

Abstract
A second-generation (“Gen 2”) device capable of surface-induced dissociation (SID) and collision-induced dissociation (CID) for Fourier transform ion cyclotron resonance mass spectrometry of protein complexes has been designed, simulated, fabricated, and experimentally compared to a first-generation device (“Gen 1”). The primary goals of the redesign were to 1) simplify SID by reducing the number of electrodes, 2) increase CID and SID sensitivity by lengthening the collision cell, and 3) increase the mass range of the device for analysis of larger multimeric proteins, all while maintaining the normal instrument configuration and operation. Compared to Gen 1, Gen 2 exhibits an approximately 10x increase in sensitivity in flythrough mode, 7x increase in CID sensitivity for protonated leucine enkephalin (m/z 556) and 14x increase of CID sensitivity of 53 kDa streptavidin tetramer. It also approximately doubles of the useful mass range (from m/z 8,000 to m/z 15,000) using a rectilinear ion trap with a smaller inscribed radius or triples it (to m/z 22,000) using a hexapole collision cell and yields a 3–10x increase in SID sensitivity. We demonstrate the increased mass range and sensitivity on a variety of model molecules spanning nearly 3 orders of magnitude in absolute mass and present examples where the high resolution of the FT-ICR is advantageous for deconvoluting overlapping SID fragments.
Keywords: native mass spectrometry, surface-induced dissociation, FT-ICR, protein complex
Graphical Abstract
Introduction
Mass spectrometry is emerging as a powerful tool for structural biology.1–3 The observation that the tertiary and quaternary structure of proteins and their complexes can be kinetically trapped after transfer from solution to the gas phase4–6 through a combination of careful sample preparation (exchanging into a volatile buffer, e.g. ammonium acetate) and soft electrospray ionization7 led to the eventual coining of the term “native mass spectrometry” (nMS). This was followed by the emergence of improved mass spectrometric technologies (e.g. ion mobility,8–10 surface-induced dissociation,11–13 ultraviolet photodissociation,14 Q-IM-TOFs,15 FT-ICR,16–20 and high mass Orbitraps21,22) for studying high mass species.
Upon transfer to the gas phase, the intact mass of protein complexes can be measured with high accuracy (for Orbitraps and FT-ICRs), but complexes must be further probed in order to gain insight into complex stoichiometry, intersubunit interaction strength and arrangement, and possible ligand binding, all of which influence protein function. Activation methods are useful for dissecting intact complexes into subunits. Electron-transfer dissociation23 and ultraviolet photodissociation14,24 tend to dissociate proteins into small fragments (“top-down” covalent fragmentation) which are useful for sequencing and locating post-translational modifications and ligand binding sites but don’t typically provide overall connectivity/topology information. Collision-induced dissociation (CID), the most common form of activation due to its widespread commercial availability, generally produces highly unfolded/restructured, highly charged monomers with complementary (n-1)mers.10,25,26 While CID can provide direct stoichiometric (and mass) information, it generally does not give fragments consistent with inter-subunit connectivity and interaction strength, and the unfolding/restructuring of monomers is undesirable because native structure is not retained.
Surface-induced dissociation (SID), in which the collision target is a rigid, high-mass surface (usually coated with a fluorocarbon self-assembled monolayer) rather than a small gas molecule, has been shown to generate more symmetrically charged fragments that are reminiscent of the three-dimensional structure of protein complexes,11,27,28 in part because SID consists of a single collision with higher energy conversion vs. many low energy collisions in CID. SID can provide a wealth of information, including overall conformational changes,28–30 subunit interconnectivity30–35 and interaction strength36 (which can be used to constrain computer models36,37), and subunit-ligand interactions.38
To date, SID has been implemented on various instrument platforms. Small molecule SID has been performed on sector instruments,39,40 hybrid sector/quadrupole spectrometers,41 a 3D quadrupole ion trap,42 tandem quadrupoles,43,44 tandem time-of-flight systems,45 FT-ICRs,46,47 and in conjunction with quadrupole time-of-flight analyzers.48 More recently, we have implemented SID on instruments better equipped for nMS, namely Q-IM-TOFs (Waters Synapts),48 Thermo Scientific EMR and UHMR Orbitraps,49 and a Bruker 15 T FT-ICR.19 The legacy Synapt SID design, placed just after a truncated trap cell and consisting of 10 independently controlled DC electrodes48has been distributed to several other laboratories (Russell,50 Prell,51 Sharon,52 etc.) as part of our goal to disseminate native MS technology to the community. The Orbitrap and FT-ICR designs are more recent innovations but are especially promising because of the high resolution and mass accuracy of the respective analyzers. The 15 T FT-ICR, in particular, is a state-of-the-art mass spectrometer with ultra-high resolution, useful for resolving overlapping species in product-rich, often-convoluted SID spectra. Other groups have demonstrated the use of FT-ICR for analysis of protein complexes through a combination of intact mass analysis, CID, and other complementary activation techniques (infrared multiphoton dissociation and electron based dissociation).16–18
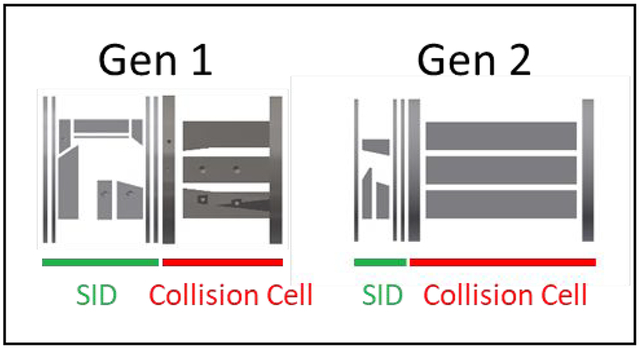
In this study we iterated on the design of the first-generation SID/CID device for FT-ICR mass spectrometry in order to improve its performance. In order to increase its sensitivity in full MS, CID, and SID modes, the SID region was shortened and the collision cell was lengthened by ~67% compared to Gen 1. This was accomplished, in part, by reducing the number of independently controlled SID electrodes from 10 to 6. Additionally, the mass range of Gen 2 was approximately doubled by decreasing the inscribed radius of the collision cell and further increased by adopting a hexapole design, allowing us to perform SID and successfully trap larger protein complexes. Even with these modifications, the device maintains its low mass capabilities for users of OSU’s mass spectrometry facility.
