an improvement over treatment with genotoxic agent, a cure for MCL has been rarely achieved even with the use of those molecular targeted agents, indicating that single molecular targeting is not sufficient for eradicating MCL cells. BRD4 targeting is an attractive strategy, because it simultaneously regulates multiple genes associated with both disease development and progression. Several basic studies have supported this notion and found that BRD4 inhibitors can synergistically augment the anti-MCL effect of ibrutinib or lenalidomide even in bortezomib-resistant MCL cells (29). Moreover, our study also revealed that the combination of ibrutinib and lenalidomide increased anti-proliferative efficacy in MCL cells. Although we initially expected that BRD4 screening may uncover novel molecular targets other than the already known therapeutic targets, namely BCR pathway associated genes and the IKZF gene family, this was not the case. Indeed, gene knockdown experiments on several other BRD4 targets, such as MYB or PAX5, did not show therapeutic efficacy for proliferation, survival, or sensitivity to genotoxic agents (data not shown). However, our results collectively suggest the functional significance of dual or compensative activation of the BCR signaling pathway and IKZF in MCL. Although the single blockade of either pathway was not sufficient as a therapeutic approach, our data also imply the importance of concomitant blockade of those targets.
Our study also gained insight into the mechanism of resistance to currently available molecular targeted approaches in MCL. While the in vitro efficacies of ibrutinib and lenalidomide were limited in our study, I-BET151 demonstrated high efficacy against all four MCL-derived cell lines examined. This result was consistent with a previous study using broad pharmacological profiling which demonstrated that a subset of MCL cell lines was refractory to the BCR pathway inhibitors, ibrutinib and sotrastaurin, due to recurrent alterations in genes of the alternative NF-ĸB pathway that are downstream of BCR signaling (30). Resistance to ibrutinib likely involves the BTK C481S mutation at the ibrutinib binding site in MCL cells. This mutation can enhance the activation of both BTK and AKT, and also promotes tissue-specific proliferation by CDK4 activation (31). In the MCL lines studied, lenalidomide reduced the protein expression of IKZF1 and IKZF3, but had no effect on IRF4 or MYC levels. Since the antitumor effect of the BRD4 inhibitor is not hampered by BTK mutational status and BRD4 regulates multiple genes in the BCR signaling pathway in addition to MYC, BRD4 inhibitors may be relevant in overcoming resistance to current MCL treatments. We previously identified the higher potency of a novel bivalent BRD4 inhibitor AZD51513 on BCLs concomitantly expressing MYC and BCL2 compared with that of I-BET151, the classical monovalent BRD4 inhibitor utilized in this study. AZD5153 also concomitantly blocks multiple genes involved in the BCR signaling pathway in addition to MYC (32).
There are concerns about BRD4-targeting therapy as an anticancer strategy. In this study, PAX5 transcription was directly regulated by BRD4 at promoter regions, and constituted a super-enhancer. PAX5, a transcription factor, is essential for B-cell differentiation. Interestingly, PAX5 has been reported to act as a tumor suppressor in MCL, and we also confirmed that silencing PAX5 led to increased cell proliferation in MCL cell lines (data not shown), indicating that a subset of BRD4 target genes are not responsible for tumor progression, and may be tumor suppressors (33). These findings may be translated into future studies on the mechanism of sensitivity and resistance to BRD4 inhibitors in BCLs, including MCL. It is also anticipated that the inhibition of BRD4 may lead to unnecessary off-target molecular effects which may potentially cause unexpected adverse events.
In conclusion, BRD4 regulates transcription of multiple genes by binding to promoter regions, partly including super-enhancers and, thus, simultaneously regulates various pathways responsible for disease development and progression. Our study disclosed the therapeutic importance of simultaneous blockade of the BCR signaling and IKZF axis in MCL, indicating that BRD4 inhibitors constitute promising candidates for MCL treatment.
Conflicts of Interest
The Authors do not have any potential conflict of interest to disclose.
