

Abstract
OBJECTIVES
The adenoid pad, which is located between the orifice of the Eustachian tube (ET) and posterior nasal cavity, can affect the development of otitis media with effusion (OME) because of its anatomical location. The aim of the present study was to evaluate adenoid microbial colonization through 16S ribosomal RNA (rRNA) pyrosequencing, an advanced molecular technique, and to document the relationship with OME.
MATERIALS and METHODS
Adenoid samples were collected using sterile cotton from 32 children during ventilation tube insertion. Sixteen children with OME who underwent tonsillectomy and adenoidectomy due to obstructive symptoms were assigned to the OME group and sixteen children without OME were assigned to the control group. We performed a 16S rRNA-based culture-independent survey of bacterial communities using the MiSeq platform.
RESULTS
The diversity index, mean operational taxonomic units, and Shannon index were lower in the OME group than those in the control group. A taxonomic analysis showed differences in microbiota distribution between the OME and control groups at the phylum, genus, and species levels. The analysis, which was based on weighted UniFrac distances, revealed differences in microbial composition between the two groups.
CONCLUSION
Bacterial community analysis using 16S rRNA pyrosequencing allows us to understand the relationship between the microbial communities of adenoids and the development of OME better.
Keywords: Otitis media, microbiome, adenoid
INTRODUCTION
Otitis media with effusion (OME) is one of the most common diseases in early infancy and childhood and represents a major disease burden worldwide. OME is defined as the presence of middle ear effusion without acute inflammation. In the United States, approximately 90% of children develop OME before reaching school age. Unlike acute otitis media (AOM), OME has no subjective symptoms, and it is difficult to determine the exact incidence. OME can cause acquired conductive hearing loss and subsequent developmental delays in childhood [1]. Causative host factors of OME are associated with age, sex, Eustachian tube (ET) dysfunction, allergies, abnormalities of the systemic mucosal immune system for the inflammatory response, and breastfeeding. Recently, gastroesophageal reflux and the role of biofilms have been recognized as causes of OME [2]. Among these factors, adenoid vegetation or hypertrophy is associated with ET dysfunction, which causes mechanical obstruction of the ET orifice and problems with the regulation of middle ear pressure and mucus drainage [1]. In addition, microorganisms in the adenoids are potential sources of infection and may spread infection to the adjacent middle ear [3].
Detection of bacteria and virus in the middle ear fluid is essential for the effective treatment of OME. Although there are differences in the bacterial and viral species identified in culture- and PCR-based assays by host factors, Moraxella catarrhalis, Haemophilus influenzae, and Streptococcus pneumoniae (among bacteria) and rhinovirus, respiratory syncytial virus, and human coronavirus (among viruses) are the most commonly identified microbiome in the middle ear fluid [4].
A microbiological association between adenoids and OME has also been demonstrated in culture-based studies. These results suggest that bacteria go through the ET and that adenoidectomy is an effective treatment for recurrent OME. However, culture- and PCR-based studies do not represent the diversity of bacteria because they focus on a limited number of bacteria and viruses [5].
The 16S ribosomal RNA (rRNA) pyrosequencing assay is a diagnostic technique that can identify a broader spectrum of bacteria than culture-based bacterial profiles by analyzing the nucleotide sequences of prokaryotes [6]. The National Institutes of Health Human Microbiome Project has initiated research into the human microbiome, which has been linked to various diseases, including infectious and immune diseases and cancer [7]. Additionally, several studies have reported that the microbiome is associated with infectious and inflammatory diseases as well as non-infectious diseases such as obesity and diabetes [8]. Although OME is controversial in that it is an infectious disease, the occurrence of OME can be predicted by the direct identification of pathogens from the middle ear, as well as by altering the composition and distribution of the microbiome in the adjacent adenoids.
Microbiome studies of the adenoids using 16S rRNA pyrosequencing have recently begun but have shown varying results [9, 10].
In this prospective study, we analyzed the microbiome of the adenoids in Korean patients with OME requiring ventilator tube insertion and in those without OME requiring tonsillectomy and adenoidectomy for snoring using 16S rRNA pyrosequencing. Thus, we investigated whether the adenoid microbiome is associated with the development of OME by comparing the bacterial communities between the two groups.
MATERIALS AND METHODS
Ethical Approval
We received informed consents from the parents of patients. The study was approved by the Institutional Review Board (2016-158-I). All procedures were conducted in accordance with the Declaration of Helsinki on biomedical studies involving human subjects.
