

SUMMARY
Movement is an essential behavior requiring the assembly and refinement of spinal motor circuits. However, the mechanisms responsible for circuit refinement and synapse maintenance are poorly understood. Similarly, the molecular mechanisms by which gene mutations cause dysfunction and elimination of synapses in neurodegenerative diseases that occur during development are unknown. Here, we demonstrate that the complement protein C1q is required for the refinement of sensory-motor circuits during normal development, as well as for synaptic dysfunction and elimination in spinal muscular atrophy (SMA). C1q tags vulnerable SMA synapses, which triggers activation of the classical complement pathway leading to microglia-mediated elimination. Pharmacological inhibition of C1q or depletion of microglia rescues the number and function of synapses, conferring significant behavioral benefit in SMA mice. Thus, the classical complement pathway plays critical roles in the refinement of developing motor circuits, while its aberrant activation contributes to motor neuron disease.
In Brief
Vukojicic et al. show that complement protein C1q is required for the refinement of spinal sensory-motor circuits during normal development, as well as for synaptic elimination in spinal muscular atrophy (SMA). Pharmacological inhibition of C1q or depletion of microglia rescues vulnerable synapses, yielding significant behavioral benefit in SMA mice.
Graphical Abstract
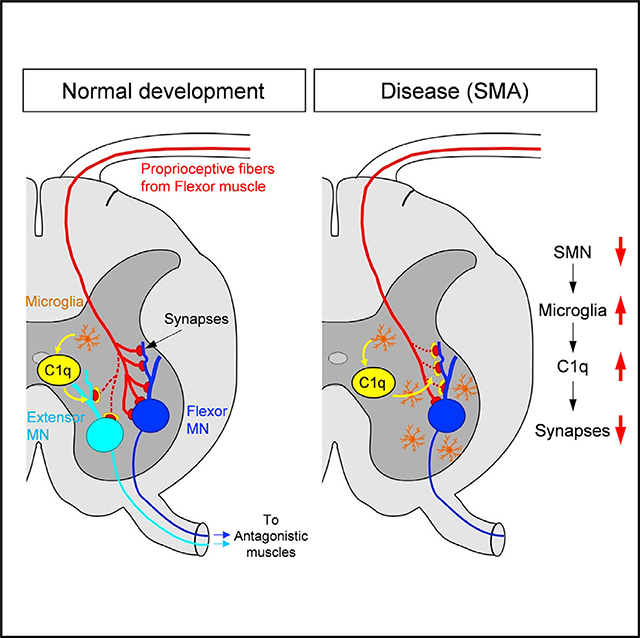
INTRODUCTION
Movement is a simple but essential behavior requiring the precise assembly and function of spinal sensory-motor circuits. During early development, neuronal circuits are specified by molecular mechanisms regulating the formation and maintenance of synapses (Arber, 2012; Riccomagno and Kolodkin, 2015). Within spinal motor circuits, motor neurons innervate and activate select muscles, while they receive instructive information from the periphery via sensory feedback pathways (Burke, 1979) and descending commands from the brain via direct contacts or spinal interneuron circuits (Ferreira-Pinto et al., 2018; Goulding and Pfaff, 2005; Kiehn, 2016). One of the earliest formed spinal sensory-motor circuits is the proprioceptive-motor neuron reflex arc (Arber, 2012; Mears and Frank, 1997). In mature circuits, each motor neuron receives proprioceptive synapses originating from the homonymous muscle that is innervated by this motor neuron (Eccles et al., 1957), as well as from muscles that serve a synergistic function (Mears and Frank, 1997; Mendelsohn et al., 2015), but more important, not from antagonistic muscles. However, during early development, motor neurons do receive proprioceptive inputs from antagonistic muscles that need to be eliminated for proper motor function (Poliak et al., 2016; Seebach and Ziskind-Conhaim, 1994). The molecular mechanisms responsible for this sensory-motor refinement are currently unknown.
Disruption of neuronal networks through synaptic elimination leading to compromised neuronal output underlies neurodegenerative diseases such as Alzheimer, frontotemporal dementia (FTD), and spinal muscular atrophy (SMA) (Lui et al., 2016; Tisdale and Pellizzoni, 2015; Verret et al., 2012). However, unlike Alzheimer disease and FTD, SMA is a disease that occurs during early development in both humans and animal models (Montes et al., 2009; Tisdale and Pellizzoni, 2015), implying that neuronal circuits are immature and possibly more vulnerable to synaptic perturbations. SMA patients have homozygous deletions or mutations in the survival motor neuron 1 (SMN1) gene, but retain at least one copy of a nearly identical hypomorphic gene, SMN2 (Lefebvre et al., 1995), resulting in a ubiquitous deficiency of the SMN protein (Tisdale and Pellizzoni, 2015). Although the hallmarks of SMA are motor neuron death and muscle atrophy, sensory-motor dysfunction is one of the earliest manifestations of the disease in mouse models (Mentis et al., 2011). Furthermore, we demonstrated that SMN deficiency in proprioceptive synapses decreases presynaptic glutamate release onto motor neurons, resulting in the reduction of their firing ability (Fletcher et al., 2017). The inability of motor neurons to sustain high-frequency firing contributes to deficits in muscle contraction and limb movement. However, the molecular mechanisms involved in proprioceptive synaptic dysfunction and their reduction in SMA are not well understood.
It is becoming increasingly evident that dysfunction and the elimination of synaptic connections is an early pathogenic event triggering a cascade of network changes that contribute to the neurodegenerative disease process (Palop and Mucke, 2010). Some studies have highlighted an essential role for glial cells in synaptic elimination (Paolicelli et al., 2011; Schafer et al., 2012; Stephan et al., 2013; Stevens et al., 2007). A strong emerging candidate is the anomalous opsonization, or “tagging,” of synapses by complement proteins. Complement-mediated synaptic elimination has been proposed as a mechanism for the removal of select synapses during the development of the retinogeniculate system (Stevens et al., 2007). Complement proteins have been implicated in Alzheimer disease (Hong et al., 2016) and FTD (Lui et al., 2016). Whether complement proteins are involved in synapse elimination in sensory-motor circuits, either during normal development or in disease, is poorly understood.
Here, we report that the complement proteins of the classical pathway, C1q and C3, are expressed early during normal spinal cord development and aberrantly upregulated in SMA. We further show that C1q is responsible for the proper assembly and refinement of sensory-motor circuits during development in wild-type (WT) mice. Moreover, the aberrant activation of classical complement cascade is involved in the dysfunction and is responsible for the selective elimination of synapses in SMA. Inhibition of C1q deposition on synapses with an anti-C1q antibody in vivo or pharmacological depletion of microglia confers both structural and functional rescue of synapses in SMA mice, resulting in significant behavioral benefit, suggesting that inhibition of this pathway may be a potential therapeutic target. Our study demonstrates that the classical complement cascade plays critical roles in the refinement of developing spinal motor circuits, while its aberrant activation is involved in SMA pathology.
RESULTS
Proprioceptive Synapses Are Eliminated at the Onset of SMA
Proprioceptive synapses contact motor neurons at late embryonic stages (Chen et al., 2003) and proliferate during postnatal development (Kudo and Yamada, 1987; Li and Burke, 2002; Mentis et al., 2011). In SMA, proprioceptive synapses are reduced compared to age-matched WT mice (Mentis et al., 2011). However, it is unknown whether SMA synapses are developmentally arrested and fail to increase in number during postnatal development as with their WT counterparts or they are physically eliminated during the disease process. To address this question, we performed a temporal analysis of the synaptic density of proprioceptive synapses on SMA motor neurons in the L1 spinal segment. We focused on proximal dendrites of motor neurons, since proprioceptive synapses expressing the vesicular glutamate transporter 1 (VGluT1) are highly populated (>80%) on these sites (Fletcher et al., 2017; Rotterman et al., 2014). At birth (postnatal day 0 [P0]), there is no significant difference in the number of proprioceptive synapses between WT and SMA mice (Figures 1A–1C), suggesting that sensory synapses are formed normally in SMA. However, at later ages, there is a significant progressive loss of SMA VGluT1+ synapses at P11 and P13 compared to P4 in proximal dendrites, measured at both 0–50 and 50–100 μm dendritic compartments from the soma (Figures 1B and 1C). There was no significant difference in synaptic number on the soma of motor neurons at different ages in SMA (Figure S1A). This result indicates that SMN-deficient synapses are formed normally at birth and subsequently eliminated, starting at the onset of the disease phenotype.
Figure 1. Elimination of Vulnerable Synapses Coincides with Increased C1q Expression in SMA.

(A) z stack projection of confocal images from retrogradely labeled L1 motor neurons (MNs) (blue) and VGluT1 synapses (green) in WT and SMA mice at P11 (total distance in z axis: 6 μm).
(B) VGluT1+ synaptic density (number of synapses per 50 μm of dendritic length) in proximal dendritic compartments (0–50 μm) in WT (blue) and SMA (red) MNs at P0, P4, P11, and P13 (N ≥ 3 mice per group). In WT (number of dendrites): n = 11 (P0), n = 51 (P4), n = 35 (P11), and n = 18 (P13); in SMA: n = 9 (P0), n = 42 (P4), n = 33 (P11), and n = 10 (P13).
(C) VGluT1+ synaptic density in 50–100 μm proximal dendrites in WT (blue) and SMA (red) MNs at P0, P4, P11, and P13 (N ≥ 3 mice per group). In WT (number of dendrites): n = 15 (P0), n = 44 (P4), n = 36 (P11), and n = 15 (P13); in SMA: n = 9 (P0), n = 34 (P4), n = 31 (P11), and n = 11 (P13). *p < 0.05, **p < 0.001, ***p < 0.001; one-way ANOVA with Tukey’s test.
(D) z stack projection of confocal images with ChAT (blue) and C1q (red) immunoreactivity from P0, P4, and P11 WT and SMA MN pools in the L1 segment (total distance in z axis: 4.9 μm).
(E) MNs (ChAT: blue) and C1q (red) immunoreactivity.
(F) qRT-PCR analysis of C1qA mRNA levels in the L1–L2 spinal cord segments of WT (n = 3) and SMA (n = 3) mice at P4. *p < 0.05; t test.
C1q Is Transiently Expressed during Normal Spinal Cord Development and Upregulated in SMA
To probe into the molecular mechanisms responsible for the synaptic loss in SMA, we explored the involvement of C1q, the initiating protein of the classical complement pathway that has been previously implicated in synapse elimination in glaucoma (Howell et al., 2011; Stevens et al., 2007), Alzheimer disease models (Hong et al., 2016), and FTD models (Lui et al., 2016). Immunoreactivity experiments against C1q revealed the presence of this protein at similar levels in both WT and SMA mice at birth (Figure 1D). However, in striking contrast, there is a strong upregulation of C1q in SMA spinal cord both at P4 (onset of SMA) and at P11 (late stage of disease) compared to their age-matched WT counterparts (Figure 1D). C1q immunoreactivity revealed an increased signal around but not within motor neurons (Figure 1E). The specificity of C1q antibody was validated in C1q knockout (C1q−/−) mice (Figure S1B). In addition, we found increased C1q immunoreactivity in human post-mortem spinal cord from a type I SMA patient compared to a control (Figure S1C). qRT-PCR experiments from mouse spinal cords revealed that C1qA mRNA was significantly increased at P4 in SMA compared to WT L1–L2 spinal cords (Figure 1F). This increase of C1q expression in SMA was not associated with the activation of other inflammatory pathways since there were no differences in the mRNA levels of the proinflammatory cytokines interleukin 1β (IL-1β), IL-6, and tumor necrosis factor (TNF) in WT and SMA L1–L2 spinal cords at P4 (Figure S1D). These results suggest that C1q was produced locally in the vicinity of the motor neurons and demonstrate that C1q is transiently expressed during early postnatal development in WT spinal cords and is aberrantly upregulated at the onset of SMA.
C1q and C3 Tag Vulnerable Synapses in SMA
To determine whether C1q is associated with proprioceptive sensory synapses, we investigated putative interactions between C1q and VGluT1+ synapses with high-resolution confocal microscopy followed by deconvolution and image analysis in WT and SMA spinal cords at P4. We discovered that C1q is associated with or “tags” VGluT1 synapses in both WT and SMA mice (Figures 2A–2D). However, a significantly higher percentage (~25%) of SMA proprioceptive synapses on the proximal dendrites of L1 motor neurons were tagged compared to WT counterparts (~4%) (Figure 2E). Each tagging event was confirmed using intensity line profiling (Figures 2C and 2D). The preferential tagging of proximally located proprioceptive synapses in SMA was not evident at birth (P0), which correlated with the lack of any difference between the number of sensory-motor synapses on WT and SMA motor neurons (Figure S2A). These results indicate that aberrant C1q tagging occurs after birth in SMA mice.
