

Abstract
Phospholipids are found throughout the natural world, including the lung surfactant (LS) layer that reduces pulmonary surface tension and enables breathing. Fibrinogen, a protein involved in the blood clotting process, is implicated in LS inactivation and the progression of disorders such as acute respiratory distress syndrome. However, the interaction between fibrinogen and LS at the air-water interface is poorly understood. Through a combined microrheological, confocal and epifluorescence microscopy approach we quantify the interfacial shear response and directly image the morphological evolution when a model LS monolayer is penetrated by fibrinogen. When injected into the subphase beneath a monolayer of the phospholipid dipalmitoylphosphatidylcholine (DPPC, the majority component of LS), fibrinogen preferentially penetrates disordered liquid expanded (LE) regions and accumulates on the boundaries between LE DPPC and liquid condensed (LC) DPPC domains. Thus, fibrinogen is line active. Aggregates grow from the LC domain boundaries, ultimately forming a percolating network. This network stiffens the interface compared to pure DPPC and imparts the penetrated monolayer with a viscoelastic character reminiscent of a weak gel. When the DPPC monolayer is initially compressed beyond LE-LC coexistence, stiffening is significantly more modest and the penetrated monolayer retains a viscous-dominated, DPPC-like character.
Graphical Abstract
The shear response of a phospholipid monolayer penetrated by fibrinogen is determined by the formation of a domain-templated protein network.
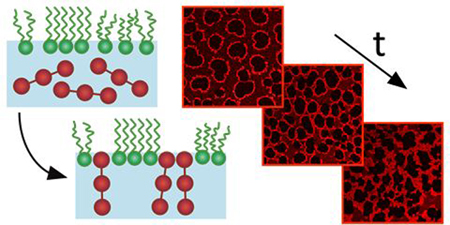
1. Introduction
The adsorption of a soluble surface-active species into a pre-existing insoluble Langmuir monolayer is known as monolayer penetration.1,2 Understanding this phenomenon is of central importance to diverse industries as well as lung physiology. Foodstuffs, for instance, often contain mixtures of surface active proteins and lipids3,4 whose interactions at oil-water or air-water interfaces determine the stability and texture of the emulsions and foams that are ubiquitous in processed foods.5–7 In a biological context, penetration of bilayer membranes by amphiphilic molecules such as peptides or saponins can render a vesicle or cell leaky, facilitating drug release or transmembrane transport.8–10 Phospholipid monolayers are commonly studied as a proxy for a single bilayer leaflet11–13 in order to understand the effect of soluble surfactant molecules on the mechanical, structural and chemical properties of biomembranes.14–18
Phospholipids compose the majority of the lung surfactant (LS) monolayer at the alveolar hypophase interface. This multicomponent mixture of lipids and proteins reduces pulmonary surface tension, lowering the energetic cost of breathing.19–21 Following lung trauma, LS can become inactivated and unable to sufficiently reduce surface tension, resulting in the onset and progression of Acute Respiratory Distress Syndrome (ARDS).19,20,22 The effects of ARDS include impaired gas exchange, decreased lung volume, alveolar collapse and, ultimately, systemic oxygen starvation.23–27 ARDS is fatal in 30–40% of cases.28
Many physicochemical factors contribute to LS inactivation, and it is unlikely that a single “cause” of ARDS can be identified.26,29 However, the alveoli of ARDS patients typically contain elevated levels of various surface-active contaminants compared to healthy lungs.22,30,31 The inflammation that accompanies ARDS increases capillary permeability, allowing the influx of serum proteins such as fibrinogen and albumin.19,20,22 Many serum proteins are surface-active and compete or interact with LS, altering its ability to lower surface tension and affecting lung compliance, thereby making a mechanical contribution to the development of ARDS.31–35 In particular, fibrinogen, a surface-active blood protein responsible for clot formation, is implicated in LS inhibition32,34,36–40. We have recently shown that fibrinogen can penetrate model LS monolayers, altering their mechanical properties and morphology.41 The rheological response of a material is often determined by its mesoscopic structure,42 and so here we examine penetration of model LS monolayers by fibrinogen using a combination of epifluorescence and laser scanning confocal microscopy. The structural insight gained through microscopy allows us to interpret differences in the rate-dependent interfacial shear response pre- and post-penetration. These new rheological measurements are consistent with our previous work. Building upon these results, the primary focus and novelty of the present work lies in confocal microscopy revealing the formation of a interfacial protein gel during monolayer penetration, and the interpretation of the rheological measurements in terms of this structure.
2. Methods
The most abundant molecule in LS is the phospholipid dipalmitoylphosphatidylcholine (DPPC),21 and this serves as our model lung surfactant. DPPC (Avanti Polar Lipids Ltd.) is diluted in HPLC grade chloroform (Sigma Aldrich) and monolayers are prepared by spreading these solutions at the air-saline interface in Langmuir troughs mounted on either a custom made brightfield/epifluorescence microscope or a commercial laser scanning confocal microscope (Nikon Eclipse 80i fitted with a C1 confocal scan head). The subphase is phosphate buffered saline (PBS, Sigma Aldrich) containing 0.01 M phosphate buffer, 0.0027 M potassium chloride and 0.137 M sodium chloride. After spreading DPPC from chloroform solution, 20 minutes are allowed for solvent evaporation and equilibration before the monolayer is compressed at maximum rate of 0.08 cm2 s–1. The surface pressure, 𝛱, the reduction in surface tension compared to the clean buffer-air interface, is monitored using a filter paper Wilhelmy plate tensiometer (Riegler and Kirstein). Fibrinogen from bovine plasma (Sigma Aldrich) is purchased as powder and dissolved in PBS. Depending on the experiment, fluorescence contrast is provided by either a labelled lipid [Texas Red DHPE (Texas Red 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine) supplied by Life Technologies,43,44 mixed with DPPC at 0.5 wt %], or labelled Alexa Fluor 594 conjugated fibrinogen from human plasma, supplied by ThermoFisher Scientific. The labelled fibrinogen is mixed with unlabelled bovine fibrinogen solutions at 1 wt %. There are an estimated 15 Alexa Fluor fluorescent groups per fibrinogen molecule. The molecular weight of fibrinogen is ~ 340 kDa, so labelling is not expected to change fibrinogen solubility or interactions. All experiments are performed at room temperature.