Study Design and Inclusion Criteria
The present study was carried out as a prospective study from May 2016 to April 2017. Children who required ventilation tube insertion alone or ventilation tube insertion and adenoidectomy were selected as the experimental group. The inclusion criteria of ventilation tube insertion were as follows: (1) chronic OME persisting for >3 months from the date of onset or diagnosis, (2) an irreversible change in the tympanic membrane (retraction pocket, ossicular abnormalities, and/or adhesive otitis), (3) a speech or developmental delay, and (4) a threshold of air-conduction hearing >40 dB. Criteria (2) through (4) were included because ventilation tube insertion is necessary even if the duration of OME is less than 3 months. If the children who had a history of previous ventilation tube insertion with recurrent OME had radiologic or physical evidence of adenoid vegetation, adenoidectomy was performed with ventilation tube insertion. The control group consisted of children undergoing tonsillectomy and adenoidectomy due to obstructive symptoms such as snoring and mouth breathing.
Sample Collection
All adenoid samples were collected during surgery under general anesthesia at the Dongtan Sacred Heart1 Hospital. To prevent contamination of the adenoid samples, we observed the adenoids with direct vision using a 70° rigid telescope (KARL STORZ, Tuttlingen, Germany) during ventilation tube insertion. Swabs were obtained through an intraoral approach using sterile cotton (3M™, Maplewood, MN, USA). When the sterile cotton touched the adenoid surface, the cotton was rotated 180° and removed. When adenoidectomy was performed with ventilation tube insertion or tonsillectomy, samples were obtained by placing and rotating the sterile cotton in the removed adenoid tissues. After the adenoid samples were collected, the cotton was immediately placed in the broth and transferred to a −80 °C freezer after the operation was complete.
DNA Extraction
DNA was extracted using a DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany). The contents of the swab tube were carefully poured into a 1 mL sterile tube and centrifuged for 3 min. Next, total DNA from all collected samples was extracted using an enzymatic and mechanical protocol. The concentration and purity of DNA were measured using a UV-VIS spectrophotometer (Quawell, San Jose, CA, USA). The extracted DNA was stored at −70 °C until sequencing.
PCR Amplification of the 16S rRNA Gene and Sequencing
Each sequenced sample was prepared according to the Illumina 16S Metagenomic Sequencing Library protocol. The quantity and quality of the DNA were measured by PicoGreen (PerkinElmer, Waltham, MA, USA) and NanoDrop spectrophotometry, respectively. The 16S rRNA genes were amplified using 16S V3–V4 primers for 1–155 samples and ITS3–ITS4 primers for 153–156 samples. The primer sequences were as follows:
-
16S Amplicon PCR Forward Primer
5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG-3′;
-
16S Amplicon PCR Reverse Primer
5′-TCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC-3′;
-
ITS3 Amplicon PCR Forward Primer
5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGGCATCGATGAAGAACGCAGC-3′; and
-
ITS4 Amplicon PCR Forward Primer
5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGTCCTCCGCTTATTGATATGC-3′.
Input genomic DNA (12.5 ng) was amplified with the 16S V3–V4 or ITS3–ITS4 primers, and a subsequent limited-cycle amplification step was performed to add multiplexing indices and Illumina sequencing adapters. The final products were normalized and pooled using a PicoGreen (PerkinElmer) kit, and the size of the library was verified using a LabChip GX HT DNA High Sensitivity kit (PerkinElmer). Samples were sequenced using the MiSeq™ platform (Illumina, San Diego, CA, USA).
Bioinformatic Pipeline and Statistical Analyses
MiSeq™ raw data for 32 samples were clustered among sequences with more than 97% sequence similarity after eliminating sequences considered to be errors to form an operational taxonomic unit (OTU) at the species level. A representative sequence of each OTU was obtained for taxonomic assignment using a reference database (NCBI 16S). OTU abundance and taxonomy information were used to obtain the Shannon index (SI) and Simpson index using QIIME v1.8.0, and diversity information was confirmed by generating a rarefaction curve and the Chao1 value. Based on the weighted UniFrac distance, we confirmed the beta diversity between samples. The relationships and differences in bacterial composition between the two groups were visualized by reconstruction using principal coordinates analysis (PCoA) and the unweighted pair group method with an arithmetic mean (UPGMA) tree.
Statistical Analysis
Statistical tests were carried out using R version 3.1.2. The Wilcoxon rank-sum test was performed at the phylum and species levels to confirm differences in the microbiomes between the two groups. p<0.05 were considered statistically significant.
