

Abstract
Objectives
To compare efficacy and safety of ixekizumab (IXE) to adalimumab (ADA) in biological disease-modifying antirheumatic drug-naïve patients with both active psoriatic arthritis (PsA) and skin disease and inadequate response to conventional synthetic disease-modifying antirheumatic drug (csDMARDs).
Methods
Patients with active PsA were randomised (1:1) to approved dosing of IXE or ADA in an open-label, head-to-head, blinded assessor clinical trial. The primary objective was to evaluate whether IXE was superior to ADA at week 24 for simultaneous achievement of a ≥50% improvement from baseline in the American College of Rheumatology criteria (ACR50) and a 100% improvement from baseline in the Psoriasis Area and Severity Index (PASI100). Major secondary objectives, also at week 24, were to evaluate whether IXE was: (1) non-inferior to ADA for achievement of ACR50 and (2) superior to ADA for PASI100 response. Additional PsA, skin, treat-to-target and quality-of-life outcome measures were assessed at week 24.
Results
The primary efficacy endpoint was met (IXE: 36%, ADA: 28%; p=0.036). IXE was non-inferior for ACR50 response (IXE: 51%, ADA: 47%; treatment difference: 3.9%) and superior for PASI100 response (IXE: 60%, ADA: 47%; p=0.001). IXE had greater response versus ADA in additional PsA, skin, nail, treat-to-target and quality-of-life outcomes. Serious adverse events were reported in 8.5% (ADA) and 3.5% (IXE) of patients.
Conclusions
IXE was superior to ADA in achievement of simultaneous improvement of joint and skin disease (ACR50 and PASI100) in patients with PsA and inadequate response to csDMARDs. Safety and tolerability for both biologicals were aligned with established safety profiles.
Keywords: ixekizumab, adalimumab, psoriatic arthritis, head-to-head, clinical trial
Key messages.
What is already known about this subject?
Many patients with psoriatic arthritis and active skin and joint disease do not achieve satisfactory clinical response with conventional synthetic disease-modifying antirheumatic therapy in both important domains of the disease simultaneously.
In this patient group, biological disease-modifying antirheumatic drugs (bDMARDs) offer additional treatment options, but the comparative efficacy and safety of bDMARDs is not known.
What does this study add?
The findings of this study demonstrate that ixekizumab was superior to adalimumab for simultaneous achievement of American College of Rheumatology 50 (ACR50) and Psoriasis Area and Severity Index (PASI100), was non-inferior to adalimumab for achievement of ACR50 and was superior to adalimumab for achievement of PASI100 at week 24.
Response with ixekizumab was significantly greater than adalimumab for Minimal Disease Activity, Very Low Disease Activity, Disease Activity in Psoriatic Arthritis remission (≤4), change from baseline in modified Composite Psoriatic Disease Activity Index, resolution of enthesitis (Spondyloarthritis Research Consortium of Canada Enthesitis Index=0), PASI75, PASI90 and Dermatology Life Quality Index (0 or 1) and was at least similar to adalimumab for all other psoriatic arthritis, treat-to-target, skin, nail and quality of life endpoints.
How might this impact on clinical practice or future developments?
The findings of this study increase awareness of current treatment options and informs evidence-based treatment decisions for patients with active psoriatic arthritis and active psoriatic skin disease.
Introduction
The goal of treatment in patients with active psoriatic arthritis (PsA) is to simultaneously improve the manifestations of the disease, including arthritis and skin disease. Improvements in both joint and skin disease are necessary to achieve optimal improvement in health-related quality of life in patients with PsA, an important indicator of treatment success.1 Treatment options for patients with PsA include non-pharmacological intervention, symptomatic treatment, conventional synthetic disease-modifying antirheumatic drugs (csDMARDS), biological DMARDs (bDMARDs) and other immunomodulatory therapies.2–6
Among patients who fail to achieve adequate response to csDMARDs, bDMARDs targeting inflammatory cytokines such as tumour necrosis factor α (TNF), interleukin (IL)-12/23 or IL-17A offer an alternative either as a combination therapy with csDMARDs or as monotherapy. Some evidence suggests that combination therapy with csDMARDs such as methotrexate may inhibit development of antidrug antibodies to bDMARDs, and some studies observed better treatment persistence with combination therapy.7 Concomitant methotrexate has been associated with greater serum concentration of adalimumab (ADA) versus patients receiving ADA monotherapy.8
The objective of the current study is to determine whether ixekizumab (IXE), a high-affinity monoclonal antibody that selectively targets IL-17A, is superior to ADA, a TNF inhibitor, as measured by a combined arthritis and skin endpoint in bDMARD-naïve patients with active PsA and inadequate response to csDMARDs. Concomitant use of a stable dose of csDMARDs was permitted during the study.
Methods
Participants
Eligible participants had an established diagnosis of PsA for at least 6 months, fulfilled the Classification for Psoriatic Arthritis criteria with at least 3/66 swollen and 3/68 tender joints, had previous inadequate response to ≥1 csDMARD, had active plaque psoriasis affecting ≥3% of body surface area (BSA) and had not previously received bDMARD or Janus kinase inhibitor therapy.9 Patients on csDMARDs at screening were allowed to continue a stable dose of csDMARD therapy.
Study design
This study is a 52 week, phase IIIb/IV, multicentre, randomised, open-label, blinded-assessor, parallel-group study evaluating the efficacy and safety of IXE versus ADA in bDMARD-naïve, csDMARD-inadequate-responder patients (based on medical history) with active PsA. Following a 28-day screening period, participants were randomised 1:1 to open-label IXE or ADA during a 52-week open-label treatment period (weeks 0–52). Randomisation was stratified by concomitant csDMARD use at baseline and moderate-to-severe plaque psoriasis involvement (Psoriasis Area and Severity Index (PASI)≥12, BSA ≥10% and static physician’s global assessment (sPGA) ≥3). Study visits occurred at screening, baseline and postbaseline at weeks 1, 4, 8, 12, 16, 24, 32, 40 and 52. Treatment allocation was revealed after randomisation to sponsors, investigators, patients and all study staff except for blinded assessors. Blinded assessors evaluated tender joint count, swollen joint count, PASI, % BSA, enthesitis, Leeds Dactylitis Index–Basic (LDI-B), Nail Psoriasis Severity Index (NAPSI) fingernails and sPGA.
Participants received approved-label dosing of assigned treatments by subcutaneous injection. All patients randomised to IXE received a 160 mg starting dose (two 80 mg injections) at week 0. IXE-treated patients received 80 mg IXE every 4 weeks from week 4 onwards (seven doses up to week 24) unless they met criteria for moderate-to-severe psoriasis, in which case they received 80 mg IXE every 2 weeks from week 2 to week 12, followed by IXE every 4 weeks (10 doses up to week 24, three additional doses). Patients randomised to ADA received a 40 mg starting dose followed by 40 mg ADA every 2 weeks starting at week 2 (12 doses up to week 24), or if they met criteria for moderate-to-severe psoriasis, they received an 80 mg starting dose of ADA (two 40 mg injections) at week 0, followed by 40 mg ADA every 2 weeks starting at week 1 (14 doses up to week 24, two additional doses). Thus, among patients with moderate-to-severe psoriasis, the IXE dosing regimen resulted in one more additional dose relative to those receiving ADA.