Experimental
Experimental details can be found in the supplemental information. Briefly, nanoelectrospray ionization generated ions for analysis by a Bruker solariX XR 15 T FT-ICR mass spectrometer.
Device design
The dual-purpose SID/CID cell is designed to replace Bruker’s commercially supplied collision cell, which is approximately 62 mm in length and is preceded by an electrospray (and MALDI) ion source, two ion funnels and skimmers, a transport multipole, and a quadrupole mass filter that can isolate up to m/z 6,000 (Figure 1a). The first generation SID/CID cell (Table 1, column 2, “Gen 1”)19 consisted of a 33.3 mm SID region with 10 independent DC electrodes coupled to a 28.7 mm rectilinear ion trap collision cell for CID and for trapping, cooling, and accumulating ions in SID, CID, and flythrough modes. The second-generation cell (“Gen 2”) presented in this work iterates on the Gen 1 design to improve its performance for native mass spectrometry. In this iteration, we focused on improving sensitivity and mass range while also reducing the number of SID electrodes to simplify tuning.
Figure 1:

Second-generation dual-purpose SID/CID cell for Fourier transform ion cyclotron resonance mass spectrometry. The SID/CID cell is designed to replace the Bruker collision cell in (a) the original instrument configuration. (b) Second generation SID/CID cell (electrodes only) and cross section of the rectilinear and hexapole designs, (c) 3D CAD drawing of the device with holders and collision cell housing, (d) fabricated design, and (e) SIMION simulation of SID of +10 streptavidin (53 kDa) at 85 V acceleration potential after exiting the quadrupole, showing the high efficiency of ion capture in the collision cell. In this simulation, only 10 ions are shown (black and red are the same mass and charge before and after collision, respectively), but for most simulations a population of 1,000 ions was used. The inset steel surface shows, via discoloration, where the ions are colliding with the surface experimentally, in good agreement with the simulation. Note that for SID of protein complexes, a gold-SAM surface was used.
Table 1:
Comparison of Gen 1 and Gen 2 characteristics.
| Characteristic | Gen 1 | Gen 2 |
|---|---|---|
| SID cell length | 33.3 mm | 14.0 mm |
| Number of SID Electrodes | 10 | 6 |
| Surface orientation | Level | 10° tilt |
| Surface material | Fluorinated SAM (C12) on gold | Fluorinated SAM (C12) on gold |
| CID cell length | 28.7 mm | 48.0 mm |
| Collision cell r0 | 5.28 mm | (Rectilinear) 4.5 mm (Hexapole) 4.35 mm |
| Collision cell rod shape | Square, 6.35 × 6.35 mm | (Rectilinear) Square, 6.35 × 6.35 mm (Hexapole) Circular, 4.75 × 4.75 mm |
| Collision cell voltage | 2,000 Vpp, two-phase | 2,000 Vpp, two-phase |
| Collision cell frequency (switchable) | 1.58 MHz or 2.63 MHz | (Rectilinear) 1.54 MHz or 2.54 MHz (Hexapole) 1.5 MHz or 2.46 MHz |
| Front endcap aperture diameter | 5.0 mm | 5.0 mm |
| Rear endcap aperture diameter | 2.4 mm | 2.5 mm |
| Asymptotic rods | Yes | Optional (not used here) |
The design of Gen 2 is shown in Figure 1b and its characteristics are given in Table 1, column 3. The SID region is considerably shorter than in Gen 1 – 14 mm compared to 33.3 mm - and also consists of 4 fewer electrodes (6 instead of 10). The front lens and rear two lenses control ion kinetic energies prior to and after surface collision and have inner apertures 5.0 mm in diameter. The surface in Gen 2 is tilted at 10 degrees to improve ion extraction after surface collision, as demonstrated previously in designs from Stiving et al.53,54 In our simulations of Gen 2, reflected ions were more likely to turn around and return to the surface (which is held at a highly attractive negative voltage) if the surface orientation was level, whereas they were less likely to do so when the surface was slightly tilted. The simplification of the SID region from 10 to 6 electrodes allowed us to extend the CID/trapping region to 48.0 mm from 28.7 mm, an increase of 67%. In doing so, the trapping capacity – and hence sensitivity – of the cell improved by approximately an order of magnitude, discussed later. Gen 2 otherwise retains the favorable trapping characteristics of the Gen 1 rectilinear ion trap collision cell. However, in order to increase the effective mass range of the device, the inscribed radius (r0) of the collision cell was reduced from 5.28 mm to 4.5 mm. As discussed later, this change resulted in a doubling of the device’s mass range due to increased radial pseudopotential well depth.55,56 In particular, any “hot” high m/z ions that are reflected off the surface with relatively high radial kinetic energy (e.g. a +20 ion accelerated through a 100 V potential difference keeps ~5% of its pre-collision kinetic energy, i.e. 100 eV) were not trapped in a sufficiently deep well in Gen 1’s collision cell, even when using the 2,000 Vpp maximum voltage (at 1.58 MHz) of the Bruker rf driver. By decreasing the half distance between the rods by 0.78 mm, the radial pseudopotential well depth is increased by approximately 37% (all else being equal). A second design incorporated a hexapole of the same length with r0 = 4.35 mm which further increased the ions’ pseudopotential well depth and led to a further increase in mass range.