RESULTS
Patient Demographics
Thirty-two patients were recruited for this study. Their ages ranged from 19 months to 15 years (mean=5.8 years, standard deviation=3.2 years). The OME and control groups were distributed equally (16 patients each). The mean duration of disease in the OME group was 12.5 weeks. Ten children in the OME group had bilateral OME and six had unilateral OME. No patient received an antibiotic within 4 weeks prior to surgery. All patients received full pneumococcal vaccination before starting the study. In the control group, patients with tonsillitis more than four times per year and symptoms of an upper respiratory infection within 4 weeks before surgery were excluded.
Bacterial Diversity and Composition in the Adenoids
PCR and 16S rRNA sequencing were performed on adenoid samples of 32 patients. A total of 1,367,587 high-quality reads were obtained after excluding low-quality reads (657,313), chimeric reads (167,142), ambiguous reads (81,498), and other errors (2,583,793). The average read count was 42,737, and the gamma diversity (alpha+beta diversity) was 1,203. The OTU value per sample was at least 18; the maximum was 339.
The diversity index of the OME group was lower than that of the control group. The mean OTU value was 75 in the OME group and 141 in the control group. The mean SI was 2.853 in the OME group and 4.316 in the control group, indicating that the OME group had lower diversity than the control group. The inverse Simpson index was 0.694 in the OME group and 0.864 in the control group (Figure 1).
Figure 1. a–c.
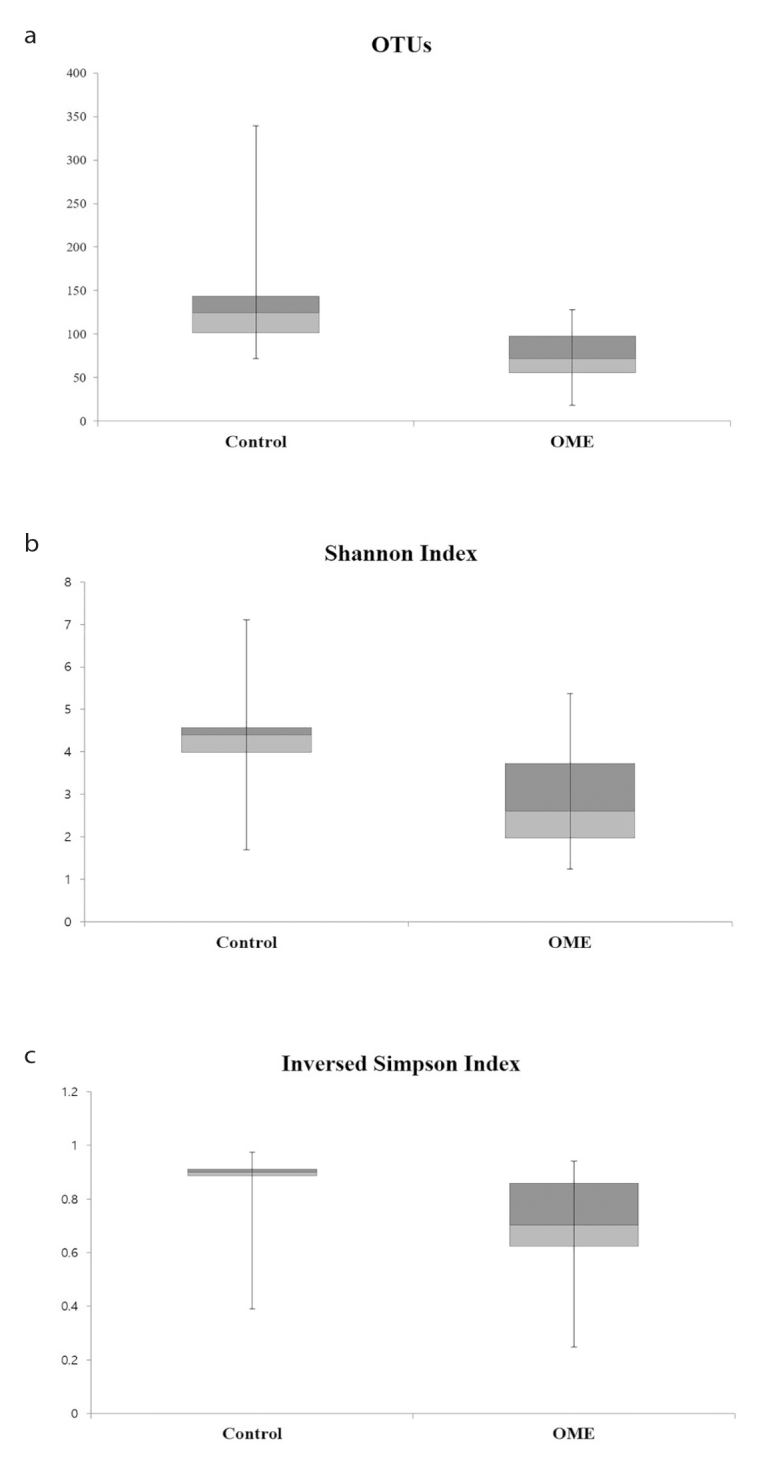
Box plot comparison of the OTUs (a), SI (b), and inverse Simpson index (c). The diversity of the bacterial microbiome and SI was lower in the OME group than that in the control group. The inverse Simpson index was lower in the OME group; therefore, the OME group had fewer species, and specific microbiomes tended to dominate in the OME group. Boxes represent third quartiles and red boxes indicate second quartiles. Whiskers represent the minimum and maximum scores of each index.