Figure 2. C1q Tags VGluT1 SMA Synapses and Triggers the Classical Complement Pathway.

(A and B) Confocal images of ChAT (blue), C1q (red), and VGluT1 synapses (green) in WT and SMA mice at P4. Arrows indicate the tagged synapses. Dotted-line box is shown in higher magnification in (B).
(C) Higher magnification of the dotted-line box in (B), indicating the line in which the fluorescence profile is measured.
(D) Fluorescence intensity profile (in arbitrary units) for C1q (red) and VGluT1 (green) signal across the line shown in (C).
(E) Percentage of VGluT1 synapses tagged by C1q on the somata and dendrites of L1 WT (2,170 synapses from 76 MNs) and SMA (1,580 synapses from 86 MNs) MNs at P4. ***p < 0.001; t test.
(F) qRT-PCR analysis of C3 mRNA levels in the L1–L2 spinal cord segments of WT (n = 3) and SMA (n = 3) mice at P4. *p < 0.05; t test.
(G) Confocal images of ChAT (blue), C3 (red), and VGluT1 synapses (green) in WT and SMA mice at P4. The arrows indicate C3-deposited synapses.
(H) Single optical plane of a higher magnification of a C3 (red) deposited VGluT1 synapse (green) indicated by the dotted-line box in (G; SMA image)
(I) Percentage of VGluT1 C3-deposited synapses on the somata and the proximal dendrites of L1 WT (1,155 synapses from 27 MNs) and SMA (1,305 synapses from 22 MNs) MNs at P4. *p < 0.05; t test.
We next addressed the possibility that C1q randomly tags synapses in SMA, irrespective of the neurotransmitter used, since experiments with synaptophysin antibodies, a pan-synaptic marker, revealed a widespread tagging of SMA synapses contacting motor neurons (Figures S2B and S2C). Using antibodies against the vesicular GABAergic transporter (VGAT), which labels GABAergic synapses, and the antibody against C1q, we found that there was no difference in the extent of tagging between WT and SMA mice (Figures S2D and S2E). These results indicate that increased C1q tagging is specific for proprioceptive synapses, which are reduced in SMA.
To test whether C1q tagging triggers the activation of the classical complement pathway, we investigated the expression and localization of the complement component C3, a downstream and central protein of the classical pathway. qRT-PCR experiments revealed a significant increase in C3 mRNA in SMA spinal cords compared to WT controls at P4 (Figure 2F). To determine the localization of C3 in vulnerable synapses in SMA, immunohistochemistry experiments against C3 were performed, followed by high-resolution confocal microscopy, deconvolution, and image analysis. The specificity of C3 immunoreactivity was validated in C3 knockout (C3−/−) mice (Figure S2F). Our analysis revealed that C3 was associated with both WT and SMA VGluT1+ synapses (Figures 2G and 2H). However, a significantly higher number of proprioceptive synapses were associated with C3 in SMA mice compared to WT controls (Figure 2I). These results indicate that the percentage of synapses associated with C3 on proximal dendrites was similar to those tagged by C1q, suggesting the activation of the classical complement pathway early in the disease process.
Microglia Are Responsible for Synaptic Removal in the Spinal Cord
We next sought to examine the cell type responsible for VGluT1+ synaptic elimination. Since microglia have been implicated in synaptic pruning via the receptor for cleaved C3 protein (Wu et al., 2015), we examined their presence by immunohistochemistry using Iba1 as a microglia marker. We observed an increase in the presence of Iba1+ microglia within the motor neuron nucleus in SMA mice compared to age-matched controls at P4 (Figure 3A). Remnants of VGluT1+ synapses were observed within microglia in both WT and SMA motor neuron pools (insets in Figure 3A). Quantification of the number of remnants of VGluT1+ synapses within microglia revealed a significantly higher incidence in SMA mice (Figure 3B). The large extent of VGluT1 remnants within microglia were localized to the internal lysosomal compartments marked by CD68 antibody (Figure S3A). In addition, every Iba1+ cell—in both WT and SMA—revealed a low expression of CD45, while no CD45+/Iba1− cells were observed, suggesting that there is no infiltration of leukocytes (Figure S3B). These results suggest phagocytosis of VGluT1 synapses by microglia, the resident macrophage within the CNS, upon activation of the classical complement cascade.
Figure 3. Microglia Mediate Synaptic Elimination.

(A) z stack projections of confocal images of ChAT (blue), VGluT1 (green), and Iba1 (red) immunoreactivity in WT and SMA L1 MN pools at P4 (total distance: 10 μm). Insets show single optical planes in which VGluT1 remnants (arrows) are found within Iba1+.
(B) Percentage of VGluT1 synapses engulfed by microglia in WT and SMA mice at P4. *p < 0.001; t test (n = 3).
(C) z stack projections of confocal images of ChAT (blue) and VGluT1 synapses (white) from untreated SMA and PLX5622-treated SMA mice at P11 (total distance: 20 μm).
(D) VGluT1 synaptic density (number of synapses in 50-μm dendritic compartments from the soma) for P11 WT (n = 41), WT+PLX5622 (n = 17), SMA (n = 43), and SMA+PLX5622 (n = 9) dendrites. *p < 0.05, ***p < 0.001; one-way ANOVA with Tukey’s test.
(E) Righting times for WT (n = 10), WT+PLX5622 (n = 9), SMA (n = 8), and SMA+PLX5622 (n = 18) mice. *p < 0.05, **p < 0.01, ***p < 0.001; t test (SMA versus SMA+PLX5622) for individual ages.
(F) Total number of L1 MNs at P11 for WT (n = 3), SMA (n = 4), and SMA+PLX5622 (n = 3) mice. **p < 0.01; one-way ANOVA with Tukey’s test.
(G) z stack projection of confocal images of retrogradely labeled MNs (blue), VGluT1 synapses (green), and C1q (red) from an SMA::PvCre MN pool at P4 (total distance: 4.2 μm).
(H) Percentage of VGluT1 C1q-tagged synapses on the somata and dendrites of L1 WT (2,170 synapses from 76 MNs), SMA (1,580 synapses from 86 MNs), and SMA::PvCre (921 synapses from 52 MNs) MNs at P4. ***p < 0.001; one-way ANOVA with Tukey’s test.
To see whether other components of the classical complement cascade become activated, we tested gene expression levels (by qRT-PCR) of the downstream C5 and its receptor C5aR1 in WT and SMA mice at the onset of disease at P4, when the complement tagging of synapses occurs. We found no difference in C5 or C5aR1 mRNA levels between WT and SMA L1–L2 spinal cords (Figures S3C and S3D). We also tested for the potential difference between WT and SMA mice in the expression of the membrane attack complex (MAC) inhibitor CD59b, a key complement regulator in mice (Qin et al., 2003), since studies in Alzheimer disease and amyotrophic lateral sclerosis reported high levels of downstream complement components, coupled with a low expression of CD59 (Lee et al., 2013; Yang et al., 2000). We found no difference between WT and SMA L1–L2 spinal cords at P4 (Figure S3E). Although our results cannot directly rule out a role for the downstream components of the classical complement cascade, the gene expression levels of C5, C5aR1, and CD59 suggest that activation of the membrane attack complex is unlikely to be involved in synaptic removal in SMA.
If microglia play a critical role in synaptic elimination in SMA mice, then the depletion of microglia should protect vulnerable synapses in SMA mice. To test this idea, we treated WT and SMA mice from P0 until P10 twice daily with 50 mg/kg of PLX5622, a pharmacological inhibitor of the colony-stimulating factor 1 receptor (CSF1R) (Elmore et al., 2018). We found a significantly higher number of microglia in the ventral horn of SMA mice compared to WT age-matched controls (Figures S3F and S3G). The administration of PLX5622 during the first 10 days of the postnatal period resulted in an ~90% reduction of microglia in both WT and SMA mice (Figure S3G). There was a significant rescue of proprioceptive synapses (VGluT1+) on motor neuron proximal dendrites in PLX5622-treated SMA mice compared to untreated SMA mice (Figures 3C and 3D). In addition, there was significant behavioral benefit in SMA mice depleted of microglia, as shown by the gradual temporal improvement in righting time (Figure 3E). WT mice treated with PLX5622 did not exhibit any significant differences in behavioral changes in righting (Figure 3E) or body weight gain (Figure S3H). The treatment of SMA mice with PLX5622 did not result in the rescue of motor neuron death (Figure 3F), which is consistent with the intrinsic mechanisms of motor neuron degeneration previously reported (Simon et al., 2017; Van Alstyne et al., 2018). These results indicate that microglia, which are aberrantly increased in SMA mice, are responsible for synaptic removal in SMA.
C1q Tagging of Proprioceptive Synapses Is Driven by SMN Deficiency in Proprioceptive Neurons
To identify the trigger for C1q deposition at proprioceptive synapses, we restored SMN protein expression selectively in proprioceptive neurons in SMA mice. For this, we used a mouse model of SMA harboring a single targeted mutation and two transgenic alleles, resulting in the following genotype: Smn+/Res;SMN2+/+;SMNΔ7+/+ (Fletcher et al., 2017). The allele carrying the targeted mutation (SmnRes) is engineered to revert to a fully functional Smn allele upon Cre-mediated recombination (Cre+/−;SmnRes/−;SMN2+/+;SMNΔ7+/+) (Lutz et al., 2011); SMN2 is the human gene and SMNΔ7 corresponds to the human SMN cDNA lacking exon 7. In the absence of the Cre recombinase (Cre−/−;SmnRes/−;SMN2+/+;SMNΔ7+/+), the phenotype of these mice is similar to that of the SMNΔ7 SMA mice (Fletcher et al., 2017). The restoration of SMN protein in proprioceptive neurons was achieved by crossing these conditional inversion SMA mice with parvalbumin::Cre (PvCre) mice, known to label exclusively proprioceptive neurons during the first 10 postnatal days, as we have previously shown (Fletcher et al., 2017; Mendelsohn et al., 2015). We found a significant reduction in C1q-tagged VGluT1+ proprioceptive synapses in the proximal dendrites of vulnerable SMA motoneurons in SMA::PvCre mice (Figures 3G and 3H). This result indicates that C1q tagging of proprioceptive synapses is driven by the effects of SMN deficiency in proprioceptive neurons.
Microglia Are the Local Source of C1q in the Spinal Cord
We next addressed the source of C1q in the spinal cord. Since we did not observe C1q within motor neurons and the large accumulations of the C1q resembled the microglia shape, we performed immunohistochemistry experiments in which the C1q antibody was combined with the antibodies against Iba1 and TMEM119, a specific microglial marker that is not expressed by either macrophages or other immune and neuronal cell types (Bennett et al., 2016). We found that all Iba1+/TMEM119+ cells (n = 128 from 3 WT mice; n = 215 from 3 SMA mice) expressed a strong signal against C1q in both WT and SMA mice (Figures 4A and S4A). In addition, we found that all Iba1+ cells were CD68+ (n = 135 from 3 WT mice; n = 252 from 3 SMA mice), suggesting that there is no infiltration of monocytes or macrophages. Fluorescence in situ hybridization (FISH) combined with immunohistochemistry revealed the presence of C1qA mRNA within microglia labeled by the Iba1 antibody, but not inside motor neurons labeled by the choline acetyltransferase (ChAT) antibody (Figure S4B). These results demonstrate that microglia are the major source of C1q in the developing spinal cord.
Figure 4. Treatment with Neutralizing C1q Antibody Rescues VGluT1 Synapses and Improves Lifespan and Motor Behavior in SMA Mice.

(A) z stack projections of confocal images of C1q (red), TMEM119 (blue), and Iba1 (green) in SMA in the L1 spinal ventral horn at P4 (total distance: 5.25 μm). The circled area indicates the MN nucleus. The dotted-line box indicates the area shown in the insets. The insets show at higher magnification the colocalization of microglia (Iba1+/TMEM119+ cells) and C1q.
(B) z stack projections of confocal images of retrogradely labeled MNs (blue) and VGluT1 (white) in SMA treated with isotype control Ab (left) and with anti-C1q Ab (right) mice at P11 (total distance in the z axis: 22.4 μm). Higher-magnification images of individual MN dendrites with VGluT1 synapses are shown at right.
