

Abstract
Aberrant regulation of myocardial force production represents an early biomechanical defect associated with sarcomeric cardiomyopathies, but the molecular mechanisms remain poorly defined. Here, we evaluated the pathogenicity of a previously unreported sarcomeric gene variant identified in a pediatric patient with sporadic dilated cardiomyopathy, and we determined a molecular mechanism. Trio whole-exome sequencing revealed a de novo missense variant in TNNC1 that encodes a p.I4M substitution in the N-terminal helix of cardiac troponin C (cTnC). Reconstitution of this human cTnC variant into permeabilized porcine cardiac muscle preparations significantly decreases the magnitude and rate of isometric force generation at physiological Ca2+-activation levels. Computational modeling suggests that this inhibitory effect can be explained by a decrease in the rates of cross-bridge attachment and detachment. For the first time, we show that cardiac troponin T (cTnT), in part through its intrinsically disordered C terminus, directly binds to WT cTnC, and we find that this cardiomyopathic variant displays tighter binding to cTnT. Steady-state fluorescence and NMR spectroscopy studies suggest that this variant propagates perturbations in cTnC structural dynamics to distal regions of the molecule. We propose that the intrinsically disordered C terminus of cTnT directly interacts with the regulatory N-domain of cTnC to allosterically modulate Ca2+ activation of force, perhaps by controlling the troponin I switching mechanism of striated muscle contraction. Alterations in cTnC–cTnT binding may compromise contractile performance and trigger pathological remodeling of the myocardium.
Keywords: cardiac muscle, contractile protein, protein dynamic, structure–function, cardiovascular disease, cardiac muscle, cardiomyopathy, cross-bridges, NMR, troponin
Introduction
Dilated cardiomyopathy (DCM)2 is a leading cause of heart failure and the most frequent indication for heart transplantation in both children and adults (1–3). DCM is characterized clinically by ventricular wall thinning, chamber volume enlargement, and contractile (systolic) dysfunction in the absence of coronary artery disease (4, 5). Primary DCM can result from genetic mutations in proteins related to diverse cellular functions, ranging from muscle contraction to RNA splicing and gene transcription (6, 7). Although pathways that contribute to the DCM phenotype are complex and heterogeneous, sarcomeric protein mutations associated with DCM have been shown to alter the force-generating capacity of the myocardium by directly impacting myofilament-based regulation of contraction (8).
Genetic variants account for a significant proportion of cardiomyopathies in both children and adults, which underscores the importance of fully evaluating sequence variations associated with cases of cardiomyopathy in humans (9, 10). However, establishing the pathogenicity for sarcomere gene variants can be challenging in the absence of genetic segregation and linkage analyses (4). This is often the case for rare variants that arise de novo, which are often associated with lethality at early stages of development. Fortunately, structural and functional investigations into cardiomyopathy-associated variants can aid in establishing the potential genotype–phenotype relation in the absence of genetic linkage analysis, as well as provide mechanistic insights into physiological muscle regulation (11–15). Moreover, these studies bear the potential to unveil novel therapeutic targets to improve contractile performance in disease, and perhaps aging, by directly targeting the contractile machinery (16).
The sarcomere is a fundamental structural unit of striated muscle cells that is responsible for contractile function. This macromolecular complex is primarily composed of overlapping thick (myosin) and thin (actin, tropomyosin, and troponin) myofilaments that, seen in a cross-section, form a double-hexagonal lattice (8). Cardiac troponin (cTn) is a heterotrimeric complex that plays a central role in regulating muscle contraction by functioning as the Ca2+ sensor in the contractile apparatus. It is composed of a Ca2+-binding subunit, cardiac/slow skeletal muscle troponin C (cTnC); an inhibitory subunit, cardiac troponin I (cTnI); and a tropomyosin-binding subunit, cardiac troponin T (cTnT) (17). cTn regulates contraction–relaxation through a series of conformational rearrangements and myofilament protein–protein interactions in response to cardiomyocyte intracellular Ca2+ transients (18).
During the contractile (systolic) period of the cardiac cycle, elevated intracellular free Ca2+ results in Ca2+ binding to a single, low-affinity (∼105 m−1) regulatory site in cTnC (19). In this Ca2+-activated state, an interaction between cTnI and cTnC is stabilized, which shifts tropomyosin into a position that exposes myosin-binding sites on actin to allow cross-bridge cycling and force generation (18). As free intracellular Ca2+ levels fall during the relaxation phase (diastole), Ca2+ dissociates from cTnC, and cTnI and tropomyosin are repositioned on actin filaments to inhibit strong myosin binding (18). Although it is well-established that cTnI–cTnC interactions are critical for cTn function, little is known about the potential functional significance of cTnT–cTnC interactions.
The TnT subunit has been viewed primarily as the subunit that anchors cTn to the thin filament via its highly-extended N terminus (20). However, there is evidence that cTnT might also have a regulatory role. Analysis of the crystal structure of the cTn core domain and results from small-angle neutron–scattering experiments indicate putative interactions between cTnC and C-terminal region of cTnT (21, 22). In addition, it was previously shown that skeletal muscle TnT is essential to confer Ca2+ sensitivity of tension and ATPase activity independent of cTnT–cTnI interactions (23). Functional studies on a human cardiomyopathic variant that results in deletion of the last 14 C-terminal residues in cTnT have further highlighted the potential importance of cTnT in cardiac muscle regulation (24–28). Taken together, these observations raise the possibility that cTnT directly interacts with cTnC for Ca2+-dependent control of myocardial force generation, alterations that may trigger contractile dysfunction and pathological remodeling of the heart.