OTU: operational taxonomic unit; SI: Shannon index; OME: otitis media with effusion.
Taxonomic analysis revealed that Proteobacteria was the most common phylum in both groups. The OME group showed a 10.05% higher rate than the control group, although the difference was not significant (p=0.239). Proteobacteria (45.06%), Cyanobacteria (13.81%), Bacteroidetes (12.85%), and Firmicutes (7.23%) were frequently detected in the control group, whereas Proteobacteria (55.11%), Firmicutes (15.20%), Bacteroidetes (15.06%), and Actinobacteria (5.75%) were frequently detected in the OME group (Figure 2).
Figure 2.
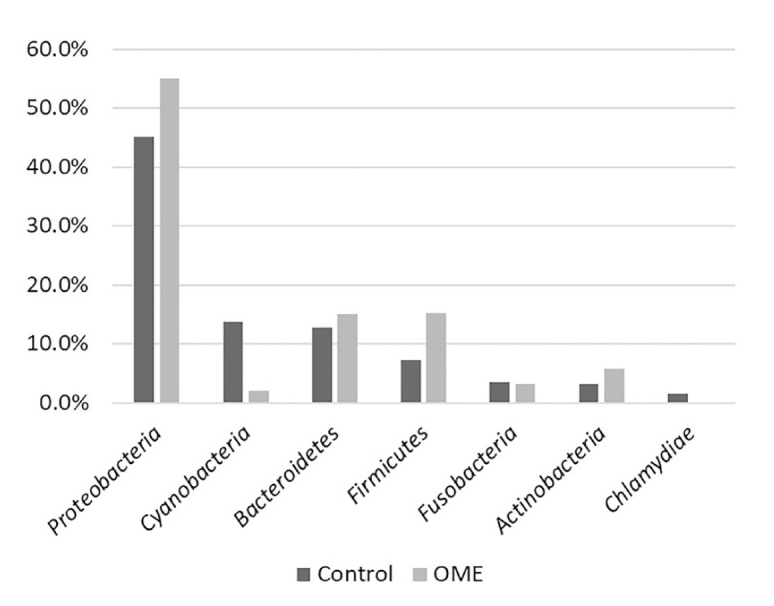
The phylum with more than 1% of all strains. Proteobacteria accounted for the highest percentage in both groups and was 10.5% higher in the OME group than in the control group. Cyanobacteria, Bacteroidetes, and Firmicutes were also identified in the control group. Firmicutes and Bacteroidetes were detected at the next highest level in the OME group. There was no significant difference in phylum between the two groups.
OME: otitis media with effusion.
Taxonomic analysis at the genus level identified 286 taxa in the control group and 127 taxa in the OME group. Haemophilus was the most frequently identified genus in both the control (13.7%) and OME (28.4%) groups, and there were 13 taxa in each group, accounting for more than 1% in both groups. Among the 13 taxa that accounted for more than 1% of the OME group, the genera that were significantly higher than those in the control group were Haemophilus (p=0.014), Prevotella (p=0.032), Streptococcus (p=0.017), Delftia (p=0.000), Corynebacterium (p=0.029), Veillonella (p=0.021), Niastella (p=0.011), and Streptobacillus (p=0.024). In the control group, Loriellopsis (p=0.000), Burkholderia (p=0.000), Pantoea (p=0.000), Chlamydia (p=0.000), Pseudomonas (p=0.023), and Xanthomonas (p=0.006), were significantly higher than those in the OME group (Figure 3).
Figure 3. a, b.