(C) The total number of VGluT1 synapses on the entire soma of WT (n = 10), SMA (n = 10), SMA+Ctr Ab (n = 10), and SMA+a-C1q Ab (n = 6) L1 MNs. *p < 0.05, **p < 0.01, ***p < 0.001; one-way ANOVA with Tukey’s test.
(D) Synaptic density in 50-μm dendritic compartments from the soma for WT (n = 41), SMA (n = 43), SMA+Ctr Ab (n = 31), and SMA+a-C1q Ab (n = 54) L1 MNs. ***p < 0.001; one-way ANOVA with Tukey’s test.
(E) Total number of L1 MNs in WT (n = 3), SMA (n = 4), SMA+Ctr Ab (n = 5), and SMA+a-C1q Ab (n = 8) mice at P11. ***p < 0.001; one-way ANOVA with Tukey’s test.
(F) z stack projections of confocal images of synaptophysin (green), neurofilament (blue) and α-bungarotoxin (red) to label pre- and postsynaptic elements of neuromuscular junctions in the QL muscle in WT, SMA, SMA+Ctr Ab, and SMA+a-C1q Ab mice at P11. The arrows indicate denervated NMJs (total distance: 7 μm).
(G) Percentage extent of innervation of the QL muscle in WT (n = 3 mice), SMA (n = 3), SMA+Ctr Ab (n = 3), and SMA+a-C1q Ab (n = 5). ***p < 0.001; one-way ANOVA with Tukey’s test.
(H) Ventral root responses to stimulation of the L1 dorsal root in WT, SMA, SMA+Ctr Ab, and SMA+a-C1q Ab at P10. The arrowheads indicate stimulus artifacts, the dotted line indicates baseline, the arrowheads point to stimulus artifacts, and the arrows indicate the maximal amplitude of the monosynaptic responses.
(I) The maximum amplitude of the monosynaptic response in WT (n = 6), SMA (n = 9), SMA+Ctr Ab (n = 7), and SMA+a-C1q Ab (n = 7) mice. *p < 0.05, ***p < 0.001; one-way ANOVA with Tukey’s test.
(J) Percentage amplitude change of the 5th response (normalized to the 1st) following 20-Hz stimulation in WT (n = 6), SMA (n = 9), SMA+Ctr Ab (n = 7), and SMA+a-C1q Ab (n = 7) mice at P11. ***p < 0.001; one-way ANOVA with Tukey’s test.
(K) Lifespan of SMA+Ctr Ab (n = 8) and SMA+a-C1q Ab (n = 10) mice. **p < 0.01; t test.
(L) Righting times of WT (n = 10), SMA (n = 9), SMA+Ctr Ab (n = 15), and SMA+a-C1q Ab (n = 24). *p < 0.05, **p < 0.01; t test for individual ages.
Postnatal Block of C1q Tagging Confers Synaptic and Behavioral Benefit in SMA Mice
To confirm and expand on the role of C1q, we used a mouse monoclonal anti-C1q antibody (a-C1q) (Lansita et al., 2017) to neutralize C1q in SMA mice. The C1q globular domain responsible for its recognition properties has a compact, almost spherical heterotrimeric assembly of C1qA, C1qB, and C1qC, which appears to be behind the versatile recognition properties of C1q (Gaboriaud et al., 2003). The exact target of C1q binding on a synapse is unknown. In addition, it is not known which subunits of C1q drive the synaptic localization of C1q. The neutralizing antibody binds to a conformational epitope spanning two of the three subunits that make up the globular head groups of C1q. The antibody blocks the interaction of C1q with diverse ligands, including immunoglobulin M (IgM), phosphatidyl serine, and C-reactive protein (CRP). Results from Lansita and colleagues (Lansita et al., 2017) demonstrated that a-C1q antibody can fully block the functional activation of the classical cascade in multiple animal species. It has also been shown to be active in animal models of antibody-mediated disease (McGonigal et al., 2016) and antibody-independent neurodegeneration (Hong et al., 2016; Dejanovic et al., 2018). SMA mice were treated with either a-C1q or isotype (mIgG1) control antibody from birth (P0) every other day at a dose of 100 mg/kg.
The binding affinity and specificity of the anti-C1q antibody versus its isotype control were validated by the C1q-binding assay (Figure S4C). We found a significant increase in VGluT1+ synapses on the somata and proximal dendrites of L1 motor neurons at P10 in SMA mice treated with a-C1q antibody (SMA+a-C1q Ab), but not in the control treated SMA mice (SMA+Ctr Ab) (Figures 4B–4D). Furthermore, there were no differences in VGluT1 synaptic density between untreated mice and isotype control treated SMA mice (Figures 4C and 4D). There was a nearly complete rescue of VGluT1+ synapses on the proximal dendrites of motor neurons in a-C1q-treated SMA mice (Figure 4D). Treatment with a-C1q antibodies did not upregulate SMN in the spinal cord (Figure S4D), nor in dorsal root ganglions (DRGs), since there was no increase in Gems, which are nuclear structures containing SMNs (Liu and Dreyfuss, 1996), in proprioceptive neurons (Figures S4E and S4F). a-C1q-treated SMA mice did not show any rescue of SMA motor neurons (Figure 4E), further supporting that C1q is not involved in motor neuron death induced by SMN deficiency. To test whether the blocking of C1q has any effects on neuromuscular junction (NMJ) denervation, we investigated the extent of denervation in the vulnerable quadratus lumborum (QL) muscle. We found no difference between untreated SMA, isotype control-treated SMA, and a-C1q-treated SMA mice (Figures 4F and 4G). This is likely due to the absence of C1q around NMJs in the QL muscle in SMA mice (Figure S4G). In addition, the compound muscle action potentials (CMAPs) tested in the QL muscle following stimulation of the L1 ventral root did not show any improvement compared to untreated or isotype control treated SMA mice (Figure S4H). These results demonstrate the involvement of C1q in central but not peripheral synapse elimination in SMA.
To test whether the rescued VGluT1+ synapses in a-C1q-treated SMA mice were functional, we used the ex vivo spinal cord preparation and performed dorsal-to-ventral root reflexes in the L1 segment at P11. We found a significant increase in the amplitude of the reflex response in a-C1q-treated SMA mice compared to controls (Figures 4H and 4I). Moreover, the synaptic reliability of the rescued synapses was similar to that of WT mice (Figure 4K), following high-frequency stimulation (20 Hz). The synaptic reliability test is independent of the number of synapses present and the number of motor neurons present, since the amplitude of motor neuron response at high frequency is normalized to the first stimulus. These results demonstrate that the neutralization of C1q by in vivo treatment with an a-C1q antibody from birth confers an increase in the number and function of proprioceptive synapses in SMA mice, suggesting complement involvement early in synaptic dysfunction, besides its role in selective synaptic elimination. To test whether this observation translates into any behavioral improvement in SMA mice, we monitored daily the righting time, body weight, and lifespan in untreated SMA mice, isotype control treated, and a-C1q antibody-treated SMA mice. a-C1q antibody treatment provided a significant improvement in the survival of SMA mice by extending their lifespan by ~25% (Figure 4L). We found that although the body weight gain was minimal in all groups (Figure S5A), there was a significant improvement in the righting time in a-C1q-treated SMA mice compared to controls (Figure 4M). The improvement in righting time was progressive over time, which was markedly defined by the ability of SMA mice to right and even walk (see Video S1 for WT and SMA mice, and Video S2 for a-C1q-treated SMA mice). These results demonstrate that postnatal treatment with anti-C1q antibody in SMA mice after birth rescues both the number and the function of sensory synapses, which provides remarkable behavioral benefits that emphasize the significance of this approach as a potential combinatorial therapeutic strategy in SMA patients.
Genetic Deletion of C1q Rescues Proprioceptive Synapses in SMA Mice
To determine the direct involvement of C1q in the removal of proprioceptive synapses in SMA, we genetically removed C1q from SMA mice by crossing SMA mice with mice harboring a ubiquitous deletion of C1qa (Botto et al., 1998). We found a significant rescue of proprioceptive synapses both on the somata and the proximal dendrites of vulnerable L1 motor neurons in SMA::C1q−/− mice compared to SMA mice at P10 (Figures 5A–5C). As expected (Simon et al., 2017; Van Alstyne et al., 2018), we did not observe any significant rescue of vulnerable L1 motor neurons in SMA::C1q−/− mice (Figure 5D). To test whether the rescued synapses were functional, we used the ex vivo spinal cord preparation to measure the amplitude of monosynaptic reflex recorded in the L1 ventral root evoked by stimulation of the homonymous dorsal root (Figure 5E). We found a significant increase in the amplitude of the reflex in SMA::C1q−/− mice compared to SMA age-matched mice (Figure 5F), but no difference in the latency of the reflex (Figure 5G). These results demonstrate that genetic deletion of C1q from SMA mice rescues the number and function of proprioceptive synapses, indicating that C1q-mediated synaptic removal is an aberrant pathogenic process.
Figure 5. Genetic Deletion of C1q Rescues Synapses and Their Function in SMA Mice.

(A) z stack projections of confocal images of ChAT (blue) and VGluT1 (white) in SMA and SMA::C1q−/− MNs at P11 (total distance: 22.4 μm). Inset, higher-magnification MN dendrites with VGluT1 synapses.
(B) The total number of VGluT1 synapses on the entire soma of WT (n = 10), SMA (n = 10), and SMA::C1q−/− (n = 10) L1 MNs. *p < 0.05, **p < 0.01, ***p < 0.01; one-way ANOVA with Tukey’s test.
(C) VGluT1 synaptic density (number of synapses in 50-μm dendritic compartments from the soma) for WT (n = 41), SMA (n = 43), and SMA::C1q−/− (n = 29) MNs. *p < 0.05, **p < 0.01, ***p < 0.01; one-way ANOVA with Tukey’s test.
(D) Total number of L1 MNs in WT (n = 3), SMA (n = 4), and SMA::C1q−/− (n = 4) mice. ***p < 0.01; one-way ANOVA with Tukey’s test.
(E) Schematic illustration of the ex vivo spinal cord physiological assessment, in which stimulation of the L1 dorsal root elicits a response on the homonymous ventral root. The monosynaptic response (arrows) in SMA and SMA::C1q−/− mice at P10 are shown. The arrowheads indicate the stimulus artifacts.
(F) The amplitude of the monosynaptic reflex responses in SMA (n = 9) and SMA::C1q−/− (n = 4) mice at P10. **p < 0.01; t test.
(G) The latency of the onset of the monosynaptic response for the same groups.
C1q Mediates the Refinement of Sensory-Motor Circuits during Normal Development
Genetic deletion of C1q did not confer any behavioral benefit in SMA mice, unlike postnatal C1q inhibition with anti-C1q antibody, despite the similar rescue of proprioceptive synapses. Both body weight and righting time across all ages were not improved compared to those in SMA mice (Figures S5B and S5C). These results raised the possibility that C1q may be involved in synaptic elimination as part of the refinement of spinal sensory-motor circuits during prenatal development. To address this, we investigated the synaptic density of VGluT1+ proprioceptive synapses on motor neurons in C1q−/− mice. We discovered ~25% excessive VGluT1 synapses, both on the somata and the proximal dendrites of motor neurons in C1q−/− mice compared to WT counterparts at P11 (Figures 6A–6C). We next tested whether these excessive synapses were functional by measuring dorsal root-to-ventral root reflexes ex vivo in the L1 lumbar segment at P11 (Figure 6D). We found that the amplitude of the monosynaptic reflex was significantly greater compared to that of WT counterparts (Figures 6D and 6F). In addition, to test the synaptic efficacy of the supernumerary synapses in C1q−/− mice, we challenged synaptic neurotransmission under high-frequency (20 Hz) stimulation. We found that the amplitude change of the reflex was similar to that observed in WT mice (Figures 6F and 6G). Since there were also no differences in motor neuron numbers between C1q−/− and WT mice (Figures S6A and S6B), these results indicate that the supernumerary number of synapses in C1q−/− mice is functional.
Figure 6. Genetic Deletion of C1q Results in an Increase in Proprioceptive Synapses during Normal Development.

(A) z stack projections of confocal images of ChAT (blue) and VGluT1 synapses (white) in WT and C1q−/− mice at P11 (total distance in z axis: 25.2 μm). Circled areas indicate MN nuclei. Higher magnification (right) of z stack projections from single MNs in WT and C1q−/− mice (total distance in z axis: 2.1 μm).
(B) The total number of VGluT1 synapses on the entire soma from WT (n = 10) and C1q−/− (n = 12) L1 MNs. **p < 0.01; t test.