SPIRIT-H2H (Clinicaltrials.gov: NCT03151551) was conducted in accordance with the ethical principles of the Declaration of Helsinki. All patients provided written informed consent, and the study protocol was approved by the ethical review board prior to the start of study-related procedures.
Patient and public involvement
Patients were not involved in the design or conduct of the study, development of outcomes or dissemination of study results.
Efficacy endpoints
The primary and two major secondary endpoints were tested using a sequential hierarchical testing procedure in the order presented below. There were no adjustments for multiple comparisons for any other analyses.
Primary endpoint (simultaneous achievement of ACR50 and PASI100)
The primary endpoint assessed superiority of IXE versus ADA at week 24, as measured by the proportion of patients who simultaneously achieved an American College of Rheumatology 50 (ACR50) response and PASI100 response. After the week 24 database lock and initial analysis run, a medical inconsistency in baseline PASI data was identified (PASI=0 but BSA ≥3%) in nine patients. This scenario was not anticipated or described in the protocol or statistical analysis plan. The inconsistency was resolved using medical judgement. The impacted patients met baseline criteria for active psoriasis. In the final primary analysis, patients with baseline PASI=0 and BSA ≥3 were considered PASI100 responders if, and only if, an absolute PASI=0 and BSA=0 was achieved at week 24. Multiple analyses to assess the robustness of this approach were conducted (see online supplementary table 1).
annrheumdis-2019-215386supp001.pdf (311.9KB, pdf)
Major secondary endpoint 1 (ACR50)
Major secondary endpoint 1 assessed whether IXE was non-inferior to ADA at week 24 as measured by the proportion of patients achieving ACR50.
Major secondary endpoint 2 (PASI100)
Major secondary endpoint 2 assessed whether IXE was superior to ADA at week 24 as measured by the proportion of patients achieving PASI100.
Other secondary endpoints
Additional prespecified outcomes included the proportion of patients achieving ≥20% or ≥70% improvement from baseline in ACR criteria (ACR20/70), ≥75% or ≥90% improvement from baseline in PASI (PASI75/90), resolution of fingernail psoriasis (NAPSI fingernails=0), PsA minimal disease activity (MDA), a minimal clinically important difference (MCID) of ≥0.35-point improvement from baseline in Health Assessment Questionnaire–Disability Index (HAQ-DI) among patients with ≥0.35 at baseline, a Dermatology Life Quality Index score of 0 or 1 (DLQI (0 or 1)), resolution of enthesitis as measured by the Spondyloarthritis Research Consortium of Canada Enthesitis Index (SPARCC Enthesitis Index=0) or Leeds Enthesitis Index (LEI=0) among patients with enthesitis at baseline (SPARCC Enthesitis Index >0 or LEI >0) and resolution of dactylitis as measured by the Leeds Dactylitis Index–Basic (LDI-B=0) among patients with dactylitis at baseline (LDI-B >0). Prespecified continuous outcomes included the mean change from baseline in NAPSI and the modified Composite Psoriatic Disease Activity Index (mCPDAI) (see online supplementary table 2).
Post hoc continuous analyses included mean change from baseline in Disease Activity in Psoriatic Arthritis (DAPSA) and in the psoriatic arthritis disease activity score (PASDAS). Post hoc categorical analyses included the percentage of patients achieving DAPSA ≤4 (remission), DAPSA ≤14 (low disease activity or remission), PASDAS ≤3.2 (low disease activity), PASDAS ≤1.9 (near remission) and meeting 7/7 MDA criteria (very low disease activity (VLDA)).
Safety
Treatment-emergent adverse events (TEAEs) were defined as events that first occurred or worsened in severity after the first dose of study treatment and on or prior to the date of the last visit within the treatment period. AEs of special interest included infections, injection-site reactions, cytopaenias, liver function test changes/enzyme elevations, allergic reactions/hypersensitivity, cerebrocardiovascular events, malignancies, depression and suicide/self-injury, interstitial lung disease and inflammatory bowel disease (IBD). Data relating to cerebrocardiovascular events and suspected IBD were adjudicated by external clinical events committees.
Statistical analyses
Analyses of efficacy were performed at the week 24 primary database lock for the intent-to-treat population, consisting of all randomised patients according to treatment assigned at week 0. A hierarchical multiple testing procedure for the primary and two major secondary endpoints was implemented to control the family-wise type I error rate at a two-sided α level of 0.05. The first test in the statistical hierarchy was a superiority test of the primary endpoint (simultaneous ACR50 and PASI100). If IXE was determined to be statistically superior to ADA for the primary endpoint, a non-inferiority test of IXE versus ADA was performed for secondary endpoint 1 (ACR50). If the test for major secondary endpoint 1 was successful (indicating IXE was non-inferior to ADA for achieving ACR50 at Week 24), a superiority test was conducted for major secondary endpoint 2 (PASI100). If a test in this sequence was not successful, all subsequent tests were considered unsuccessful.
A fixed-margin approach was used for non-inferiority testing of ACR50 response, where IXE was deemed non-inferior to ADA if the lower bound of the two-sided 95% CI for the difference in proportions of ACR50 responders on IXE minus ADA was greater than the prespecified margin of −12.0%. This non-inferiority margin represents an approximately 50% preservation of the ADA treatment effect observed in historical phase III studies per Food and Drug Administration (FDA)/European Medicines Agency (EMA) non-inferiority study design guidelines.10–13
Categorical efficacy and health outcome variables were analysed based on treatment success/failure using a logistic regression model with treatment, concomitant csDMARD use at baseline and moderate-to-severe plaque psoriasis involvement as factors. Patients were considered treatment failures (or non-responders) if they did not meet the clinical response criteria or had missing clinical response data at a particular time point of analysis.
Continuous variables were analysed using a mixed effects model of repeated measures analysis, which included treatment group, concomitant csDMARD use at baseline, moderate-to-severe plaque psoriasis involvement and visit as fixed factors; baseline value as covariate; and baseline-by-visit and treatment-by-visit interaction terms. Missing data were imputed using a modified baseline observation carried forward method.
Descriptive safety analyses were performed on all randomised patients according to assigned treatment who received ≥1 dose of study treatment and included all data available up to the time of database lock.
Results
Participants
Of 684 patients screened, 566 were randomised between 24 August 2017 and 24 May 2018, to either ADA (n=283) or IXE (n=283); 269 (95%) patients randomised to ADA and 262 (93%) patients randomised to IXE completed the week 24 study visit (figure 1). Baseline demographics and disease characteristics were balanced between treatment groups (table 1). All patients had active plaque psoriasis with BSA ≥3%.
Figure 1.
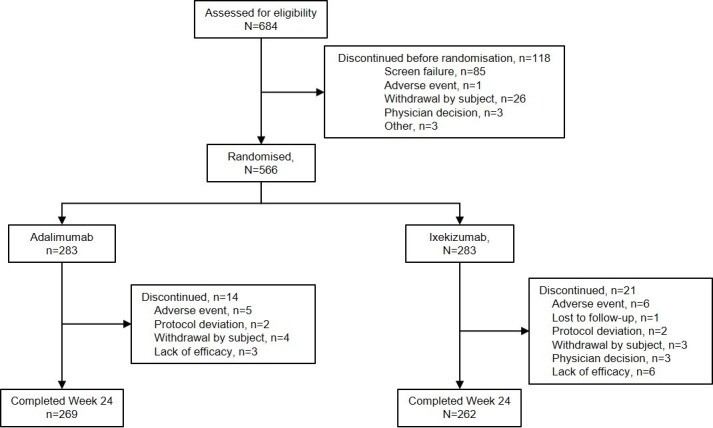
Participant flow diagram up to week 24.