TList of genera with more than 1% of all strains in each group. (a) In the control group, 13 taxa with more than 1% were detected, and Loriellopsis, Burkholderia, Pantoea, Chlamydia, Pseudomonas, and Xanthomonas were more abundant than those in the OME group. (b) In the OME group, 13 taxa with more than 1% were detected, and Haemophilus, Prevotella, Streptococcus, Delftia, Corynebacterium, Veillonella, Niastella, and Streptobacillus were more abundant than those in the control group (*p<0.05, **p<0.01).
OME: otitis media with effusion.
At the species level, Haemophilus aegyptius (27.6%), Moraxella nonliquefaciens (13.7%), and Streptococcus pseudopneumoniae (6.4%) were the most abundant in the OME group, and Haemophilus aegyptius (13.0%), Loriellopsis cavernicola (12.3%), and Burkholderia plantarii (10.2%) were abundant in the control group. Fifty-five species showed a significant difference when comparing the microbiome composition of the OME group with that of the control group. The microbiomes that were significantly higher in the OME group were Dolosigranulum pigrum (p=0.035), Megasphaera micronuciformis (p=0.004), Clostridium disporicum (p=0.039), and Stomatobaculum longum (p=0.019). Of the 51 species that were significantly higher in the control group than in the OME group, five had more than 1% of the species on average; these were Loriellopsis cavernicola (p=0.000), Burkholderia plantarii (p=0.000), Pantoea deleyi (p=0.000), Curtobacterium albidum (p=0.000), and Xanthomonas theicola (p=0.000) (Figure 4).
Figure 4. a, b.

Taxonomic analysis at the species level (microbiomes showing a significant difference in each group). Fifteen species with more than 1% in each group are listed in order. (a) Dolosigranulum pigrum, Megasphaera micronuciformis, Clostridium disporicum, and Stomatobaculum longum were significantly higher in the OME group than in the control group. However, Dolosigranulum pigrum was the only species with more than 1% composition. (b) Of the 51 species that were significantly higher in the control group, five species (Loriellopsis cavernicola, Burkholderia plantarii, Pantoea deleyi, Curtobacterium albidum, and Xanthomonas theicola) showed more than 1% composition on average (*p<0.05, **p<0.01).
OME: otitis media with effusion.
The diversity and association of bacterial composition between the OME and control groups were reconstructed by various methods. Using PCoA, it is possible to determine whether there is a tendency to show a difference between groups depending on the degree of grouping of samples located in the coordinate plane. These three components (PC1–3) can explain 67% of the variance among samples. The similarity difference between the two groups can be seen on the PC2 axis, but they did not form completely separate clusters (Figure 5). The UPGMA tree shows a hierarchical structure by forming a cluster of samples with high similarity using the distance between samples. The UniFrac distance is used in UPGMA tree analysis to calculate the distance between samples considering not only the quantitative comparison of OTUs, but also the phylogenetic differences of the microbiome. Of 16 samples in the control group, 15 formed cluster branches. In the OME group, 10 samples clustered on an independent branch and four samples constituted the cluster but belonged to another branch. Although some samples were located close to other groups, similar to the PCoA analysis, most samples were closer together within each group (Figure 6).
Figure 5.
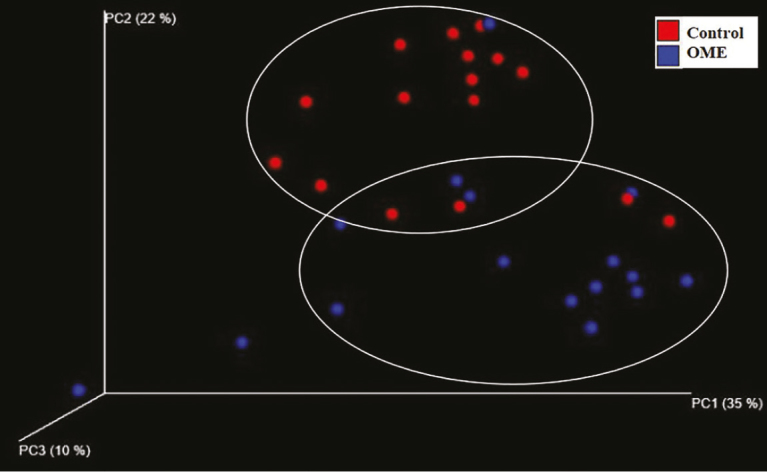
A three-dimensional PCoA based on the weighted UniFrac distance matrix. These three components explain 67% of the variance among samples. The differences in similarity between the two groups can be observed on the PC2 axis but are not completely separated. Red spheres indicate the control group and blue spheres indicate the OME group. PC1-3 indicate the percent similarity explained by each axis.