In this report, we describe a previously unreported de novo, nonsynonymous variant (p.I4M) in the cTnC gene (TNNC1) that was identified in a pediatric patient with severe DCM. The objective of this study was to ascertain whether or not there is sufficient evidence to establish this variant as pathogenic and to elucidate an underlying molecular mechanism for its potential pathogenicity. By combining in silico, physiological, and biophysical approaches, we demonstrate this variant satisfies the American College of Medical Genetics and Genomics (ACMG) criteria for strong evidence of pathogenicity (29). We also identify a novel intersubunit interaction between cTnC and the C terminus of cTnT, which is disrupted by this cardiomyopathic variant. Our findings suggest that cTnC–cTnT interactions are potentially critical for modulation of contractility.
Results
Clinical evaluation
The proband was a female neonate who presented with a heart murmur shortly after birth and was subsequently recommended for a follow-up. Medical records noted difficulty feeding and severe failure to thrive since birth. At 6 weeks of age, the proband underwent an extensive clinical work-up and was diagnosed with sporadic DCM. Complete blood count and comprehensive metabolic panel were reported to be normal. Enzymatic activity testing for Pompe disease was negative. On physical examination, severe hypotonia, global developmental delay, and nondysmorphia were noted. One parent is an Ashkenazi Jew and the other parent is Ashkenazi and Moroccan Jew. The proband had a negative family history for cardiomyopathy. Echocardiographic imaging revealed left ventricular hypertrabeculation and overt left ventricular and left atrial dilation (Fig. 1A). Electrocardiography indicated sinus rhythm with diagnostic criteria for biventricular hypertrophy, right atrial enlargement, and diffuse ST-T wave changes (Fig. 1B). Based on the clinical presentation, the proband and parents underwent genetic testing on the basis of a suspected genetic variant.
Figure 1.

Clinical presentation of the proband. A, representative echocardiographic images demonstrating left ventricular dilation and hypertrabeculation (top); parasternal long axis view showing dilation of the left atrium and left ventricle (bottom). B, representative electrocardiogram tracings showing sinus rhythm with criteria satisfied for biventricular hypertrophy, right atrial enlargement, and diffuse STT changes.
Molecular genetic analysis
Trio whole-exome sequencing revealed a previously unreported heterozygous variant of uncertain significance in exon 1 of TNNC1, in which a cytosine (C) to guanine (G) transversion at nucleotide position 12 was detected in the cDNA from the proband (c.12C→G) (Fig. S1). This codon alteration (ATC → ATG) encodes for a nonsynonymous substitution, whereby isoleucine is replaced by methionine at position 4 in cTnC (cTnC–I4M). Both parents were genotype-negative for this variant (Fig. 2A) and appeared to be clinically unaffected, suggesting that it arose de novo. No other genetic variants were identified in the sequencing. At 1 year old, the proband succumbed to acute viral infection with exacerbation of heart function (Fig. 2A). This variant is absent in the Genome Aggregation database, suggesting it is not merely a benign polymorphism found in the general population of diverse racial and ethnic backgrounds (30). Furthermore, TNNC1 p.I4M was absent in the on-line database, ClinVar, at the time of this report (31). The I4M variant is located in the N-helix, which is positioned between the N- and C-domain in the tertiary structure of Ca2+-saturated cTnC (Fig. 2B). This affected residue, like most of the primary sequence of cTnC, is highly conserved across species (Fig. S2), suggesting that any protein-altering variant would not be tolerated. Indeed, 2 out of 4 in silico prediction programs revealed that TNNC1 p.I4M is predicted to be pathogenic (Table S1) (32–35). Based on these collective findings, we carried out structural and functional studies to evaluate the putative pathogenicity and underlying mechanism of this variant.
Figure 2.
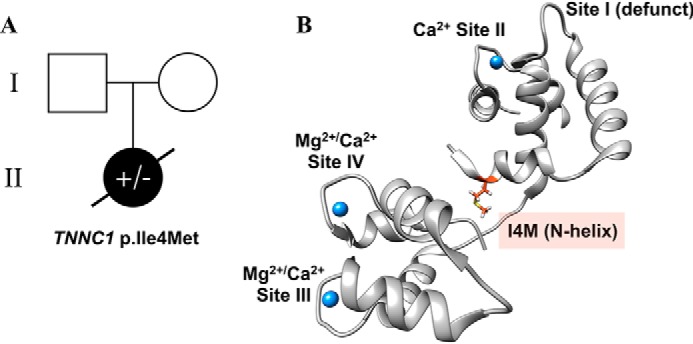
Molecular genetics analysis. A, pedigree analysis based on the trio whole-exome sequencing results. Open square and circle (I) represent the genotype-negative father and mother, respectively, of the genetically affected female proband (filled circle, II). B, molecular visualization of cTnC (gray ribbon) in the Ca2+-saturated state (blue spheres) showing the location of I4M (orange stick). Image was rendered with PyMOL using the crystal structure of the cTn core domain (PDB code 1J1E).
Impact on cardiac myofilament function in situ
To assess the direct effects of cTnC–I4M on the Ca2+-dependent contractile properties in cardiac muscle, endogenous cTnC was extracted from permeabilized cardiac muscle preparations (CMP) and reconstituted with recombinant cTnC–WT control or cTnC–I4M. Incorporation of cTnC–I4M into CMP resulted in statistically significant lower myofilament Ca2+ sensitivity of a steady-state isometric force compared with cTnC–WT (cTnC–WT pCa50 = 5.83 ± 0.04 versus cTnC–I4M pCa50 5.70 ± 0.04) (Fig. 3, A and B, and Table 1). This decrease in myofilament Ca2+ sensitivity is indicated by the rightward shift in the pCa-tension relation (ΔpCa50 = −0.13) (Fig. 3A and Table 1). Reconstitution of CMP with cTnC–I4M tended to increase cooperativity of thin filament activation (nHill = 1.3 ± 0.1) compared with cTnC–WT control (nHill = 1.1 ± 0.1), but the difference was not statistically significant (Fig. 3C and Table 1). There was no significant difference in maximal tension per cross-sectional area for CMP reconstituted with cTnC–WT (19.7 ± 2.9 mN/mm2) or cTnC–I4M (21.7 ± 4.2 mN/mm2) (Fig. 3D). There was also no significant difference in maximal restored tension between cTnC–WT (89.6 ± 8.3%) and cTnC–I4M (88.0 ± 10.0%), suggesting that the observed effects cannot be explained by differences in incorporation of exogenous cTnC–WT or cTnC–I4M (Table 1).
