

Abstract
GM1 gangliosidosis is a lysosomal storage disorder caused by mutations in GLB1 encoding a lysosomal β-galactosidase. This disease is a continuum from the severe infantile form with rapid neurological decline to the chronic adult form, which is not life-limiting. The intermediate or type 2 form can be further classified into late infantile and juvenile forms. The frequency and severity of skeletal outcomes in late infantile and juvenile patients have not been characterized. Our goals are to describe the radiological skeletal abnormalities, bone mineral density (BMD), and frequency of fractures in patients with intermediate GM1 gangliosidosis. We evaluated 13 late infantile and 21 juvenile patients as part of an ongoing natural history study. Average time from onset of symptoms to diagnosis was 1.9 and 6.3 years for late infantile and juvenile patients, respectively. All late infantile patients had odontoid hypoplasia and pear-shaped vertebral bodies, the frequency of which was significantly different than in patients with juvenile disease (none and 14%, respectively). Juvenile patients had irregular endplates of the vertebral bodies (15/21), central indentation of endplates (10/21), and squared and flat vertebral bodies (10/21); all allowed radiographic differentiation from late infantile patients. Lumbar spine, femoral neck, and total hip BMD were significantly decreased (−2.1, −2.2, and −1.8 Z-scores respectively). Lumbar spine BMD peaked at 19 years, while distal forearm BMD peaked at 30 years. Despite low BMD, no patients exhibited fractures. We have demonstrated that all late infantile patients have some degree of odontoid hypoplasia suggesting the need for cervical spine evaluation particularly prior to anesthesia, whereas juvenile patients had variable skeletal involvement often affecting activities of daily living. Type 2 GM1 gangliosidosis patients have skeletal abnormalities that are both an early indication of their diagnosis, and require monitoring and management to ensure the highest possible quality of life.
Keywords: GM1 gangliosidosis type 2, lysosomal storage disease, skeletal dysplasia, beta-galactosidase deficiency
1. INTRODUCTION
First described as Tay-Sachs disease with visceral involvement [1], GM1 gangliosidosis is a lysosomal storage disorder caused by decreased activity of acid hydrolase β-galactosidase encoded by GLB1. This gene product is required for removal of the terminal galactose in the first step of sphingolipid degradation. The deficiency results in storage of GM1 ganglioside particularly in the central nervous system–where ganglioside synthesis is the greatest–with consequent progressive neurodegeneration. GM1 gangliosidosis represents a clinical spectrum with severity determined by the amount of residual enzyme activity a result of the combination of specific GLB1 biallelic mutations. Traditionally, the disease is classified as type 1 (infantile/acute) (OMIM 230500), type 2 (juvenile/sub-acute) (OMIM 230600), and type 3 (adult/chronic) (OMIM 230650). Recently, the type 2 patient population has been further delineated into late infantile and juvenile forms, based on age of onset and rate of disease progression [2]. Infantile GM1 gangliosidosis is the most common and most severe form of the disease, and is characterized by onset of developmental regression by one year of age, cherry red maculae, coarse facial appearance, hepatosplenomegaly, and often death by three years of age. Furthermore, these infants routinely have severe skeletal dysplasia identified after the neurological symptoms have led to a diagnosis. Intervention for these skeletal findings is often limited due to the short life span of these infants [3]. The adult form is the mildest of the disease spectrum and presents in late childhood to adulthood with primarily movement disorders, including dystonia, Parkinsonism, or ataxia. Adult GM1 gangliosidosis patients have few skeletal findings, usually not requiring surgical interventions. Patients with type 2 disease are more heterogeneous. Late infantile patients achieve appropriate developmental milestones at 12 months but generally have plateauing then loss of skills beginning at 12 to 18 months of age. These children may learn to stand, cruise, and may achieve walking but the skills are rapidly lost and by 2 years most are non-ambulatory. They can develop a 10–20 single word vocabulary but have difficulty putting words together before becoming anarthric as the disease progresses. They do not have the characteristic cherry red macula of infantile patients. Juvenile patients generally have symptom onset by 3 to 5 years. They meet early developmental milestones and learn to walk, run, and often pedal a tricycle. Speech develops normally to full sentences and the first sign of disease may be “stuttering” followed by progressive dysarthria. The children may remain ambulatory for several years yet some are limited by the pain of avascular necrosis of the hip. Most, but not all, are wheelchair-bound by mid-teens. Relentless CNS progression leads to significant impairment by early childhood and early adulthood for late infantile and juvenile patients, respectively [2].
GLB1 is causal not only for GM1 gangliosidosis but also for mucopolysaccharidosis type IVB (MPS IVB or Morquio B disease) [4]. MPS IVB is clinically indistinguishable from MPS IVA, caused by mutations in GALNS, encoding lysosomal enzyme N-acetylgalactosamine-6-sulfate sulfatase. Both MPS IVA and IVB result in accumulation of keratan sulfate, a galactose-containing glycosaminoglycan, in the periphery. Thus, their phenotype is indistinguishable [5]. The GLB1 mutations causal for MPS IVB alter the substrate specificity of the mutant towards keratan sulfate, whereas the affinity of β-galactosidase against keratan sulfate remains intact in GM1 gangliosidosis [6,7]. Skeletal abnormalities represent the predominant features of MPS IV, and the dysostosis multiplex seen in these patients is a direct result of the accumulation of keratan sulfate in the skeleton.
Previous reports have described skeletal changes in infantile and adult forms of GM1 gangliosidosis. Rabinowitz and Sacher reviewed the findings of an infant with widening of the bone marrow spaces with thinning of the cortices of the upper extremities, and lumbar kyphosis with notching of L1 and L2 [8]. Mogilner et al. and Denis et al. separately documented elevated serum alkaline phosphatase levels in the neonatal period in children with infantile GM1 gangliosidosis, suggesting abnormal bone physiology [9,10]. Owman et al. reviewed fifteen patients with GM1 gangliosidosis type 2, ten of whom demonstrated one or more of the following: flat vertebral bodies, excavation of the dorsal margin of the vertebral bodies, anterior hypoplasia and beaking of vertebrae, moderate scoliosis, horizontalization of the ribs, rarefaction of the lower arm, delayed skeletal maturation, and/or iliac dysplasia [11]. Muthane et al. reviewed 40 patients with GM1 gangliosidosis type 3 and found that 69.7% had flat vertebrae, 3% had scoliosis, and 3% anterior beaking [12].