PCoA: principal coordinates analysis; OME: otitis media with effusion.
Figure 6.

UPGMA tree between the OME and control groups. The degree of similarity between the groups was visualized, and the similarity among samples was confirmed by the phylogenetic differences and diversity. Similarity within the group between samples was observed but was not completely separated. The number between each tree indicates the UniFrac distance.
UPGMA: unweighted pair group method with an arithmetic mean.
DISCUSSION
Our study differs from other studies in that we performed sampling of adenoid tissue without contamination by using an endoscope to ensure a clear visual field and greater accessibility under general anesthesia. Our samples were not mixed with the microbiome from the nasal cavity, nasopharynx, or oral cavity and thus serve as a basis for obtaining reliable results.
The adenoid microbiomes observed in this study were more complex and diverse than the culture-based reports known to date [3, 5]. A culture-based microbial analysis of adenoid samples at the genus level showed that Haemophilus, Streptococcus, and Staphylococcus were the most abundant [11]. Streptococcus is common in infectious cases, whereas Staphylococcus is a nasopharyngeal commensal bacterium; therefore, it is highly identified in non-infectious cases. In this study, Haemophilus was the most common genus in both groups, and the proportion of Streptococcus in the OME group was higher than that in the control group, but that of Staphylococcus was low.
The microbial diversity of adenoids associated with infection, such as adenoiditis, sinusitis, and tonsillitis, was lower than that of non-infectious adenoids [11]. The SI and inverse Simpson index indicate that the dominance of specific microbiomes in the control group was higher than that in the OME group. Changes in the microbiome diversity for inflammation and infection are observed not only in adenoids, but also in other organs. It has been reported that the diversity of the fecal microbiome in Crohn’s disease and ulcerative colitis is lower than that in healthy controls [12]. A similar study showed that nasopharyngeal microbial diversity in children with AOM evaluated via 16S pyrosequencing is lower than non-infectious nasopharyngeal microbiomes [13].
The reason for the low OTU in the OME group can be explained by the bacterial interference hypothesis. Bacterial interference is an important mechanism used to maintain the normal flora of the skin and mucosal surface. Commensal bacteria interfere with bacterial pathogens by competing for resources or by producing antagonistic nutritional substances [14]. Therefore, if various commensal bacteria are not present, pathogen-induced inflammation may be likely to occur.
The microbial composition at the phylum level was slightly different between the two groups, and Actinobacteria identified in the OME group was highly dominant only in one specimen (61.75%); thus, it cannot be regarded as a high proportion in the entire OME group. The microbial composition of the adenoids in this study differed from previous studies in that the most abundant phylum species were different and Cyanobacteria was the second-most identified phylum in the control group [15].
Liu et al. [9] and Ren et al. [15] reported that the microbiomes identified in the adenoids were in the order of Firmicutes, Proteobacteria, and Fusobacteria. Bogaert et al. [16] reported that Proteobacteria was the most abundant in the nasopharynx, followed by Firmicutes and Bacteroidetes. Because Cyanobacteria are difficult to identify using conventional culture methods, their roles in the human body have not yet been studied. Cyanobacteria are beginning to be detected in the mammalian gut through culture-independent 16S rRNA gene surveys [17]. A small percentage of Cyanobacteria has been identified in previous adenoid microbiome studies, but not as much as in this study [15]. To define them as commensal bacteria of the adenoids, it will be necessary to analyze pyrosequencing data from a greater number of adenoid samples in the future.
The four dominant species in the OME group were strains that were rarely cultured in a traditional culture-based study of adenoids. However, D. pigrum was the only species that accounted for more than 1% of the OME group, and the remaining three species were rare in both groups. Therefore, it is difficult to conclude that M. micronuciformis, C. disporicum, and S. longum are dominant pathogenic species in the OME group. Of the 51 species that were more frequently identified in the control group, 15 taxa were present only in the control group. All 15 species were found after the initiation of 16S rRNA pyrosequencing and have not been identified in the nasopharynx or oral cavity until now. However, species with more than 1% of all strains among the 15 taxa identified in the control group were Chlamydia pneumoniae and Streptococcus dysgalactiae.
At the genus level, Haemophilus was the most abundant in the OME group. In addition, Prevotella, Delftia, and Corynebacterium were the dominant genera in the OME group. This is noteworthy because this result was not observed in a traditional culture-based study.