Figure 1c shows a 3D rendering (in Autodesk Inventor) of Gen 2 with holders and gas-tight collision cell housing. Figure 1d is a picture of the final fabricated device fitted into two plastic holders which mount the device to the ion optic rails in the FT-ICR. The SID and trap endcap holders are PEEK, while the electrodes and collision cell housing are stainless steel. The ion trap holders are ceramic Macor™. The collision cell is pressurized with argon using the same gas supply and fittings as the Bruker collision cell. DC voltages are applied to the six SID electrodes from a 10-channel Ardara DC power supply, which is connected to the electrodes via 1 mm diameter dowel pins (not shown). The two phases of rf voltage are inserted into the pins on top and on the side of the collision cell. The commercial Bruker rf pins for the ion trap and dc pins for the endcap voltages fit directly into Gen 2 without any adapters. Although the rectilinear and hexapole designs use unique housings, rods, and ceramic rod holders, all other parts (SID electrodes and holders as well as endcaps and their holders) were interchangeable between the two designs, simply requiring removal and reattachment of 8 screws bolting the endcap and SID holders to the collision cell.
Typical voltages applied in transmission and SID modes are given in Table S1. The quadrupole and its pre- and post-filters, which directly precede the SID device, are generally held at 1 V DC bias. In transmission mode, balanced voltages are applied to electrodes #2–4 to transmit ions from the quadrupole mass filter to the collision cell, where the ions are accumulated (and dissociated, if desired) until they are pulsed into the FT-ICR analyzer. In SID mode, the front bottom SID electrode (#2) deflects the ion beam coming from the quadrupole into the surface (#3), the reflected ions are extracted by electrodes #5 and #6, their outgoing angles are adjusted with electrode #4, and the beam is directed into the collision cell for accumulation. The voltage of the front trap endcap dictates the acceptance of the collision cell in terms of kinetic energy (assuming the kinetic energy does not exceed the pseudopotential well depth in the radial dimension). The SID acceleration voltage is varied by ΔV as shown in Table S1. For small molecule MS/MS, isolation of the precursor species prior to activation was possible; however, the vendor-provided quadrupole and RF generator cannot select protein complexes > m/z 4,000 without substantial loss in signal intensity. As a consequence, MS/MS spectra of most protein complexes in this work were obtained without precursor isolation. We are currently working to resolve this issue by lowering the frequency of the quadrupole rf driver. That change is not necessary to illustrate the increased performance of the Gen 2 device.
Simulations
All simulations were conducted in SIMION 8.1. For most simulations the initial ion distribution as it was created in the quadrupole mass filter consisted of the following: number of particles, 1,000; precursor mass, 53,220 Da; precursor charge, +10; source position, Gaussian distribution with 0.2 mm standard deviations in the radial dimension and 1 mm standard deviation in the axial dimension; kinetic energy distribution, Gaussian with mean of 35 eV and 5 eV FWHM; direction, cone distribution with 7 degree half angle (filled); time-of-birth, uniform distribution between 0 and 5 μs. Note that black trajectories consist of ions prior to surface collision and red trajectories consist of ions after surface collision. The ion kinetic energy in the FT-ICR is set in the partitioned octupole region prior to the quadrupole and was determined using simple stopping curves.
The simulation used in this work consisted of 27 electrodes: 1 quadrupole entrance lens, 12 quadrupole electrodes (4 pre-filter, 4 analyzer, 4 post-filter), 1 quadrupole exit lens and 1 focusing element, 6 SID electrodes, and 6 collision cell electrodes (4 rectangular trap rods with front and rear endcaps). A second simulation using the hexapole cell was also constructed but did not differ significantly from the rectilinear design. More details about the collision cell and SID electrodes can be found in the Device Design section. The quadrupole simulated here is a Bruker/Extrel 9.5 mm Tri model operated at 880 kHz.
In the simulation (Figure 1e), ions that are deflected into the surface by the front bottom electrode are stopped just before collision and they are reflected at a specular angle (with respect to the surface normal), while retaining 5% of their kinetic energy (measured just before surface collision). We assume here that the precursor ions do not fragment in the SID device but instead fragment later in the collision cell, an assumption supported by prior studies with leucine enkephalin.57 Hence, the mass and charge of the ion do not change during the simulation, and fragmentation is not explicitly induced. The reflected precursor ions are then extracted by a rear SID lens (#5), their axial angle is corrected by a second deflection electrode (#4), and they are captured in the rf-only collision cell, which is simulated with approximately 50 mtorr of argon as buffer gas. The simulation shown in Figure 1e shows ten ions of mass 53.220 kDa and charge +10 being accelerated by an 85 V potential difference. After surface collision the ions are brought back on axis and collected in the collision cell. In this particular simulation, all ten ions were successfully collected, but in a typical simulation approximately 60–70% of the ions were collected, with some lost to the rear SID electrodes due to large reflection angles or large deviations in kinetic energy after collision. In the simulation, charge reduction on the surface was not considered; neutralization of these large multiply-charged ions is thought to be minimal. The inset in the simulation is an image of a stainless steel surface after several hours of surface collisions. The impact location is consistent with the simulation results, indicating a closeness of fit between the simulated device and actual experimental conditions. Note that for this experiment a stainless steel surface was used; for all other experiments a fluorocarbon self-assembled monolayer was used.