We present the frequency and severity of skeletal outcomes in a cohort of type 2 GM1 gangliosidosis patients, further classified into late infantile or juvenile subtypes. Our goals are to describe the radiological skeletal abnormalities, bone mineral density (BMD), and frequency of fractures in this cohort of patients.
2. PATIENTS AND METHODS
2.1. Subjects
Patients with GM1 gangliosidosis documented by β-galactosidase enzyme deficiency and biallelic mutations in GLB1 were enrolled in clinical protocol 02-HG-0107 “Investigation of Neurodegeneration in Glycosphingolipid Storage Disorders” (ClinicalTrials.gov identifier: ). The parents gave written informed consent for their children. Due to the cognitive dysfunction of this patient population, all patients under and over 18 years of age were consented by their parents or legal guardians, respectively. All participants were evaluated for their ability to assent; however, none were deemed cognitively capable.
Cases were evaluated using clinical data from the referring physicians and confirmed on examination during visits to the NIH Clinical Center. The 13 late infantile GM1 gangliosidosis and 21 juvenile GM1 gangliosidosis patients were classified based on onset of symptoms and clinical findings [2]. Affected siblings were assigned to the subgroup of the proband. Enzymatic activity was confirmed for at least one patient per family. Each participant underwent GLB1 sequencing either before or during evaluation at the National Institutes of Health Clinical Center in Bethesda, Maryland.
2.2. Analysis of Skeletal Radiographs
Skeletal radiographs were available for all patients, and were evaluated longitudinally (19 patients) or at a single time point (14 participants). All images were obtained at the NIH Clinical Center. Radiographs were reviewed by two of the authors (CRF, JS). Ages were provided to allow for age-related bone spectrum. Bone age was determined according to the Greulich and Pyle standard [13].
A Fisher’s exact test was used to compare the probability of a specific radiographic finding being found in the late infantile vs juvenile cohort. Statistical significance was assigned to a two-tailed p-value <0.05. Analysis was performed with Prism version 6.0c (Graphpad Software Inc, La Jolla, CA).
2.3. Analysis of Bone Mineral Density and Fracture Risk
The incidence of pathologic fractures in the GM1 gangliosidosis patients was ascertained by clinical history. Dual Energy Absorptiometry (DXA) was used to determine bone mineral density (BMD). BMD was compared to children of the same age, gender, and ethnicity. Ambulatory children had full bone density scanning performed (femoral neck, total hip, lumbar spine L1-L4, and 1/3 forearm). Children with decreased ability to ambulate were evaluated using the lateral distal femur reference data [14]. Regions 1, 2, and 3 were identified according to published standards; predicted mean BMD values for age were calculated using the previously published reports [14]. Eleven late infantile patients were evaluated; not all patients in this group qualified for Z-score analysis due to patient age. Nineteen juvenile patients were evaluated. Low bone mass for age was defined as a Z-score <−2 in one or more sites.
A D’Agostino-Pearson normality test was performed for all seven sites were BMD was evaluated. Given a normal distribution, an unpaired t test was performed comparing the mean Z-scores at each site against a mean Z-score of 0 for the control population. For patients with serial BMD measurements, only the most current BMD value was used for calculation of the mean Z-score. Statistical significance was assigned to a two-tailed p-value <0.05. Analysis was performed with Prism version 6.0c (Graphpad Software Inc, La Jolla, CA).
The relationship between BMD and age was analyzed cross-sectionally using simple and multivariable regression models (IBM SPSS Statistics Subscription for Windows, IBM Corp., Armonk, New York USA). Lumbar spine, distal forearm, total hip, and femoral BMD were evaluated individually. The effects of gender and ethnicity (black vs. non-black) were also assessed. Longitudinal data on BMD was available for a subset of subjects; generalized mixed-effects models were used to assess change in BMD over time, accounting for unbalanced serial observations, with an unstructured covariance structure for inter-subject random intercepts (IBM SPSS Statistics).
3. RESULTS
The subtype, age of onset, age at diagnosis, and genotype of all subjects is summarized in supplemental table 1. The mean age of onset for the late infantile cohort was 1 year 4 months, with a mean age at diagnosis of 3 years 3 months (mean diagnostic delay: 1 year 11 months). For the juvenile group, the mean age of onset was 3 years 7 months, with a mean age at diagnosis of 9 years 10 months, and a mean diagnostic delay of 6 years 3 months.
3.1. Radiographic findings
All children with late infantile GM1 gangliosidosis had abnormal bone findings (Table 1), including odontoid hypoplasia in 6/6 (Figure 1). These patients also demonstrated multiple spine anomalies including pear-shaped vertebral bodies (12/12 patients), anterior hypoplasia of L1 or L2 (9/12), and scoliosis (6/12). Spine disease progression was noted in patients in whom serial imaging was available (Figure 2A–D). Frequent findings in the pelvis and femoral neck included hypoplasia of the lower ilia (5/13) and acetabulae (6/12), subluxation of the femoral heads (6/12), coxa valga (11/11), and short femoral necks (6/11) (Figure 3). Pelvic changes over time are noted in a patient with serial imaging at 4 years (Figure 3D) and 9 years 10 months (Figure 3E).