(C) VGluT1 synaptic density (number of synapses in 50-μm dendritic compartments from the soma) in WT (n = 41) and C1q−/− (n = 35) MNs. **p < 0.01, ***p < 0.001; t test.
(D) Ventral root responses to supramaximal stimulation of the homonymous L1 dorsal root in WT and C1q−/− spinal cords at P10. The arrowheads indicate the stimulus artifacts.
(E) The amplitude of the monosynaptic reflex responses for WT (n = 6) and C1q−/− (n = 7) mice at P11. *p < 0.05; t test.
(F) Superimposed traces of the 1st (black) and 5th (red) responses following 20-Hz stimulation of the dorsal root in WT and C1q−/− mice.
(G) Percentage amplitude change of the 5th response normalized to the 1st response, following 20-Hz stimulation in WT (n = 6) and C1q−/− (n = 7) mice.
C1q−/− mice were mildly underweight compared to their WT controls (Figure S7A). They exhibited stark differences in the righting time (Figure 7A). C1q−/− mice exhibited a marked delay in righting, which was very prominent at birth and progressively improved over the second postnatal week (Figure 7A). The increased number of proprioceptive synapses on motor neurons, coupled with the inability of mice to right themselves, raised the possibility that sensory-motor circuits may have been inappropriately established during late embryonic and early postnatal development. We therefore investigated the pattern of connectivity between two antagonistic motor pools, the tibialis anterior (TA) and the gastrocnemius (Gastro) muscles. We used an assay that exploits the transganglionic transport ability of cholera toxin β-subunit (CTb) conjugated to Alexa 555. When injected in a muscle, CTb accumulates in ~40% of the proprioceptive sensory synapses, which can be marked in an independent and selective manner by the presynaptic expression of VGluT1 (Mendelsohn et al., 2015). We injected the TA muscle with CTb-Alexa 555 to label both the TA motor neurons and the proprioceptive synapses and the Gastro muscle with fluorescein dextran (Dext) that labels only motor neurons (Figures 7B and 7C). This distinction allows a binary comparison of the density of CTb-labeled proprioceptive synapses in contact with homonymous CTb-labeled or antagonistic Dext-labeled motor neurons (Mendelsohn et al., 2015). We analyzed the synaptic density ~6–7 days post injection, the minimum time required for both motor neuron and synaptic labeling (Mendelsohn et al., 2015). We found ~22% of Gastro motor neurons receiving inappropriate sensory synapses emanating from proprioceptive neurons in the antagonistic TA muscle in C1q−/− mice, but none in WT mice (Figures 7D and 7E). These results indicate that C1q plays a major role in spinal sensory-motor circuit refinement during normal development and may also explain the lack of motor improvement observed in SMA::C1q−/− mice.
Figure 7. C1q Mediates Sensory-Motor Synaptic Refinement during Normal Development.

(A) Righting times in WT (n = 10) and C1q−/− (n = 18) mice during the first 12 postnatal days. ***p < 0.001; t test for the individual ages.
(B) Schematic illustration for retrograde labeling of TA MNs and their proprioceptive synapses with CTb-555 (red) and gastrocnemius MNs only with fluorescinated dextran (green). The dotted red line indicates inappropriate TA synapses onto gastrocnemius MNs.
(C) TA and gastrocnemius MNs labeled with CTb-555 and fluorescinated dextran correspondingly.
(D) Single optical planes of confocal images with the tracers fluorescinated dextran (green), CTb-555 (red) and VGluT1 (blue) immunoreactivity from gastrocnemius MNs in a WT and a C1−/− mouse at P4. Inset shows a TA-originating proprioceptive synapse (arrow) on a gastrocnemius MN.
(E) Percentage of MNs receiving inappropriate synapses in WT (n = 3) and C1q−/− mice (n = 4). *p < 0.05; t test.
DISCUSSION
Our study demonstrates the involvement of C1q, the initiating protein of the classical complement pathway, in the refinement of spinal sensory-motor circuits during normal development and identifies its involvement in dysfunction and its role in the selective elimination of proprioceptive synapses in the neurodegenerative disease SMA. Under SMN-deficient conditions, there is an aberrant upregulation of C1q protein by microglia. Dysfunction of vulnerable synapses on motor neurons is coupled with the deposition of C1q and subsequent activation of the classical complement pathway, leading to their progressive microglia-mediated elimination in SMA. Postnatal in vivo inhibition of the classical complement cascade with an anti-C1q antibody rescues the number and function of synapses, extends the lifespan, and confers significant behavioral benefit in SMA mouse models. Therefore, protecting vulnerable synapses by blocking aberrant C1q may represent a viable therapeutic approach that is complementary to SMN upregulation. We have uncovered previously unknown roles of the classical complement pathway in the refinement of developing spinal motor circuits and synaptic pathology in SMA.
Aberrant Activation of the Classical Complement Pathway Induces Loss of Vulnerable Synapses in SMA
SMA has emerged as a model disease to study the role of dysfunction of synaptic contacts in neurodegenerative diseases (Shorrock et al., 2019; Tisdale and Pellizzoni, 2015). We have previously shown that motor neuron loss follows sensory synapse dysfunction (Mentis et al., 2011). We have also shown that selective restoration of SMN in proprioceptive sensory neurons restored the number of sensory synapses and corrected their dysfunction, which resulted in improvement in the spiking frequency of SMA motor neurons, while it had no effect on motor neuron survival. These findings revealed that synaptic dysfunction and motor neuron survival are two independent events in SMA (Fletcher et al., 2017). The restoration of SMN in sensory and spinal interneurons was also reported to improve the behavioral phenotype in Drosophila models of SMA (Imlach et al., 2012). All of these studies identified sensory synapses as a primary neuronal target contributing to the disease phenotype, but the mechanisms by which sensory synapses in SMA are actively eliminated remained elusive. Here, we demonstrate that proprioceptive sensory synapses on SMA motor neurons are tagged by the aberrantly expressed classical complement proteins C1q and C3. While C1q and C3 localize to proprioceptive synapses in both WT and SMA mice, they are significantly higher in SMA. Furthermore, the presence of VGluT1 remnants within microglia suggests that C1q deposition on these synapses triggers the activation of the classical complement cascade, leading to their subsequent microglia-mediated elimination, likely via complement receptor 3 (CR3). In addition, we show another important role for microglia. C1q is produced exclusively by microglia in the spinal cord of both WT and SMA mice, which is consistent with a recent report in the brain (Fonseca et al., 2017), but not by motor neurons, as previously suggested (Zhang et al., 2013). Genetic ablation or pharmacological inhibition of C1q in SMA mice rescued vulnerable synapses, indicating that the activation of the classical complement cascade is the major mechanism for synaptic elimination in SMA. In addition, postnatal in vivo depletion of microglia with PLX5622 resulted in a significant rescue of synapses and phenotypic benefit in SMA mice. Furthermore, since VGluT2+ synapses have also been reported to be vulnerable and reduced in SMA (Ling et al., 2010; Simon et al., 2016), complement and microglia could also be implicated in their removal. Our study therefore demonstrates that the aberrant activation of the classical complement pathway triggers synaptic loss and indicates that microglia are the major cell type responsible for synaptic elimination in SMA.
The trigger for the selective elimination of proprioceptive glutamatergic synapses in SMA is unknown. It is unlikely that C1q preferentially tags glutamatergic synapses for elimination in SMA due to the neurotransmitter type, since inhibitory synapses in a mouse model of FTD are selectively removed by C1q (Lui et al., 2016). This raises the possibility that neuronal activity may be a determining factor for the selective synaptic elimination. We have previously shown that proprioceptive synapses in SMA exhibit dysfunction to a certain extent early (~P2) in the disease process (Fletcher et al., 2017). It is possible that C1q “senses” early synaptic changes and further propagates synaptic dysfunction and the demise of motor function. Furthermore, selective restoration of SMN in proprioceptive neurons or a pharmacologically induced increase in neuronal activity rescues synaptic function and elimination in SMA mice (Fletcher et al., 2017). In this study we show that the selective restoration of SMN in proprioceptive neurons in SMA mice abrogates C1q tagging to nearly WT levels, supporting the idea that C1q tagging is mediated by synaptic mechanisms that originate within proprioceptive neurons. CD47, a transmembrane protein also known as a “do not eat me” signal, is localized at synapses (Mi et al., 2000; Toth et al., 2013). It was also demonstrated that CD47 in the developing lateral geniculate nucleus (LGN) preferentially localizes in more active synapses, where it may serve as a protective signal against complement proteins, thereby directing microglia to eliminate weak or dysfunctional synapses (Lehrman et al., 2018). Vulnerable SMA proprioceptive synapses may have reduced CD47 expression and become available to microglia-mediated elimination by the complement cascade. Although several genes have recently been implicated in sensory neuron dysfunction in SMA (Ackermann et al., 2013; Shorrock et al., 2018), a mechanistic link that connects SMN loss with the increased C1q expression we report here remains unknown. It is possible that ubiquitous SMN deficiency, which leads to motor neuron dysfunction causes motor neurons to release yet unknown factor(s) that either directly or through other glial cell(s) signal to microglia and trigger the overexpression of C1q.
Requirement of C1q for the Normal Development of Sensory-Motor Circuits
The monosynaptic spinal sensory-motor arc reflex consists of proprioceptive sensory neurons and motor neurons innervating the homonymous muscle. During embryonic development, ~30% of motor neurons are innervated by proprioceptive afferents from antagonistic muscles (Seebach and Ziskind-Conhaim, 1994), which persists during the first postnatal week (Poliak et al., 2016). However, proprioceptive afferents connecting motor neurons supplying antagonistic muscles are removed during postnatal development (Mears and Frank, 1997; Mendelsohn et al., 2015). Here, we show that the molecular mechanisms responsible for the elimination of these inappropriate synapses that had long remained elusive require C1q and activation of the classical complement pathway.
We demonstrate that the genetic removal of C1q in WT mice results in a ~25% increase in proprioceptive synapses on both the somata and the proximal dendrites of motor neurons. Furthermore, our tracing experiments demonstrate that C1q is involved in the refinement of proprioceptive synapses on motor neurons originating from antagonistic muscles, revealing a C1q-mediated mechanism responsible for synaptic refinement in the developing spinal cord. During this period, proprioceptive synapses have been shown to play a major role in righting behavior (Fletcher et al., 2017), whereas descending pathways such as vestibulospinal inputs play a more prominent role than proprioceptive in this behavior after the first postnatal week (Bignall, 1974; Pellis and Pellis, 1994; ten Donkelaar, 2000). The supernumerary synapses in C1q−/− mice are functional, as demonstrated by the increase in amplitude of the homonymous dorsal-to-ventral root reflex. However, the lack of pruning of inappropriate synapses could explain the deficits in motor behavior, illustrated by the impairment of righting reflex during the first postnatal week.
How does C1q eliminate inappropriate proprioceptive synapses? The assembly of selective sensory-motor connections has been shown to depend on the position at which motor neurons settle in the ventral spinal cord, as well as their surface recognition features (Fukuhara et al., 2013; Pecho-Vrieseling et al., 2009; Sürmeli et al., 2011). Microglia are known to exhibit an ability to interact with synapses in response to neurotransmitter release or sensory experience (Nimmerjahn et al., 2005; Ransohoff and Perry, 2009; Wake et al., 2009). In addition, during early development, microglia engulf synapses in the LGN, which is dependent upon neural activity, complement tagging, and the microglia-specific CR3 (Schafer et al., 2012). It is possible that inappropriate proprioceptive synapses are less active, which may be sensed by microglia, leading to C1q deposition, activation of the classical pathway, and microglia-mediated synaptic elimination via CR3. In support of this, we observed C3 localized on VGluT1 synapses in early postnatal WT mice as well as VGluT1 synaptic remnants within microglia. These data suggest that at least a portion of C1q-tagged inappropriate proprioceptive synapses are eliminated via the classical complement pathway and microglia, revealing a previously unsuspected role in the refinement of spinal sensory-motor circuits during normal development.
In summary, here, we show that sensory-motor circuit refinement occurs through C1q-mediated mechanisms during early development, while aberrant activation of the classical complement pathway contributes to synaptic dysfunction and causes elimination of proprioceptive synapses at the onset of SMA, which propagates the demise of motor function. In the future, it will be interesting to determine whether other synapses in the spinal motor circuit are pruned by similar mechanisms as part of their developmental refinement. Based on our findings, it will also be important to unravel how C1q is upregulated in SMA and to establish the therapeutic potential of its inhibition for treatment of this neurodegenerative disease.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact George Z. Mentis (gzmentis@columbia.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Mice were housed under pathogen-free conditions and all surgical procedures were performed on postnatal mice in accordance with the National Institutes of Health Guidelines on the Care and Use of Animals and approved by the Columbia animal care and use committee (IACUC). Animals of both sexes were used in this study since previous published work in mouse models of SMA as well as patient data do not show any sex-related difference in the onset or severity of SMA.