Results & Discussion
Mass Range
A critical limitation of the Gen 1 SID design was the limited mass range (< m/z 8,000), evident in the mass spectrum of CsPFHA clusters in Figure S1, orange. The highest detected m/z is approximately m/z 8,859. This is a direct consequence of using a large inscribed radius in the collision cell, 5.28 mm in Gen 1. The truncated mass range is further evidenced by decreasing the collision cell voltage systematically until 50% of the CsPFHA intensity (at each m/z) is lost compared to the maximum intensity observed at higher rf voltage. Figure S2 (orange) shows the resulting plot which indicates that the Gen 1 collision cell is only capable of efficiently trapping up to ~m/z 7,000, in agreement with the mass restrictions observed by Jing et al.19,20 To increase the mass range for native mass spectrometry of high molecular weight protein complexes, the inscribed radius of the rectilinear ion trap was reduced from 5.28 mm to 4.5 mm, resulting in an approximate doubling of the mass range to ~m/z 15,000 (Figure S1, blue), allowing, for example, observation of higher m/z species in the native mass spectrum of 115 kDa C-reactive protein (CRP, Figure S3). The species observed around m/z 7,000 are various charge states of the pentameric form of the protein complex, and the higher m/z species near m/z 10,000 correspond to the nonspecific CRP decamer, which is observed in Gen 2 but not Gen 1 (even when spraying the same solution under identical conditions). The second notable feature of Figure S3 is the increased signal-to-noise ratio, which is discussed next. Adoption of a hexapole design with slightly smaller inscribed radius (4.35 mm) further increased the mass range of the device to >m/z 22,000, as discussed later. Because the hexapole rods are identical to those from the original Bruker collision cell, the mass range of Gen 2 is now approximately equal to the original collision cell.
Sensitivity
The sensitivity of Gen 2 compared to Gen 1 was profiled in 1) full MS, 2) CID, and 3) SID modes using a variety of large and small molecules. For this comparison the rectilinear cell was used. Although in Gen 2 the collision cell inscribed radius was reduced from 5.28 mm to 4.5 mm, the trap was lengthened by 67%, more than making up for any trap capacity lost in the radial dimension. For CsPFHA clusters analyzed in transmission mode at moderate accumulation times (< 0.5 s), the absolute ion signal increased by 2–3.5x when using Gen 2 (Figure S4). At higher accumulation times, where the ion trap was purposely saturated to find relative capacities, Gen 2 exhibited approximately 30% higher signal. For charge-reduced streptavidin tetramer (53 kDa, mostly 10+ charge state), the gains in signal were more apparent (Figure S5), especially at longer accumulation times. For accumulation times less than 1 s, a gain in signal intensity of approximately 3x was observed, whereas a gain of more than an order of magnitude (up to 14x) was observed when the accumulation time was >1s. The increased performance was particularly evident in the mass spectrum of C-reactive protein already discussed (Figure S3), where a S/N value of ~110 was observed for the 18+ charge state on Gen 2 using a 1 s accumulation time, whereas S/N of ~28 was observed for the 17+ charge state (the most intense peak) on Gen 1. These gains are likely due to the increased trapping capacity, but improved collisional cooling in the longer Gen 2 collision cell also likely plays a role.
The CID signal intensity was profiled using protonated leucine enkephalin ([YGGFL+H]+). At 0.5 s accumulation time, the isolated [YGGFL+H]+ signal (5 m/z isolation width) was 20x higher for the second-generation cell (Figure S6, Isolation). Using 25 V of accelerating potential and argon as collision gas, the absolute CID signal for leucine encephalin product ions was 7x higher in Gen 2. CID of streptavidin tetramer to produce highly charged monomer, charge-stripped tetramer, and a small amount of trimer (Figure S7), results in an up to 20x increase in signal intensity for Gen 2. Gen 2 was tested with 65 V of potential difference and Gen 1 with 35 V because Gen 1 returned no signal at 65 V. Because the collision energies are different, only the saturated collision cells should be compared. Even so, when the cells were fully saturated, Gen 2 accumulated up to 20x higher signal intensity. Compared to the Bruker collision cell, Gen 2 has approximately the same sensitivity for protein complexes.
High-resolution SID of Protein Complexes
The increased sensitivity and mass range of Gen 2 yielded far better signal-to-noise than the previous design and further allowed us to work with higher mass macromolecules, where SID is particularly useful for dissecting topology and stoichiometry of protein complexes29,32,58 as well as for determining intersubunit interaction strength.36 Streptavidin is a D2-symmetric tetrameric protein complex that is a dimer of dimers. SID of the intact protein complex should yield fragments consistent with this observation.31 Although streptavidin has been investigated thoroughly by SID previously, we use it here to demonstrate the signal-to-noise ratio (S/N) achievable on Gen 2.
A full scan – using the Gen 2 rectilinear cell - of charge-reduced streptavidin is shown in Figure S8a. The dominant charge states under charge-reducing conditions are 9+ through 11+. The S/N of the 10+ tetramer in this scan is approximately 470. Note that the coldness of the FT-ICR’s source and the lack of an effective in-source activation method results in salt adducts being observed. CID of the tetrameric complex using an accelerating voltage of 65 V (Figure S8b) yields mostly highly charged monomers and a small amount of complementary trimers which are not indicative of the dimer of dimers arrangement of the subunits. In contrast, SID (Figure S8c) yields mostly symmetrically charged dimers and monomers which are consistent with the D2 molecular symmetry.31 The high resolution of the FT-ICR allows us to isotopically resolve overlapping +6 dimer and 3+ monomer at m/z 4424 (not shown) and 4+ dimer and 2+ monomer at m/z 6636 (Figure S8d). At lower SID energies the 6+ and 4+ dimers are more apparent (Figure S8d), whereas at higher SID energies mostly monomer is observed since the dimers further dissociate at higher collision energy. Even so, the charge states of the monomers are significantly lower than those of the unfolded monomers produced through CID, and both dimers and monomers take an amount of charge approximately proportional to their mass, a characteristic of SID spectra of protein complexes. The S/N of the most intense peak in panel (c) is approximately 1,000, illustrating a substantial gain in sensitivity. Compared to Gen 1, Gen 2 provided up to an order of magnitude higher signal in SID mode using streptavidin as a model protein complex (Figure S9), especially at low-to-moderate accumulation times. At full trap capacity (high accumulation times), the signal from Gen 2 was approximately 3.6x that of Gen 1, even though the length of the rectilinear collision cell was only increased by 67%.