Table 1:
Summary of skeletal findings in patients with GM1 gangliosidosis. Ages are given in years with a range given if more than one was radiograph evaluated. Positive (+), borderline ((+)), and negative (−) evaluation are shown for the skull, spine, and pelvis radiographs.
| Patient | Age (years) | Spine | Pelvis | Proximal femur | Hands | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Odontoid hypoplasia | Scoliosis | Pear-shaped vertebrae | Anterior vertebral hypoplasia | Central end plate indentation | Irregular endplates | Squared vertebral bodies | Hypoplastic lower ileum | Hypoplastic fossa acetabulae | Subluxed femoral heads | Abnormal epiphyses | Short femoral neck | Coxa valga | Proximal metacarpal tapering | Short phalanges | Dysplastic carpal bones | ||
| Late infantile | |||||||||||||||||
| GSL007 | 5–10 | + | + | + | + | − | − | − | + | + | + | − | + | + | − | − | − |
| GSL013 | 4–11 | + | + | + | + | − | − | (+) | + | + | + | Flat | + | + | − | − | − |
| GSL022 | 2–3 | ND | − | + | + | − | − | − | − | − | − | − | − | + | − | − | − |
| GSL023 | 5 | ND | − | + | + | − | − | − | − | − | + | Flat | − | + | − | − | − |
| GSL024 | 1–4 | ND | + | + | + | − | − | − | ND | ND | ND | ND | N D | ND | ND | ND | ND |
| GSL030 | 3 | + | − | (+) | − | − | − | − | − | − | − | − | − | + | − | − | − |
| GSL035 | 4 | + | − | (+) | + | − | − | − | + | + | + | − | − | + | − | − | − |
| GSL062 | 6 | ND | − | + | + | − | − | − | − | − | − | − | + | + | − | − | − |
| GSL063 | 3 | ND | N D | ND | ND | ND | ND | ND | − | − | − | − | N D | ND | ND | ND | ND |
| GSL072 | 8–9 | + | + | (+) | + | − | − | − | + | + | + | − | + | + | − | − | − |
| GSL073 | 5–9 | + | + | + | − | − | − | − | − | + | − | − | + | + | − | − | −19 |
| GSL084 | 3–4 | ND | − | (+) | + | − | − | − | − | − | − | − | − | + | − | − | − |
| GSL085 | 6 | ND | + | (+) | − | − | − | − | + | + | + | − | + | + | − | − | − |
| Juvenile | |||||||||||||||||
| GSL003 | 18–30 | − | + | − | + | + | + | + | + | + | (+) | Dysplastic | + | + | − | − | − |
| GSL004 | 12 | − | − | − | − | + | + | + | − | − | − | − | + | + | − | − | − |
| GSL009 | 13–23 | − | + | − | − | + | + | + | − | − | − | Dysplastic | − | − | − | − | − |
| GSL010 | 12–22 | − | + | − | + | − | + | + | − | − | − | − | + | − | − | − | − |
| GSL011 | 9–19 | − | + | (+) | + | + | + | − | − | − | − | Flat | − | + | − | − | − |
| GSL020 | 10–15 | − | − | − | − | + | + | − | − | − | − | − | − | − | − | − | − |
| GSL025 | 12–17 | − | − | − | − | + | + | + | − | − | − | − | − | − | − | − | − |
| GSL026 | 11–16 | − | − | − | − | − | + | − | − | − | − | − | − | − | − | − | − |
| GSL027 | 7–11 | − | − | − | − | − | + | − | − | − | − | − | − | − | − | − | − |
| GSL028 | 17–25 | − | − | − | − | − | + | − | − | − | − | − | + | − | − | − | − |
| GSL034 | 23 | − | + | − | − | + | + | − | − | − | − | − | − | − | ND | ND | ND |
| GSL053 | 15 | − | − | − | − | − | + | − | − | − | − | − | (+) | − | − | − | − |
| GSL054 | 7–9 | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − | − |
| GSL058 | 9–17 | − | − | (+) | (+) | + | + | + | − | − | + | Dysplastic | − | − | − | − | − |
| GSL064 | 11 | ND | − | − | − | + | + | + | − | + | − | Dysplastic | − | + | − | − | − |
| GSL069 | 7–8 | − | − | (+) | + | − | − | + | − | − | − | − | − | + | − | − | − |
| GSL071 | 12–13 | − | + | − | − | − | − | + | − | − | − | − | − | − | − | − | − |
| GSL082 | 17 | − | − | − | − | − | + | + | − | − | − | − | + | − | − | − | − |
| GSL093 | 9 | − | − | − | − | − | − | − | − | − | + | − | − | − | − | − | − |
| GSL094 | 9 | − | + | − | + | − | − | − | + | + | − | − | − | − | − | − | − |
| GSL095 | 1 | ND | − | − | − | + | − | − | − | − | − | − | − | − | ND | ND | ND |
Figure 1:
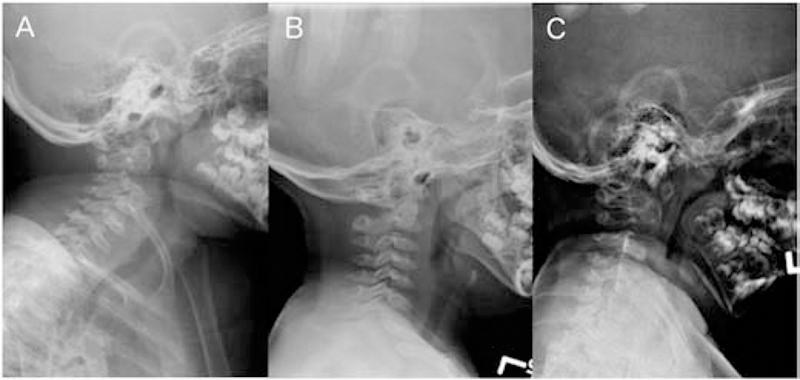
Cervical spine radiographs in late infantile patients. A. Patient GSL013, 9 years. B. Patient GSL030, 3 years. C. GSL035, 4 years. In all patients, the odontoid process is underossified.