Table 1.
Baseline demographics and disease characteristics
| IXE (n=283) | ADA (n=283) | |
| Baseline demographics | ||
| Age, years | 47.5 (12.0) | 48.3 (12.3) |
| Sex, n (%) | ||
| Male | 162 (57) | 150 (53) |
| Female | 121 (43) | 133 (47) |
| Race, n (%) | ||
| White | 222 (78) | 211 (75) |
| Asian | 29 (10) | 33 (12) |
| Weight, kg | 85.3 (19.8) | 81.9 (18.3) |
| Body mass index, kg/m2 | 30.0 (6.9) | 29.7 (8.3) |
| Duration of symptoms since PsA diagnosis, years | 6.6 (7.4) | 5.9 (6.4) |
| Duration of symptoms since psoriasis diagnosis, years | 16.1 (13.1) | 14.7 (12.6) |
| Concomitant csDMARD use, n (%) | 193 (68) | 199 (70) |
| Concomitant methotrexate use, n (%) | 167 (59) | 169 (60) |
| Baseline disease scores | ||
| Tender joint count | 19.1 (12.7) | 21.3 (15.4) |
| Swollen joint count | 10.1 (7.5) | 10.7 (8.1) |
| Patient pain VAS | 59.7 (21.9) | 62.4 (21.1) |
| Patient’s global assessment of disease activity VAS, mm | 62.4 (20.3) | 65.2 (20.7) |
| Physician’s global assessment of disease activity VAS, mm | 58.9 (17.5) | 59.4 (18.2) |
| HAQ-DI | 1.2 (0.6) | 1.3 (0.7) |
| C-reactive protein, mg/L | 9.8 (13.7) | 10.5 (19.3) |
| SPARCC Enthesitis Index >0, n (%) | 189 (67) | 171 (60) |
| SPARCC Enthesitis Index* | 4.9 (3.5) | 5.7 (3.8) |
| LEI >0, n (%) | 159 (56) | 147 (52) |
| LEI† | 2.5 (1.4) | 2.7 (1.5) |
| LDI-B >0, n (%) | 42 (15) | 58 (21) |
| LDI-B‡ | 40.1 (42.4) | 55.8 (128.4) |
| PASDAS | 5.8 (0.9) | 5.8 (1.0) |
| DAPSA | 42.7 (20.6) | 45.8 (23.5) |
| Moderate-to-severe psoriasis, n (%) | 49 (17) | 51 (18) |
| PASI ≥12, n (%) | 55 (19) | 57 (20) |
| sPGA ≥3, n (%) | 173 (61) | 181 (64) |
| BSA ≥3%, n (%) | 283 (100) | 283 (100) |
| BSA ≥10%, n (%) | 113 (40) | 104 (37) |
| PASI | 7.9 (8.7) | 7.7 (7.3) |
| Percentage BSA | 14.8 (18.4) | 12.9 (15.6) |
| DLQI | 9.8 (7.6) | 9.8 (7.6) |
| NAPSI fingernails >0, n (%) | 191 (68) | 177 (63) |
| NAPSI fingernails§ | 19.7 (18.5) | 19.1 (16.3) |
Unless indicated otherwise, data are presented as mean (SD).
*Assessed in patients with SPARCC Enthesitis Index >0 at baseline.
†Assessed in patients with LEI >0 at baseline.
‡Assessed in patients with LDI-B >0 at baseline.
§Assessed in patients with NAPSI >0 at baseline.
ADA, adalimumab; BSA, body surface area; csDMARD, conventional synthetic disease-modifying antirheumatic drug; DAPSA, Disease Activity in Psoriatic Arthritis; DLQI, Dermatology Life Quality Index; HAQ-DI, Health Assessment Questionnaire–Disability Index; IXE, ixekizumab; LDI-B, Leeds Dactylitis Index–Basic; LEI, Leeds Enthesitis Index; NAPSI, Nail Psoriasis Area and Severity Index; PASDAS, psoriatic arthritis disease activity score; PASI, Psoriasis Area and Severity Index; PsA, psoriatic arthritis; SPARCC, Spondyloarthritis Research Consortium of Canada; sPGA, static physician’s global assessment; VAS, visual analogue scale.
Efficacy
Efficacy outcomes at week 24 are summarised in table 2. The primary and all major secondary endpoints of the study were met. The proportion of patients simultaneously achieving ACR50 and PASI100 was significantly (p=0.036) greater for patients receiving IXE (36%) than ADA (28%); significant differences were observed as early as week 8 (figure 2A). IXE was non-inferior to ADA as measured by ACR50 response (IXE: 50.5%, ADA: 46.6%, IXE vs ADA treatment difference: 3.9% (95% CI −4.3% to 12.1%)); there were no statistically significant differences in ACR50 response between treatment arms (figure 2B). PASI100 response was significantly (p=0.001) greater in the IXE (60%) versus ADA (47%) group; statistically significant differences were observed as early as the first PASI assessment (week 4) and persisted through week 24 (figure 2c).
Table 2.