Figure 3.
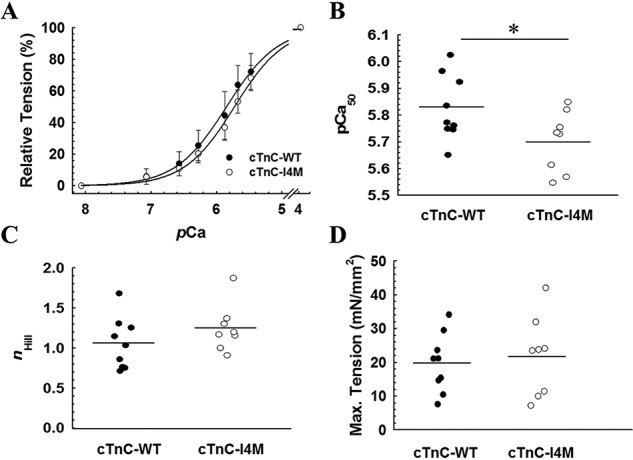
Effects of cTnC–I4M on Ca2+-activated tension at SL 2.1 μm in CMP. A, normalized steady-state isometric tension as a function of pCa recorded at 30 °C. Quantitative comparison of pCa50 (B), nHill (C), and maximum steady-state isometric tension (D) values for CMP reconstituted with cTnC–WT or -I4M. The force values were normalized to the maximum force for each CMP. Data are shown as mean ± S.D. n = 9 CMP for cTnC–WT and n = 8 CMP for cTnC–I4M. Horizontal bars represent the mean, and * denotes statistical significance between the means (p < 0.05) determined by an unpaired Student's t-test.
Table 1.
Summary of contractile parameters for reconstituted CMP
The number of independent experiments for restored tension, pCa50, and nHill was n = 9 CMP for cTnC–WT and n = 8 CMP for cTnC–I4M. For kTR, max, n = 8 CMP for cTnC–WT and n = 5 CMP for cTnC–I4M. Exclusion criteria for certain CMPs are described under “Experimental procedures.” Data are represented as mean ± S.D.
| Reconstituted CMP | pCa50 | ΔpCa50 | nHill | Restored tension | kTR, max |
|---|---|---|---|---|---|
| % | s−1 | ||||
| cTnC–WT | 5.83 ± 0.12 | 1.1 ± 0.3 | 89.6 ± 24.8 | 7.0 ± 1.5 | |
| cTnC–I4M | 5.70 ± 0.11a | −0.13 | 1.3 ± 0.3 | 88.0 ± 28.2 | 4.6 ± 0.8a |
aData denote statistical significance between the means of cTnC–WT versus cTnC–I4M (p < 0.05) as determined by unpaired Student's t-test.
The rate constant of isometric tension redevelopment (kTR) provides insight into both actin-myosin cross-bridge kinetics and Ca2+-dependent dynamics of thin filament regulatory units. Therefore, we hypothesized that this Ca2+-desensitizing mutation would impact kTR. CMP reconstituted with cTnC–14m exhibited a slower rate of isometric tension redevelopment compared with cTnC–WT at all tested levels of Ca2+ activation (Fig. 4, left). Noteworthy, this decrease in kTR occurred at both submaximal and maximal Ca2+ activation (Fig. 4, left). The difference in the mean maximum kTR between cTnC–WT (7.0 ± 0.54 s−1) and cTnC–I4M (4.64 ± 0.4 s−1) was statistically significant (Table 1), suggesting that this variant has an inhibitory effect on cross-bridge cycle kinetics in cardiac muscle, in addition to its influence on Ca2+ sensitivity, an effect that is typically associated with changes in individual regulatory unit dynamics.
Figure 4.
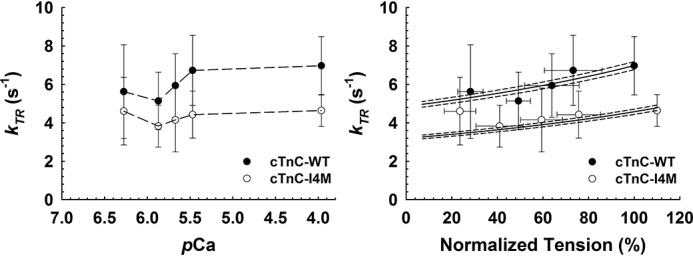
Effect of cTnC–I4M on Ca2+-activated rate of isometric tension redevelopment at SL 2.1 μm in CMP. Left, pCa-dependence of kTR at 30 °C. Right, relation between kTR and normalized steady-state isometric tension. The lines represent solutions to the three-state model of striated muscle contraction (36). Solid lines indicate the solutions from the model based on the parameter values in Table 2, and the dashed lines indicate the solutions from the sensitivity analysis whereby all four parameters were increased (upper traces) or decreased (lower traces) by 2%. Force values were normalized to the maximum force of cTnC–WT–reconstituted CMP. Data are represented as mean ± S.D.