Figure 2:
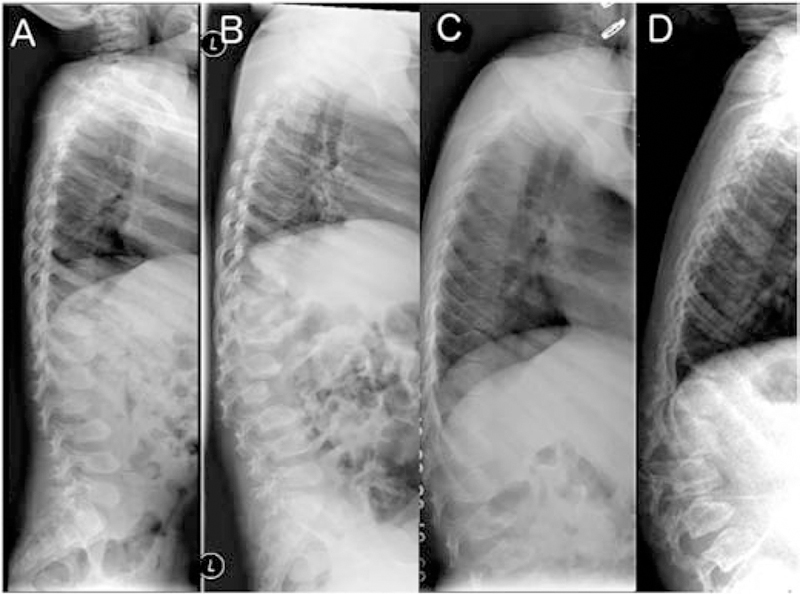
Lumbar spine radiographs in late infantile patients. A, B. Patient GSL0013 at 4 years and 9 years. The vertebral bodies have a pear-shaped ovoid appearance. Ossification of the anterior segments of the vertebral bodies is defective in the thoraco-lumbar junction leading to their hook-shape or an aspect of dorsal displacement. Note progression in 5 years consistent with clinical disease course C, D. Patient GSL007 at 6 and 10 years. Similar changes and progression are seen over time.
Figure 3:
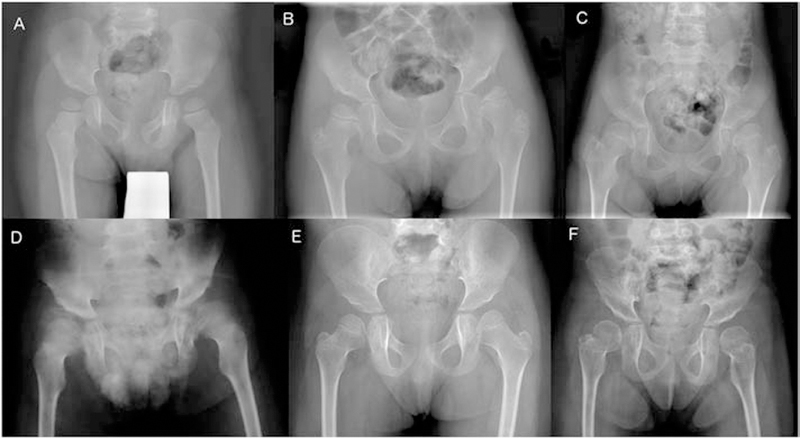
Pelvic radiographs in late infantile patients show hypoplasia of the infero-lateral portions of the ilia resulting in slanted acetabular roofs and wide ileo-acetabular angles. A. Patient GSL022, 3 years. The femoral necks are in valgus. The capital femoral epiphyses are well rounded and slightly large. B. Patient GSL023, 5 years. C. Patient GSL007, 6 years. D. Patient GSL072, 8 years. E. Patient GSL013, 4 years. The capital femoral epiphyses are dysplastic due to deficient ossification of their medial portions. F. Patient GSL013, 9 years. Compared to E, epiphyseal ossification has normalized, and the femoral necks are shorter.
Children with juvenile GM1 gangliosidosis had flat and squared vertebral bodies (10/21), irregular endplates of the vertebral bodies (15/21), central indentation of endplates (10/21), or scoliosis (7/19) (Figure 4). The juvenile GM1 gangliosidosis patients had a lower rate of pelvic anomalies (Figure 5). Radiographic features allowing differentiation of late infantile vs juvenile patients included odontoid hypoplasia (only seen in late infantile patients), and irregularity and central indentation of the vertebral body endplates (only seen in juvenile patients). The finding of pear-shaped vertebral bodies favors late infantile GM1 gangliosidosis, while the presence of flat and squared vertebral bodies favors juvenile GM1 gangliosidosis (supplemental table 2).
Figure 4:
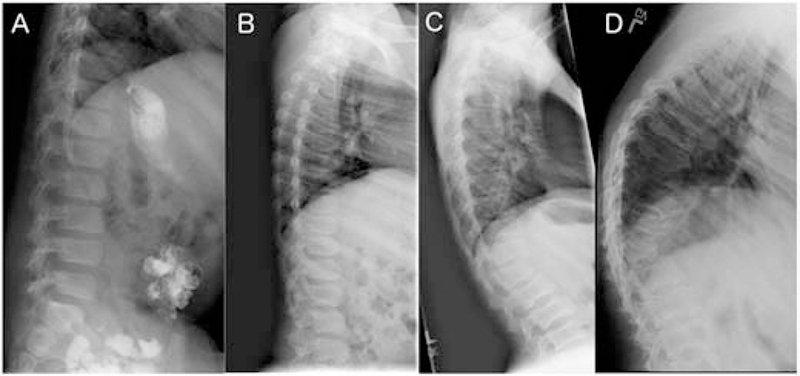
Lumbar spine radiographs in juvenile patients. A. Patient GSL027, age 7 years. Normal spine. B. Patient GSL020, 12 years. The vertebral bodies are dorsally flat with irregular upper and lower margins. C. Patient GSL004, 12 years. The upper and lower endplates show central indentations. D. Patient GSL003, 18 years. The lumbar vertebral bodies are deformed with central indentations. The body of T12 is triangular resulting in an acute dorsolumbar kyphosis.