Efficacy and health outcomes at week 24
| IXE (n=283) | ADA (n=283) | Treatment difference IXE versus ADA (95% CI) |
IXE versus ADA P value |
|
| Primary endpoint | ||||
| ACR50+PASI100 | 102/283 (36.0) 30.4% to 41.6% |
79/283 (27.9) 22.7% to 33.1% |
8.1% (0.5% to 15.8%) |
0.036 |
| Major secondary endpoints | ||||
| ACR50* | 143/283 (50.5) 44.7% to 56.4% |
132/283 (46.6) 40.8% to 52.5% |
3.9% (-4.3% to 12.1%) |
0.338 |
| PASI100 | 170/283 (60.1) 54.4% to 65.8% |
132/283 (46.6) 40.8% to 52.5% |
13.4% (5.3% to 21.6%) |
0.001 |
| PsA endpoints | ||||
| MDA | 135/283 (47.7) 41.9% to 53.5% |
100/283 (35.3) 29.8% to 40.9% |
12.4% (4.3% to 20.4%) |
0.003 |
| VLDA† | 49/283 (17.3) 12.9% to 21.7% |
29/283 (10.2) 6.7% to 13.8% |
7.1% (1.4% to 12.7%) |
0.015 |
| DAPSA remission (≤4)† | 75/283 (26.5) 21.4% to 31.6% |
51/283 (18.0) 13.5% to 22.5% |
8.5% (1.7% to 15.3%) |
0.016 |
| DAPSA low disease activity or remission (≤14)† | 174/283 (61.5) 55.8% to 67.2% |
171/283 (60.4) 54.7% to 66.1% |
1.1% (-7.0% to 9.1%) |
0.737 |
| DAPSA, LSM change from baseline (SE)† | −31.74 (0.94) | −30.10 (0.94) | −1.64 (-3.94 to 0.66) |
0.161 |
| PASDAS low disease activity (≤3.2)† | 164/283 (58.0) 52.2% to 63.7% |
147/283 (51.9) 46.1% to 57.8% |
6.0% (-2.2% to 14.2%) |
0.153 |
| PASDAS near remission (≤1.9)† | 82/283 (29.0) 23.7% to 34.3% |
55/283 (19.4) 14.8% to 24.0% |
9.5% (2.5% to 16.6%) |
0.009 |
| PASDAS, LSM change from baseline (SE)† | −3.08 (0.10) | −2.94 (0.10) | −0.14 (-0.38 to 0.10) |
0.260 |
| mCPDAI, LSM change from baseline (SE) | −3.98 (0.14) | −3.46 (0.13) | −0.53 (-0.85 to −0.20) |
0.002 |
| ACR20 | 195/283 (68.9) 63.5% to 74.3% |
204/283 (72.1) 66.9% to 77.3% |
−3.2% (-10.7% to 4.3%) |
0.403 |
| ACR70 | 90/283 (31.8) 26.4% to 37.2% |
73/283 (25.8) 20.7% to 30.9% |
6.0% (-1.4% to 13.5%) |
0.111 |
| SPARCC Enthesitis Index=0‡ | 107/189 (56.6) 49.5% to 63.7% |
77/171 (45.0) 37.6% to 52.5% |
11.6% (1.3% to 21.9%) |
0.019 |
| LEI=0§ | 95/159 (59.7) 52.1% to 67.4% |
81/147 (55.1) 47.1% to 63.1% |
4.6% (-6.4% to 15.7%) |
0.432 |
| LDI-B=0¶ | 37/42 (88.1) 78.3% to 97.9% |
54/58 (93.1) 86.6% to 99.6% |
−5.0% (-16.8% to 6.8%) |
0.658 |
| Skin and nail psoriasis endpoints | ||||
| PASI75 | 227/283 (80.2) 75.6% to 84.9% |
195/283 (68.9) 63.5% to 74.3% |
11.3% (4.2% to 18.4%) |
0.002 |
| PASI90 | 203/283 (71.7) 66.5% to 77.0% |
158/283 (55.8) 50.0% to 61.6% |
15.9% (8.1% to 23.7%) |
<0.001 |
| NAPSI fingernails=0** | 111/191 (58.1) 51.1% to 65.1% |
88/177 (49.7) 42.4% to 57.1% |
8.4% (-1.8% to 18.6%) |
0.082 |
| NAPSI, LSM change from baseline (SE) | −15.89 (0.82) | −12.53 (0.82) | −3.37 (-5.40 to −1.33) |
0.001 |
| Quality of life endpoints | ||||
| HAQ-DI ≥0.35†† | 168/252 (66.7) 60.8% to 72.5% |
166/254 (65.4) 59.5% to 71.2% |
1.3% (-6.9% to 9.6%) |
0.741 |
| DLQI (0, 1) | 174/283 (61.5) 55.8% to 67.2% |
147/283 (51.9) 46.1% to 57.8% |
9.5% (1.4% to 17.7%) |
0.020 |
Unless otherwise indicated, values are presented as n/N (%), 95% CI.
*The treatment difference of IXE minus ADA was 3.9% (95% CI −4.3% to 12.1%). The lower bound of the 95% CI (−4.3%) was greater than −12%, thus meeting noninferiority criteria.
†Post hoc analysis.
‡Assessed for patients with SPARCC Enthesitis Index score >0 at baseline.
§Assessed for patients with LEI score >0 at baseline.
¶Assessed for patients with LDI-B score >0 at baseline.
**Assessed for patients with NAPSI fingernails score >0 at baseline.
††Assessed for patients with HAQ-DI score ≥0.35 at baseline. A response of ≥0.35 change from baseline is the minimal clinically important difference in HAQ-DI.
ACR, American College of Rheumatology; ADA, adalimumab; DAPSA, Disease Activity in Psoriatic Arthritis; DLQI, Dermatology Life Quality Index; HAQ-DI, Health Assessment Questionnaire–Disability Index; IXE, ixekizumab; LDI-B, Leeds Dactylitis Index–Basic; LEI, Leeds Enthesitis Index; LSM, least squares mean; mCPDAI, modified Composite Psoriatic Disease Activity Index; MDA, minimal disease activity; NAPSI, Nail Psoriasis Area and Severity Index; PASDAS, psoriatic arthritis disease activity score; PASI, Psoriasis Area and Severity Index; SPARCC, Spondyloarthritis Research Consortium of Canada; VLDA, very low disease activity.
Figure 2.
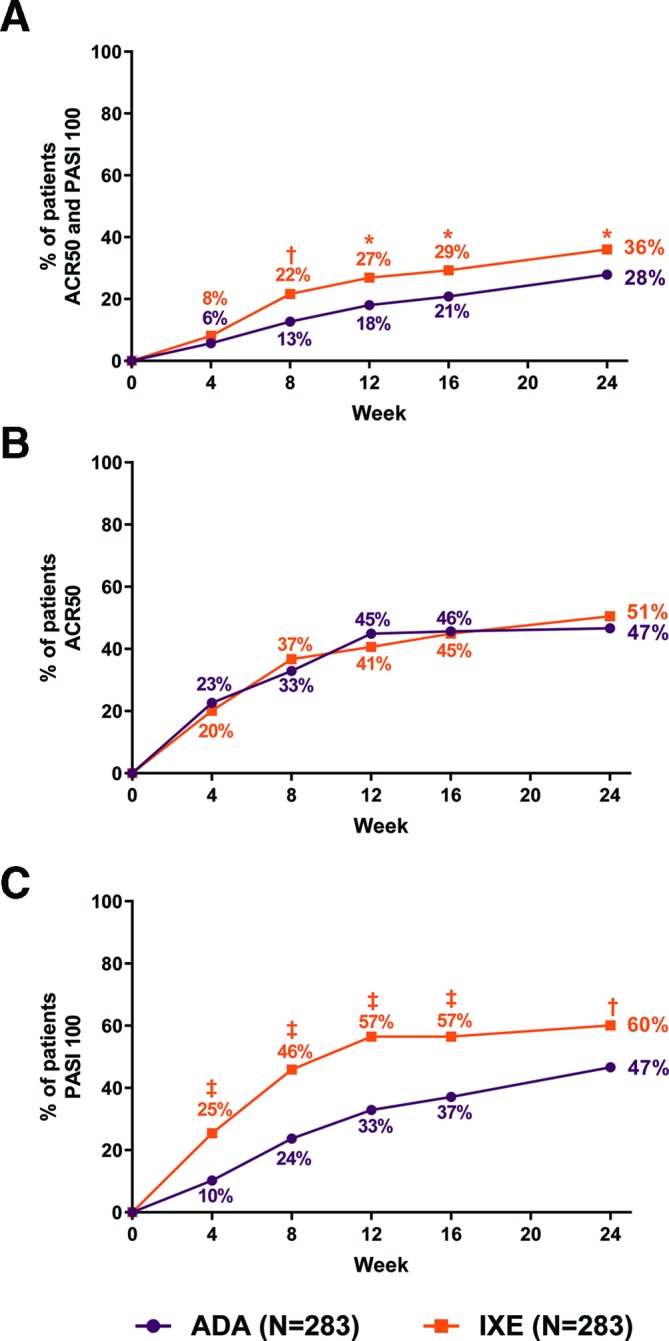
Clinical response rates for primary and major secondary outcomes through week 24 (non-responder imputation). (A) Percentage of patients simultaneously achieving ACR50 and PASI100 (primary endpoint). (B) Percentage of patients achieving ACR50 (major secondary endpoint). The treatment difference of IXE minus ADA was 3.9% (95% CI −4.3% to 12.1%). The lower bound of the 95% CI (−4.3%) was greater than −12%, thus meeting noninferiority criteria. (C) Percentage of patients achieving PASI100. IXE versus ADA: *P<0.05, †p<0.01, ‡p<0.001. ACR, American College of Rheumatology; ADA, adalimumab; IXE, ixekizumab; PASI, Psoriasis Area Severity Index.