Pentameric C-reactive protein (CRP, ~115 kDa) was also tested as a model protein complex on Gen 1 and Gen 2. The full scan of charge-reduced CRP using Gen 2 (using the hexapole design) is shown in Figure 2a. The pentameric complex with charge states ranging from 15+ to 19+ was observed in transmission mode using both the rectilinear cell (not shown) and the hexapole. The species near m/z 9,000 and above correspond to nonspecific CRP decamers which are not of interest here but cannot be excluded due to quadrupole limitations. As stated before, approximately an order of magnitude higher signal was observed on Gen 2 compared to Gen 1 in full MS mode (Figure S3). CID and SID of CRP on Gen 2 using the hexapole collision cell gave the spectra shown in Figure 2b and 2c for an average of only ten scans. The rectilinear cell gave similar spectra (not shown). The CID spectrum shows highly charged monomer with charge states ranging from 8+ to 12+ as well as complementary tetramer (9+ to 13+) and unfragmented pentamer. SID yields more moderately charged monomers with charge states from 3+ to 5+ and a small amount of dimer, trimer, and tetramer with a proportional amount of charge. The observation of monomers, dimers, trimers, and tetramers is consistent with the known ring pentameric structure.
Figure 2:

SID of pentameric C-reactive protein using gen 2 (hexapole design). (a) Mass spectrum of charge-reduced CRP (some decamer is also observed above m/z 9,000), (b) CID spectrum using 70 V acceleration potential, and (c) SID spectrum using 65 V acceleration potential.. P = pentamer, Q = tetramer, Tr = trimer, D = dimer, and M = monomer.
Cholera toxin B (CTB) is also a ring-structured homopentameric protein complex (58 kDa); under moderate SID conditions CTB fragments to monomers, dimers, trimers, and tetramers (in agreement with our previous results on FT-ICR and Orbitrap instruments19,38) and therefore provides a suitable example of the utility of the FT-ICR’s resolution. The full scan of CTB (Figure S10a) using the rectilinear cell gives 10+ through 12+ charge states as the prevalent charge reduced species. The SID spectrum in Figure S10b give more diverse fragmentation than that of CRP, with monomer through tetramer all represented in the spectrum. The high resolving power of the FT-ICR (approximately 366,000 at m/z 5803 using FWHM in this spectrum) allows us to distinguish the overlapping 8+ tetramer from 6+ trimer, 4+ dimers, and 2+ monomer (Figure S10b, inset). Both the full MS and SID spectrum were collected using an average of 10 scans, whereas similar spectra were collected previously on Gen 1 using 30 scans for the full MS and 181 scans for the SID spectrum.19
Figure 3 is a further example of the utility of the FT-ICR for resolving overlapping species using Hfq65. Hfq proteins are bacterial homohexameric chaperones that aid in binding of regulatory RNA to target mRNAs.59,60 The ring-like hexameric structure assists with RNA interactions with three types of binding of RNA to the protein complex. Hfq65 is a 43 kDa variant with truncation after the 65th residue. Figure 3a shows the mass spectrum of the isolated 9+ hexamer using the rectilinear cell. In this case, where the low mass of the complex yields precursor m/z values lower than 4000 that can be mass-selected with the quadrupole, isolation of a single charge state allowed us to resolve even the carbon isotopes of the intact complex – separated by 0.11 m/z – by reducing space charge interactions in the FT-ICR. The resolution of the peaks is approximately 197,000. Resolved carbon isotopes were not observed for other protein complexes due to salt adduction, heterogeneity, and possible space charging, whereas Hfq65 (even with only one buffer exchange) had very little adduction even prior to isolation. The SID spectrum of the homohexamer, shown in Figure 3b, gives abundant monomer through pentamer with high S/N, as expected for a cyclic hexameric complex. The peak at m/z 7186 consists of monomer through tetramer (and perhaps pentamer of low abundance), with each species easily resolved (Figure 3c) using an 8M transient with low m/z set to 1,000 (~16 s transient). The resolution observed at m/z 7186 in panel (c) is approximately 148,000.
Figure 3:

SID of hexameric Hfq65 (43 kDa) RNA chaperone (using rectilinear design). (a) Mass spectrum of isolated 9+ hexamer, (b) SID spectrum using 85 V of acceleration, yielding overlapping monomers, dimers, trimers, tetramers, and pentamers, and (c) resolution of the overlapping species at m/z 7186. The resolution observed in panel (a) is approximately 197,000 and in panel (c) is approximately 148,000.
Extended mass range
The increased mass range and sensitivity of Gen 2 allowed us to probe even larger protein complexes such as 333 kDa hexameric glutamate dehydrogenase (GDH). Previously the largest complex we were able to work with on FT-ICR was a 211 kDa multicopper oxidase protein complex.20 However, at such a high m/z and with limited trapping capacity, the analysis required 300 scans each. Gen 2, on the other hand, is capable of working with larger complexes due to higher trap capacity, higher pseudopotential well depth, and improved collisional cooling, and even >300 kDa ions can be successfully collided with the SID surface and collected in the collision cell.