Figure 5:
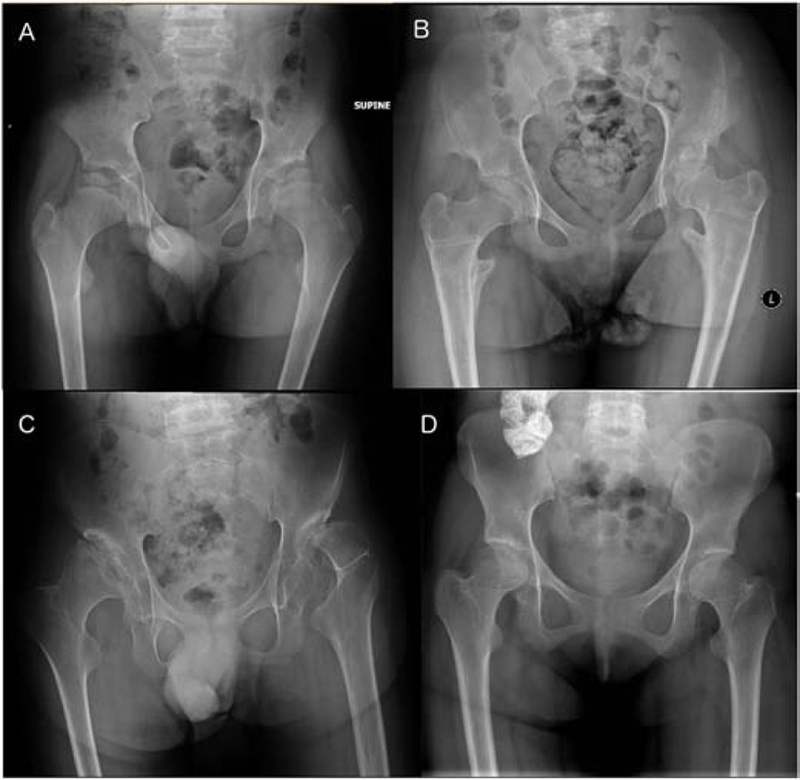
Pelvic radiographs in juvenile patients. A, B, C. Varying degrees of over-constricted lower ilia are seen with small acetabulae and subluxated femoral heads. The epiphyses are flat. A, patient GSL009, age 13 years. B, patient GSL010, 15 years. C, patient GSL003, 18 years. D. Patient GSL028, 19 years. In this patient the ilia are well developed with full acetabular coverage of the slightly flat femoral heads.
Bone age was normal in both late infantile and juvenile patients. Importantly and unlike other disorders with dysostosis multiplex, they demonstrated no anomalies of the carpal bones, metacarpals, or phalanges (supplemental figure 1).
3.2. Bone Mineral Density
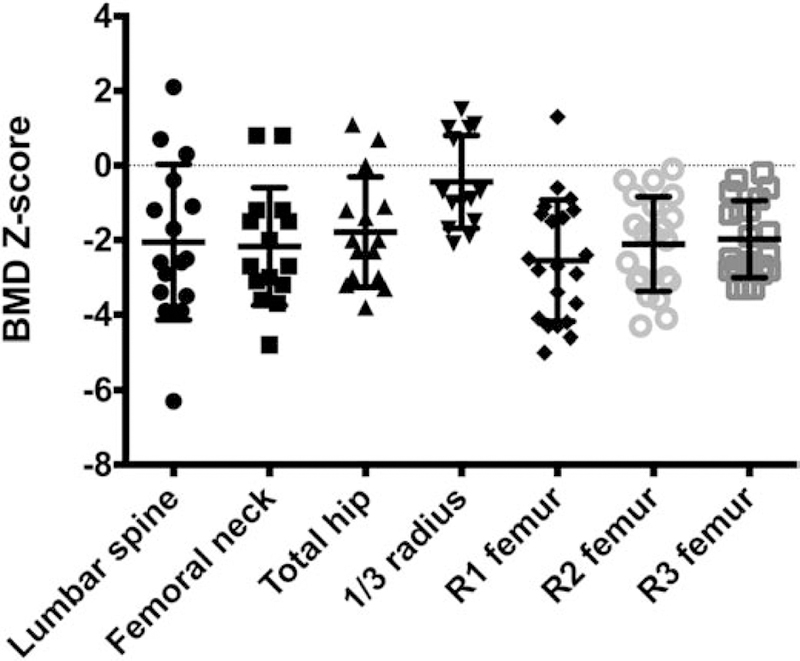
Primary densitometry data can be found in Supplemental Table 3. Z-scores showed decreased density from age, gender, and race-matched controls (Table 2; Figure 6). Despite these decreased Z-scores, no pathologic fractures were reported in the late infantile or juvenile cohort.
Table 2:
Summary of bone mineral density findings in patients with GM1 gangliosidosis.
| Site | Number | Mean age | Mean BMD (Z-score) | Standard error of mean | P-value |
|---|---|---|---|---|---|
| Lumbar spine | 16 | 15y 9m | −2.1 | 0.5 | 0.0013 |
| Femoral neck | 15 | 15y 5m | −2.2 | 0.4 | 0.0001 |
| Total hip | 15 | 15y 5m | −1.8 | 0.4 | 0.0004 |
| 1/3 forearm | 14 | 17y 0m | −0.4 | 0.3 | 0.2061 |
| R1 femur | 21 | 11y 0m | −2.5 | 0.4 | < 0.0001 |
| R2 femur | 21 | 11y 0m | −2.1 | 0.3 | < 0.0001 |
| R3 femur | 21 | 11y 0m | −2.0 | 0.2 | < 0.0001 |
Figure 6:
BMD Z-scores in patients with GM1 gangliosidosis.