Significantly more patients achieved PsA MDA (treatment difference: 12.4%, 95% CI 4.3% to 20.4%) and VLDA (treatment difference: 7.1%, 95% CI 1.4% to 12.7%) at week 24 in the IXE versus ADA groups (figure 3A,B). Although there were no significant differences between treatment groups in DAPSA change from baseline (treatment difference: −1.64, 95% CI −3.94 to 0.66) or DAPSA low disease activity, including remission (DAPSA ≤14) (treatment difference: 1.1%, 95% CI −7.0% to 9.1%) at week 24 (figure 3C), significantly more patients achieved the more stringent DAPSA remission (DAPSA ≤4) in the IXE versus ADA group (treatment difference: 8.5%, 95% CI 1.7% to 15.3%) (figure 3D). There were no significant differences between treatment groups in PASDAS change from baseline (treatment difference: −0.14, 95% CI −0.38 to 0.10) or PASDAS low disease activity (PASDAS ≤3.2) (treatment difference: 6.0%, 95% CI −2.2% to 14.2%), but PASDAS near remission (PASDAS ≤1.9) was achieved by significantly more patients in the IXE than ADA group (treatment difference: 9.5%, 95% CI 2.5% to 16.6%). No significant differences were observed in ACR20 (treatment difference: −3.2%, 95% CI −10.7% to 4.3%) or ACR70 (treatment difference: 6.0%, 95% CI −1.4% to 13.5%). Change from baseline in mCPDAI was significantly greater in the IXE versus ADA group at week 24 (treatment difference: −0.53, 95% CI −0.85 to −0.20), with statistically significant improvements as early as the first assessment at week 12.
Figure 3.

Clinical response rates for treat-to-target outcomes through week 24. (A) Percentage of patients achieving minimal disease activity. (B) Percentage of patients achieving very low disease activity. (C) Percentage of patients achieving a DAPSA score of ≤14 (LDA or remission). (D) Percentage of patients achieving a DAPSA score ≤4 (remission). IXE versus ADA: *P<0.05, †p<0.01, ‡p<0.001. ADA, adalimumab; DAPSA, Disease Activity in Psoriatic Arthritis; IXE, ixekizumab; LDA, low disease activity.
SPARCC Enthesitis Index=0 was achieved by significantly more patients in the IXE versus ADA group at week 24 (treatment difference: 11.6%, 95% CI 1.3% to 21.9%). Both IXE and ADA were efficacious as measured by LDI-B=0 response, but there were no statistically significant differences between treatment groups in LDI-B=0 response up to week 24 (treatment difference: −5.0%, 95% CI −16.8% to 6.8%).
Significantly more patients achieved PASI75 (treatment difference: 11.3%, 95% CI 4.2% to 18.4%) and PASI90 (treatment difference: 15.9%, 95% CI 8.1% to 23.7%) in the IXE versus ADA group. Significant differences in PASI75 and PASI90 response were observed as early as the first assessment at week 4. No significant differences were observed in NAPSI fingernails=0 response between treatment groups (treatment difference: 8.4%, 95% CI −1.8% to 18.6%). However, NAPSI fingernails change from baseline was significantly greater with IXE than ADA at week 24 (treatment difference: −3.37, 95% CI −5.40 to −1.33), with significant improvements as early as the first assessment at week 12.
DLQI (0, 1) response was significantly greater at week 24 in the IXE versus ADA group (treatment difference: 9.5%, 95% CI 1.4% to 17.7%), with significant differences as early as the first assessment at week 4. There were no statistically significant differences between groups in HAQ-DI MCID response (treatment difference: 1.3%, 95% CI −6.9% to 9.6%).
Safety
TEAEs were more common in the IXE versus ADA group (table 3); most were mild or moderate in severity. Discontinuations due to AEs and serious AEs (SAEs) were numerically lower in the IXE versus ADA group. No deaths occurred during the study.
Table 3.
Safety outcomes
| IXE (n=283) | ADA (n=283) | |
| Extent of exposure, mean days (total patient-years) | 236.8 (183.5) | 228.9 (117.3) |
| Treatment-emergent adverse events | 197 (69.6) | 173 (61.1) |
| Mild | 97 (34.3) | 87 (30.7) |
| Moderate | 91 (32.2) | 71 (25.1) |
| Severe | 9 (3.2) | 15 (5.3) |
| Serious adverse events | 10 (3.5) | 24 (8.5) |
| Deaths | 0 | 0 |
| Discontinuations due to adverse events | 7 (2.5) | 13 (4.6) |
| Adverse events of special interest | ||
| Infections | 102 (36.0) | 87 (30.7) |
| Serious infections | 4 (1.4) | 8 (2.8) |
| Candida infections | 7 (2.5) | 2 (0.7) |
| Injection-site reactions | 27 (9.5) | 9 (3.2) |
| Allergic/hypersensitivity reactions | 7 (2.5) | 11 (3.9) |
| Potential anaphylaxis | 0 | 0 |
| Cytopaenias | 5 (1.8) | 11 (3.9) |
| Cerebrocardiovascular events* | 3 (1.1) | 5 (1.8) |
| Malignancies | 0 | 3 (1.1) |
| Depression | 3 (1.1) | 7 (2.5) |
| Inflammatory bowel disease | 2 (0.7)† | 0 |
| Ulcerative colitis | 1 (0.4)‡, | 0 |
| Crohn’s disease | 1 (0.4)§ | 0 |
Safety data were analysed in the safety population at the time of database lock. Of the 566 randomized patients, n=70 completed, n=52 discontinued, and n=444 were ongoing in the open label treatment period at the time of database lock.
*Of eight treatment-emergent cerebrocardiovascular events reported, four (IXE: n=2 (0.7%); ADA: n=2 (0.7%)) were adjudicated.
†EPIdemiologique des Maladies de l’Appareil Digestif (EPIMAD) criteria for adjudication of suspected inflammatory bowel disease define ‘probable’ and ‘definite’ classifications as confirmed cases. Only one case met the EPIMAD criteria of confirmed inflammatory bowel disease.
‡Event was reported as colitis ulcerative and was adjudicated as possible ulcerative colitis.
§Event was reported as colitis and was adjudicated as probable Crohn’s disease.
ADA, adalimumab; IXE, ixekizumab.
Safety data were analysed in the safety population at the time of database lock. Of the 566 randomised patients, n=70 completed, n=52 discontinued and n=444 were ongoing in the open-label treatment period at the time of database lock.
Most infection-related TEAEs were mild or moderate in severity. Serious infections were more frequent in the ADA versus IXE group (see online supplementary table 3). Three patients discontinued due to infection-related AEs, including two in the ADA group (lymph node tuberculosis, pneumonia legionella) and one in the IXE group (arthritis bacterial). There were no confirmed cases of pulmonary tuberculosis. TEAEs of Candida infections were more frequent in the IXE group (n=7; four oral and three genital Candida infections) than the ADA group (n=2; one oral and one genital Candida infection). All Candida-related TEAEs resolved except one (IXE, oral Candida) that was ongoing at the week 24 database lock; none resulted in discontinuation.