The interfacial shear response is measured using magnetic microbutton microrheometry, as described at length elsewhere41,45–47 and illustrated schematically in Fig. 1 (a) and (b). A lithographically fabricated probe of radius a = 50μm, consisting of layers of photoresist, nickel, gold and thiol, is placed at the buffer-air interface, where it remains pinned as a result of the hydrophobic/hydrophilic properties of its thiol and photoresist faces. The nickel layer renders the probe ferromagnetic, and so it rotates to align itself with an applied magnetic field. Using a pair of electromagnets connected in series, a sinusoidal magnetic torque is applied, and the corresponding rotational probe response is measured by tracking its “buttonholes” in brightfield microscopy. The Boussinesq number, Bo = 𝜂s/𝜂a, quantifies the relative contributions of interfacial and subphase drag, where 𝜂 is the subphase viscosity and 𝜂s is the interfacial viscosity. For Bo ≫ 1, interfacial drag dominates and the amplitude and phase lag of the probe motion relative to a known magnetic field give the interfacial moduli G’s and G”s characterising the interfacial elastic and viscous response, respectively.41,45,46
Figure 1.

(a) Schematic representation of magnetic microbutton microrheology. A sinusoidal torque is applied to a 100μm diameter amphiphilic, ferromagnetic probe by a pair of electromagnets (EM1 & EM2) connected in series. Not to scale. (b) Side-view schematic showing layered structure of microbutton probe adsorbed at buffer-air interface within a phospholipid monolayer. Not to scale. (c) Schematic representation of ordering on compression. LC domains nucleate within the LE continuum, appearing dark in fluorescence micrographs. At high surface pressure, monolayer consists of tightly packed condensed domains (d-f) Fluorescence micrographs showing DPPC monolayer in LE state (d), LE-LC coexistence (e) and LC state (f) The residual fluorescent dye accumulates at the LC grain boundaries. Scale bars are 50μm.
The rheological measurements presented here follow our previously established protocol.41 The microbutton is deposited at the clean buffer-air interface and its magnetic moment is calibrated before DPPC is spread or fibrinogen is injected. During fibrinogen adsorption or monolayer penetration, the magnetic torque amplitude is manually controlled to maintain visible but small probe oscillation. Subsequently, in frequency sweep experiments, the magnetic torque amplitude is maintained such that the angular strain amplitude is <1.5°.
3. Results
In the absence of fibrinogen, the morphology of a DPPC monolayer is well established.48,49 Figure 1c to 1f illustrate the evolution of a DPPC monolayer upon compression with cartoons (Fig. 1c) and a series of epifluorescence micrographs (Fig. 1d, to 1f), where fluorescence contrast is provided by Texas red DHPE. At a molecular area of ~100 Å2 at room temperature (the morphological behaviour of DPPC depends strongly on temperature50–52), DPPC causes a measurable increase in surface pressure, 𝛱, (decrease in surface tension) and exists in a liquid expanded (LE) state characterised by translational and orientational disorder. The fluorescently labelled lipid distributes homogenously throughout this disordered phase so LE regions appear uniformly bright in epifluorescence micrographs (Fig. 1d). Upon compression, surface pressure rises and liquid condensed (LC) DPPC domains nucleate within the LE continuum. Molecules in these domains have local positional and long-ranged orientational order with a regular headgroup packing and registration between adjacent tails.53 This crystalline packing excludes the fluorophore and so LC domains appear dark (Fig. 1e), adopting a characteristic “bean” shape which is a signature of the underlying chirality of the DPPC molecule.48,53,54 Further compression increases the area fraction of condensed domains and packs them together, creating a mesoscopically granular texture (Fig. 1f). The fluorescent lipid is expelled to the LC domain boundaries, delineating the shapes and sizes of the LC domains.
Previous work has shown that the extent to which fibrinogen can penetrate a DPPC monolayer and the consequent modification of its interfacial shear response depends strongly on the degree to which the initial DPPC monolayer is compressed, and therefore, presumably, the initial monolayer morphology.41 Through monitoring the evolution of surface pressure and the interfacial viscoelastic moduli at 1 Hz, we have previously shown that fibrinogen has a larger impact on the rheology of monolayers at lower surface pressures in which there is an appreciable fraction of LE phase. The post-penetration, mixed monolayer retains a DPPC-like, viscous dominated shear response at 1 Hz when the initial monolayer is at high surface pressure, in its LC state. However, a material’s shear response is typically rate-dependent,42 so here we extend our previous investigation by shearing the post-penetration, end-state monolayer at range of frequencies and measuring its response. We further augment previous insights by directly visualising fluorescently labelled fibrinogen during monolayer penetration. Through comparing micrographs of fluorescent fibrinogen acquired via confocal microscopy with epifluorescence micrographs showing the labelled lipid, similar to those in Fig. 1d to 1f, we reveal the evolution of monolayer morphology during penetration and relate this to the rheological properties of the end-state.
A DPPC monolayer is formed at the buffer-air interface and compressed to initial surface pressure 𝛱i, after which, trough surface area, A, is held constant. Initial surface pressures for which the DPPC monolayer is at LE-LC coexistence, at the onset of the coexistence plateau in the pressure-area isotherm (~4 to 6 mN/m at room temperature), and initial surface pressures well above coexistence, where the DPPC monolayer consists of closely packed LC domains (~15 mN/m) are considered separately. 0.5 mL of fibrinogen solution is injected into the subphase to give a concentration c ≈ 0.007 mg/mL. Penetration is allowed to proceed for more than 5 hours, after which time the surface pressure reaches a plateau at 𝛱f and the end-state is established. The end-state rheological response is measured at frequencies between 0.1 and 4 Hz, where the upper limit in frequency is set by the frame rate of video acquisition. This frequency sweep rheometry distinguishes these measurements from our previous work41 which focused exclusively on the shear response at 1Hz.