Authors’ Contributions
T.T., S.N. R.S., A.K., M.N., Y.C., S.M.M., Y.M.K, T.T.S., Y.M., Y.K., and O.K.S. performed experiments and analysed the data. T.T. and J.K. drafted the article. J.K., and K.M. designed the research and analyzed the data. M.T., T.I. and K.T. designed the experiments and supervised the research.
Acknowledgements
The Authors are grateful to Ms Ikawa T, Ms Sakamoto-Inada N, and Ms Yamane Y for excellent scientific support. Part of this work was carried out at the Joint Usage/Research Center (RIRBM), Hiroshima University. This work was supported in part by a Grant-in-Aid for Scientific Research from The Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT KAKENHI 16K09856) (MT); by the National Cancer Center Research and Development Fund (26-A-4, 29-A-3); by a grant (Practical Research for Innovative Cancer Control) from the Japan Agency for Medical Research and Development (AMED) (JP16ck0106077h003, JP17ck0106348h0001, JP18ck0106348h0002 and JP19ck0106348h0003); by Astra Zeneca R & D grant 2014; and by a Grant-in-Aid by Takeda Science Foundation (JK).
References
- 1.Lenz G, Dreyling M, Hoster E, Wormann B, Duhrsen U, Metzner B, Eimermacher H, Neubauer A, Wandt H, Steinhauer H, Martin S, Heidemann E, Aldaoud A, Parwaresch R, Hasford J, Unterhalt M, Hiddemann W. Immunochemotherapy with rituximab and cyclophosphamide, doxorubicin, vincristine, and prednisone significantly improves response and time to treatment failure, but not long-term outcome in patients with previously untreated mantle cell lymphoma: Results of a prospective randomized trial of the German Low Grade Lymphoma Study Group (GLSG) J Clin Oncol. 2005;23(9):1984–1992. doi: 10.1200/JCO.2005.08.133. PMID: 15668467. DOI: 10.1200/JCO.2005.08.133. [DOI] [PubMed] [Google Scholar]
- 2.Rummel MJ, Niederle N, Maschmeyer G, Banat GA, von Grünhagen U, Losem C, Kofahl-Krause D, Heil G, Welslau M, Balser C, Kaiser U, Weidmann E, Dürk H, Ballo H, Stauch M, Roller F, Barth J, Hoelzer D, Hinke A, Brugger W. Bendamustine plus rituximab versus chop plus rituximab as first-line treatment for patients with indolent and mantle-cell lymphomas: An open-label, multicentre, randomised, phase 3 non-inferiority trial. Lancet. 2013;381(9873):1203–1210. doi: 10.1016/S0140-6736(12)61763-2. PMID: 23433739. DOI: 10.1016/s0140-6736(12)61763-2. [DOI] [PubMed] [Google Scholar]
- 3.Vogt N, Dai B, Erdmann T, Berdel WE, Lenz G. The molecular pathogenesis of mantle cell lymphoma. Leuk Lymphoma. 2017;58(7):1530–1537. doi: 10.1080/10428194.2016.1248965. PMID: 27894215. DOI: 10.1080/10428194.2016.1248965. [DOI] [PubMed] [Google Scholar]
- 4.Jares P, Colomer D, Campo E. Molecular pathogenesis of mantle cell lymphoma. J Clin Invest. 2012;122(10):3416–3423. doi: 10.1172/JCI61272. PMID: 23023712. DOI: 10.1172/JCI61272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153(2):320–334. doi: 10.1016/j.cell.2013.03.036. PMID: 23582323. DOI: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lockwood WW, Zejnullahu K, Bradner JE, Varmus H. Sensitivity of human lung adenocarcinoma cell lines to targeted inhibition of BET epigenetic signaling proteins. Proc Natl Acad Sci USA. 2012;109(47):19408–19413. doi: 10.1073/pnas.1216363109. PMID: 23129625. DOI: 10.1073/pnas.1216363109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ott CJ, Kopp N, Bird L, Paranal RM, Qi J, Bowman T, Rodig SJ, Kung AL, Bradner JE, Weinstock DM. BET bromodomain inhibition targets both c-MYC and IL7R in high-risk acute lymphoblastic leukemia. Blood. 