Injection-site reactions were more frequent in the IXE versus ADA group; most were mild in severity. One severe injection-site reaction (injection site hypersensitivity) occurred in the ADA group, and one SAE (injection-site rash) occurred in the IXE group. Discontinuations due to injection-site reactions occurred in one IXE-treated and three ADA-treated patients. Most treatment-emergent allergic/hypersensitivity events were mild or moderate in severity, all were nonanaphylactic and none were SAEs. One ADA-treated patient discontinued due to an allergic/hypersensitivity event (hypersensitivity).
One serious treatment-emergent cerebrocardiovascular event occurred in each treatment group (IXE: atrial fibrillation; ADA: myocardial ischaemia). One IXE-treated patient discontinued due to a treatment-emergent cerebrocardiovascular event of bradycardia. One major adverse cerebrocardiovascular event of moderate haemorrhagic stroke occurred in the ADA group; this event was an SAEand did not result in discontinuation.
No treatment-emergent malignancies occurred in the IXE group, and three occurred in the ADA group, two of which were considered by the investigator as SAEs (basal cell carcinoma and rectal neoplasm). No patients discontinued due to malignancy. No TEAEs of cytopaenia were SAEs, and none resulted in discontinuation. No patients had a worsening to grade 3 or 4 neutropenia. There were no suicide or self-injury-related TEAEs in either group. There were no depression-related SAEs, and no patients discontinued due to depression-related TEAEs.
Suspected IBD-related events were adjudicated by an expert panel as defined by the EPIdemiologique des Maladies de l’Appareil Digestif (EPIMAD) criteria for adjudication of suspected IBD, where ‘probable’ and ‘definite’ classifications are considered as confirmed cases.14 Three TEAEs were identified in two IXE-treated patients as suspected IBD. One IXE-treated patient had an event reported as ‘colitis’ that was sent for adjudication, but there was insufficient information to make a definitive classification. The same patient also had an event of ‘colitis ulcerative’, adjudicated as possible ulcerative colitis, which resulted in study discontinuation. Another IXE-treated patient with no prior medical history of IBD had an event reported as ‘colitis’ that was adjudicated as probable Crohn’s disease and was the only case that met EPIMAD criteria for confirmed IBD. No SAEs of IBD occurred, and no TEAEs of potential IBD were reported in the ADA group.
Discussion
Treatment choices for PsA in clinical practice are made between medications that have shown efficacy and sufficient safety in clinical trials. Because comparative clinical trials are rare in PsA, indirect comparisons are often made using meta-analyses. However, head-to-head trials where active agents are compared, rather than an active agent and placebo, offer the highest level of evidence.15–18 The SPIRIT-P1 and OPAL trials (which compared IXE or tofacitinib, respectively, with placebo) included an ADA active reference arm but were not powered for head-to-head comparisons with ADA.11 19 A study (EXCEED 1) comparing replacement of csDMARDs with secukinumab or adalimumab monotherapy is ongoing (NCT02745080). Although both SPIRIT-H2H and EXCEED 1 included bDMARD-naïve patients with inadequate response to csDMARDs, key differences between the studies include blinding (double-blind in EXCEED 1 vs open-label in SPIRIT-H2H) and concomitant csDMARD use (not allowed in EXCEED 1). SPIRIT-H2H is the first completed head-to-head trial comparing two bDMARDs in patients with active PsA and inadequate response to csDMARDs.
Although skin involvement is usually mild in patients with PsA, clinicians and patients judge the impact of a PsA treatment by effects on all domains affected by the disease, in particular joints and skin.20–22 Furthermore, achievement of optimal health-related quality of life, the ultimate treatment goal in PsA, requires improvements in both joint and skin manifestations of the disease.1 Therefore, a combination of two validated and well-established outcome measures, a relatively stringent endpoint for articular disease (ACR50) and a very stringent endpoint for skin disease (PASI100) was employed as the primary endpoint. The 24-week efficacy data from the present study demonstrate that IXE was superior to ADA in simultaneously leading to an ACR50 and PASI100 response, was non-inferior to ADA for achieving ACR50 and was superior to ADA for achieving PASI100. Furthermore, significantly more patients achieved DAPSA remission (which does not include a measure of skin response) with IXE than ADA, suggesting that skin changes were not the only domain contributing to differences between biologics. IXE further demonstrated significantly higher clinical response rates than ADA at week 24 for SPARCC Enthesitis Index, psoriasis (PASI75/90), fingernail psoriasis (NAPSI fingernails change from baseline), mCPDAI and treat-to-target endpoints of MDA, VLDA and DAPSA remission.23 Rapid and significantly greater improvements in skin-related quality of life were also observed (DLQI (0 or 1)). No significant differences were observed between treatment groups for ACR20/50/70, suggesting IXE had similar speed and level of response compared to ADA for joint improvement.
SAEs, especially those related to infections, were numerically higher in the ADA group. Infections were more frequent in the IXE group than the ADA group. Injection site reactions (including injection site pain) were numerically higher in the IXE arm, although most were mild in both groups. Overall, the safety profiles of both bDMARDs were consistent with those described in the prescriber information.
A key strength of the SPIRIT-H2H study is its relevance to real-world clinical settings. The open-label study design and absence of a placebo arm was modelled after real-world clinical settings where patients receive active treatments and are aware of which treatment they receive. Patients were treated with the approved dosing regimens of both IXE and ADA (according to presence/absence of moderate-to-severe psoriasis), as monotherapy or in combination with csDMARDs. Approximately 82% of patients did not meet criteria for moderate-to-severe psoriasis, consistent with the patient population typically seen by rheumatologists.20–22
Although comparisons between clinical studies are limited by differences in design and study population, joint and skin responses for both IXE and ADA were higher in SPIRIT-H2H than in historical studies.10 11 24 The use of two efficacious treatments, open-label study design and lack of a placebo arm may have contributed to increased responses in SPIRIT-H2H, since all patients knew they would receive active therapy. To minimise bias, key outcomes were measured by blinded assessors. However, an expectation of different rates of improvement (especially in skin outcomes) with IXE versus ADA could potentially influence blinded assessors. However, this limitation also exists for double-blind, placebo-controlled studies, where greater response is expected with an active treatment versus a placebo comparator. SPIRIT-H2H is ongoing through 52 weeks of treatment, and the current report is limited to 24 weeks. Thus, it is currently unknown how clinical responses will compare over longer treatment periods. An additional study limitation was the absence of imaging or structural joint damage assessments. Though the patient population in this study is similar to other clinical trials in PsA, it may not represent all patients with PsA in daily clinical practice (eg, patients in this study predominantly had polyarthritis).
In conclusion, IXE was associated with greater improvement of a combined articular and cutaneous endpoint in PsA compared with ADA over a 24-week period and had numerically lower incidence of SAEs compared to ADA.
Acknowledgments
The authors would like to thank Clint Bertram, PhD, a medical writer and employee of Eli Lilly and Company, for writing and editorial support.