These data are shown as red points in Fig. 2a and 2b. Triangles represent the interfacial storage modulus, G’s, characterising the elastic response, and squares show the interfacial loss modulus, G”s, encoding viscous behaviour. Figure 2a shows the frequency sweep rheology of the end-state mixed monolayer when the initial DPPC monolayer is in its LE-LC coexistence region at 𝛱i = 4 mN/m. Figure 2b shows the same data for a DPPC monolayer initially in the LC phase at 𝛱i = 15 mN/m. In both cases, the post-penetration data are compared to the response of pure DPPC monolayers at the post-penetration surface pressure, 𝛱f. Figure 2c and 2d show the ratio G’s/G”s (the inverse of the loss tangent) corresponding to the data plotted in 2a and 2b. G’s/G”s ≪ 1 (≫ 1) represents a viscous-dominated (elastic-dominated) shear response while G’s/G”s ≈ 1 indicates a mixed viscoelastic character.
Figure 2.

Interfacial shear rheology. (a,b) Interfacial viscoelastic moduli, G’s (triangles) and G”s (squares), as a function of frequency, for pure DPPC monolayers (open green) and DPPC monolayers penetrated by fibrinogen (solid red), 5 hours after fibrinogen injection. Solid (dashed) lines show linear or power law fits to G”s (G’s). (a) Monolayer penetration initiated at 𝛱i = 4 mN/m (LE-LC coexistence), resulting in end-state 𝛱f = 9.5 mN/m, compared with pure DPPC at 𝛱 = 9.4 mN/m. (b) Monolayer penetration initiated at 𝛱i = 15 mN/m (LC DPPC), resulting in end-state 𝛱f = 20.3 mN/m, compared with pure DPPC at 𝛱 = 20 mN/m. Inset shows |Gs| at 1 Hz as a function of 𝛱 in these 4 experiments. (c) and (d) Ratios G’s/G”s for data in (a) and (b). All points are the average of ≥10 measurements and error bars on G’s and G”s represent standard deviation.
The rheology of DPPC monolayers without fibrinogen is qualitatively similar at both 𝛱 = 9.4 mN/m (Fig. 2a) and 𝛱 = 20 mN/m (Fig. 2b). G’s is relatively constant over the frequency range considered while G”s increases linearly with frequency (solid green lines show linear fits to G”s for pure DPPC). Moreover, at both surface pressures, G”s ≫ G’s indicating that DPPC is viscous dominated. The similarity is unsurprising when one appreciates that the pure DPPC monolayer is in its LC state at both surface pressures. The constant surface shear elasticity G’s and surface shear viscosities 𝜂s = G”s /ω are consistent with previous measurements of DPPC surface rheology, which was argued by analogy with compressed emulsions. The constant surface elasticity was attributed to the line tension λ surrounding close-packed domains of characteristic size R via G’ ~λ/R, and the surface viscosity 𝜂s reflected Newtonian viscous dissipation within domains as they deformed.46,49
By contrast, the addition of fibrinogen increases both G’s and G”s relative to pure DPPC at both surface pressures. This increase is far larger when the initial monolayer is at coexistence. Fig. 2a shows the fibrinogen penetrated DPPC monolayer moduli (red) are ~3 orders of magnitude larger than the pure DPPC moduli at 𝛱 = 9 mN/m (green). When the initial DPPC monolayer is fully in the LC phase, the relative increase in the moduli is more modest - only around half an order of magnitude (Fig. 2b). If the relative magnitude of each data set is taken as a measure of interfacial stiffness, increasing the surface pressure increases the stiffness of the pure DPPC monolayer, which is the case for all other lipid monolayers examined.46,49,55 However, for the penetrated DPPC monolayers, increasing the initial surface pressure decreases the stiffness of the end-state mixed monolayer (Fig. 2b). The mixed monolayer formed by penetrating DPPC at coexistence (and low surface pressure) is stiffer than that formed by penetrating DPPC in the single LC phase at higher surface pressure. This is illustrated in the inset to Fig. 2b where the magnitude |GS| at 1 Hz is shown as a function of surface pressure. G’s/G”s ≪ 1 for all monolayers except for the fibrinogen penetrated DPPC monolayer at 9.5 mN/m, in which G’s/G”s ≈ 1. In both cases, the observed increase in moduli is connected to the presence of fibrinogen. LE DPPC is more penetrable to fibrinogen than LC DPPC and we hypothesize that more fibrinogen enters the interface when the initial monolayer is at coexistence and thus the two monolayers penetrated under different initial conditions have distinct molecular compositions in their end states.
Fig. 3 shows that fibrinogen, by itself, adsorbs from solution to a pristine interface to form monolayers that are too stiff to measure using the microbutton rheometer (|GS|> 103 μN/m).41 However, the inset to Fig. 3 shows that, except in the very early stages of adsorption, the fibrinogen monolayer has a strongly elastic character with G’s/G”s ≫ 1. Therefore, a mixed monolayer containing a greater proportion of fibrinogen is likely to be both stiffer and to have an enhanced elastic response.
Figure 3.

(a) Interfacial viscoelastic moduli G’s (open symbols) and G”s (filled symbols) measured at 1 Hz as a function of surface pressure during fibrinogen adsorption to the clean saline/air interface from solution at concentration 0.004 ± 0.001 mg/mL in 3 nominally identical experiments, each indicated by a different symbol. Time proceeds in direction of blue arrow. In all experiments, the maximum measurable modulus, Gmaxs ~ 103 μN/m, is eventually exceeded. Inset shows the evolution of the ratio G’s/G”s as a function of 𝛱.