2012;120(14):2843–2852. doi: 10.1182/blood-2012-02-413021. PMID: 22904298. DOI: 10.1182/blood-2012-02-413021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dawson MA, Prinjha RK, Dittmann A, Giotopoulos G, Bantscheff M, Chan WI, Robson SC, Chung CW, Hopf C, Savitski MM, Huthmacher C, Gudgin E, Lugo D, Beinke S, Chapman TD, Roberts EJ, Soden PE, Auger KR, Mirguet O, Doehner K, Delwel R, Burnett AK, Jeffrey P, Drewes G, Lee K, Huntly BJ, Kouzarides T. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478(7370):529–533. doi: 10.1038/nature10509. PMID: 21964340. DOI: 10.1038/nature10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, Taylor MJ, Johns C, Chicas A, Mulloy JC, Kogan SC, Brown P, Valent P, Bradner JE, Lowe SW, Vakoc CR. RNAi screen identifies BRD4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478(7370):524–528. doi: 10.1038/nature10334. PMID: 21814200. DOI: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takimoto-Shimomura T, Nagoshi H, Maegawa S, Fujibayashi Y, Tsukamoto T, Matsumura-Kimoto Y, Mizuno Y, Chinen Y, Mizutani S, Shimura Y, Horiike S, Taniwaki M, Kobayashi T, Kuroda J. Establishment and characteristics of a novel mantle cell lymphoma-derived cell line and a bendamustine-resistant subline. Cancer Genomics Proteomics. 2018;15(3):213–223. doi: 10.21873/cgp.20080. PMID: 29695404. DOI: 10.21873/cgp.20080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25(14):1754–1760. doi: 10.1093/bioinformatics/btp324. PMID: 19451168. DOI: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9(9):R137. doi: 10.1186/gb-2008-9-9-r137. PMID: 18798982. DOI: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, Brown GD, Gojis O, Ellis IO, Green AR, Ali S, Chin SF, Palmieri C, Caldas C, Carroll JS. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 2012;481(7381):389–393. doi: 10.1038/nature10730. PMID: 22217937. DOI: 10.1038/nature10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, Young RA. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell. 2013;153(2):307–319. doi: 10.1016/j.cell.2013.03.035. PMID: 23582322. DOI: 10.1016/j.cell.2013.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhu LJ, Gazin C, Lawson ND, Pages H, Lin SM, Lapointe DS, Green MR. ChIPpeakAnno: A Bioconductor package to annotate ChIPseq and ChIP-chip data. BMC Bioinformatics. 2010;11:237. doi: 10.1186/1471-2105-11-237. PMID: 20459804. DOI: 10.1186/1471-2105-11-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu G, Haw R. Functional interaction network construction and analysis for disease discovery. Methods Mol Biol. 2017;1558:235–253. doi: 10.1007/978-1-4939-6783-4_11. PMID: 28150241. DOI: 10.1007/978-1-4939-6783-4_11. [DOI] [PubMed] [Google Scholar]