The 33% decrease in maximum kTR observed for cTnC–I4M could be explained by changes in the apparent cross-bridge attachment rate (fapp) and/or detachment rate (gapp), since maximal kTR ∝ fapp + gapp. Therefore, we used a computational approach to estimate the best-fit values for fapp, gapp, and koff based on the three-state model of striated muscle contraction (36). We found 23 and 33% slower fapp and gapp, respectively, for cTnC–I4M compared with cTnC–WT (Fig. 4, right and Table 2). These data suggest that the slower kTR values observed for this variant derive from decreases in both cross-bridge attachment (fapp) and detachment (gapp) rates. Taken together, these results indicate that reconstitution of cTnC–I4M into CMP causes deficits in both the rate and magnitude of isometric tension generation at a given level of Ca2+ activation.
Table 2.
Three-state model parameters for reconstituted CMP
Values were obtained as described to fit the three-state model to the force-kTR data in Fig. 4 along with the average pCa50 values (Fig. 3 and Table 1) for CMP reconstituted with cTnC–WT or -I4M.
| Reconstituted CMP | Three-state model-fitted parameters |
Three-state model predictions |
|||||
|---|---|---|---|---|---|---|---|
| f | g | kON (90) | kOFF | pCa50 | Max force (norm) | kTR, max | |
| s−1 | s−1 | m−1 s−1 | s−1 | s−1 | |||
| cTnC–WT | 2.1 | 4.9 | 1.84 × 108 | 403.3 | 5.83 | 1.00 | 7.0 |
| cTnC–I4M | 1.6 | 3.2 | 1.84 × 108 | 576.9 | 5.70 | 1.10 | 4.6 |
Evidence for direct cTnT–cTnC binding
Recent studies suggest that the last 14 residues at the C terminus of cTnT are important for limiting thin-filament activation in the presence of Ca2+, but the mechanism is unknown. Therefore, we hypothesized that this region of cTnT directly interacts with cTnC to control Ca2+ activation of the myofilament, the alterations in which could mechanistically explain the reduced myofilament Ca2+ sensitivity and kTR value observed for cTnC–I4M.
To determine whether the C terminus of cTnT interacts with cTnC, we performed chemical cross-linking followed by MS analysis on reconstituted WT cTn in the presence of Ca2+. Prior to cross-linking, successful ternary complex formation was verified by native gel analysis (Fig. S3). Strikingly, we identified high-confidence cross-linked peptides corresponding to intersubunit interactions between Lys-282 of cTnT and Lys-6 in the N-helix of cTnC in the “light band” (Fig. 5A and Fig. S4). This finding is based on the corresponding mass spectrum in Fig. 5B, where a 4.0272-Da change in mass is characteristic of peptide cross-linking by BS3-d0/d4. Consistent with the crystal structure of the core domain of cTn, we also identified cross-linked peptides corresponding to intersubunit cTnT and cTnI interactions in the rigid I–T arm of the ternary complex in the “dark band” (Fig. 5A and Table S2).
Figure 5.
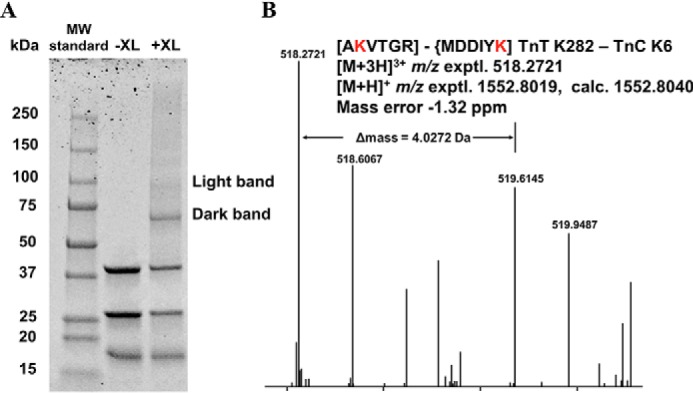
In vitro BS3 cross-linking of WT cTn followed by bottom-up MS analysis. A, Coomassie-stained denaturing gradient gel of uncross-linked (−XL) and cross-linked (+XL) products. B, observed precursor MS of the cross-linked peptides between cTnT–Lys-282 and TnC–Lys-6. Observed precursor ion of [M + 3H]3+ has m/z 518.2721 with −1.32 ppm mass error. Peptide AKVTGR corresponds to cTnT 281–286. Peptide MDDIYK corresponds to cTnC 1–6. The 4.0272-Da mass shift between the two isotopic clusters indicates the presence of BS3-d0/d4. The red text denotes the cross-linked Lys residues.
Based on the chemical cross-linking results, we hypothesized that the C terminus of cTnT directly interacts with cTnC. To address this question, we titrated cTnC–WT or cTnC–I4M to a synthetic peptide (Fig. 6A) corresponding to the last 14 amino acids at the C terminus of cTnT and monitored intrinsic tryptophan (Trp-287) fluorescence using steady-state fluorescence spectroscopy. Titration of cTnC–WT or cTnC–I4M with the cTnT C-terminal peptide resulted in a concentration-dependent increase in tryptophan fluorescence and a blue-shifted emission wavelength maximum (Fig. 6, B and C). Normalized tryptophan fluorescence was plotted against free cTnC concentration to calculate Kd values for cTnC–WT and cTnC–I4M (Fig. 6D). There was a statistically significant difference between the binding constants for cTnC–WT (Kd = 14.7 ± 0.5 μm) and cTnC–I4M (Kd = 10.9 ± 0.4 μm) (Table 3). These results show, for the first time, that the intrinsically disordered C terminus of cTnT interacts with cTnC—an interaction that is strengthened in the case of cTnC–I4M.
Figure 6.