The original breeding pairs for the SMA mice used in our study (Smn+/−/SMN2+/+/SMNΔ7+/+) were purchased from Jackson Mice (Jax stock #005025; FVB background). C1qa−/− mice (C57BL6 background) were provided by Dr. M. Botto (Imperial College London, UK) as previously reported (Botto et al., 1998) under an MTA agreement. C1qa-deficient mice (C57BL/6) have no C1q protein and have disrupted classical complement activity (Botto et al., 1998). C3−/− mice (C57BL/6 background), which are deficient in C3 protein, were obtained from The Jackson Laboratory (B6 129S4-C3 < tmCrr > /J; stock No. 003641). C1qa−/− mice were bred with SMA mice to generate Smn−/−;SMN2+/+;SMNΔ7+/+;C1qa−/−. PvCre (Jax stock #008069) mice were bred with SMA mice expressing a Smn Cre-inducible allele (SmnRes; JAX stock #007951; (Lutz et al., 2011) to generate Pv-Cre+/−/SMN2+/+/SMNΔ7+/+, expressing mice. Cre- SMA mice were null for the Smn allele, Cre absent and carrying the SmnRes allele (SmnRes/−/SMN2+/+/SMNΔ7+/+). Cre+ SMA animals were carrying one allele of the SmnRes, absent of endogenous mouse SMN and heterozygous for PvCre (PV-Cre+/−/SmnRes/−/SMN2+/+/SMNΔ7+/+). WT mice were those homozygous for the wild-type allele in the absence of Cre (Smn+/+/SMN2+/+/SMNΔ7+/+).
Genotyping
Tail DNA PCR genotyping protocols for SMA-Δ7, C3−/− and PvCre mice were followed as described on the Jackson website (https://www.jax.org). For the Smn, PvCre and C3 alleles universal PCR reaction was used as follows: 12.5 μL of GoTaq Hot Start Green Master Mix (Promega), 0.5 μL of each primer (25 μM; Sigma), and 4 μL of 1:20 diluted lysed tail DNA in a final volume of 25 μl using ddH2O. For the Smn alleles products were amplified using the following thermal cycling method: 95°C for 2 mins, followed by 35 cycles of 95°C for 1 min, 55°C for 1 min, 72°C for 1 min and followed by 72°C for 5 mins. For the PvCre alleles products were amplified using the following thermal cycling method: 94°C for 3 mins, followed by 34 cycles of 94°C for 30 s, 58°C for 30 s, 72°C for 1 min and followed by 72°C for 5 mins. For the C3 alleles products were amplified using the following thermal cycling method: 94°C for 3 mins, followed by 12 cycles of 94°C for 20 s, 64°C for 30 s (−0.5°C per cycle), 72°C for 35 s, followed by 25 cycles of 94°C for 20 s, 58°C for 30 s and 72°C for 35 s, and followed by 72°C for 2 mins. For the C1q genotyping, tail DNA extraction was performed using KAPA Biosystems Express Extract Kit (cat #KK7103). PCR reaction was used as follows: 25 μL 2× Master Mix (2 mM; Promega #: M750B), MgCl2 (25 mM; Promega #: A351F), 2 μL of Neo3′ primer, 1 μL of mC1qIN/1− and mC1qA/5′+ primer, 0.5 μL of lysed tail DNA and 16.5 μL of ddH2O. For the C1q alleles products were amplified using the following thermal cycling method: 95°C for 4 mins, 94°C for 1 min, followed by 35 cycles of 94°C for 1 min, 57°C for 30 s and 72°C for 30 s, followed by 70°C for 10 mins. Customized primers used are listed in the Key Resources Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat polyclonal anti-ChAT | Millipore | Cat# AB144P; RRID:AB_2079751 |
| Rabbit monoclonal anti-C1q | Abcam | Cat# ab182451; RRID:AB_2732849 |
| Rabbit polyclonal anti-human C1q | Agilent | Cat# A013602 RRID:AB_578496 |
| Rabbit polyclonal anti-C3d | Dako | Cat# A006302; RRID:AB_578478 |
| Rat monoclonal anti-CD45 | Millipore | Cat# 05–1416, RRID:AB_10562966 |
| Rat monoclonal anti-CD68 | AbD Serotec | Cat# MCA1957GA; RRID:AB_324217 |
| Rabbit polyclonal anti-GFAP | Dako | Cat# Z0334; RRID:AB_10013382 |
| Mouse monoclonal anti-GFAP | Sigma-Aldrich | Cat# G3893; RRID:AB_477010 |
| Rabbit polyclonal anti-Iba1 | Wako | Cat# 019–19741; RRID:AB_839504 |
| Goat polyclonal anti-Iba1 | Abcam | Cat# ab5076; RRID:AB_2224402 |
| Rabbit polyclonal anti-Neurofilament | Millipore | Cat# AB1987; RRID:AB_91201 |
| Chicken polyclonal anti-Parvalbumin | This paper | N/A |
| Mouse monoclonal anti-SMN | BD Biosciences | Cat# 610646; RRID:AB_397973 |
| Guinea pig polyclonal anti-Synaptophysin | Synaptic Systems | Cat# 101 004; RRID:AB_1210382 |
| Mouse monoclonal anti-TMEM119 | Bennett lab | N/A |
| Mouse monoclonal Anti-α-Tubulin | Sigma-Aldrich | Cat# T9026; RRID:AB_477593 |
| Guinea pig polyclonal anti-VGAT | Synaptic Systems | Cat# 131 004; RRID:AB_887873 |
| Guinea pig polyclonal anti-VGluT1 | This paper | N/A |
| Alexa Fluor 488 donkey anti-goat | Jackson ImmunoResearch | Cat# 705–545–003, RRID:AB_2340428 |
| Alexa Fluor 488 donkey anti-rabbit | Jackson ImmunoResearch | Cat# 711–545–152, RRID:AB_2313584 |
| Alexa Fluor 488 donkey anti-guinea pig | Jackson ImmunoResearch | Cat# 706–545–148; RRID:AB_2340472 |
| Alexa Fluor 488 donkey anti-chicken | Jackson ImmunoResearch | Cat# 703–545–155; RRID:AB_2340375 |
| Alexa Fluor 488 donkey anti—mouse | Jackson ImmunoResearch | Cat# 715–545–150; RRID:AB_2340846 |
| Donkey anti-guinea pig-Cy5 IgG | Jackson ImmunoResearch | Cat# 706–175–148; RRID:AB_2340462 |
| Donkey anti-rabbit-Cy5 IgG | Jackson ImmunoResearch | Cat# 711–175–152; RRID:AB_2340607 |
| Donkey anti-mouse-Cy5 IgG | Jackson ImmunoResearch | Cat# 715–175–151; RRID:AB_2340820 |
| Donkey anti-rabbit-Cy3 IgG | Jackson ImmunoResearch | Cat# 711–165–152; RRID:AB_2307443 |
| Donkey anti-rat-Rhodamine Red-X IgG | Jackson ImmunoResearch | Cat# 712–295–153; RRID:AB_2340676 |
| Donkey anti-goat-Cy3 IgG | Jackson ImmunoResearch | Cat# 705–165–147; RRID:AB_2307351 |
| Donkey anti-mouse-Cy3 IgG | Jackson ImmunoResearch | Cat# 715–165–150; RRID:AB_2340813 |
| Donkey anti-guinea pig-Cy3 IgG | Jackson ImmunoResearch | Cat# 706–165–148; RRID:AB_2340460 |
| Peroxidase AffiniPure Goat Anti-Mouse IgG | Jackson ImmunoResearch | Cat# 115–035–003, RRID:AB_10015289 |
| Alkaline Phosphatase AffiniPure Goat Anti-Mouse IgG, Fcγ fragment specific (C1q binding assay, 1:2000) | Jackson ImmunoResearch | Cat# 115–055–071; RRID:AB_2338535 |
| TSA Plus Cyanine 5 System | PerkinElmer | Cat# #NEL745E001KT |
| Mouse anti-C1q antibody – ANX-M1 (C1q neutralizing antibody, in vivo injections) | Annexon Biosciences | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| α-Bungarotoxin, Alexa Fluor 555 conjugate (NMJ staining, 1:500) | Thermo Fisher Scientific | Cat# B35451; RRID:AB_2617152 |
| Cholera Toxin Subunit B (Recombinant), Alexa Fluor 555 Conjugate (muscle injections, | Invitrogen, Thermo Fisher Scientific | Cat# C34776 |
| Dextran, Alexa Fluor 488; 10,000 MW, Anionic, Fixable (muscle injections, | Invitrogen, Thermo Fisher Scientific | Cat# D22910 |
| Dextran, Texas Red, 10,000 MW, Neutral | Invitrogen, Thermo Fisher Scientific | Cat# D1828 |
| DAPI stain (IHC, 1:10000) | Thermo Fisher Scientific | Cat# D3571; RRID:AB_2307445 |
| Mouse on Mouse (M.O.M.) Blocking Reagent | Vector Laboratories | Cat# MKB-2213 |
| Human C1q | Complement Technology Inc. | Cat# A099 / A100 |
| CDP-star | Applied Biosystems, Thermo Fisher Scientific | Cat# T2214 |
| Power SYBR Green PCR Master Mix | Applied Biosystems, Thermo Fisher Scientific | Cat# 4367659 |
| Amersham ECL Prime Western Blotting Detection Reagent | GE Healthcare Life Sciences | Cat# RPN2232 |
| ProLong Gold Antifade Mountant with DAPI | Invitrogen, Thermo Fisher Scientific | Cat# P36931 |
| PLX5622 (in vivo injections) | Plexxikon | N/A |
| Critical Commercial Assays | ||
| RNAqueous-Micro Total RNA Isolation Kit | Invitrogen, Thermo Fisher Scientific | Cat# AM1931 |
| RevertAid First Strand cDNA Synthesis Kit | Invitrogen, Thermo Fisher Scientific | Cat# K1622 |
| KAPA Express Extract Kit | KAPA Biosystems | Cat# KK7103 |
| RNAscope Multiplex Fluorescent Reagent Kit V2 | Advanced Cell Diagnostics | Cat# 323110 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | Cat# JAX:000664; RRID:IMSR_JAX:000664 |
| Mouse: Smn+/−/SMN2+/+/SMNΔ7+/+: FVB.Cg-Grm7Tg(SMN2)89Ahmb Smn1tm1Msd Tg(SMN2*delta7)4299Ahmb/J | The Jackson Laboratory | Cat# JAX:005025; RRID:IMSR_JAX:005025 |
| Mouse: PVcre: B6;129P2-Pvalbtm1(cre)Arbr/J | The Jackson Laboratory | Cat# JAX:008069; RRID:IMSR_JAX:008069 |
| Mouse: C1qKO | Dr. M. Botto (Imperial College London, UK) | N/A |
| Mouse: C3KO: B6;129S4-C3tm1Crr/J | The Jackson Laboratory | Cat# JAX:003641; RRID:IMSR_JAX:003641 |
| Oligonucleotides | ||
| C1q knock-out allele forward primer: GGGGATCGGCAATAAAAAGAC | N/A | N/A |
| C1q wild-type allele forward primer: GGGGCCTGTGATCCAGACAG | N/A | N/A |
| C1q wild-type allele reverse primer: ACCAATCGCTTCTCAGGACC | N/A | N/A |
| uninverted SmnRes forward primer: ACGCGTACCGTTCGTATAGC | N/A | N/A |
| uninverted SmnRes reverse primer: TGAGCACCTTCCTTCTTTTTG | N/A | N/A |
| inverted SmnRes forward primer: CAACCAGTTAAGTATGAGAATTC | N/A | N/A |
| inverted SmnRes reverse primer: GTTTGCAGAAGCGGTGGG | N/A | N/A |
| PVCre wild-type allele reverse primer: CGAGGGCCATAGAGGATGG | N/A | N/A |
| PVCre allele reverse primer: GCGGAATTCTTAATTAATCAGCG | N/A | N/A |
| PVCre common forward primer: GGATGCTTGCCGAAGATAAGAGTG | N/A | N/A |
| Primers for Real time RT-qPCR can be found in Table S1. | N/A | N/A |
| C1qa probes channel 3 | Advanced Cell Diagnostics | Cat# 441221-C3 |
| Software and Algorithms | ||
| LAS X (Leica Application Suite X) | Leica Microsystems | https://www.leica-microsystems.com/products/microscope-software/details/product/leica-las-x-ls |
| ImageJ | NIH | https://imagej.net/Welcome |
| Clampex V10 | Molecular Devices | https://www.moleculardevices.com/ |
| Clampfit V10 | Molecular Devices | https://www.moleculardevices.com/ |
| GraphPad Prism 6.04 | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
METHOD DETAILS
Behavioral Analysis
Mice from all experimental groups were monitored daily, weighed, and three righting reflex tests were timed and averaged as described in Fletcher et al. (2017). Mice with a body weight reduction of 25% from the previous day and an inability to right were euthanized with carbon dioxide to comply with IACUC guidelines. Righting time was defined as the time for the pup to turn to an upright position after being placed on its back. The cut-off test time for the righting reflex was 60 s to comply with IACUC guidelines. For systemic administration of C1q neutralizing antibody, anti-C1q (ANX-M1, mouse anti-C1q antibody) or isotype control (mouse IgG1), from Annexon Biosciences were used under an MTA agreement. WT and SMA mice were injected intraperitoneally (IP) every second day starting at birth (P0) until death with a dose of 100 mg/kg and monitored daily for body weight and righting times. The investigator was blinded to the treatment either by anti-C1q or the isotype control antibodies. For systemic administration of PLX5622 IP solution, mice were injected intraperitoneally twice daily starting at birth (P0) until P10/11 (tissue collection) with a dose of 50 mg/kg and monitored daily for body weight and righting times. This treatment resulted in maximal microglia depletion but at low survival rate in both WT and SMA mice.