For example, the full MS of 333 kDa GDH obtained with Gen 2 is shown in Figure 4a. Although both the rectilinear and hexapole cells were capable of analyzing GDH, the hexapole yielded higher quality spectra and was thus used for this experiment. Prominent charge states (without charge reduction) are 36+ through 42+. The SID spectrum using 135 V of acceleration potential is given in Figure 4b. The dominant product ions are trimers with approximately half the charge of the precursors (16+ to 24+) and overlapping unfragmented and charge-stripped hexamer. The observation of trimers rather than pentamers, tetramers, or dimers is consistent with the dimer of trimers arrangement previously proposed61 and observed by SID on two separate instrument platforms.32,49 Note that the SID spectrum was recorded using a 1 M transient while averaging ten scans. This experiment illustrates the extended mass range of the new collision cell design and the ability to work with sturdy complexes that are difficult to fragment, as GDH is known to resist fragmentation even by 200 V (7800 eV for the 39+ charge state) CID.32
Figure 4:

Extended mass range using Gen 2 SID for analysis of 330 kDa glutamate dehydrogenase hexamer (using hexapole design): (a) full MS of GDH, (b) SID spectrum of GDH – without isolation – using 135 V acceleration potential. Note the overlap between trimer and hexamer. Each spectrum is an average of 10 scans.
Two other large complexes were tested on Gen 2: 230 kDa pyruvate kinase (a tetramer) and 801 kDa GroEL (14mer). Figure S11 shows the full scan and SID spectrum of pyruvate kinase. As expected for a tetramer with dimer-of-dimers configuration (like streptavidin) the dominant product ions in the SID spectrum (Figure S11b) are dimers, with some monomer and a small amount of trimer. Note that the apparent peak splitting is caused by adduction of several 324 Da molecules of 2,5-anhydro-d-glucitol, 1,6 bisphosphate, which has been observed to be stable toward activation previously by Kelleher and coworkers.62 The splitting is observed in the inset full scan (obtained with less spectral smoothing) as well as in the SID spectrum (inset shows an adducted monomer, for example).
Gen 2 is also capable of trapping and fragmenting 801 kDa GroEL by SID, a substantial increase in effective mass range compared to Gen 1. The full scan in Figure S12 shows +70 as the average and most abundant charge state under non-charge reducing conditions in 200 mM ammonium acetate buffer. The SID spectrum in panel (b) shows contribution from all oligomer fragments, with 7mer (grey circles) as a prominent species due to the 14mer’s dimers of 7mers arrangement. The peaks noted by red diamonds could consist of all possible oligomers and so they are labeled as ‘overlap’ peaks. This spectrum is in agreement with the SID spectrum previously recorded on a Synapt platform.63 One unfavorable feature of the ICR SID spectra of higher m/z complexes such as pyruvate kinase and GroEL is that the baseline rises at higher m/z and the product ions are poorly resolved, in part because of the poorer desalting and limited in source activation capabilities of the instrument as commercially supplied, limitations we are actively working to resolve.
SID of small molecules
Although the main goal of this work was to improve the ability of the FT-ICR to analyze large noncovalent protein complexes, the Gen 2 SID/CID cell is equally capable of conducting small molecule CID and SID studies. Figure 5 shows (left) CID and (right) SID spectra of the proton-bound dimer of leucine enkephalin (m/z 1111) using acceleration potentials from 15V to 65 V (15 eV to 65 eV for these singly charged ions) on the rectilinear cell. Because these were small molecules, we were able to conduct isolation (3 m/z width) prior to both CID and SID. The spectra are markedly similar with regard to the product ions observed and the relative intensities of those product ions, a typical result for activation of small, easy-to-fragment molecules by CID and SID. In both cases, the main ions detected are b and y ions, characteristic of lower-energy fragmentation pathways.24 The spectra are also similar to those we have previously observed via SID experiments.64 For example, the a4 and b3 ions are the most abundant on the FT-ICR and were reported as the most abundant species on a tandem quadrupole SID instrument. Internal F, Y, and GGF fragments are also observed. On the FT-ICR, we can resolve the y2 fragment ion from the carbon isotope of the b3 ion, as shown in the insets. SID of the yet smaller protonated leucine enkephalin (m/z 556) yielded similar fragments and intensities (Figure S13) and demonstrates the versatility of Gen 2 for studies of both small and large molecules by use of a combination CID/SID collision cell. This is particularly important to satisfy the demands of OSU’s mass spectrometry user facility, of which the FT-ICR is an integral component, so that collision cells need not be swapped between experiments.
Figure 5:

MS/MS of the proton-bound dimer of leucine enkephalin (m/z 1111) (rectilinear cell): (a) collision-induced dissociation, and (b) SID. No fragments above m/z 556 were observed.
Conclusion
An improved dual-purpose SID/CID cell has been developed for native mass spectrometry on a 15 T FT-ICR mass spectrometer while retaining low mass capabilities for OSU campus users. Compared to the previous generation, the new design boasts a 2–3x expanded mass range, 2–3x higher trap capacity, ~10x increase in signal intensity noncovalent protein complexes at moderate accumulation times, and simplification via reduction of the number of SID electrodes. Both rectilinear and hexapole collision cells were utilized; the latter could analyze protein complexes up to 801 kDa in absolute mass. The Gen 2 design couples well with the high mass resolution and mass accuracy of the FT-ICR and maintains efficient CID capabilities, which are characteristics critical to daily operation of the FT-ICR for analyzing core facility samples from OSU principal investigators.
With improved SID/CID capabilities implemented, it is now critical to optimize other aspects of the FT-ICR for native mass spectrometry. In particular, it should be a top priority to make the ion source more native MS friendly by decreasing salt adduction and increasing the versatility of in source activation capabilities so that the high resolution can be successfully utilized in combination with the many ion activation methods currently available.