- 17.Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, Haw R, Jassal B, Korninger F, May B, Milacic M, Roca CD, Rothfels K, Sevilla C, Shamovsky V, Shorser S, Varusai T, Viteri G, Weiser J, Wu G, Stein L, Hermjakob H, D’Eustachio P. The Reactome pathway knowledgebase. Nucleic Acids Res. 2018;46(D1):D649–D655. doi: 10.1093/nar/gkx1132. PMID: 29145629. DOI: 10.1093/nar/gkx1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J, Rahl PB, Sun HH, Yeda KT, Doench JG, Reichert E, Kung AL, Rodig SJ, Young RA, Shipp MA, Bradner JE. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B-cell lymphoma. Cancer Cell. 2013;24(6):777–790. doi: 10.1016/j.ccr.2013.11.003. PMID: 24332044. DOI: 10.1016/j.ccr.2013.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, Hoke HA, Young RA. Super-enhancers in the control of cell identity and disease. Cell. 2013;155(4):934–947. doi: 10.1016/j.cell.2013.09.053. PMID: 24119843. DOI: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Christensen CL, Kwiatkowski N, Abraham BJ, Carretero J, Al-Shahrour F, Zhang T, Chipumuro E, Herter-Sprie GS, Akbay EA, Altabef A, Zhang J, Shimamura T, Capelletti M, Reibel JB, Cavanaugh JD, Gao P, Liu Y, Michaelsen SR, Poulsen HS, Aref AR, Barbie DA, Bradner JE, George RE, Gray NS, Young RA, Wong KK. Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell. 2014;26(6):909–922. doi: 10.1016/j.ccell.2014.10.019. PMID: 25490451. DOI: 10.1016/j.ccell.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun B, Shah B, Fiskus W, Qi J, Rajapakshe K, Coarfa C, Li L, Devaraj SG, Sharma S, Zhang L, Wang ML, Saenz DT, Krieger S, Bradner JE, Bhalla KN. Synergistic activity of BET protein antagonist-based combinations in mantle cell lymphoma cells sensitive or resistant to ibrutinib. Blood. 2015;126(13):1565–1574. doi: 10.1182/blood-2015-04-639542. PMID: 26254443. DOI: 10.1182/blood-2015-04-639542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wong RWJ, Ngoc PCT, Leong WZ, Yam AWY, Zhang T, Asamitsu K, Iida S, Okamoto T, Ueda R, Gray NS, Ishida T, Sanda T. Enhancer profiling identifies critical cancer genes and characterizes cell identity in adult T-cell leukemia. Blood. 2017;130(21):2326–2338. doi: 10.1182/blood-2017-06-792184. PMID: 28978570. DOI: 10.1182/blood-2017-06-792184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shaffer AL, Emre NC, Lamy L, Ngo VN, Wright G, Xiao W, Powell J, Dave S, Yu X, Zhao H, Zeng Y, Chen B, Epstein J, Staudt LM. IRF4 addiction in multiple myeloma. Nature. 2008;454(7201):226–231. doi: 10.1038/nature07064. PMID: 18568025. DOI: 10.1038/nature07064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kronke J, Udeshi ND, Narla A, Grauman P, Hurst SN, McConkey M, Svinkina T, Heckl D, Comer E, Li X, Ciarlo C, Hartman E, Munshi N, Schenone M, Schreiber SL, Carr SA, Ebert BL. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Science. 2014;343(6168):301–305. doi: 10.1126/science.1244851. PMID: 24292625. DOI: 10.1126/science.1244851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pott S, Lieb JD. What are super-enhancers. Nat Genet. 2015;47(1):8–12. doi: 10.1038/ng.3167. PMID: 25547603. DOI: 10.1038/ng.3167. [DOI] [PubMed] [Google Scholar]
- 26.Wang ML, Rule S, Martin P, Goy A, Auer R, Kahl BS, Jurczak W, Advani RH, Romaguera JE, Williams ME, Barrientos JC, Chmielowska E, Radford J, Stilgenbauer S, Dreyling M, Jedrzejczak WW, Johnson P, Spurgeon SE, Li L, Zhang L, Newberry K, Ou Z, Cheng N, Fang B, McGreivy J, Clow F, Buggy JJ, Chang BY, Beaupre DM, Kunkel LA, Blum KA. Targeting BTK with ibrutinib in relapsed or refractory mantle-cell lymphoma. N Engl J Med. 2013;369(6):507–516. doi: 10.1056/NEJMoa1306220. PMID: 23782157. DOI: 10.1056/NEJMoa1306220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fisher RI, Bernstein SH, Kahl BS, Djulbegovic B, Robertson MJ, de Vos S, Epner E, Krishnan A, Leonard JP, Lonial S, Stadtmauer EA, O’Connor OA, Shi H, Boral AL, Goy A. Multicenter phase II study of bortezomib in patients with relapsed or refractory mantle cell lymphoma. J Clin Oncol. 2006;24(30):4867–4874. doi: 10.1200/JCO.2006.07.9665. PMID: 17001068. DOI: 10.1200/JCO.2006.07.9665. [DOI] [PubMed] [Google Scholar]