For the initially LC monolayer shown in Fig. 2b, the end-state rheology remains viscous dominated across the entire frequency range, with G”s ≫ G’s. As with pure DPPC at comparable surface pressure, G”s varies linearly with frequency, indicating a constant (Newtonian) surface shear viscosity in this frequency range. The surface elastic modulus G’s is approximately constant in pure DPPC, unlike in the penetrated monolayer, where G’s appears to increase as a power law with frequency. Fibrinogen causes a small increase in the elastic response, although the monolayer remains primarily viscous. This is more clearly visible in Fig. 2d. These data support our previous claim41 that penetration of a LC DPPC monolayer by fibrinogen results in a mixed monolayer that retains a primarily DPPC-like rheological response. The small increases in stiffness and elastic response are consistent with a small amount of fibrinogen having entered the monolayer. However, we note that caution must be exercised when the phase angle of probe response with respect to applied torque is close to 90° as small changes in response are interpreted as large changes in G’s.
Conversely, when the initial DPPC monolayer is at coexistence (Fig. 2a), the end-state G’s intersects G”s in the vicinity of 0.5 Hz, and G’s ~ G”s across the full range of frequencies. Both the elastic and viscous response of the monolayer increase by orders of magnitude compared to the pure DPPC monolayer. In this mixed monolayer, G”s is no longer linear in frequency, but instead increases less steeply, following G”s ~ ω0.7. The surface shear elastic modulus G’s exhibits a weaker power-law dependence on frequency G’s ~ ω0.3. Unlike pure LC-DPPC and penetrated LC-DPPC, which act like concentrated 2D emulsions46,49 (meaning both surface shear viscosity and surface shear elasticity are constant), the rheology of LE/LC coexistence DPPC changes qualitatively following fibrinogen penetration. Penetrated LE/LC coexistence becomes more significantly viscoelastic, with a power-law dependence on frequency that is also seen in aggregating colloidal gels,56–58 with the caveat that our measurements are limited to only 1–2 decades in frequency. Fibrinogen penetration has far more profound consequences for LC/LE coexistence DPPC (Fig. 2a) than for LC DPPC (Fig. 2b), as reflected in the ratio G’s/G”s (Fig. 2c). These rheological data are consistent with our previous measurements made at a single frequency41.
We hypothesize that disordered LE DPPC is more easily penetrated by fibrinogen than ordered LC DPPC, so that mixed monolayers formed by penetrating DPPC at coexistence contains proportionally more fibrinogen than mixed monolayers formed by penetrating LC DPPC. The interfacial area occupied by fibrinogen following penetration may be estimated by assuming the lipid and protein interact purely by area exclusion, and that fibrinogen effectively compresses the insoluble DPPC along its single-component pressure-area isotherm.41 The area occupied by each species is then a function of the initial and final surface pressures. Under this approximation, fibrinogen is estimated to occupy 35% of the end-state mixed monolayer area in Fig. 2a and only 4% in Fig. 2b. Given that pure fibrinogen forms very stiff, elastic-dominated monolayers, this excess of fibrinogen may explain the differences between the two penetration scenarios.
Direct imaging using confocal fluorescence microscopy confirms this hypothesis. Figure 4 shows the evolution of 𝛱 (at constant interfacial area) and morphology when fibrinogen is injected beneath a DPPC monolayer in the LE-LC coexistence region in three experiments. Two of these experiments use fluorescently labelled fibrinogen (red curves in Fig. 4a and micrographs in 4c to 4f and 4g to 4j). A complementary experiment uses fluorescently labelled lipid (teal curve in 4a and micrographs in 4k to 4n). The micrographs shown in 4c to 4n correspond to the labelled points on the (t) curves in 4a.
Figure 4.
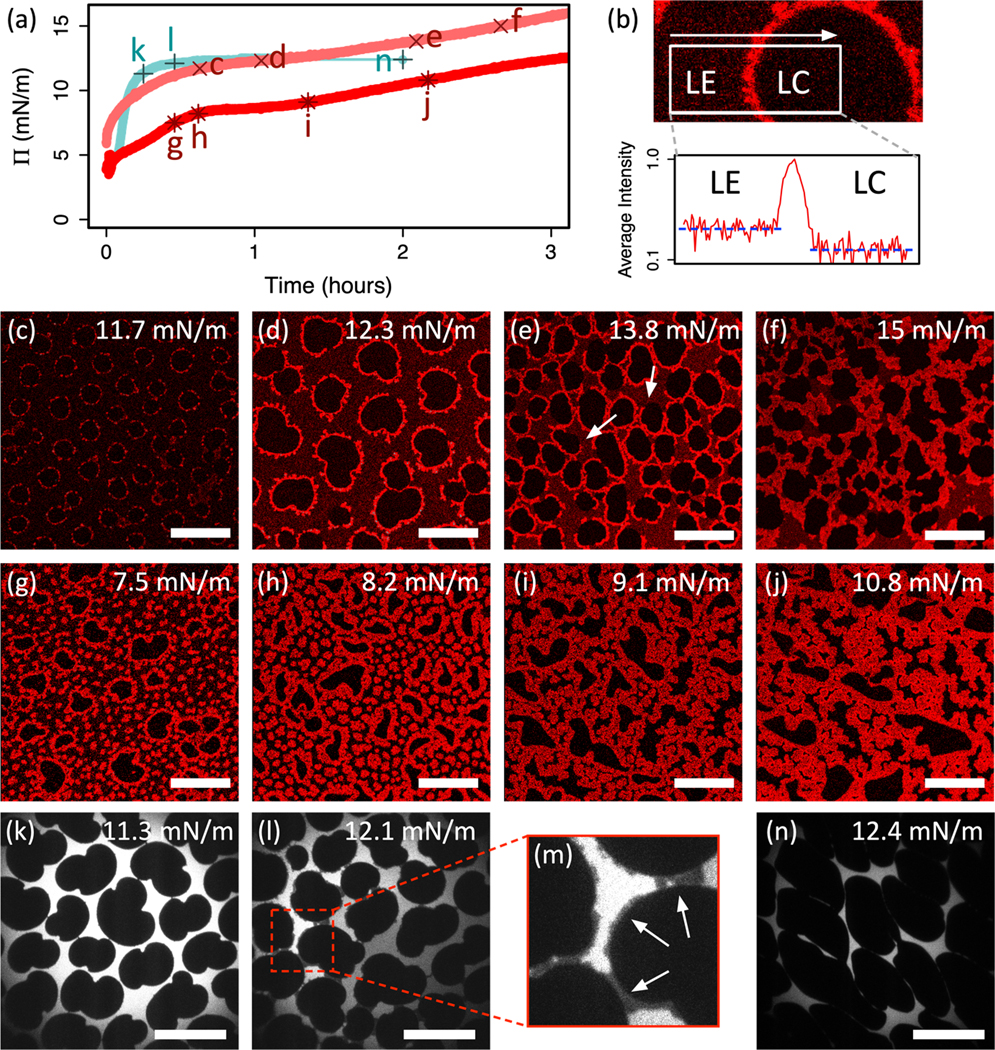
(a) Surface pressure as a function of time for 3 DPPC monolayers initially at coexistence penetrated by fibrinogen. Fibrinogen injected into subphase at t = 0. Red curves represent confocal imaging experiments. Teal curve represents epifluorescence imaging experiment. Pluses, crosses and stars indicate points corresponding to micrographs in (c) to (n) and are labelled accordingly. (b) Section of confocal micrograph focusing on interface between LE and LC regions. Plot shows average horizontal intensity profile (logarithmic scale) within the white box, crossing the LE-LC interface. (c) to (f) Confocal micrographs showing labelled fibrinogen corresponding to red crosses c-f in (a). (g) to (j) Confocal micrographs showing labelled fibrinogen corresponding to red stars g-j in (a). (k) to (n) Epifluorescence micrographs showing labelled lipid corresponding to teal stars k-n in (a). (m) is enlarged section of (l) highlighting interdomain bridging (arrows). Scale bars are 50 μm.