Physiology Using the Ex Vivo Spinal Cord Preparation
Experimental protocols used in this study have been described in previous publications (Fletcher et al., 2017; Mentis et al., 2011). Animals were decapitated, the spinal cord was dissected and isolated in cold (~12°C) oxygenated (95%O2/5%CO2) artificial cerebrospinal fluid (aCSF) containing in mM: 128.35 NaCl, 4 KCl, 0.58 NaH2PO4.H20, 21 NaHCO3, 30 D-Glucose, 1.5 CaCl2.H20, and 1 MgSO4.7H20. The spinal cord was then transferred to a customized recording chamber. The chamber was perfused continuously with oxygenated aCSF (~10 ml/min) at room temperature (~21–24°C). Dorsal and ventral roots were placed into suction electrodes for stimulation and recording, respectively.
Ventral root recordings were obtained from the L1 ventral root in response to the stimuli from the homonymous L1 dorsal root. We defined the stimulus threshold as the minimal current necessary to evoke a response in the homonymous ventral root in 3 out of 5 trials. Dorsal roots were stimulated at supramaximal intensity (0.2ms, 2–5× threshold) delivered by a constant current stimulus isolator (A365 - WPI, Sarasota, FL). The recordings were amplified 1000× (Cyberamp, Molecular Devices), fed to an A/D interface (Digidata 1320A, Molecular Devices) and acquired at 50kHZ using Clampex (V10, Molecular Devices). Data were then analyzed offline using Clampfit (Molecular Devices). Amplitude measurements were performed by averaging the responses of 10 single stimulations at 0.1 Hz. In some experiments, the tissues were collected for immunohistochemistry. Following the recording session, the spinal cord was immersion-fixed with 4% PFA overnight and subsequently transferred to PBS and processed for immunohistochemistry staining.
Functional Assessment of the NMJ
To functionally assess neuromuscular junctions of the Quadratus Lumborum (QL) muscle at P11, we developed a technique by which motor neurons axons in the ventral root L2 (innervating the QL muscle) were stimulated by drawing the ventral root into a suction electrode - having removed the spinal cord. The L2 ventral root was stimulated with a single stimulus (0.2 ms, supramaximal intensity of stimulation) at different frequencies, ranging between 0.1 Hz to 50 Hz to emulate the physiological range of neonatal motor neurons’ firing (Fletcher et al., 2017). The compound muscle action potentials (CMAPs) of the QL were recorded using a concentric bipolar electrode inserted in the belly of the muscle. The maximum CMAP amplitude (baseline-to-peak) was averaged from 10 single stimulations at 0.1 Hz. In some experiments, the tissues were collected for immunohistochemistry. Following the recording session, QL muscles were fixed in 4% PFA for 20 minutes and subsequently transferred to PBS and processed for neuromuscular junction (NMJ) staining.
Tracing and Immunohistochemistry
Experimental protocols used in this study have been described before (Fletcher et al., 2017; Mentis et al., 2011). Mice at birth (P0), P4, or P11 were used in tracing and immunohistochemistry experiments [WT, SMA, SMA+Ctr Ab, SMA+a-C1q Ab, SMA+PLX5622, WT+PLX5622, C1q−/−, SMA+C1q−/−].
Somatodendritic Labeling of Motor Neurons
The dissected ex vivo spinal cord was transferred to a dissection chamber and hemisected to improve oxygenation (for P11 spinal cords). The chamber was perfused with cold (~10°C), oxygenated (95% O2 / 5% CO2) aCSF (containing in mM: 128.35 NaCl, 4 KCl, 0.58 NaH2PO4.H2O, 21 NaHCO3, 30 D-Glucose, 0.1 CaCl2.H2O, and 2 MgSO4.7H2O). The L1 ventral root was placed in a suction electrode containing a fluorescent dextran dye to retrogradely fill motor neurons. After 14 – 20 hours the spinal cord was immersion-fixed in 4% paraformaldehyde (4 hours to overnight) and washed in 0.01M phosphate buffer saline (PBS). The spinal cord was thereafter processed for immunohistochemistry as described below.
Retrograde Labeling of Muscle-Identified Motor Neurons
WT and C1q−/− motor neurons supplying the Gastrocnemius (GA) and Tibialis Anterior (TA) muscles were retrogradely labeled in vivo by intramuscular injection of CTb conjugated to Alexa 555 or Fluorescinated Dextran. Newborn (P0) mice were anesthetized by isoflurane inhalation. A small incision in the right or left TA and GA area was made to access the muscles. TA muscles were injected with ~1 μL of 1% CTb-Alexa 555 in PBS, and GA muscles were injected with dextran-488. Injections were done using a finely pulled glass micropipette. CTb and Dextran were delivered by pressure to an adapted microsyringe. Incisions were closed with sutures. Spinal cords were collected at P7 and processed for immunohistochemistry.
Tissue Collection
Mice were anesthetized with 1.2% avertin (intraperitoneal injection, 300mg/kg) and transcardially perfused initially with the ice-cold PBS to remove the blood until tissue was completely cleared of blood. If tissue fixation was required, animals were additionally perfused with 4% PFA. After transcardial perfusion spinal cord tissue was carefully dissected, post-fixed by immersion in 4% paraformaldehyde (4 hours to overnight) and washed in PBS.
Immunohistochemistry
Spinal cords were embedded in warm 5% Agar and serial transverse sections from L1 spinal segment were cut on a Vibratome (75 μm thickness). For experiments involving anti-mouse antibodies, sections were pre-incubated for 1 hour in M.O.M blocker (Vector Laboratories) in PBS-T to block endogenous antigens. Sections were blocked with 10% normal donkey serum in 0.01M PBS with 0.1% Triton X-100 (PBS-T; pH 7.4) and incubated overnight (24 hours for the C1q antibody) at room temperature in different combinations of primary antibodies in PBS-T. The following day, sections were washed in PBS-T and secondary antibody incubations were performed for 3 hours with the appropriate species-specific antiserum diluted in PBS-T. Sections were subsequently washed in PBS, mounted on glass slides using 30% glycerol in PBS and coverslipped.
All antibodies, except Parvalbumin (custom made), VGluT1 (custom made), and TMEM119 (Barres lab), were obtained from commercial sources. Parvalbumin anti-chicken antibody was produced by Covance, designed against a full-length bacterial fusion protein. VGluT1 anti-guinea pig antibody was produced by Covance, designed against the epitope (C)GATHSTVQPPRPPPP which lies within the n-terminus of mouse VGluT1. The VGluT1 antibody was validated in VGluT1 knock out tissue (Fletcher et al., 2017). The following primary antibodies were used at the indicated dilutions: goat polyclonal anti-ChAT (1:100; Millipore, AB144P), rabbit monoclonal anti-C1q (1:1000; Abcam, ab182451), rabbit polyclonal anti-human C1q (1:500; Agilent, A013602), rabbit polyclonal anti-C3d (1:500; Dako, A006302), rat monoclonal anti-CD45 (1:1000; Millipore, 05–1416), rat monoclonal anti-CD68 (1:200; AbD Serotec, MCA1957GA), rabbit polyclonal anti-GFAP (1:500; Dako, Z0334), mouse monoclonal anti-GFAP (1:500; Sigma-Aldrich, G3893), rabbit polyclonal anti-Iba1 (1:500; Wako 019–19741), goat polyclonal anti-Iba1 (1:500; Abcam, ab5076), guinea pig polyclonal anti-Synaptophysin (1:500; Synaptic Systems, 101 004), mouse monoclonal anti-TMEM119 (1:3; Barres lab), guinea pig polyclonal anti-VGAT (1:200; Synaptic Systems, 131 004), guinea pig polyclonal anti-VGluT1 (1:2000; custom made). The following secondary antibodies were used at the 1:250 dilution: Alexa Fluor 488 donkey anti-goat (Jackson ImmunoResearch, 705–545-003), Alexa Fluor 488 donkey anti-rabbit (Jackson ImmunoResearch, 711–545-152), Alexa Fluor 488 donkey anti-guinea pig (Jackson ImmunoResearch, 706–545-148), Alexa Fluor 488 donkey anti—mouse (Jackson ImmunoResearh, 715–545-150), Donkey anti-guinea pig-Cy5 IgG (Jackson ImmunoResearch, 706–175-148), Donkey anti-rabbit-Cy5 IgG (Jackson ImmunoResearch, 711–175-152), Donkey anti-mouse-Cy5 IgG (Jackson ImmunoResearch, 715–175-151), Donkey anti-rabbit-Cy3 IgG (Jackson ImmunoResearch, 711–165-152), Donkey anti-rat-Rhodamine Red-X IgG (Jackson ImmunoResearch, 712–295-153), Donkey anti-goat-Cy3 IgG (Jackson ImmunoResearch, 705–165-147), Donkey anti-mouse-Cy3 IgG (Jackson ImmunoResearch, 715–165-150), Donkey anti-guinea pig-Cy3 IgG (Jackson ImmunoResearch, 706–165-148).
Human Post-Mortem Tissue Staining
Fresh frozen SMA type I (7 months old male) and control patient (3 years old male) spinal cord tissue was a kind gift from Dr. Wandy Chang and the Brain Center at Columbia University respectively. Spinal cords were cut on a cryostat (20 μm thickness). Sections were fixed for 2 minutes in 4% PFA and washed with PBS. Sections were then treated as described above and incubated overnight with antibodies against ChAT - goat polyclonal (1:100; Millipore, AB144P) and rabbit polyclonal anti-human C1q (1:500; Dako, A013602–1). Sections were washed and further incubated with the secondary antibodies: Alexa Fluor 488 donkey anti-goat (Jackson ImmunoResearch, 705–545-003) and Donkey anti-rabbit-Cy3 (Jackson ImmunoResearch, 711–165-152). Sections were washed and mounted in 30% glycerol in PBS.
SMN Staining of Dorsal Root Ganglions (DRGs)
P11 mice were transcardially perfused with 2% PFA in a sodium acetate buffer, pH 6. Dorsal root ganglia were dissected from L1-L3 segments and immersion fixed further for 2 hours. The tissue was cut immediately using a vibratome into 100 μm sections and blocked in PBS-T containing M.O.M. blocker (Vector Laboratories) for an hour to prevent the secondary antibody from binding to non-specific mouse antigens. Sections were then treated as described for the previous immunohistochemistry protocol and incubated with antibodies against Parvalbumin - chicken polyclonal anti-Parvalbumin (1:2000; custom made) and SMN - mouse monoclonal anti-SMN (1:50; BD Biosciences, 610646). Sections were washed and further incubated with the secondary antibodies: Alexa Fluor 488 donkey anti-chicken (Jackson ImmunoResearch, 703–545-155), donkey anti-mouse-Cy5 IgG (Jackson ImmunoResearch, 715–175-151) and counter-stained with DAPI. Sections were washed and mounted in 30% glycerol in PBS.