- 28.Ruan J, Martin P, Shah B, Schuster SJ, Smith SM, Furman RR, Christos P, Rodriguez A, Svoboda J, Lewis J, Katz O, Coleman M, Leonard JP. Lenalidomide plus rituximab as initial treatment for mantle-cell lymphoma. N Engl J Med. 2015;373(19):1835–1844. doi: 10.1056/NEJMoa1505237. PMID: 26535512. DOI: 10.1056/NEJMoa1505237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moros A, Rodriguez V, Saborit-Villarroya I, Montraveta A, Balsas P, Sandy P, Martinez A, Wiestner A, Normant E, Campo E, Perez-Galan P, Colomer D, Roue G. Synergistic antitumor activity of lenalidomide with the BET bromodomain inhibitor CPI203 in bortezomib-resistant mantle cell lymphoma. Leukemia. 2014;28(10):2049–2059. doi: 10.1038/leu.2014.106. PMID: 24721791. DOI: 10.1038/leu.2014.106. [DOI] [PubMed] [Google Scholar]
- 30.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, Berger MF, Monahan JE, Morais P, Meltzer J, Korejwa A, Jane-Valbuena J, Mapa FA, Thibault J, Bric-Furlong E, Raman P, Shipway A, Engels IH, Cheng J, Yu GK, Yu J, Aspesi P Jr., de Silva M, Jagtap K, Jones MD, Wang L, Hatton C, Palescandolo E, Gupta S, Mahan S, Sougnez C, Onofrio RC, Liefeld T, MacConaill L, Winckler W, Reich M, Li N, Mesirov JP, Gabriel SB, Getz G, Ardlie K, Chan V, Myer VE, Weber BL, Porter J, Warmuth M, Finan P, Harris JL, Meyerson M, Golub TR, Morrissey MP, Sellers WR, Schlegel R, Garraway LA. The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483(7391):603–607. doi: 10.1038/nature11003. PMID: 22460905. DOI: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chiron D, Di Liberto M, Martin P, Huang X, Sharman J, Blecua P, Mathew S, Vijay P, Eng K, Ali S, Johnson A, Chang B, Ely S, Elemento O, Mason CE, Leonard JP, Chen-Kiang S. Cell-cycle reprogramming for PI3K inhibition overrides a relapse-specific C481S BTK mutation revealed by longitudinal functional genomics in mantle cell lymphoma. Cancer Discov. 2014;4(9):1022–1035. doi: 10.1158/2159-8290.CD-14-0098. PMID: 25082755. DOI: 10.1158/2159-8290.CD-14-0098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Takimoto-Shimomura T, Tsukamoto T, Maegawa S, Fujibayashi Y, Matsumura-Kimoto Y, Mizuno Y, Chinen Y, Shimura Y, Mizutani S, Horiike S, Taniwaki M, Kobayashi T, Kuroda J. Dual targeting of bromodomain-containing 4 by AZD5153 and BCL2 by AZD4320 against B-cell lymphomas concomitantly overexpressing c-MYC and BCL2. Invest New Drugs. 2018;37(2):210–222. doi: 10.1007/s10637-018-0623-8. PMID: 29931583. DOI: 10.1007/s10637-018-0623-8. [DOI] [PubMed] [Google Scholar]
- 33.Teo AE, Chen Z, Miranda RN, McDonnell T, Medeiros LJ, McCarty N. Differential PAX5 levels promote malignant B-cell infiltration, progression and drug resistance, and predict a poor prognosis in MCL patients independent of CCND1. Leukemia. 2016;30(3):580–593. doi: 10.1038/leu.2015.140. PMID: 26073757. DOI: 10.1038/leu.2015.140. [DOI] [PMC free article] [PubMed] [Google Scholar]