Figure 4a shows a rapid initial increase in 𝛱 on injecting fibrinogen. This subsequently decelerates and approaches a plateau at 𝛱f ~ 10 – 15 mN/m over a timescale of hours. We attribute the difference in adsorption timescales between the confocal (red) and epifluorescence (teal) experiments to Langmuir troughs with different surface area-to-volume ratios. After ~30 minutes, a fluorescent signal is detected in confocal experiments using labelled fibrinogen. At this early stage, fibrinogen accumulates at the boundaries of dark LC DPPC domains (Fig. 4c) along with a lower concentration homogeneously distributed throughout the LE phase, which is faintly red as compared to the LC domains that remain black (Fig. 4b). In addition to the fibrinogen accumulating at the LE-LC boundaries, aggregates of fibrinogen (bright red circular spots) nucleate within the LE phase and co-exist with the fibrinogen distributed throughout the LE phase (faint red) (Figs. 4g and 4h). No fibrinogen appears within the LC domains, which remain black in the images.
As penetration proceeds, fibrinogen increases the surface pressure at constant trough area, and consequently, DPPC domains grow (note the increase in domain size between Fig. 4c and 4d), as they do when surface pressure is increased by decreasing the trough area for pure DPPC monolayers (Figs. 1d to 1f). However, domain growth competes with fibrinogen adsorption in the LE phase, until eventually the LC domains are entirely enclosed in bright fibrinogen-rich layers (Fig. 4d and 4g). The dark regions in Fig. 4d exhibit the characteristic bean-shape of pure DPPC LC domains (compare to Fig. 1e), and the fluorescence micrographs showing labelled lipid in Fig. 4k and 4l clearly show characteristic LC domain morphologies, supporting the assertion that the dark regions in confocal micrographs are LC DPPC domains with minimal fibrinogen.
Fibrinogen continues to accumulate at the domain boundaries, which grow dendritically until they contact and begin to merge, forming inter-domain bridges (Fig. 4e). Bridging is also visible as darker grey regions in fluorescence micrographs of labelled lipid (see Fig. 4l and detail in 4m). In Fig. 4l, distinct LE regions have different intensities, indicating that the fluorescent lipid cannot exchange between these regions. Fig. 4e and 4j show that the fibrinogen bridges the domains at higher surface pressures, thereby disconnecting the previously continuous LE phase (Fig. 4l). In the experiment shown in Fig. 4g to 4j, interdomain bridging is accompanied by the growth of the isolated fibrinogen-rich spots in the LE regions, and in some cases boundary-accumulated fibrinogen merges with these isolated aggregates (Fig. 4i). Furthermore, at the arrows in Fig. 4e, boundary-accumulated fibrinogen partially unwraps from LC domains, indicating that the fibrinogen aggregates bind more strongly to one another than they do to the LE-LC boundary line.
After sufficient penetration, a percolating fibrinogen network is formed throughout the continuous LE phase. Regions of intermediate brightness persist between the bright fibrinogen network and the dark LC DPPC domains, presumably composed of a mixture of LE DPPC and fibrinogen (Figs. 4e and 4i). Following percolation, the mixed monolayer ages and the fibrinogen network coarsens as LC domains deform and the interfacial area occupied by mixed LE-DPPC and fibrinogen decreases (Fig. 4f, 4j and 4n). This is the end-state in which a fully formed fibrinogen network has filled the LE phase regions.
Confocal imaging of fluorescently labelled fibrinogen penetrating an initially LC DPPC monolayer at 𝛱i = 15 mN/m proved more challenging, with significantly lower light intensity and low contrast images that suggest very little fibrinogen penetrates into the condensed monolayer. Imaging thus requires increased laser power and high gain, which results in fast photobleaching and low contrast images. For completeness, sample micrographs showing penetration of an initially LC DPPC monolayer are included in the Supplementary Material.
Figure 4 reveals an affinity between interfacially-adsorbed fibrinogen and LE-LC domain boundaries. Fibrinogen might therefore acts as a ‘lineactant’59 in LE-LC DPPC monolayers. The preference of certain molecules for a one-dimensional phase boundary in a two-dimensional system is well documented. For instance, cholesterol exhibits line activity when mixed with DPPC, producing elongated and spiralled domains of significantly increased perimeter:area ratio compared to pure DPPC.55,60 Similarly, the protein annexin A1 preferentially adsorbs at LE-LC DPPC phase boundaries, and forms percolating networks in an analogous manner to that shown in Fig. 4.61 Myelin Basic Protein adsorbs at the boundaries of lipid monolayers and acts to fill in defects in the monolayer62. If fibrinogen does function as a lineactant, however, it does not appear to lower the LC-LE line tension, at least insofar as the 2D emulsion analogy would predict. In particular, we would expect the surface shear elasticity Gs’ ~λ/R to decrease with reduced line tension, whereas Gs’ increases with penetration, in all cases reported here. Even if penetration does impact line tension, its dominant mechanical effect appears to be the elastic network it forms, which overwhelms the line tension contributions of the pure DPPC monolayer.