Lumbar spine, distal forearm, total hip, and femoral BMD correlated with age. Lumbar spine and distal forearm BMD were fitted to quadratic models (Figure 7); however, total hip and femoral BMD were fitted using linear models, based on lower corrected Akaike’s Information Criteria (Figure 8). Lumbar spine BMD demonstrated an initial increase of 0.07 g/cm2 (95% CI=0.03 to 0.11, p=0.001) that decreased by 0.002 g/cm2 (95% CI=−0.003 to −0.001, p=0.004) with each additional year of age, peaking at 19 years. Similarly, distal forearm BMD demonstrated an initial increase of 0.04 g/cm2 (95% CI=0.02 to 0.06, p<0.001) that decreased by 0.001 g/cm2 (95% CI=−0.001 to −0.00007, p=0.03) with each additional year of age, peaking at 30 years. Total hip BMD demonstrated an increase of 0.02 g/cm2 per year (95% CI=0.006 to 0.03, p=0.002) and femoral BMD demonstrated an increase of 0.01 g/cm2 per year (95% CI=0.003 to 0.02, p=0.01). Gender and ethnicity did not have significant effects on BMD.
Figure 7:
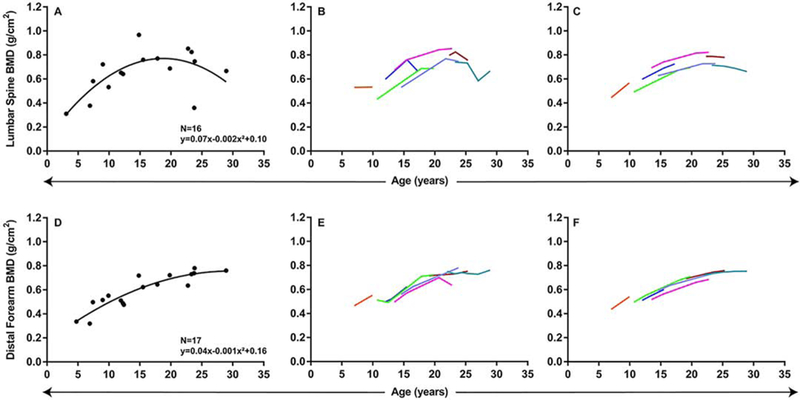
Relationship between lumbar spine and distal forearm BMD and age: quadratic models. A, D. Cross-sectional regression models. B–C, E–F. Longitudinal changes in BMD with age. B, Change in lumbar spine BMD with age raw data and (C) predicted change in BMD based on mixed-effects model regression. E, Change in distal forearm spine BMD with age raw data and (F) predicted change in BMD based on mixed-effects model regression.
Figure 8:
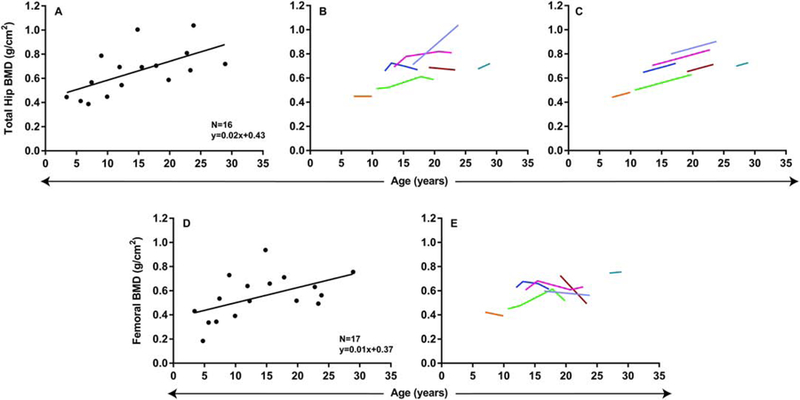
Relationship between total hip and femoral BMD and age: linear models. A, D. Cross-sectional regression models. B–C, E. Longitudinal changes in BMD with age. B, Change in total hip BMD with age raw data and (C) predicted change in BMD based on mixed-effects model regression. E, Change in femoral BMD with age raw data.
Serial BMD measurements were available for 7 patients (mean number per patient=3, range 2–6). When examined longitudinally (Supplemental Table 4), patients demonstrated an increase in lumbar spine BMD during childhood and adolescence, which then decreased after the second decade of life (Figure 7B–C). Distal forearm BMD demonstrated a similar trend, but decreased after the third decade of life (Figure 7E–F). Total hip BMD demonstrated an increase with age during the follow-up period (Figure 8B–C). Femoral BMD did not demonstrate a significant change with age in the longitudinal analysis (Figure 8D–E).
4. DISCUSSION
In this cohort of patients with Type 2 GM1 gangliosidosis, two sub-groups were identified: late infantile and juvenile. While the classification was established based on age at onset of symptoms, the skeletal radiographic findings support these groupings. The presence of odontoid hypoplasia and pear-shaped vertebral bodies was statistically higher in late infantile patients, while irregularity and central indentation of the vertebral body endplates and squared vertebral bodies were statistically more common in juvenile patients. The finding of odontoid hypoplasia in all late infantile patients is especially important for clinical management. The cervical instability and anesthesia risk for children with odontoid hypoplasia has been well characterized in the MPS IV patient population [15]. In our late infantile cohort, sedated procedures (15 in late-infantile patients, 48 in juvenile patients) have been performed successfully with close monitoring by the Clinical Center pediatric anesthesia team.
Features of dysostosis multiplex are present in patients with both late infantile and juvenile GM1 gangliosidosis. In the former they affect a more immature skeleton and combine with signs of delayed ossification such as odontoid hypoplasia, ovoid vertebral bodies, hypoplasia of the iliac bodies with slanted acetabular roofs, and abnormal capital femoral epiphyses. In juvenile GM1gangliosidosis, the vertebral bodies are more rectangular with irregular endplates, often with central indentations. In addition, the pelvic and femoral anomalies are less severe than in the late infantile type. Notably, and in contrast to the mucopolysaccharidoses, the short tubular bones are not affected in either late infantile or juvenile GM1 gangliosidosis patients, which distinguish them from other causes of dysostosis multiplex.