Chonmaitree et al. [18] found that the fluctuation of nasopharyngeal microbiota in infants could involve viral respiratory tract infection and AOM. They suggested that the stability of the microbiome in nasopharynx during asymptomatic viral infection might play a role in the prevention of disease progression.
In a case-controlled study, Lappan et al. reported that Corynebacterium and Dolosigranulum are significantly higher in the control group than in patients with recurrent AOM and these two genera are characteristic of a healthy nasopharyngeal microbiome [19].
The composition of the dominant adenoid microbiome in OME and control groups in our study was similar to those seen in studies performed in other countries and on other races [9, 10, 20]. However, the non-dominant microbiomes detected in the adenoids of the control group varied from study to study. This might be due to differences in race and age, and environmental factors would affect it.
In the present study, it was difficult to determine whether the predominant bacteria in the OME group caused the occurrence of otitis media or whether the predominant bacteria in the control group inhibited the occurrence of otitis media. However, the results of the sequence similarity-based analysis study such as PCoA and UPGMA tree analysis revealed differences in microbial composition between the OME and control groups. The results of this study suggest that the compositional variability of the microbiome in adenoids is associated with the development of OME.
In addition, because the microbiota protects the host from incoming pathogens, changes in the microbiota might lead to the dysregulation of immune homeostasis and increased susceptibility to disease [21].
It is necessary to investigate the influence of dominant species on mucosal or systemic immunity in various otitis media conditions, including AOM, COM, and recurrent otitis media in future studies.
CONCLUSION
We identified a difference in bacterial diversity and microbial composition between the OME and control groups through 16S rRNA pyrosequencing. Our results provide basic data for identifying a new pathologic mechanism and will aid in the treatment of otitis media.
Footnotes
This study was presented at the “32th Politzer Society meeting’, 28 May–1 June 2019, Warsaw, Poland.
Ethics Committee Approval: Ethics Committee Approval was received for this study from the Ethics Committee of Hallym University Dongtan Sacred Heart Hospital (IRB No. 2016-158-I).
Informed Consent: Written informed consent was obtained from the parents of the patients who participated in this study.
Peer-review: Externally peer-reviewed.
Author Contributions: Concept - S.M.H, S.K.K; Design S.M.H, S.J.H; Supervision - K.H.P; Resource S.J.H; Materials - S.M.H., S.K.K.,; Data Collection and/or Processing - S.M.H., S.K.K.,; Analysis and/or Interpretation - S.K.K., S.M.H..; Literature Search - S.K.K., S.M.H., K.H.P., S.J.H..; Writing - S.K.K., S.M.H..; Critical Reviews - S.M.H., S.J.H., K.H.P.
Conflict of Interest: The authors have no conflict of interest to declare.
Financial Disclosure: This research was supported by Hallym University Research Fund 2016 (HURF-2016-29).
REFERENCES
- 1.Teele DW, Klein JO, Rosner B. Epidemiology of otitis media during the first seven years of life in children in greater Boston: a prospective, cohort study. J Infect Dis. 1989;160:83–94. doi: 10.1093/infdis/160.1.83. [DOI] [PubMed] [Google Scholar]
- 2.Miura MS, Mascaro M, Rosenfeld RM. Association between otitis media and gastroesophageal reflux: a systematic review. Otolaryngol Head Neck Surg. 2012;146:345–52. doi: 10.1177/0194599811430809. [DOI] [PubMed] [Google Scholar]
- 3.Brook I, Yocum P, Shah K, Feldman B, Epstein S. Microbiology of serous otitis media in children: correlation with age and length of effusion. Ann Otol Rhinol Laryngol. 2001;110:87–90. doi: 10.1177/000348940111000116. [DOI] [PubMed] [Google Scholar]
- 4.Pitkaranta A, Jero J, Arruda E, Virolainen A, Hayden FG. Polymerase chain reaction-based detection of rhinovirus, respiratory syncytial virus, and coronavirus in otitis media with effusion. J Pediatr. 1998;133:390–4. doi: 10.1016/S0022-3476(98)70276-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fekete Szabo G, Berenyi I, Gabriella K, Urban E, Nagy E. Aerobic and anaerobic bacteriology of chronic adenoid disease in children. Int J Pediatr Otorhinolaryngol. 2010;74:1217–20. doi: 10.1016/j.ijporl.2010.07.013. [DOI] [PubMed] [Google Scholar]