NMJ Staining
Neuromuscular junctions (NMJs) were analyzed in the QL muscle. Muscles from P11 mice from each genotype were immersion fixed in 4% PFA for 20 mins and washed in PBS. Single fibers were teased using fine forceps and washed for 30 mins in PBS supplemented with 0.1M glycine. To stain the postsynaptic part of neuromuscular endplates fibers were incubated with alpha-bungarotoxin-555 antibody for 20 mins and washed in PBS before permeabilization with ice-cold methanol at −20°C for 2 mins. Fibers were then washed in PBS and incubated in a blocking solution containing 10% Donkey Serum in 0.3% PBS-T for an hour before exposure to antibodies against neurofilament - rabbit polyclonal anti-Neurofilament (1:500; Millipore) and synaptophysin - guinea pig polyclonal anti-synaptophysin (1:500; Synaptic Systems, 101 004) at 4°C overnight. Samples were washed in PBS prior to one-hour long incubation with the following secondary antibodies: Alexa Fluor 488 donkey anti-rabbit (Jackson ImmunoResearch, 711–545-152) and Donkey anti-guinea pig-Cy5 IgG (Jackson ImmunoResearch, 706–175-148). Fibers were washed and mounted in 30% glycerol in PBS.
QUANTIFICATION AND STATISTICAL ANALYSIS
Imaging and Analysis
Sections were imaged using the SP8 Leica confocal microscope and analyzed using LASX (Leica Microsystems) and ImageJ (NIH) software. For all immunohistochemical analysis, at least three animals were used for each genotype. For motor neuron counts, we analyzed z stacks images (acquired at 3 μm intervals) collected for each section that contained a fluorescent signal from L1 retrogradely labeled motor neurons and/or ChAT immunostained, as previously described (Fletcher et al., 2017; Mentis et al., 2011). Sections were scanned using a ×20 objective. Only motor neurons (ChAT+) that contained the nucleus were counted in order to avoid double counting of neurons from adjoining spinal cord sections.
Quantitative analysis of VGluT1 immunoreactive synaptic densities on L1 motor neurons at P11 were performed on z stacks of optical sections scanned using an ×40 objective throughout the whole section thickness (acquired at 0.35 μm z-steps) to include the entire motor neuron cell body and proximal dendrites of retrogradely labeled or/and ChAT+ motor neurons. To obtain density estimates, we counted all VGluT1+ contacts on dendritic segments at 50 μm sequential distances (0–50 and 50–100) from the motor neuron cell body and divided this number by the total linear length of all dendritic segments in each compartment as described previously (Fletcher et al., 2017; Mentis et al., 2011). For VGluT1 motor neuron soma counts, only motor neurons with a whole cell body present within the z stack were included. To exclude gamma motor neurons from the analysis, only the motor neurons with a soma perimeter larger than 400 μm2 were considered.
SMN Quantification
Sections were scanned using ×40 oil objective throughout the whole section at 0.5 μm z-steps. The number of gem positive and negative proprioceptive neurons was counted for each genotype. Three animals were used for each genotype and a minimum of 30 neurons were counted for each animal.
NMJ Quantification
To determine the extent of NMJ innervation, NMJ synapses were acquired using an ×20 objective and z stack images were scanned at 2 μm z-steps. Images were analyzed offline using LASAF software. Innervated and denervated synapses were counted. NMJs were only considered innervated if the presynaptic nerve terminal completely co-localized with the postsynaptic endplate.
Fluorescent In Situ Hybridization Combined with Immunohistochemistry
Transcardially perfused spinal cords were collected, post-fixed in 4% PFA for 24 hours at 4°C, and cryopreserved overnight in sequential 10%, 20% and 30% sucrose solutions. Spinal cord L1-L3 segments were then sectioned at 20 μm thickness using a cryostat (Leica) and mounted on superfrost slides. Fluorescent in situ hybridization for C1qa was performed using RNAscope probe from Advanced Cell Diagnostics (ACD, #441221-C3) and the RNAscope Multiplex Fluorescent Reagent Kit V2 (ACD, #323110) following the manufacturer’s recommendations. TSA Plus Cyanine 5 System (PerkinElmer, #NEL745E001KT) was used for developing HRP-C1 signal. The sample slides were briefly washed with PBS after the last wash in the RNAscope protocol, subsequently blocked in 10% NDS, and stained overnight at room temperature in a wet chamber with antibodies against ChAT (1:100; Millipore, AB144P) and against Iba1 (1:500; Wako 019–19741). The following day, sections were washed in PBS-T and secondary antibody incubation was performed for 2 hours. Sections were subsequently washed in PBS, mounted on glass slides using ProLong Gold Antifade Mountant (#P36931) and coverslipped.
Quantification of Tagged Synapses
Analysis of C1q and C3 deposition on synapses was performed from single optical plane images acquired with an ×63 or ×40 oil objective at 4096×4096 dpi resolution using the SP8 Leica confocal microscope and LASX software (Leica Microsystems) for deconvolution and subsequent offline image analyses. Only synapses contacting motor neurons on soma and on dendrites were included in analysis. Synapse was determined as “tagged” if C1q or C3 signal colocalized with the presynaptic signal, which was confirmed using the intensity line profiling tool, where synaptic markers and C1q (or C3) intensity line profiles overlapped. We counted all synaptic contacts on soma and on dendrites against the tagged synapses on soma and on dendrites respectively, to obtain the percentage of tagged synapses.
C1q Binding Assay
Black Costar 3925 plate (Corning) was coated with human C1q (Complement Technology Inc) at 2 μg/ml in bicarbonate buffer (100 μl/well) and incubated overnight at 4°C. Plate was washed 3× with dPBS (200 μl/well). Blocking was done with 200 μl/well of 3% BSA in PBS for 1 hour at room temperature. Neutralizing anti-C1q antibody or control isotype antibody (Annexon Biosciences) were added to plate in triplicate serial dilutions 200–400 ng/ml (8–12 point titration) in dPBS with 0.3% BSA, 0.1% Tween buffer (50 μL/well) and incubated 1 hour at room temperature. Goat anti-mouse-Fc-AP (Jackson ImmunoResearch) was added at 1:2000 dilution (50 μl/well) in dPBS with 0.3% BSA, 0.1% Tween. Plate was washed 3 times, 10 minutes each wash, with 200 μ of dPBS 0.05%Tween. AP substrate (CDP-Star) was incubated at room temperature 20 min and plate was read using Infinite 200 PRO plate reader (Tecan Life Sciences).
RNA Analysis
For RT-qPCR experiments spinal cords containing the L1-L2 segments, were collected from WT and SMA mice at P4 and P11. The RNAqueous-Micro Kit was used for phenol-free total RNA isolation. RNA was reverse transcribed using RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific) and triplicate reactions were performed using Power SYBR Green PCR master mix (Applied Biosystem) in a Realplex4 Mastercycler (Eppendorf).
Western Blot Analysis
For western blot analysis, proteins from spinal cord tissue (L1–L3 segments) of WT, SMA and SMA+a-C1q mice (n = 2) at P11, were homogenized in lysis buffer (150 mM NaCl, 1% Triton, 2 mM EDTA, 50 mM Tris, pH 7.4) and quantified using Bio-Rad protein assay. Protein extracts (30 μg) were run on 12% SDS–PAGE gels and transferred to PVDF membranes for probing. Blocking was done at room temperature, for 1h, in blocking buffer (5% milk in TBS/0.2% Tween). Membranes were probed with primary antibodies, either mouse monoclonal anti-SMN (1:10000; BD Biosciences, 610646) or mouse monoclonal Anti-α-Tubulin (1:10000; Sigma-Aldrich, T9026) diluted in blocking buffer, and incubated overnight at 4°C. Subsequently, membranes were washed 3 times with TBS/0.2% Tween and incubated for 1 h at room temperature with Peroxidase AffiniPure Goat Anti-Mouse IgG (H+L) (WB, 1:10000; Jackson ImmunoResearch, 115–035-003) secondary antibody diluted in TBS/0.2% Tween. After three sequential 10-min washes, enhanced chemiluminescence (GE Healthcare, Lifesciences) was used for visualization.
Statistics
Statistical analysis was performed using PRISM (GraphPad Software, Prism Version 6.04.). Comparisons were performed by either Student’s t test or One-Way ANOVA when conditions of normality and homoscedasticity were respected. If violated, Mann–Whitney-Wilcoxon and Kruskal-Wallis tests were used as non-parametric tests. Post hoc multiple comparison methods are indicated in the Results and figure legends when necessary. A statistical significance was considered p < 0.05. Results are expressed as means ± standard error of the mean (SEM).
DATA AND CODE AVAILABILITY
This study did not generate any unique datasets or code.
Supplementary Material
Highlights.
C1q refines spinal sensory-motor circuits during normal development
Upregulated C1q tags SMA synapses for elimination via classical cascade and microglia
Pharmacological inhibition of C1q or depletion of microglia confer benefit in SMA mice
C1q deletion results in an excessive number of synapses, leading to behavioral deficits
ACKNOWLEDGMENTS
We thank Dr. Marina Botto (Imperial College London) for providing the C1qa−/− mice under a Materials Transfer Agreement, Plexxikon for providing PLX5622 under an MTA agreement, and Dr. Wandy Chang (Columbia University) for providing human SMA post-mortem tissue. We also thank Drs. Livio Pellizzoni and Francisco Alvarez for comments on the manuscript. G.Z.M. is supported by the National Institute of Neurological Disorders and Stroke (NINDS), NIH (R01-NS078375), the NIH Blueprint for Neuroscience Research, National Institute of Alcohol Abuse and Alcoholism (NIAAA) and NINDS (R01-AA027079), the Department of Defense (GR.10235006), the SMA Foundation, and Project ALS. We would like to dedicate this study to the memory of our dear friend and colleague, Ben Barres.
Footnotes
DECLARATION OF INTERESTS
T.A.Y. is the chief scientific officer of and S.S. is a senior director at Annexon Biosciences. All of the other co-authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.11.013.