Footnotes
Handling editor: David S Pisetsky
Collaborators: The SPIRIT H2H study group are collaborators. The SPIRIT H2H study group includes: Leonardo Naftal, Rodolfo Ariel Pardo Hidalgo, Eduardo Mario Kerzberg, Veronica Gabriela Savio, Alicia Lazaro, Benito Jorge Velasco, Norma Beatriz Verzero, Cecilia Adma Asnal, Eduardo Fabian Mysler, Alberto Berman, Federico Javier Ariel, Maureen Rischmueller, Jane Margaret Zochling, Paul A Bird, Stephen Hall, Andrew Ostor, Evange Romas, Georg Stummvoll, Klaus Machold, Peter Spellitz, Ursula Hanusch, Marc Vanden Berghe, Marc Leon, Johan Louis Magda Vanhoof, Filip Eduard Jeanne Van den Bosch, Kurt Leo Francois De Vlam, Frederic Morin, Louis Bessette, Derek A Haaland, Annette Margrethe Schlemmer, Lars Erik Kristensen, Pentti Jarvinen, Ritva Liisa Peltomaa, Laura Pirila, Jorma Vuotila, Eric Lespessailles, Philippe Goupille, Bernard Combe, Arnaud Constantin, Gregoire Cormier, Celine Cozic, Cyrille B Confavreux, Hendrik Schulze-Koops, Andrea Everding, Micheala Kohm, Siegfried Wassenberg, Magdolna Nagy, Noemi Bakos, Eva Melegh, Edit Drescher, Lalit Duggal, Piyush Narayanbhai Joshi, Srabani Ghosh Zoha, A Ramakrishnam Naidu, Puja Pushkarkumar Srivastava, Vishnu Devkinandan Sharma, Ajit Bapurao Nalawadae, Jyotsna Laxmikant Oak, Sarath Chandra Mouli Veeravalli, Jugal Kishore Kadel, Manisha Ashwin Daware, Yogesh S Marfatia, Sumeet Agarwal, Yolanda Braun Moscovici, Ori Elkayam, Yair Molad, Tatiana Reitblat, Moshe Tishler, Yair Levy, Merav Lidar, Devy Zisman, Antonio Costanzo, Paolo Amerio, Riccardo Meliconi, Rosario Foti, Roberto Perricone, Maurizio Rossini, Elisa Gremese, Beatriz Elena Zazueta Montiel, Juan Manuel Miranda Limon, Marco Antonio Maradiaga Cecena, Sandra Araceli Sicsik Ayala, Miguel Angel Alvarez Guerrero, Jorge Rojas Serrano, Efren Antonio Canul Novelo, Francisco Fidencio Cons-Molina, Ed N Griep, Andrzej Kaszuba, Jolanta Bogna Węgłowska, Barbara Bazela, Maria Rell-Bakalarska, Malgorzata Miakisz, Catherine Elizabeth Spargo, Elsa Margaretha Van Duuren, Asokan Naidoo, Nomawethu Cleopatra Matsiliza, Johannes Breedt, Gareth Scott Tarr, Jenny Potts, Savithree Nayiager, Mahmood Moosa Tar Mahomed Ally, Francisco Javier Blanco Garcia, Antonio Mera Varela, Jose Rosas, Juan Miguel Sanchez Burson, Javier Garcia Miguel, Jon Lampa, Milad Rizk, Giovanni Cagnotto, Per Larsson, Guozhong Fei, Ruediger Mueller-Mar, Andrea Rubbert-Roth, Michael John Nissen, Oleh Iaremenko, Svitlana Anatoliivna Trypilka, Dmytro Rekalov, Mykola Stanislavchuk, Viktoriia Viktorivna Vasylets, Olena Garmish, Svitlana Smiyan, David Walker, Deepak Jadon, Stuart Ralston, Stephen Boyle, Pippa Anne Watson, James E Dale, Sara Carty, Karen Douglas, Christopher J Edwards, David Hutchinson, Nick Barkham.
Contributors: All authors contributed to critical revision of the manuscript and gave final approval for submission for publication. PJM, JSS, FB, EK and HL-S contributed to interpretation of data. PN contributed to study conception, data acquisition and interpretation of data. SLL contributed to study conception, analysis of data and interpretation of data. LL and PE contributed to analysis of data and interpretation of data. HT, MG, SP and PSH contributed to acquisition of data and interpretation of data.
Funding: This study was funded by Eli Lilly and Company, which contributed to study design, data collection, data analysis, data interpretation, manuscript preparation and publication decisions.
Competing interests: PJM reports research grants and personal fees from AbbVie, Amgen, Bristol Myers Squibb, Celgene, Janssen and Lilly; and personal fees from Boehringer Ingelheim, Galapagos, Genentech and Gilead. JSS reports research grants from AbbVie, Astra-Zeneca, Janssen, Lilly, MSD, Novartis, Pfizer and Roche; and personal fees from AbbVie, Amgen, Astra-Zeneca, Astro, BMS, Celgene, Celltrion, Chugai, Gilead, ILTOO, Janssen, Lilly, MSD, Novartis-Sandoz, Pfizer, Roche, Samsung, Sanofi and UCB. FB reports research grants from Pfizer, Janssen, Chugai, Celgene and Roche; personal fees from Pfizer, AbbVie, Sanofi, Lilly, Novartis, Genzyme, Boehringer, Janssen, MSD, Celgene, Roche and Chugai; and investigator fees from Lilly. PN reports research grants and personal fees from AbbVie, BMS, Janssen, Lilly, MSD, Novartis, Pfizer, Celgene, Gilead, Sanofi, UCB and Roche. HT reports research grants and non-financial support from Lilly. MG reports personal fees from AbbVie, Actelion Pharmaceuticals, Akros Pharma Inc, AMGEN Inc, Arcutis Pharmaceuticals Inc, Boehringer Engelheim International GmbH, Bristol-Myers Squibb Company, Celgene corporation, Dermira Inc, Eli Lilly and Company, Galderma, GlaxoSmithKline, Glenmark, Jannsen Inc, LEO Pharma, MedImmune, Merck and Co, Novartis Pharmaceuticals, Pfizer Inc, Regeneron Pharmaceuticals Inc, Roche Laboratories, Sanofi Genzyme, UCB and Valeant Pharmaceuticals Inc. PE has undertaken clinical trials and provided expert advice to Pfizer, MSD, Abbvie, BMS, UCB, Roche, Novartis, Samsung, Sandoz and Lilly; has received consultant fees from BMS, AbbVie, Pfizer, MSD, Novartis, Roche and UCB; and has received research grants paid to his employer from AbbVie, BMS, Pfizer, MSD and Roche. PSH reports research grants, personal fees and non-financial support from AbbVie; research grants from Amgen, Janssen, Pfizer and UCB; and personal fees from Lilly and Galapagos. SLL, LL, EK, HL-S and SP are employees of, and own stock in, Eli Lilly and Company.
Patient consent for publication: Not required.
Provenance and peer review: Not commissioned; externally peer reviewed.
Data availability statement: Lilly provides access to all individual participant data collected during the trial, after anonymization, with the exception of pharmacokinetic or genetic data. Data are available to request 6 months after the indication studied has been approved in the US and EU and after primary publication acceptance, whichever is later. No expiration date of data requests is currently set once data are made available. Access is provided after a proposal has been approved by an independent review committee identified for this purpose and after receipt of a signed data sharing agreement. Data and documents, including the study protocol, statistical analysis plan, clinical study report, blank or annotated case report forms, will be provided in a secure data sharing environment. For details on submitting a request, see the instructions provided at www.vivli.org.