The BMD Z-scores for patients were statistically lower in the lumbar spine, femoral neck, total hip, and distal femur. No children were reported to have fractures within the cohort; thus, the change in bone density does not appear to have obvious clinical implications for quality of life. It is unclear if the low BMD is related to the underlying disease, secondary to the severity of decreased mobility from their neurodegenerative disease, or a combination of the two. Based on the young age at which the bone density and radiographic findings presented in the patients (even during the first natural history visit), it would be reasonable to assume that these are primary changes. Additionally, the presence of skeletal changes, namely retarded endochondral ossification and osteoporosis, in canine models of GM1 gangliosidosis also favors a primary etiology [16]. Osteoporosis and fracture were previously described in 1 out of 5 patients with infantile GM1 gangliosidosis [17]; to the best of our knowledge, however, there has been no systematic study of bone mineral density in human patients until now.
Since GM1 gangliosidosis is allelic with Morquio syndrome type B, a well-described skeletal dysplasia, it would be expected that some patients would have more severe bone findings if they harbor pathogenic mutations known to cause MPS IVB. Together, this would raise the concern that enzyme replacement or gene therapy even at the time of diagnosis may be too late to completely ameliorate the skeletal manifestations in these patients. The line between the MPS IVB patients and GM1 gangliosidosis patients has been fairly distinct in the historical literature [3]. However, this cohort along with a single previous case report [18] suggest that the line between MPS IVB and GM1 gangliosidosis may be a continuum with regard to skeletal involvement. In fact, some patients with MPS IVB and typical keratan sulfaturia can also have progressive developmental regression, blurring the lines between these two conditions even further [19,20]. Other than the fact that patients with homozygous p.Trp273Leu mutations are not known to develop neurodegeneration [21], there is no clear genotype-phenotype correlation; although it has been stated that mutations associated with infantile GM1 gangliosidosis are concentrated in the protein core, mutations associated with intermediate or adult GM1 gangliosidosis are located on the surface, and mutations leading to MPS IVB are biased toward the ligand binding pocket and β-domain 2, this generalization is far from universal [22].While the patients with late infantile and juvenile GM1 gangliosidosis do not demonstrate the extreme skeletal dysplasia diagnosed in the MPS IVB patients, they may be at risk for the same sequelae over time. For the MPS IV patients, aggressive stabilization of the odontoid process has improved cognitive and nervous system outcomes [23]. It is unclear if late infantile patients may need this type of stabilization to optimize their neurologic outcomes. If an ongoing gene therapy trial (NCT) is successful in stabilizing or improving the neurologic disease then progression of the skeletal manifestations may emerge as a more prominent secondary phenotype.
The treatment and monitoring implications of this set of data are apparent. Based on the finding of hypoplasia of the odontoid process in all late infantile patients, careful monitor prior to and during anesthesia and evaluation after even minimal trauma is appropriate. With the blurred line between MPS IVB and GM1 gangliosidosis, it is reasonable that the anesthesiologist treating GM1 gangliosidosis patients be prepared to use the same interventions developed for safely sedating patients with MPS IV (i.e. anterior tongue placement, intubation via video laryngoscopy, intubation time limits, neurological monitoring, extubation protocols) [15]. Based on our findings the following recommendations can be made: a) children diagnosed with late infantile disease should have lateral cervical spine imaging in flexion and extension, similar to protocols for MPS IV children; and b) during anesthesia, patients should have cervical spine stabilization and care by an experienced pediatric anesthesiologist.
As therapies evolve to include disorders affecting the central nervous system, treatment options for GM1 gangliosidosis pediatric patients will likely become available. Treatment of the CNS-specific mucopolysaccharidoses are just one set of examples of progression of this field [24]. As treatments for the primary CNS phenotype become available, the secondary sequelae of skeletal disease may become more limiting and should be closely monitored to maintain an improved quality of life.
Supplementary Material
Highlights.
We describe the radiographic and densitometric progression of disease in the largest cohort of patients with intermediate GM1 gangliosidosis.
Radiographic changes allow differentiation between late infantile and juvenile patients.
Lumbar spine, femoral neck, and total hip bone mineral density were significantly decreased across the cohort.
Despite low bone mineral density, no patients exhibited fractures.
The presence of odontoid hypoplasia in all late infantile patients suggests the need for cervical spine evaluation prior to anesthesia.