Supplementary Material
Acknowledgements
The authors thank Jing Yan (Amgen) and Randy Pedder (Ardara Technologies) for helpful discussions about the Gen 1 design, Mengxuan Jia (The Ohio State University), Jorjethe Roca (Johns Hopkins University), and Sarah Woodson (Johns Hopkins University) for the Hfq65 sample, and Sophie Harvey (OSU) for sample preparation training. The authors also thank Larry Antal and the OSU machine shop for fabricating Gen 2, and Mark Ridgeway and Mel Park (Bruker) for helpful discussions and for providing parts for Gen 2. The Hfq65 sample was funded by NIH R01 GM120425. The FT-ICR was purchased using funds from NIH Award S10 OD018507. This work was supported by the NIH P41 Resource for Native Mass Spectrometry Guided Structural Biology, P41GM128577.
References
- (1).Heck AJ Nature Methods 2008, 5, 927. [DOI] [PubMed] [Google Scholar]
- (2).van den Heuvel RH; Heck AJ Curr. Opin. Chem. Biol 2004, 8, 519–526. [DOI] [PubMed] [Google Scholar]
- (3).Leney AC; Heck AJ J. Am. Soc. Mass Spectrom 2017, 28, 5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Katta V; Chait BT J. Am. Chem. Soc 1991, 113, 8534–8535. [Google Scholar]
- (5).Ganem B; Li YT; Henion JD J. Am. Chem. Soc 1991, 113, 6294–6296. [Google Scholar]
- (6).Laganowsky A; Reading E; Hopper JT; Robinson CV Nat. Protoc 2013, 8, 639–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Fenn JB; Mann M; Meng CK; Wong SF; Whitehouse CM Science 1989, 246, 64–71. [DOI] [PubMed] [Google Scholar]
- (8).Allen SJ; Eaton RM; Bush MF Anal. Chem 2016, 88, 9118–9126. [DOI] [PubMed] [Google Scholar]
- (9).Lanucara F; Holman SW; Gray CJ; Eyers CE Nat. Chem 2014, 6, 281. [DOI] [PubMed] [Google Scholar]
- (10).Ruotolo BT; Hyung S-J; Robinson PM; Giles K; Bateman RH; Robinson CV Angew. Chem. Int. Ed 2007, 46, 8001–8004. [DOI] [PubMed] [Google Scholar]
- (11).Wysocki VH; Jones CM; Galhena AS; Blackwell AE J. Am. Soc. Mass Spectrom 2008, 19, 903–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Wysocki VH; Joyce KE; Jones CM; Beardsley RL J. Am. Soc. Mass Spectrom 2008, 19, 190–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Chorush RA; Little DP; Beu SC; Wood TD; McLafferty FW Anal. Chem 1995, 67, 1042–1046. [DOI] [PubMed] [Google Scholar]
- (14).O’Brien JP; Li W; Zhang Y; Brodbelt JS J. Am. Chem. Soc 2014, 136, 12920–12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Giles K; Pringle SD; Worthington KR; Little D; Wildgoose JL; Bateman RH Rapid Commun. Mass Spectrom 2004, 18, 2401–2414. [DOI] [PubMed] [Google Scholar]
- (16).Li H; Nguyen HH; Ogorzalek Loo RR; Campuzano IDG; Loo JA Nat. Chem 2018, 10, 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Lippens JL; Nshanian M; Spahr C; Egea PF; Loo JA; Campuzano IDG J. Am. Soc. Mass Spectrom 2018, 29, 183–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Zhang H; Cui W; Wen J; Blankenship RE; Gross ML J. Am. Soc. Mass Spectrom 2010, 21, 1966–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Yan J; Zhou M; Gilbert JD; Wolff JJ; Somogyi A; Pedder RE; Quintyn RS; Morrison LJ; Easterling ML; Pasa-Tolic L; Wysocki VH Anal. Chem 2017, 89, 895–901. [DOI] [PubMed] [Google Scholar]
- (20).Zhou M; Yan J; Romano CA; Tebo BM; Wysocki VH; Paša-Tolić LJ Am. Soc. Mass Spectrom 2018, 29, 723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Fort KL; Van de Waterbeemd M; Boll D; Reinhardt-Szyba M; Belov ME; Sasaki E; Zschoche R; Hilvert D; Makarov AA; Heck AJ Analyst 2018, 143, 100–105. [DOI] [PubMed] [Google Scholar]
- (22).Rose RJ; Damoc E; Denisov E; Makarov A; Heck AJ Nature methods 2012, 9, 1084. [DOI] [PubMed] [Google Scholar]
- (23).Syka JE; Coon JJ; Schroeder MJ; Shabanowitz J; Hunt DF Proc. Natl. Acad. Sci. U.S.A 2004, 101, 9528–9533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Brodbelt JS Anal. Chem 2016, 88, 30–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Felitsyn N; Kitova EN; Klassen JS Anal. Chem 2001, 73, 4647–4661. [DOI] [PubMed] [Google Scholar]
- (26).Benesch JL; Aquilina JA; Ruotolo BT; Sobott F; Robinson CV Chem. Biol 2006, 13, 597–605. [DOI] [PubMed] [Google Scholar]
- (27).Jones CM; Beardsley RL; Galhena AS; Dagan S; Cheng G; Wysocki VH J. Am. Chem. Soc 2006, 128, 15044–15045. [DOI] [PubMed] [Google Scholar]
- (28).Zhou M; Dagan S; Wysocki VH Analyst 2013, 138, 1353–1362. [DOI] [PubMed] [Google Scholar]
- (29).Zhou M; Wysocki VH Acc. Chem. Res 2014, 47, 1010–1018. [DOI] [PubMed] [Google Scholar]