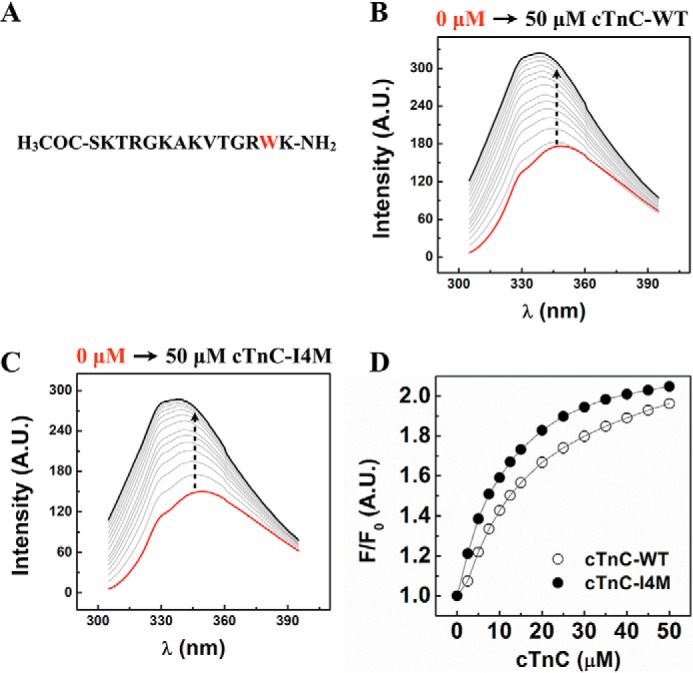
C-terminal cTnT peptide–cTnC-binding assay monitored by intrinsic tryptophan fluorescence at 21 °C. A, primary sequence of the synthetic peptide corresponding to the last 14 C-terminal amino acids of cTnT. B, changes in tryptophan fluorescence intensity spectra upon titration of cTnC–WT into cTnT peptide (n = 5). C, changes in tryptophan fluorescence spectra upon titration of cTnC–I4M into cTnT peptide (n = 5). D, normalized intensity of peak tryptophan fluorescence as a function of increasing concentrations of cTnC–WT or -I4M. A.U., arbitrary units.
Table 3.
Affinity constants for cTnT peptide binding to cTnC
Parameters were obtained by fitting the titration data in Fig. 6D to Equation 1.
| cTnC | k | nHill |
|---|---|---|
| μm | ||
| WT | 14.7 ± 0.9 | 1.2 ± 0.1 |
| I4M | 10.9 ± 0.8a | 1.2 ± 0.1 |
a Value denotes statistical significance between the means of cTnC–WT versus cTnC–I4M (p < 0.05) as determined by an unpaired Student's t-test. Data are represented as mean ± S.D.
To determine whether full-length cTnT interacts directly with cTnC and to assess potential binding alterations for this cTnC variant, we used microscale thermophoresis (MST) to quantify cTnT–cTnC binding in the presence of Ca2+. The results indicate full-length cTnT directly binds to cTnC (Fig. 7A). Calculation of the equilibrium dissociation constant (Kd) yielded 13.2 ± 1.6 μm for cTnC–WT and 4.84 ± 1.1 μm for cTnC–I4M (Fig. 7A). Note that full-saturation was not feasible due to solubility issues of full-length cTnT at low ionic strength. These results indicate that full-length cTnT interacts with cTnC–WT and that the binding affinity is enhanced 2.7-fold for the cTnC–I4M variant.
Figure 7.
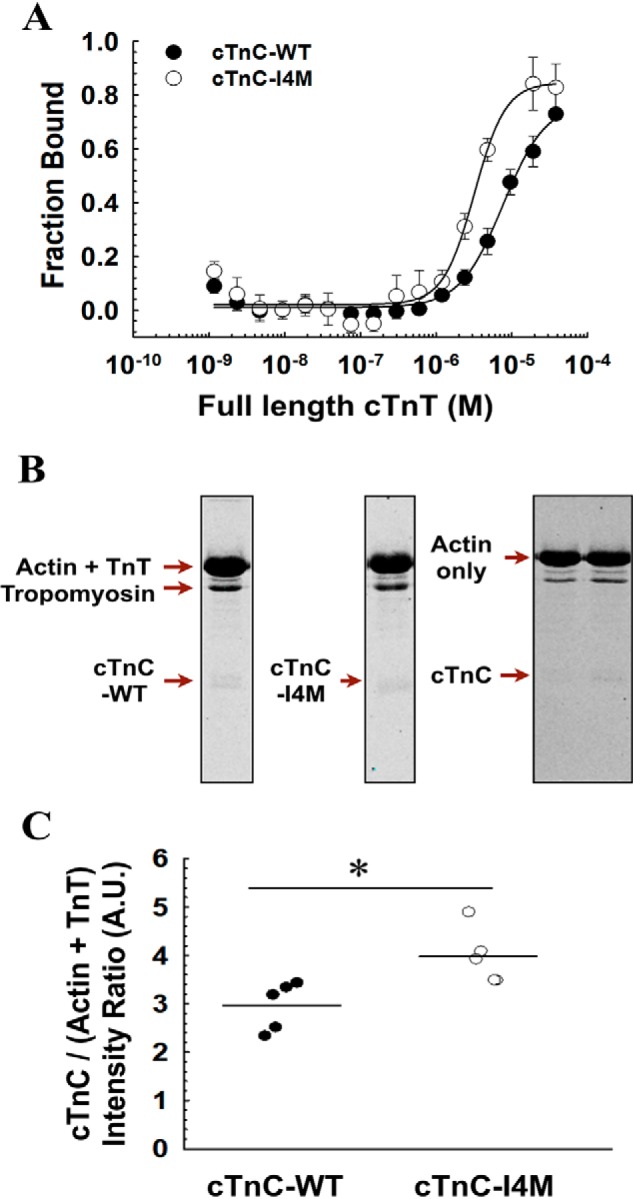
Full-length cTnT–cTnC-binding assays. A, fraction of bound cTnT versus molar concentration of cTnC–WT (n = 4) or cTnC–I4M (n = 4) measured by MST. The data points were fitted with a Hill equation for graphical representation. B, representative Coomassie-stained denaturing gels of pelleted fractions from thin filament co-sedimentation assay. C, quantitative comparison of pelleted cTnC–WT (n = 5) or cTnC–I4M (n = 5). Intensity ratio = signal intensity of cTnC–WT- or cTnC–I4M divided by the signal intensity of the loading control (actin + cTnT). Data in A are represented as mean ± S.D. Horizontal bars in C represent mean values and * denotes statistical significance between the means (p < 0.05) determined by unpaired Student's t-test. A.U., arbitrary units.