ACKNOWLEDGEMENTS
This research was funded in part by the Intramural Research Program of the NHGRI (CRF, CT), National Institutes of Health, Bethesda, MD.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES:
- [1].Norman RM, Urich H, Tingey AH, Goodbody RA, Tay-Sachs’ disease with visceral involvement and its relationship to Niemann-Pick’s disease, J Pathol Bacteriol 78 (1959) 409–421. [DOI] [PubMed] [Google Scholar]
- [2].Regier DS, Tifft CJ, GLB1-Related Disorders, in: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, Amemiya A (Eds.), GeneReviews®, University of Washington, Seattle, Seattle (WA), 1993. http://www.ncbi.nlm.nih.gov/books/NBK164500/ (accessed January 31, 2019). [PubMed] [Google Scholar]
- [3].Suzuki Y, Nanba E, Matsuda J, Higaki K, Oshima A, β-Galactosidase Deficiency (β-Galactosidosis): GM1 Gangliosidosis and Morquio B Disease, in: Beaudet AL, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson KM, Mitchell G (Eds.), The Online Metabolic and Molecular Bases of Inherited Disease, The McGraw-Hill Companies, Inc., New York, NY, 2014. ommbid.mhmedical.com/content.aspx?aid=1121415439 (accessed February 1, 2019). [Google Scholar]
- [4].Oshima A, Yoshida K, Shimmoto M, Fukuhara Y, Sakuraba H, Suzuki Y, Human beta-galactosidase gene mutations in morquio B disease, Am. J. Hum. Genet 49 (1991) 1091–1093. [PMC free article] [PubMed] [Google Scholar]
- [5].Regier DS, Oetgen M, Tanpaiboon P, Mucopolysaccharidosis Type IVA, in: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, Amemiya A (Eds.), GeneReviews®, University of Washington, Seattle, Seattle (WA), 1993. http://www.ncbi.nlm.nih.gov/books/NBK148668/ (accessed February 1, 2019). [Google Scholar]
- [6].van der Horst GT, Kleijer WJ, Hoogeveen AT, Huijmans JG, Blom W, van Diggelen OP, Morquio B syndrome: a primary defect in beta-galactosidase, Am. J. Med. Genet 16 (1983) 261–275. 10.1002/ajmg.1320160215. [DOI] [PubMed] [Google Scholar]
- [7].Okumiya T, Sakuraba H, Kase R, Sugiura T, Imbalanced substrate specificity of mutant beta-galactosidase in patients with Morquio B disease, Mol. Genet. Metab 78 (2003) 51–58. [DOI] [PubMed] [Google Scholar]
- [8].Rabinowitz JG, Sacher M, Gangliosidosis (GM1). A re-evaluation of the vertebral deformity, Am J Roentgenol Radium Ther Nucl Med 121 (1974) 155–158. [DOI] [PubMed] [Google Scholar]
- [9].Mogilner BM, Barak Y, Amitay M, Zlotogora J, Hyperphosphatasemia in infantile GM1 gangliosidosis: possible association with microscopic bone marrow osteoblastosis, J. Pediatr 117 (1990) 758–761. [DOI] [PubMed] [Google Scholar]
- [10].Denis R, Wayemberg JL, Vermeulen M, Gorus F, Liebaers I, Vamos E, Hyperphosphatasemia in GM1 gangliosidosis, J. Pediatr 120 (1992) 164. [DOI] [PubMed] [Google Scholar]
- [11].Owman T, Sjöblad S, Göthlin J, Radiographic skeletal changes in juvenile GM1-gangliosidosis, Rofo 132 (1980) 682–688. 10.1055/s-2008-1056643. [DOI] [PubMed] [Google Scholar]
- [12].Muthane U, Chickabasaviah Y, Kaneski C, Shankar SK, Narayanappa G, Christopher R, Govindappa SS, Clinical features of adult GM1 gangliosidosis: report of three Indian patients and review of 40 cases, Mov. Disord 19 (2004) 1334–1341. 10.1002/mds.20193. [DOI] [PubMed] [Google Scholar]
- [13].Greulich WW, Pyle SI, Todd TW, Radiographic atlas of skeletal development of the hand and wrist, Stanford University Press, 1959. [Google Scholar]
- [14].Henderson RC, Lark RK, Newman JE, Kecskemthy H, Fung EB, Renner JB, Harcke HT, Pediatric reference data for dual X-ray absorptiometric measures of normal bone density in the distal femur, AJR Am J Roentgenol 178 (2002) 439–443. 10.2214/ajr.178.2.1780439. [DOI] [PubMed] [Google Scholar]
- [15].Theroux MC, Nerker T, Ditro C, Mackenzie WG, Anesthetic care and perioperative complications of children with Morquio syndrome, Paediatr Anaesth 22 (2012) 901–907. 10.1111/j.1460-9592.2012.03904.x. [DOI] [PubMed] [Google Scholar]
- [16].Alroy J, Knowles K, Schelling SH, Kaye EM, Rosenberg AE, Retarded bone formation in GM1-gangliosidosis: a study of the infantile form and comparison with two canine models, Virchows Arch 426 (1995) 141–148. 10.1007/bf00192635. [DOI] [PubMed] [Google Scholar]
- [17].Jarnes Utz JR, Kim S, King K, Ziegler R, Schema L, Redtree ES, Whitley CB, Infantile gangliosidoses: Mapping a timeline of clinical changes, Mol. Genet. Metab 121 (2017) 170–179. 10.1016/j.ymgme.2017.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Moore T, Bernstein JA, Casson-Parkin S, Cowan TM, β-Galactosidosis in Patient with Intermediate GM1 and MBD Phenotype, JIMD Rep 7 (2013) 77–79. 10.1007/8904_2012_145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Giugliani R, Jackson M, Skinner SJ, Vimal CM, Fensom AH, Fahmy N, Sjövall A, Benson PF, Progressive mental regression in siblings with Morquio disease type B (mucopolysaccharidosis IV B), Clin. Genet 32 (1987) 313–325. 10.1111/j.1399-0004.1987.tb03296.x. [DOI] [PubMed] [Google Scholar]
- [20].Mayer FQ, Pereira F. dos S., Fensom AH, Slade C, Matte U, Giugliani R, New GLB1 mutation in siblings with Morquio type B disease presenting with mental regression, Mol. Genet. Metab 96 (2009) 148 10.1016/j.ymgme.2008.11.159. [DOI] [PubMed] [Google Scholar]
- [21].Bleier M, Yuskiv N, Priest T, Moisa Popurs MA, Stockler-Ipsiroglu S, BC Children’s Hospital, University of British Columbia, Morquio B patient/caregiver survey: First insight into the natural course of a rare GLB1 related condition, Mol Genet Metab Rep 16 (2018) 57–63. 10.1016/j.ymgmr.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ohto U, Usui K, Ochi T, Yuki K, Satow Y, Shimizu T, Crystal structure of human β-galactosidase: structural basis of Gm1 gangliosidosis and morquio B diseases, J. Biol. Chem 287 (2012) 1801–1812. 10.1074/jbc.M111.293795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ransford AO, Crockard HA, Stevens JM, Modaghegh S, Occipito-atlanto-axial fusion in Morquio-Brailsford syndrome. A ten-year experience, J Bone Joint Surg Br 78 (1996) 307–313. [PubMed] [Google Scholar]
- [24].Scarpa M, Orchard PJ, Schulz A, Dickson PI, Haskins ME, Escolar ML, Giugliani R, Treatment of brain disease in the mucopolysaccharidoses, Mol. Genet. Metab 122S (2017) 25–34. 10.1016/j.ymgme.2017.10.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.