4. Discussion & Conclusions
Combining insights from direct imaging using complementary microscopy techniques facilitates the development of a coherent picture of the morphological evolution due to fibrinogen penetrating a DPPC monolayer at LE-LC coexistence. Such complete insight would be difficult to obtain from either technique alone. Fibrinogen penetrates disordered LE DPPC regions, and, at constant interfacial area, increases the surface pressure, which in turn, causes the growth of LC domains. Fibrinogen does not penetrate LC domains at any surface pressure. The adsorbed fibrinogen preferentially accumulates at the boundary between the LC domains and the continuous LE phase. As adsorption proceeds, fibrinogen also forms isolated aggregates in the LE phase. These aggregates grow dendritically and eventually merge, forming bridges between neighbouring LC domains, ultimately developing an interconnected fibrinogen network templated around the LC domains. The later stages of evolution see this network coarsen, reducing the area occupied by the LE phase and distorting the shapes of the LC domains.
The formation of this fibrinogen network is responsible for the greatly increased elastic and viscous moduli as shown in Fig. 2a and 2b. Frequency sweep rheometry shows that the fibrinogen network results in interfacial viscoelastic moduli G’s ~ G”s, with a crossover in the vicinity of 0.5 Hz. Although G’s and G”s are of comparable magnitude across the accessible frequency range, G’s is slightly larger below and G”s is slightly larger above the crossover frequency. This is consistent with the reported rheology of gels formed from weakly interacting constituents including colloidal carbon black,56 thermoresponsive microgels57, polystyrene spheres suspended in a nematic liquid crystal,63 and interfacial aggregates of charged polystyrene colloids.64 The formation of interfacial gels of colloidal particles64,65 and proteins66–69 is well documented. What is unique about the multispecies protein-lipid system is the templating of gel structure around LC domains. Due to attractive interactions, the surface shear rheology of such materials is typically elastic-dominated with a yield stress associated with bond breaking.65 Our system will also presumably yield under sufficiently large shear stresses, although the microbutton technique cannot exert large enough torques to test this hypothesis.
Physiological lung surfactant operates at surface pressures in the range 50 mN/m to 70 mN/m,20,21 which is significantly higher than the surface pressures considered in this study. However, lung surfactant typically consists of approximately 25% unsaturated phospholipid, which are excluded from LC domains,21,70 meaning that disordered expanded regions persist even at these high surface pressures in the lung.71–74 Furthermore, physiological temperatures are elevated compared to room temperature, and LE-LC coexistence in pure DPPC occurs at higher surface pressures as temperature is increased.51,52,75 Our data suggest that it is monolayer morphology and the differential penetrability of expanded and condensed lipid regions that govern monolayer penetration and morphology evolution. Since the distinction between expanded and condensed regions persists in vivo at physiological temperatures and surface pressures, it is plausible that the mechanisms we describe are active in the lung.
In conclusion, microrheological measurements show that the shear response of a DPPC monolayer penetrated by fibrinogen depends strongly on its initial morphology. Initially LC monolayers retain a viscous dominated, DPPC-like shear response, with only a small enhancement in the elastic response compared to a pure DPPC monolayer at comparable surface pressure. By contrast, monolayers at LE-LC coexistence exhibit a much greater overall stiffening, and a more significant enhancement in their elastic response, resulting in an overall mixed viscoelastic rheological character in the end-state. Confocal and epifluorescence microscopy reveal that fibrinogen more readily penetrates LE regions than it does LC domains, accumulating preferentially at the LE-LC borders, eventually resulting in a sample-spanning fibrinogen network. The formation of this network is responsible for the greatly increased elasticity in the penetrated monolayer, which is similar to the increase in elasticity for monolayers of pure fibrinogen (Fig. 3). This mechanism of preferential penetration in disordered regions followed by aggregation and network formation templated on ordered domains, appears to be a general feature of monolayer penetration by proteins.55,59–62
This work also suggests how the presence of fibrinogen in the alveolar fluids during ARDS might negatively affect lung surfactant performance. During the rapid expansion of the lung alveolar interface, fibrinogen may adsorb to the lung surfactant monolayer as the surface pressure of the monolayer decreases. If fibrinogen adsorption persists and a network structure forms, as we show in Fig. 4, the monolayer will stiffen and become elastic and it will be difficult for the monolayer to spread to cover the expanding interface on subsequent inhalations. This likely will result in increased interfacial tension in the alveoli and decreased lung compliance, symptoms consistent with the progression of ARDS.
Supplementary Material
Acknowledgements
The authors express their gratitude to Amit K. Sachan, Sourav Barman, Cain Valtierrez-Gaytan and Benjamin L. Stottrup for valuable advice and guidance with the confocal microscope. Research reported in this publication was supported by the National Heart, Lung, & Blood Institute of the National Institutes of Health under grant numbers R01HL135065 and R01HL51177 (IW and TMS) and the National Science Foundation grant CBET 170378 (JAZ). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Conflicts of interest
There are no conflicts to declare.