References
- 1. Kavanaugh A, Gottlieb A, Morita A, et al. . The contribution of joint and skin improvements to the health-related quality of life of patients with psoriatic arthritis: a post hoc analysis of two randomised controlled studies. Ann Rheum Dis 2019;78:1215–9. 10.1136/annrheumdis-2018-215003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kavanaugh A, Helliwell P, Ritchlin CT. Psoriatic arthritis and burden of disease: patient perspectives from the population-based multinational assessment of psoriasis and psoriatic arthritis (MAPP) survey. Rheumatol Ther 2016;3:91–102. 10.1007/s40744-016-0029-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gladman DD, Antoni C, Mease P, et al. . Psoriatic arthritis: epidemiology, clinical features, course, and outcome. Ann Rheum Dis 2005;64 Suppl 2:ii14–17. 10.1136/ard.2004.032482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Singh JA, Guyatt G, Ogdie A, et al. . 2018 American College of Rheumatology/National psoriasis Foundation guideline for the treatment of psoriatic arthritis. Arthritis Care Res 2019;71:2–29. 10.1002/acr.23789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gossec L, Smolen JS, Ramiro S, et al. . European League against rheumatism (EULAR) recommendations for the management of psoriatic arthritis with pharmacological therapies: 2015 update. Ann Rheum Dis 2016;75:499–510. 10.1136/annrheumdis-2015-208337 [DOI] [PubMed] [Google Scholar]
- 6. Coates LC, Kavanaugh A, Mease PJ, et al. . Group for research and assessment of psoriasis and psoriatic arthritis 2015 treatment recommendations for psoriatic arthritis. Arthritis Rheumatol 2016;68:1060–71. 10.1002/art.39573 [DOI] [PubMed] [Google Scholar]
- 7. Elmamoun M, Chandran V. Role of methotrexate in the management of psoriatic arthritis. Drugs 2018;78:611–9. 10.1007/s40265-018-0898-2 [DOI] [PubMed] [Google Scholar]
- 8. Vogelzang EH, Kneepkens EL, Nurmohamed MT, et al. . Anti-adalimumab antibodies and adalimumab concentrations in psoriatic arthritis; an association with disease activity at 28 and 52 weeks of follow-up. Ann Rheum Dis 2014;73:2178–82. 10.1136/annrheumdis-2014-205554 [DOI] [PubMed] [Google Scholar]
- 9. Taylor W, Gladman D, Helliwell P, et al. . Classification criteria for psoriatic arthritis: development of new criteria from a large international study. Arthritis Rheum 2006;54:2665–73. 10.1002/art.21972 [DOI] [PubMed] [Google Scholar]
- 10. Mease PJ, Gladman DD, Ritchlin CT, et al. . Adalimumab for the treatment of patients with moderately to severely active psoriatic arthritis: results of a double-blind, randomized, placebo-controlled trial. Arthritis Rheum 2005;52:3279–89. 10.1002/art.21306 [DOI] [PubMed] [Google Scholar]
- 11. Mease PJ, van der Heijde D, Ritchlin CT, et al. . Ixekizumab, an interleukin-17A specific monoclonal antibody, for the treatment of biologic-naive patients with active psoriatic arthritis: results from the 24-week randomised, double-blind, placebo-controlled and active (adalimumab)-controlled period of the phase III trial SPIRIT-P1. Ann Rheum Dis 2017;76:79–87. 10.1136/annrheumdis-2016-209709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. [FDA] Food and Drug Administration Center for Drug evaluation and Research (CDER) Center for Biologics Evaluation and Research (CBER). Guidance for industry, non-inferiority clinical trials to establish effectiveness. Available: https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm202140.pdf [Accessed 12 Feb 2019].
- 13. [CHMP] Committe for Medicinal Products for Human Use. Guidelines on the Choice of the Noninferiority Margin. Available: http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2009/09/WC500003636.pdf [Accessed 12 Feb 2019].
- 14. Gower-Rousseau C, Salomez JL, Dupas JL, et al. . Incidence of inflammatory bowel disease in northern France (1988-1990). Gut 1994;35:1433–8. 10.1136/gut.35.10.1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kirson NY, Rao S, Birnbaum HG, et al. . Matching-adjusted indirect comparison of adalimumab vs etanercept and infliximab for the treatment of psoriatic arthritis. J Med Econ 2013;16:479–89. 10.3111/13696998.2013.768530 [DOI] [PubMed] [Google Scholar]
- 16. McInnes IB, Nash P, Ritchlin C, et al. . Secukinumab for psoriatic arthritis: comparative effectiveness versus licensed biologics/apremilast: a network meta-analysis. J Comp Eff Res 2018;7:1107–23. 10.2217/cer-2018-0075 [DOI] [PubMed] [Google Scholar]
- 17. Nash P, McInnes IB, Mease PJ, et al. . Secukinumab versus adalimumab for psoriatic arthritis: comparative effectiveness up to 48 weeks using a Matching-Adjusted indirect comparison. Rheumatol Ther 2018;5:99–122. 10.1007/s40744-018-0106-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu D, Yue J, Tam L-S. Efficacy and safety of biologics targeting interleukin-6, -12/23 and -17 pathways for peripheral psoriatic arthritis: a network meta-analysis. Rheumatology 2018;57:563–71. 10.1093/rheumatology/kex452 [DOI] [PubMed] [Google Scholar]
- 19. Mease P, Hall S, FitzGerald O, et al. . Tofacitinib or adalimumab versus placebo for psoriatic arthritis. N Engl J Med 2017;377:1537–50. 10.1056/NEJMoa1615975 [DOI] [PubMed] [Google Scholar]
- 20. Busquets-Pérez N, Rodriguez-Moreno J, Gómez-Vaquero C, et al. . Relationship between psoriatic arthritis and moderate-severe psoriasis: analysis of a series of 166 psoriatic arthritis patients selected from a hospital population. Clin Rheumatol 2012;31:139–43. 10.1007/s10067-011-1787-1 [DOI] [PubMed] [Google Scholar]
- 21. Feldman SR, Zhao Y, Shi L, et al. . Economic and comorbidity burden among moderate-to-severe psoriasis patients with comorbid psoriatic arthritis. Arthritis Care Res 2015;67:708–17. 10.1002/acr.22492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Möller B, Stekhoven D, Villiger P. Major differences in the pattern of joint swelling and tenderness in a large psoriatic arthritis cohort – results from an exploratory hierarchical cluster analysis. Arthritis Rheum 2013;65:S141. [Google Scholar]
- 23. Smolen JS, Schöls M, Braun J, et al. . Treating axial spondyloarthritis and peripheral spondyloarthritis, especially psoriatic arthritis, to target: 2017 update of recommendations by an international Task force. Ann Rheum Dis 2018;77:3–17. 10.1136/annrheumdis-2017-211734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nash P, Kirkham B, Okada M, et al. . Ixekizumab for the treatment of patients with active psoriatic arthritis and an inadequate response to tumour necrosis factor inhibitors: results from the 24-week randomised, double-blind, placebo-controlled period of the SPIRIT-P2 phase 3 trial. Lancet 2017;389:2317–27. 10.1016/S0140-6736(17)31429-0 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
annrheumdis-2019-215386supp001.pdf (311.9KB, pdf)