Although we showed that isolated cTnC and cTnT subunits could form an interaction, we wondered whether this interaction was maintained in the presence of other major thin filament binding partners using a label-free approach. Therefore, we tested whether or not the cTnT–cTnC interaction also occurs in the context of F-actin and tropomyosin using a co-sedimentation assay. We found that both cTnC–WT and -I4M associated with full-length cTnT, F-actin, and tropomyosin in the pellet upon ultra-centrifugation (Fig. 7B, leftmost and middle gels). To exclude the possibility that cTnC was forming an interaction with the thin filament proteins independent of cTnT, the assay was performed in the absence of cTnT, and we found that cTnC no longer associated with F-actin and tropomyosin in the pellet (Fig. 7B, rightmost gel). The co-sedimentation assay revealed that a statistically significant greater proportion of cTnC–I4M associated with cTnT–F-actin–tropomyosin in the pellet compared with cTnC–WT (Fig. 7C), further supporting the concept that this cTnC variant results in tighter binding to cTnT. These results suggest that cTnC interacts with cTnT in the setting of the thin myofilament apparatus.
Structure and backbone dynamics of cTnC
Next, we asked the question of whether perturbations in cTnC–I4M structure and/or dynamics could explain the observed effects on myofilament function and altered cTnT binding. Ca2+-induced hydrophobic exposure of cTnC is critical for activation of the thin filament and muscle contraction. To specifically examine the overall structural profiles of Ca2+-induced hydrophobic exposure in cTnC, increasing concentrations of free Ca2+ (pCa 8.0, 6.0, and 4.0) were titrated to cTnC–WT or cTnC–I4M with bis-ANS, a fluorescent probe sensitive to hydrophobic environments (Fig. 8, A and B). Examination of the bis-ANS fluorescence-emission curves revealed that both cTnC–WT and cTnC–I4M displayed identical fluorescence intensities at very low Ca2+ (pCa 8.0), suggesting that there was no difference in hydrophobic exposure prior to Ca2+ titrations (Fig. 8, A and B). Interestingly, at both sub-saturating (pCa 6.0) and saturating (pCa 4.0) Ca2+ concentrations, the magnitude of the fluorescence intensity was lower for cTnC–I4M compared with cTnC–WT (Fig. 8B). These results indicate that Ca2+-induced hydrophobic exposure is markedly blunted as a consequence of this N-helix cTnC variant.
Figure 8.
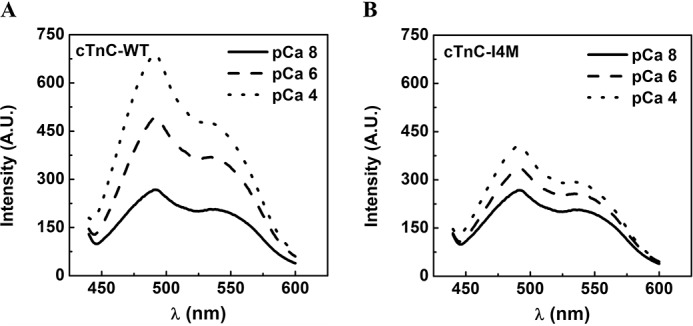
Measurement of Ca2+-induced hydrophobic exposure of cTnC monitored by bis-ANS fluorescence at 21 °C. Representative plots of bis-ANS fluorescence intensity spectra for cTnC–WT (A) and cTnC–I4M (B) upon titration of CaCl2 to achieve a final free Ca2+ concentration corresponding to pCa 8.0 (solid line), pCa 6.0 (dashed line), and pCa 4.0 (dotted line). At least three independent titrations per condition were performed. A.U., arbitrary units; λ, wavelength.
To acquire insight into potential alterations in cTnC–I4M structural dynamics, we designed experiments using solution-state nuclear magnetic resonance (NMR) spectroscopy. Because the 1H–15N HSQC of cTnC–I4M revealed only modest changes in chemical shifts compared with cTnC–WT (Fig. 9A and Fig. S5), we were able to successfully assign 1H–15N correlations to this novel variant and track the chemical-shift changes induced by the I4M substitution (Fig. 9A and Fig. S5) (37). Chemical-shift perturbation (CSP) analysis obtained by comparing cTnC–WT and cTnC–I4M 1H–15N peak assignments identified protein segments mostly affected by the mutation (Fig. 9B). CSP outliers differing by ≥2 S.D. from the average across studied residues were mapped onto the cTnC atomic model (Fig. 9C). Perturbed residues were located at the N-helix (Asp-3), and on both the defunct site I (Gly-30, Asp-33, and Glu-40) and the Ca2+-binding EF-hand regulatory site II (Glu-66 and Asp-67). In addition to perturbations in the N-domain, the end of helix-G in the C-domain (Gly-140 and Lys-142) also experienced a different chemical environment compared with cTnC–WT. These results suggest that, whereas the overall tertiary structures are similar between cTnC–WT and cTnC–I4M, discrete regions are structurally impacted by this variant.