Notes and references
- 1.Schulman JH and Rideal EK, Proceedings of the Royal Society B, 1937, 122, 29–45. [Google Scholar]
- 2.Schulman JH and Stenhagen E, Proceedings of the Royal Society B, 1938, 126, 356–369. [Google Scholar]
- 3.Dickinson E, Annual Review of Food Science and Technology, 2015, 6, 211–233. [DOI] [PubMed] [Google Scholar]
- 4.McClements DJ, Current Opinion in Colloid & Interface Science, 2017, 28, 7–14. [Google Scholar]
- 5.Bos MA and van Vliet T, Advances in Colloid and Interface Science, 2001, 91, 437–471. [DOI] [PubMed] [Google Scholar]
- 6.Carrera Sánchez C, Cejudo Fernández M, Rodríguez Niño MR and Rodríguez Patino JM, Langmuir, 2006, 22, 4215–4224. [DOI] [PubMed] [Google Scholar]
- 7.Cejudo Fernández M, Carrera Sánchez C, Rodríguez Niño MR and Rodríguez Patino JM, Food Hydrocolloids, 2007, 21, 906–919. [Google Scholar]
- 8.Jacob MC, Favre M. and Bensa J-C, Cytometry, 1991, 12, 550–558. [DOI] [PubMed] [Google Scholar]
- 9.Mayer LD, Krishna R, Webb M. and Bally M, Journal of Liposome Research, 2000, 10, 99–115. [Google Scholar]
- 10.Foerg C. and Merkle HP, Journal of Pharmaceutical Sciences, 2008, 97, 144–162. [DOI] [PubMed] [Google Scholar]
- 11.Kimelberg HK and Papahadjopoulos D, Biochimica et Biophysica Acta, 1971, 233, 805–809. [DOI] [PubMed] [Google Scholar]
- 12.Feng S, Langmuir, 1999, 15, 998–1010. [Google Scholar]
- 13.Brockman H, Current Opinion in Structural Biology, 1999, 9, 438–443. [DOI] [PubMed] [Google Scholar]
- 14.Saint-Pierre-Chazalet M, Fressigné C, Billoudet F. and Pileni MP, Thin Solid Films, 1992, 210, 743–746. [Google Scholar]
- 15.Charron JR and Tilton RD, Langmuir, 1997, 13, 5524–5527. [Google Scholar]
- 16.Hussain H, Kerth A, Blume A. and Kressler J, The Journal of Physical Chemistry B, 2004, 108, 9962–9969. [Google Scholar]
- 17.Ferri JK, Miller R. and Makievski AV, Colloids and Surfaces A: Physicochemical and Engineering Aspects, 2005, 261, 39–48. [Google Scholar]
- 18.Eeman M, Berquand A, Dufrêne Y, Paquot M, Dufour S. and Deleu M, Langmuir, 2006, 22, 11337–11345. [DOI] [PubMed] [Google Scholar]
- 19.Zuo YY, Veldhuizen RAW, Neumann AW, Petersen NO and Possmayer F, Biochimica et Biophysica Acta, 2008, 1778, 1947–1977. [DOI] [PubMed] [Google Scholar]
- 20.Zasadzinski JA, Stenger P, Shieh I. and Dhar P, Biochimica et Biophysica Acta, 2010, 1798, 801–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parra E. and Pérez-Gil J, Chemistry and Physics of Lipids, 2015, 185, 153–175. [DOI] [PubMed] [Google Scholar]
- 22.Günther A, Ruppert C, Schmidt R, Markart P, Grimminger F, Walmrath D. and Seeger W, Respiratory Research, 2001, 2, 353–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pison U, Herold R. and Schürch S, Colloids and Surfaces A: Physicochemical and Engineering Aspects, 1996, 114, 165–184. [Google Scholar]
- 24.Clements JA and Avery ME, American Journal of Respiratory and Critical Care Medicine, 1998, 157, S59–S66. [DOI] [PubMed] [Google Scholar]
- 25.Avery ME, American Journal of Respiratory and Critical Care Medicine, 2000, 161, 1074–1075. [DOI] [PubMed] [Google Scholar]
- 26.Ware LB and Matthay MA, The New England Journal of Medicine, 2000, 342, 1334–1349. [DOI] [PubMed] [Google Scholar]
- 27.Spragg RG, Lewis JF, Walmrath H-D, Johannigman J, Bellingan G, Laterre P-F, Witte MC, Richards GA, Rippin G, Rathgeb F, Häfner D, Taut FJH and Seeger W, The New England Journal of Medicine, 2004, 351, 884–892. [DOI] [PubMed] [Google Scholar]
- 28.McIntyre RC, Pulido EJ, Bensard DD, Shames BD and Abraham E, Critical Care Medicine, 2000, 28, 3314. [DOI] [PubMed] [Google Scholar]
- 29.Holm, Wang Z. and Notter RH, Pediatric Research, 1999, 46, 85–93. [DOI] [PubMed] [Google Scholar]
- 30.Nakos G, Kitsiouli EI, Tsangaris I. and Lekka ME, Intensive Care Medicine, 1998, 24, 296–303. [DOI] [PubMed] [Google Scholar]
- 31.Warriner HE, Ding J, Waring AJ and Zasadzinski JA, Biophysical Journal, 2002, 82, 835–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seeger W, Stöhr G, Wolf HRD and Neuhof H, Journal of Applied Physiology, 1985, 58, 326–338. [DOI] [PubMed] [Google Scholar]
- 33.Seeger W, Günther A. and Thede C, American Journal of Physiology, 1992, 261, L286–L291. [DOI] [PubMed] [Google Scholar]
- 34.Fuchimukai T, Fujiwara T, Takahashi A. and Enhorning G, Journal of Applied Physiology, 1987, 62, 429–437. [DOI] [PubMed] [Google Scholar]
- 35.Holm BA, Notter RH and Finkelstein JN, Chemistry and Physics of Lipids, 1985, 38, 287–298. [DOI] [PubMed] [Google Scholar]
- 36.Bratek-Skicki A, Żeliszewska P. and Ruso JM, Soft Matter, 2016, 12, 8639–8653. [DOI] [PubMed] [Google Scholar]
- 37.Muszbek L, Bagoly Z, Bereczky Z. and Katona É, Cardiovascular & Hematological Agents in Medicinal Chemistry, 2008, 6, 190–205. [DOI] [PubMed] [Google Scholar]
- 38.Hernández EM, Phang T-L, Wen X. and Franses EI, Journal of Colloid and Interface Science, 2002, 250, 271–280. [DOI] [PubMed] [Google Scholar]