- 6.Petrosino JF, Highlander S, Luna RA, Gibbs RA, Versalovic J. Metagenomic pyrosequencing and microbial identification. Clin Chem. 2009;55:856–66. doi: 10.1373/clinchem.2008.107565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peterson J, Garges S, Giovanni M, McInnes P, Wang L, Schloss JA, et al. The NIH Human Microbiome Project. Genome Res. 2009;19:2317–23. doi: 10.1101/gr.096651.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tilg H, Adolph TE. Influence of the human intestinal microbiome on obesity and metabolic dysfunction. Curr Opin Pediatr. 2015;27:496–501. doi: 10.1097/MOP.0000000000000234. [DOI] [PubMed] [Google Scholar]
- 9.Liu CM, Cosetti MK, Aziz M, Buchhagen JL, Contente-Cuomo TL, Price LB, et al. The otologic microbiome: a study of the bacterial microbiota in a pediatric patient with chronic serous otitis media using 16SrRNA gene-based pyrosequencing. Arch Otolaryngol Head Neck Surg. 2011;137:664–8. doi: 10.1001/archoto.2011.116. [DOI] [PubMed] [Google Scholar]
- 10.Jervis-Bardy J, Rogers GB, Morris PS, Smith-Vaughan HC, Nosworthy E, Leong LE, et al. The microbiome of otitis media with effusion in Indigenous Australian children. Int J Pediatr Otorhinolaryngol. 2015;79:1548–55. doi: 10.1016/j.ijporl.2015.07.013. [DOI] [PubMed] [Google Scholar]
- 11.Subtil J, Rodrigues JC, Reis L, Freitas L, Filipe J, Santos A, et al. Adenoid bacterial colonization in a paediatric population. Eur Arch Otorhinolaryngol. 2017;274:1933–8. doi: 10.1007/s00405-017-4493-z. [DOI] [PubMed] [Google Scholar]
- 12.Manichanh C, Rigottier-Gois L, Bonnaud E, Gloux K, Pelletier E, Frangeul L, et al. Reduced diversity of faecal microbiota in Crohn’s disease revealed by a metagenomic approach. Gut. 2006;55:205–11. doi: 10.1136/gut.2005.073817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hilty M, Qi W, Brugger SD, Frei L, Agyeman P, Frey PM, et al. Nasopharyngeal microbiota in infants with acute otitis media. J Infect Dis. 2012;205:1048–55. doi: 10.1093/infdis/jis024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hooper LV, Gordon JI. Commensal host-bacterial relationships in the gut. Science. 2001;292:1115–8. doi: 10.1126/science.1058709. [DOI] [PubMed] [Google Scholar]
- 15.Ren T, Glatt DU, Nguyen TN, Allen EK, Early SV, Sale M, et al. 16S rRNA survey revealed complex bacterial communities and evidence of bacterial interference on human adenoids. Environ Microbiol. 2013;15:535–47. doi: 10.1111/1462-2920.12000. [DOI] [PubMed] [Google Scholar]
- 16.Bogaert D, Keijser B, Huse S, Rossen J, Veenhoven R, van Gils E, et al. Variability and diversity of nasopharyngeal microbiota in children: a metagenomic analysis. PLoS One. 2011;6:e17035. doi: 10.1371/journal.pone.0017035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, Bircher JS. Evolution of mammals and their gut microbes. Science. 2008;320:1647–51. doi: 10.1126/science.1155725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chonmaitree T, Jennings K, Golovko G. Nasopharyngeal microbiota in infants and changes during viral upper respiratory tract infection and acute otitis media. PLoS One. 2017;12:e0180630. doi: 10.1371/journal.pone.0180630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lappan R, Imbrogno K, Sikazwe C. A microbiome case-control study of recurrent acute otitis media identified potentially protective bacterial genera. BMC Microbiol. 2018;18:13. doi: 10.1186/s12866-018-1154-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johnston J, Hoggard M, Biswas K, Astudillo-García C, Radcliff FJ, Mahadevan M, et al. Pathogen reservoir hypothesis investigated by analyses of the adenotonsillar and middle ear microbiota. Int J Pediatr Otorhinolaryngol. 2019;118:103–9. doi: 10.1016/j.ijporl.2018.12.030. [DOI] [PubMed] [Google Scholar]
- 21.Willing BP, Russell SL, Finlay BB. Shifting the balance: antibiotic effects on host-microbiota mutualism. Nat Rev Microbiol. 2011;9:233–43. doi: 10.1038/nrmicro2536. [DOI] [PubMed] [Google Scholar]