REFERENCES
- Ackermann B, Kröber S, Torres-Benito L, Borgmann A, Peters M, Hosseini Barkooie SM, Tejero R, Jakubik M, Schreml J, Milbradt J, et al. (2013). Plastin 3 ameliorates spinal muscular atrophy via delayed axon pruning and improves neuromuscular junction functionality. Hum. Mol. Genet. 22, 1328–1347. [DOI] [PubMed] [Google Scholar]
- Arber S (2012). Motor circuits in action: specification, connectivity, and function. Neuron 74, 975–989. [DOI] [PubMed] [Google Scholar]
- Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, Mulinyawe SB, Bohlen CJ, Adil A, Tucker A, et al. (2016). New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 113, E1738–E1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bignall KE (1974). Ontogeny of levels of neural organization: the righting reflex as a model. Exp. Neurol. 42, 566–573. [DOI] [PubMed] [Google Scholar]
- Botto M, Dell’Agnola C, Bygrave AE, Thompson EM, Cook HT, Petry F, Loos M, Pandolfi PP, and Walport MJ (1998). Homozygous C1q deficiency causes glomerulonephritis associated with multiple apoptotic bodies. Nat. Genet 19, 56–59. [DOI] [PubMed] [Google Scholar]
- Burke RE (1979). The role of synaptic organization in the control of motor unit activity during movement. Prog. Brain Res 50, 61–67. [DOI] [PubMed] [Google Scholar]
- Chen HH, Hippenmeyer S, Arber S, and Frank E (2003). Development of the monosynaptic stretch reflex circuit. Curr. Opin. Neurobiol 13, 96–102. [DOI] [PubMed] [Google Scholar]
- Dejanovic B, Huntley MA, De Mazière A, Meilandt WJ, Wu T, Srinivasan K, Jiang Z, Gandham V, Friedman BA, Ngu H, et al. (2018). Changes in the Synaptic Proteome in Tauopathy and Rescue of Tau-Induced Synapse Loss by C1q Antibodies. Neuron 100, 1322–1336.e7. [DOI] [PubMed] [Google Scholar]
- Eccles JC, Eccles RM, and Lundberg A (1957). The convergence of monosynaptic excitatory afferents on to many different species of alpha motoneurones. J. Physiol 137, 22–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmore MRP, Hohsfield LA, Kramár EA, Soreq L, Lee RJ, Pham ST, Najafi AR, Spangenberg EE, Wood MA, West BL, and Green KN (2018). Replacement of microglia in the aged brain reverses cognitive, synaptic, and neuronal deficits in mice. Aging Cell 17, e12832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira-Pinto MJ, Ruder L, Capelli P, and Arber S (2018). Connecting Circuits for Supraspinal Control of Locomotion. Neuron 100, 361–374. [DOI] [PubMed] [Google Scholar]
- Fletcher EV, Simon CM, Pagiazitis JG, Chalif JI, Vukojicic A, Drobac E, Wang X, and Mentis GZ (2017). Reduced sensory synaptic excitation impairs motor neuron function via Kv2.1 in spinal muscular atrophy. Nat. Neurosci 20, 905–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonseca MI, Chu SH, Hernandez MX, Fang MJ, Modarresi L, Selvan P, MacGregor GR, and Tenner AJ (2017). Cell-specific deletion of C1qa identifies microglia as the dominant source of C1q in mouse brain. J. Neuroinflammation 14, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuhara K, Imai F, Ladle DR, Katayama K, Leslie JR, Arber S, Jessell TM, and Yoshida Y (2013). Specificity of monosynaptic sensory-motor connections imposed by repellent Sema3E-PlexinD1 signaling. Cell Rep. 5, 748–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaboriaud C, Juanhuix J, Gruez A, Lacroix M, Darnault C, Pignol D, Verger D, Fontecilla-Camps JC, and Arlaud GJ (2003). The crystal structure of the globular head of complement protein C1q provides a basis for its versatile recognition properties. J. Biol. Chem 278, 46974–46982. [DOI] [PubMed] [Google Scholar]
- Goulding M, and Pfaff SL (2005). Development of circuits that generate simple rhythmic behaviors in vertebrates. Curr. Opin. Neurobiol 15, 14–20. [DOI] [PubMed] [Google Scholar]
- Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, Merry KM, Shi Q, Rosenthal A, Barres BA, et al. (2016). Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 352, 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell GR, Macalinao DG, Sousa GL, Walden M, Soto I, Kneeland SC, Barbay JM, King BL, Marchant JK, Hibbs M, et al. (2011). Molecular clustering identifies complement and endothelin induction as early events in a mouse model of glaucoma. J. Clin. Invest 121, 1429–1444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imlach WL, Beck ES, Choi BJ, Lotti F, Pellizzoni L, and McCabe BD (2012). SMN is required for sensory-motor circuit function in Drosophila. Cell 151, 427–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiehn O (2016). Decoding the organization of spinal circuits that control locomotion. Nat. Rev. Neurosci 17, 224–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo N, and Yamada T (1987). Morphological and physiological studies of development of the monosynaptic reflex pathway in the rat lumbar spinal cord. J. Physiol 389, 441–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansita JA, Mease KM, Qiu H, Yednock T, Sankaranarayanan S, and Kramer S (2017). Nonclinical Development of ANX005: A Humanized Anti-C1q Antibody for Treatment of Autoimmune and Neurodegenerative Diseases. Int. J. Toxicol 36, 449–462. [DOI] [PubMed] [Google Scholar]
- Lee JD, Kamaruzaman NA, Fung JN, Taylor SM, Turner BJ, Atkin JD, Woodruff TM, and Noakes PG (2013). Dysregulation of the complement cascade in the hSOD1G93A transgenic mouse model of amyotrophic lateral sclerosis. J. Neuroinflammation 10, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, et al. (1995). Identification and characterization of a spinal muscular atrophy-determining gene. Cell 80, 155–165. [DOI] [PubMed] [Google Scholar]
- Lehrman EK, Wilton DK, Litvina EY, Welsh CA, Chang ST, Frouin A, Walker AJ, Heller MD, Umemori H, Chen C, and Stevens B (2018). CD47 Protects Synapses from Excess Microglia-Mediated Pruning during Development. Neuron 100, 120–134.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, and Burke RE (2002). Developmental changes in short-term synaptic depression in the neonatal mouse spinal cord. J. Neurophysiol 88, 3218–3231. [DOI] [PubMed] [Google Scholar]
- Ling KK, Lin MY, Zingg B, Feng Z, and Ko CP (2010). Synaptic defects in the spinal and neuromuscular circuitry in a mouse model of spinal muscular atrophy. PLoS One 5, e15457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, and Dreyfuss G (1996). A novel nuclear structure containing the survival of motor neurons protein. EMBO J. 15, 3555–3565. [PMC free article] [PubMed] [Google Scholar]
- Lui H, Zhang J, Makinson SR, Cahill MK, Kelley KW, Huang HY, Shang Y, Oldham MC, Martens LH, Gao F, et al. (2016). Progranulin Deficiency Promotes Circuit-Specific Synaptic Pruning by Microglia via Complement Activation. Cell 165, 921–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz CM, Kariya S, Patruni S, Osborne MA, Liu D, Henderson CE, Li DK, Pellizzoni L, Rojas J, Valenzuela DM, et al. (2011). Postsymptomatic restoration of SMN rescues the disease phenotype in a mouse model of severe spinal muscular atrophy. J. Clin. Invest 121, 3029–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGonigal R, Cunningham ME, Yao D, Barrie JA, Sankaranarayanan S, Fewou SN, Furukawa K, Yednock TA, and Willison HJ (2016). C1q-targeted inhibition of the classical complement pathway prevents injury in a novel mouse model of acute motor axonal neuropathy. Acta Neuropathol. Commun 4, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mears SC, and Frank E (1997). Formation of specific monosynaptic connections between muscle spindle afferents and motoneurons in the mouse. J. Neurosci 17, 3128–3135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelsohn AI, Simon CM, Abbott LF, Mentis GZ, and Jessell TM (2015). Activity Regulates the Incidence of Heteronymous Sensory-Motor Connections. Neuron 87, 111–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentis GZ, Blivis D, Liu W, Drobac E, Crowder ME, Kong L, Alvarez FJ, Sumner CJ, and O’Donovan MJ (2011). Early functional impairment of sensory-motor connectivity in a mouse model of spinal muscular atrophy. Neuron 69, 453–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi ZP, Jiang P, Weng WL, Lindberg FP, Narayanan V, and Lagenaur CF (2000). Expression of a synapse-associated membrane protein, P84/ SHPS-1, and its ligand, IAP/CD47, in mouse retina. J. Comp. Neurol 416, 335–344. [PubMed] [Google Scholar]
- Montes J, Gordon AM, Pandya S, De Vivo DC, and Kaufmann P (2009). Clinical outcome measures in spinal muscular atrophy. J. Child Neurol 24, 968–978. [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A, Kirchhoff F, and Helmchen F (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318. [DOI] [PubMed] [Google Scholar]
- Palop JJ, and Mucke L (2010). Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat. Neurosci 13, 812–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, et al. (2011). Synaptic pruning by microglia is necessary for normal brain development. Science 333, 1456–1458. [DOI] [PubMed] [Google Scholar]
- Pecho-Vrieseling E, Sigrist M, Yoshida Y, Jessell TM, and Arber S (2009). Specificity of sensory-motor connections encoded by Sema3e-Plxnd1 recognition. Nature 459, 842–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellis SM, and Pellis VC (1994). Development of righting when falling from a bipedal standing posture: evidence for the dissociation of dynamic and static righting reflexes in rats. Physiol. Behav 56, 659–663. [DOI] [PubMed] [Google Scholar]
- Poliak S, Norovich AL, Yamagata M, Sanes JR, and Jessell TM (2016). Muscle-type Identity of Proprioceptors Specified by Spatially Restricted Signals from Limb Mesenchyme. Cell 164, 512–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin X, Krumrei N, Grubissich L, Dobarro M, Aktas H, Perez G, and Halperin JA (2003). Deficiency of the mouse complement regulatory protein mCd59b results in spontaneous hemolytic anemia with platelet activation and progressive male infertility. Immunity 18, 217–227. [DOI] [PubMed] [Google Scholar]
- Ransohoff RM, and Perry VH (2009). Microglial physiology: unique stimuli, specialized responses. Annu. Rev. Immunol 27, 119–145. [DOI] [PubMed] [Google Scholar]
- Riccomagno MM, and Kolodkin AL (2015). Sculpting neural circuits by axon and dendrite pruning. Annu. Rev. Cell Dev. Biol 31, 779–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotterman TM, Nardelli P, Cope TC, and Alvarez FJ (2014). Normal distribution of VGLUT1 synapses on spinal motoneuron dendrites and their reorganization after nerve injury. J. Neurosci 34, 3475–3492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, Ransohoff RM, Greenberg ME, Barres BA, and Stevens B (2012). Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 74, 691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seebach BS, and Ziskind-Conhaim L (1994). Formation of transient inappropriate sensorimotor synapses in developing rat spinal cords. J. Neurosci 14, 4520–4528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorrock HK, van der Hoorn D, Boyd PJ, Llavero Hurtado M, Lamont DJ, Wirth B, Sleigh JN, Schiavo G, Wishart TM, Groen EJN, and Gillingwater TH (2018). UBA1/GARS-dependent pathways drive sensory-motor connectivity defects in spinal muscular atrophy. Brain 141, 2878–2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorrock HK, Gillingwater TH, and Groen EJN (2019). Molecular Mechanisms Underlying Sensory-Motor Circuit Dysfunction in SMA. Front. Mol. Neurosci 12, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon CM, Janas AM, Lotti F, Tapia JC, Pellizzoni L, and Mentis GZ (2016). A Stem Cell Model of the Motor Circuit Uncouples Motor Neuron Death from Hyperexcitability Induced by SMN Deficiency. Cell Rep. 16, 1416–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon CM, Dai Y, Van Alstyne M, Koutsioumpa C, Pagiazitis JG, Chalif JI, Wang X, Rabinowitz JE, Henderson CE, Pellizzoni L, and Mentis GZ (2017). Converging Mechanisms of p53 Activation Drive Motor Neuron Degeneration in Spinal Muscular Atrophy. Cell Rep. 21, 3767–3780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephan AH, Madison DV, Mateos JM, Fraser DA, Lovelett EA, Coutellier L, Kim L, Tsai HH, Huang EJ, Rowitch DH, et al. (2013). A dramatic increase of C1q protein in the CNS during normal aging. J. Neurosci 33, 13460–13474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, et al. (2007). The classical complement cascade mediates CNS synapse elimination. Cell 131, 1164–1178. [DOI] [PubMed] [Google Scholar]
- Sürmeli G, Akay T, Ippolito GC, Tucker PW, and Jessell TM (2011). Patterns of spinal sensory-motor connectivity prescribed by a dorsoventral positional template. Cell 147, 653–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ten Donkelaar HJ (2000). Development and regenerative capacity of descending supraspinal pathways in tetrapods: a comparative approach. Adv. Anat. Embryol. Cell Biol 154, iii–ix, 1–145. [DOI] [PubMed] [Google Scholar]
- Tisdale S, and Pellizzoni L (2015). Disease mechanisms and therapeutic approaches in spinal muscular atrophy. J. Neurosci 35, 8691–8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth AB, Terauchi A, Zhang LY, Johnson-Venkatesh EM, Larsen DJ, Sutton MA, and Umemori H (2013). Synapse maturation by activity-dependent ectodomain shedding of SIRPα. Nat. Neurosci 16, 1417–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Alstyne M, Simon CM, Sardi SP, Shihabuddin LS, Mentis GZ, and Pellizzoni L (2018). Dysregulation of Mdm2 and Mdm4 alternative splicing underlies motor neuron death in spinal muscular atrophy. Genes Dev. 32, 1045–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verret L, Mann EO, Hang GB, Barth AM, Cobos I, Ho K, Devidze N, Masliah E, Kreitzer AC, Mody I, et al. (2012). Inhibitory interneuron deficit links altered network activity and cognitive dysfunction in Alzheimer model. Cell 149, 708–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wake H, Moorhouse AJ, Jinno S, Kohsaka S, and Nabekura J (2009). Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J. Neurosci. 29, 3974–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Dissing-Olesen L, MacVicar BA, and Stevens B (2015). Microglia: Dynamic Mediators of Synapse Development and Plasticity. Trends Immunol. 36, 605–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang LB, Li R, Meri S, Rogers J, and Shen Y (2000). Deficiency of complement defense protein CD59 may contribute to neurodegeneration in Alzheimer’s disease. J. Neurosci 20, 7505–7509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Pinto AM, Wan L, Wang W, Berg MG, Oliva I, Singh LN, Dengler C, Wei Z, and Dreyfuss G (2013). Dysregulation of synaptogenesis genes antecedes motor neuron pathology in spinal muscular atrophy. Proc. Natl. Acad. Sci. USA 110, 19348–19353. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.