- 39.Hernández EM and Franses EI, Colloids and Surfaces A: Physicochemical and Engineering Aspects, 2003, 214, 249–262. [Google Scholar]
- 40.Hassan N, Maldonado-Valderrama J, Gunning AP, Morris VJ and Ruso JM, The Journal of Physical Chemistry B, 2011, 115, 6304–6311. [DOI] [PubMed] [Google Scholar]
- 41.Williams I. and Squires TM, Journal of the Royal Society, Interface, 2018, 15, 20170895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Furst EM and Squires TM, Microrheology, Oxford University Press, 1st edn., 2017. [Google Scholar]
- 43.Lösche M. and Möhwald H, Review of Scientific Instruments, 1984, 55, 1968–1972. [Google Scholar]
- 44.Gaub HE, Moy VT and McConnell HM, Journal of Physical Chemistry, 1986, 90, 1721–1725. [Google Scholar]
- 45.Zell ZA, Mansard V, Wright J, Kim K, Choi SQ and Squires TM, Journal of Rheology, 2016, 60, 141–159 [Google Scholar]
- 46.Choi SQ, Steltenkamp S, Zasadzinski JA and Squires TM, Nature Communications, 2011, 2, 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chang C-C, Nowbahar A, Mansard V, Williams I, Mecca J, Schmitt AK, Kalantar TH, Kuo T-C and Squires TM, Langmuir, 2018, 34, 5409–5415. [DOI] [PubMed] [Google Scholar]
- 48.McConlogue CW and Vanderlick TK, Langmuir, 1997, 13, 7158–7164. [Google Scholar]
- 49.Kim K, Choi SQ, Zasadzinski JA and Squires TM, Soft Matter, 2011, 7, 7782–7789. [Google Scholar]
- 50.Lawrie GA, Gentle IR and Barnes GT, Colloids and Surfaces A: Physicochemical and Engineering Aspects, 2000, 171, 217–224. [Google Scholar]
- 51.Toimil P, Prieto G, Miñones J Jr and Sarmiento F, Physical Chemistry Chemical Physics, 2010, 12, 13323–13332. [DOI] [PubMed] [Google Scholar]
- 52.Hermans E. and Vermant J, Soft Matter, 2014, 10, 175–186. [DOI] [PubMed] [Google Scholar]
- 53.Moy VT, Keller DJ, Gaub HE and McConnell HM, Journal of Physical Chemistry, 1986, 90, 3198–3202. [Google Scholar]
- 54.Shieh IC and Zasadzinski JA, Proceedings of the National Academy of Sciences, 2015, 112, E826–E835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim K, Choi SQ, Zell ZA, Squires TM and Zasadzinski JA, Proceedings of the National Academy of Sciences, 2013, 110, E3054–E3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Trappe V. and Weitz DA, Physical Review Letters, 2000, 85, 449–452. [DOI] [PubMed] [Google Scholar]
- 57.Appel J, Fölker B. and Sprakel J, Soft Matter, 2016, 12, 2515–2522. [DOI] [PubMed] [Google Scholar]
- 58.Laurati M, Petekidis G, Koumakis N, Cardinaux F, Schofield AB, Brader JM, Fuchs M. and Egelhaaf SU, Journal of Chemical Physics, 2009, 130, 134907. [DOI] [PubMed] [Google Scholar]
- 59.Bernardini C, Stoyanov SD, Arnaudov LN and Cohen Stuart MA, Chemical Society Reviews, 2013, 42, 2100–2129. [DOI] [PubMed] [Google Scholar]
- 60.Weis RM and McConnell HM, Journal of Physical Chemistry, 1985, 89, 4453–4459. [Google Scholar]
- 61.Freites JA, Ali S, Rosengarth A, Luecke H. and Dennin MB, Langmuir, 2004, 20, 11674–11683. [DOI] [PubMed] [Google Scholar]
- 62.Hu Y, Doudevski I, Wood D, Moscarello M, Husted C, Genain C, Zasadzinski JA and Israelachvili J, Proceedings of the National Academy of Sciences, 2004, 101, 13466–13471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bukusoglu E, Pal SK, de Pablo JJ and Abbott NL, Soft Matter, 2014, 10, 1602–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reynaert S, Moldenaers P. and Vermant J, Phys Chem Chem Phys, 2007, 9, 6463–6475. [DOI] [PubMed] [Google Scholar]
- 65.Masschaele K, Fransaer J. and Vermant J, Journal of Rheology, 2009, 53, 1437–1460. [Google Scholar]
- 66.Vessely CR, Carpenter JF and Schwartz DK, Biomacromolecules, 2005, 6, 3334–3344. [DOI] [PubMed] [Google Scholar]
- 67.Sorret LL, DeWinter MA, Schwartz DK and Randolph TW, Protein Science, 2018, 27, 1191–1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Roberts SA, Kellaway IW, Taylor KMG, Warburton B. and Peters K, Langmuir, 2005, 21, 7342–7348. [DOI] [PubMed] [Google Scholar]
- 69.Freer EM, Yim KS, Fuller GG and Radke CJ, The Journal of Physical Chemistry B, 2004, 108, 3835–3844. [Google Scholar]
- 70.Takamoto DY, Lipp MM, von Nahmen A, Lee KYC, Waring AJ and Zasadzinski JA, Biophysical Journal, 2001, 81, 153–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lipp MM, Lee KYC, Zasadzinski JA and Waring AJ, Science, 1996, 273, 1196–1199. [DOI] [PubMed] [Google Scholar]
- 72.Bernardino J. de la Serna, Orädd G, Bagatolli LA, Simonsen AC, Marsh D, Lindblom G. and Perez-Gil J, Biophysical Journal, 2009, 97, 1381–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sachan A. and Zasadzinski JA, Proceedings of the National Academy of Sciences, 2018, 115, E134–E143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bernardino J. de la Serna, Perez-Gil J, Simonsen AC and Bagatolli LA, Journal of Biological Chemistry, 2004, 279, 40715–40722. [DOI] [PubMed] [Google Scholar]
- 75.Rice DK, Cadenhead DA, Lewis RNAH and McElhaney RN, Biochemistry, 1987, 26, 3205–3210. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.