- (30).Quintyn RS; Zhou M; Yan J; Wysocki VH Anal. Chem 2015, 87, 11879–11886. [DOI] [PubMed] [Google Scholar]
- (31).Quintyn Royston S.; Yan J; Wysocki Vicki H. Chem. Biol 2015, 22, 583–592. [DOI] [PubMed] [Google Scholar]
- (32).Ma X; Zhou M; Wysocki VH J. Am. Soc. Mass Spectrom 2014, 25, 368–379. [DOI] [PubMed] [Google Scholar]
- (33).Blackwell AE; Dodds ED; Bandarian V; Wysocki VH Anal. Chem 2011, 83, 2862–2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Zhou M; Dagan S; Wysocki VH Angew. Chem. Int. Ed 2012, 51, 4336–4339. [DOI] [PubMed] [Google Scholar]
- (35).Sahasrabuddhe A; Hsia Y; Busch F; Sheffler W; King NP; Baker D; Wysocki VH Proc. Natl. Acad. Sci. U.S.A 2018, 115, 1268–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Harvey SR; Seffernick JT; Quintyn RS; Song Y; Ju Y; Yan J; Sahasrabuddhe AN; Norris A; Zhou M; Behrman EJ; Lindert S; Wysocki VH Proc. Natl. Acad. Sci. U.S.A 2019, 116, 8143–8148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Song Y; Nelp MT; Bandarian V; Wysocki VH ACS Cent. Sci 2015, 1, 477–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Busch F; VanAernum ZL; Ju Y; Yan J; Gilbert JD; Quintyn RS; Bern M; Wysocki VH Anal. Chem 2018, 90, 12796–12801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Schey KL; Durkin DA; Thornburg KR J. Am. Soc. Mass Spectrom 1995, 6, 257–263. [DOI] [PubMed] [Google Scholar]
- (40).Despeyroux D; Wright AD; Jennings KR Int. J. Mass Spectrom. Ion Process 1993, 126, 95–99. [Google Scholar]
- (41).Mabud MA; Dekrey MJ; Graham Cooks R Int. J. Mass Spectrom. Ion Process 1985, 67, 285–294. [Google Scholar]
- (42).Lammert S; Cooks RJ Am. Soc. Mass Spectrom 1991, 2, 487–491. [DOI] [PubMed] [Google Scholar]
- (43).Wysocki VH; Ding J-M; Jones JL; Callahan JH; King FL J. Am. Soc. Mass Spectrom 1992, 3, 27–32. [DOI] [PubMed] [Google Scholar]
- (44).Bier M; Amy J; Cooks R; Syka J; Ceja P; Stafford G Int. J. Mass Spectrom. Ion Process 1987, 77, 31–47. [Google Scholar]
- (45).Schey K; Cooks R; Grix R; Wöllnik H Int. J. Mass Spectrom. Ion Process 1987, 77, 49–61. [Google Scholar]
- (46).Laskin J; Denisov EV; Shukla AK; Barlow SE; Futrell JH Anal. Chem 2002, 74, 3255–3261. [DOI] [PubMed] [Google Scholar]
- (47).Williams ER; Henry KD; McLafferty FW; Shabanowitz J; Hunt DF J. Am. Soc. Mass Spectrom 1990, 1, 413–416. [DOI] [PubMed] [Google Scholar]
- (48).Galhena AS; Dagan S; Jones CM; Beardsley RL; Wysocki VH Anal. Chem 2008, 80, 1425–1436. [DOI] [PubMed] [Google Scholar]
- (49).VanAernum ZL; Gilbert JD; Belov ME; Makarov AA; Horning SR; Wysocki VH Anal. Chem 2019, 91, 3611–3618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Shirzadeh M; Boone CD; Laganowsky A; Russell DH Anal. Chem 2019, 91, 2345–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Donor MT; Mroz Austin M.; Prell JS Chem. Sci 2019, 10, 4097–4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Vimer S; Ben-Nissan G; Morgenstern D; Quintyn RS; Wysocki VH; Sharon M Submitted [Google Scholar]
- (53).Stiving AQ; Gilbert JD; Wysocki VH In 66th ASMS Conference on Mass Spectrometry & Allied Topics: San Diego, CA, 2018. [Google Scholar]
- (54).Stiving AQ; Gilbert JD; Jones BJ; Wysocki VH Submitted 2019. [Google Scholar]
- (55).Dehmelt HG In Advances in Atomic and Molecular Physics; Elsevier, 1968, pp 53–72. [Google Scholar]
- (56).March RE; Todd JF Practical aspects of trapped ion mass spectrometry, volume IV: Theory and instrumentation; CRC press, 2010. [Google Scholar]
- (57).Laskin JJ Phys. Chem. A 2006, 110, 8554–8562. [DOI] [PubMed] [Google Scholar]
- (58).Stiving AQ; VanAernum ZL; Busch F; Harvey SR; Sarni SH; Wysocki VH Anal. Chem 2019, 91, 190–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Vogel J; Luisi BF Nat. Rev. Microbiol 2011, 9, 578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Sauer E; Weichenrieder O Proc. Natl. Acad. Sci. U.S.A 2011, 108, 13065–13070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Peterson PE; Smith TJ Structure 1999, 7, 769–782. [DOI] [PubMed] [Google Scholar]
- (62).Belov ME; Damoc E; Denisov E; Compton PD; Horning S; Makarov AA; Kelleher NL Anal. Chem 2013, 85, 11163–11173. [DOI] [PubMed] [Google Scholar]
- (63).Zhou M; Jones CM; Wysocki VH Anal. Chem 2013, 85, 8262–8267. [DOI] [PubMed] [Google Scholar]
- (64).Meot-Ner M; Dongre AR; Somogyi A; Wysocki VH Rapid Commun. Mass Spectrom 1995, 9, 829–836. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.