Figure 9.

Structural characterization of cTnC–I4M by solution-state NMR. A, overlaid 1H–15N HSQC spectra for cTnC–WT (red) and cTnC–I4M (blue). B, plot of CSP as a function of cTnC residue number. Solid, dashed, and dotted blue lines show average CSP values, average ± 1 S.D., and average ± 2 S.D., respectively. Residues exhibiting values greater than 2 S.D. above the average are highlighted in A and B. Secondary structure elements (αN–αH) and Ca2+-binding sites are shown above the CSP plot. C, molecular visualization of perturbed residues (>2 S.D. above the average) in cTnC–I4M. ppm, parts per million.
The diffusional properties measured by 15N longitudinal (R1) and transverse (R2) relaxation times and the 1H–15N heteronuclear NOE provide information on the local dynamic changes on a fast (picosecond–nanosecond) timescale and also report, in a qualitative manner, slower (microsecond–millisecond) conformational exchange motions. The R2/R1 plot obtained for cTnC–I4M revealed a cluster of residues close to the defunct Ca2+-binding loop presenting values above the average +1 S.D. (4.95 ± 0.93) (Fig. 10a). These data suggest a local conformational exchange at this region caused by the presence of I4M and that the N- and C-domains tumble independently with different rotational correlation times (Fig. 10a). Similar to the cTnC–WT, residues within the cTnC–I4M helix-D (Asp-73, Phe-74, Phe-77, and Val-79) also revealed increased R2/R1 values (Fig. 10a) (37). Importantly, residues close to the defunct Ca2+-binding loop in cTnC–I4M display higher R2/R1 values, which were not observed when the same experiments were carried out on cTnC–WT (37). Internal motions on a picosecond–nanosecond timescale of 1H–15N bonds can be assessed by NOE values. Except for residue Glu-32 that exhibits a higher flexibility of the H–N bond, no other changes in NOE values were observed between cTnC–WT and cTnC–I4M (Fig. 10b). Using the Lipari-Szabo approach, we also calculated the order parameter (S2) and conformational exchange ratio (Rex) for cTnC–I4M, which reflects movements of the backbone amide bonds in the picosecond–nanosecond and microsecond–nanosecond time regimes, respectively. Residues throughout the regulatory N-domain (Met-8, Glu-10, Leu-29, Asp-33, Asn-50, and Phe-74), as well as the C-domain (Lys-117, Asp-109, and Lys-158), of cTnC–I4M show contributions from exchange between alternative conformations in the relatively slow (microsecond–millisecond) timescale (Fig. 10, c and e). Lower than average S2 values, which indicate increased flexibility, are seen in the Asp/Glu linker and hinge regions of cTnC–I4M (Fig. 10, d and f). Intriguingly, residues in the N-domain, a presumed region of cTnT C-terminal binding, show higher than average S2 values, suggestive of reduced backbone flexibility in this region. Taken together, these results show that the N-helix I4M variant, in part through allosteric transmission, alters the structural dynamics of cTnC and can reasonably explain the enhanced cTnT-binding affinity.
Figure 10.

Structural dynamics of cTnC–I4M by NMR relaxation experiments. R2/R1 (a) and HetNOE rate (b) as a function of cTnC residue number. Conformational exchange rate, Rex, in Hz (c), and order parameter, S2 (d), as a function of cTnC–I4M residue number. S2 values range from 0 to 1, where values that approach 1 indicate greater structural rigidity. e, residues showing conformational exchange in C are colored in orange and the I4M mutation in red on cTnC crystal structure (PDB code 1J1E). f, Ca2+-saturated cTnC crystal structure (PDB code 1J1E) colored using the blue-white-red color gradient following S2 values shown in D.
Discussion
In this study, we identified a previously unreported cardiomyopathic variant as well as a novel molecular mechanism underlying its pathogenicity. By leveraging a broad array of experimental approaches to assess this variant, we found evidence for direct communication between the extreme C terminus of cTnT and the regulatory domain of cTnC, alterations that are expected to impact contractile force and potentially serve as a primary stimulus for pathological remodeling of the myocardium.
The first report to establish TNNC1 as a cardiomyopathic gene was published in 2004 (38). Since then, multiple distinct variants have been identified in patients with DCM, hypertrophic cardiomyopathy (HCM), and restrictive cardiomyopathy (39). The cTnC–I4M variant reported here should be added to the growing list of cardiomyopathic variants. Although genetic linkage and segregation analyses are undoubtedly important criteria for assigning pathogenicity to a variant, this information is often unavailable, especially for de novo variants that are often deleterious early in life (40). Furthermore, the variable expressivity and incomplete penetrance associated with sarcomeric cardiomyopathies can further complicate genetic analyses in cases where affected individuals reach reproductive age (4). Some investigators in the field have even questioned the relevance of studying de novo mutations. We argue that the clinical severity and frequent lethality often associated with rare TNNC1 variants only reinforces the importance of acquiring a better understanding of this essential gene. Indeed, investigating lethal cardiomyopathic variants provides insight into the regulatory mechanisms that are critical for sustaining contractile performance. As evidenced by previous reports and this specific case, it would appear that certain TNNC1 variants are incompatible with the normal human life span and therefore are not likely to be transmitted to offspring. Furthermore, the overall primary sequence of cTnC, including the affected residue in this report, is highly-conserved across species, suggesting an intolerance to mutations through the course of evolution (39).
Myofilament Ca2+ sensitivity of steady-state isometric tension can be used as an index for contractility. We found that cTnC–I4M is associated with reduced Ca2+ sensitivity of force, which is in alignment with the paradigm for sarcomeric cardiomyopathies in general and with DCM-associated variants in particular (41). Quantitatively, ∼1.3-fold more Ca2+ is required to achieve 50% of maximum isometric tension. Physiologically, the thin filament is never fully activated in the normal beating heart because cytosolic free Ca2+ concentration varies throughout the cardiac cycle within the approximate range of pCa 7 to 6 (∼0.1 to 1 μm) (42). Therefore, expression of cTnC–I4M in the heart is expected to depress myocardial contractility in vivo, despite no significant reduction in maximal restored tension for cTnC–I4M-reconstituted CMP. Thus, the importance of myofilament Ca2+ sensitivity should not be underestimated. Modulation of myofilament Ca2+ sensitivity by engineered mutants or pharmacological treatment has previously been shown to ameliorate, and sometimes prevent, pathological remodeling associated with sarcomeric cardiomyopathies (43–45).
The observed inhibitory effect of cTnC–I4M on Ca2+ activation of kTR (Fig. 4) suggests that expression of this N-helix variant in the myocardium would impair contractile function. Indeed, it has been previously suggested, and borne out by experimental evidence, that kTR reflects processes that limit the rate and magnitude of isovolumetric pressure rise in the cardiac ventricles (46, 47).
We observed a Ca2+ dependence of kTR across physiological activation, consistent with previous observations in cardiac muscle (47–50). Computational modeling suggested that the slower kTR value displayed by cTnC–I4M can be explained by the combination of slower rates of cross-bridge transition from the nonforce-bearing state to the strong, force-generating state (fapp), as well as the reverse process (gapp) because, to a first approximation, maximal kTR = fapp + gapp (51, 52). Our modeling further suggests that the decrease in Ca2+ sensitivity (Fig. 3 and Table 1) could be due to faster deactivation of thin filament regulatory units (Table 2); while this is the most likely scenario, in the absence of experimental measurements on the cTnC–I4M variant, we cannot rule out an effect on the kinetics of Ca2+ binding and regulatory unit activation.
According to Brenner's two-state model of cross-bridge cycling, the fraction of cross-bridges in strong, force-generating states and thus isometric tension are both proportional to fapp/(fapp + gapp) (51); this proportionality is valid to a first approximation in the three-state model at saturating levels of activating Ca2+ (48, 50, 53). The model-derived values of fapp and gapp (Table 1) are consistent with the lack of a significant difference in maximum isometric force between cTnC–WT and cTnC–I4M (Figs. 3 and 4 and Table 1) because we were able to use this information to constrain the parameter space examined in the modeling. Thus, in the absence of an indirect effect on Ca2+-handling in the living cardiomyocyte, the cTnC–I4M variant would be expected to reduce force production at a given level of intracellular free Ca2+ due to a smaller number of strong, force-generating cross-bridges that results from lower Ca2+ sensitivity in combination with slower kinetics of tension generation. Whereas haploinsufficiency has been observed for other cardiomyopathic variants in the sarcomere, it cannot explain the disruption in myofilament function for this cTnC variant; both cTnC–WT and cTnC–I4M were able to restore comparable levels of tension upon incorporation into CMP, supporting the concept that cTnC–I4M acts as a dominant-negative mutation in the contractile apparatus.
In contrast to the cTnC-I4M variant, we previously reported that a Ca2+-sensitizing mutation in the N-helix of cTnC (cTnC-A8V) increases kTR at all tested levels of activation, which could be explained by an increase in both fapp and gapp (48, 54). Hence, these two N-helix variants, which are associated with divergent pathways of cardiac remodeling (hypertrophy versus dilation), appear to have opposing effects on kTR. In addition to reinforcing the notion that sarcomeric cardiomyopathies are mutation-specific rather gene-specific, these observations raise the possibility that the N-helix confers specific regulatory properties to cTnC to allow for the Ca2+-dependent control of myocardial force production.
To understand how a variant in the N-helix of cTnC could influence molecular processes downstream of Ca2+ binding (i.e. cross-bridge cycle kinetics), we investigated the possibility of intersubunit communication between cTnT and cTnC. The cTnT subunit was a logical target because it directly interacts with the other key thin-filament regulatory proteins, actin and tropomyosin (18). In fact, results from in vitro motility assays have suggested that TnT may have a direct role in controlling kTR (55). This study is the first to quantify direct binding between the cardiac isoforms of TnT and TnC and show that this interaction is altered by a DCM-related variant in cTnC.
In the context of a molecular mechanism, cTnC–cTnT binding might impact contractile performance by shifting the distribution of thin filament equilibrium states (56, 57). The premise of this supposition is primarily derived from a series of reports by Chalovich and co-workers (25–27), in which they suggest that the C-terminal region of cTnT has a dual Ca2+-dependent regulatory role. Their findings indicate that the C-terminal residues of cTnT are important for both the formation of the inactive B-state at low levels of Ca2+ and limiting the active M-state at saturating Ca2+ concentrations. This cTnT region is particularly relevant because it is associated with genetic cardiomyopathy. A single missense mutation (R286H) as well as C-terminal deletions (Δ14 or Δ21) in cTnT are linked to HCM in humans (24, 58). Furthermore, preliminary results from our laboratory have suggested that this C-terminal cTnT peptide has an inhibitory effect on force generation in permeabilized cardiac muscle fiber preparations.3 Thus, one possibility is that the increased binding affinity of the cTnT C terminus for cTnC–I4M might shift the equilibrium in favor of stabilizing an inactive, nonforce-generating state of regulated actin. If true, this could reasonably explain the markedly slower rate (kTR) of transition between the nonforce-generating (C-state) and the force-generating (M-state) states that we observed for this variant upon reconstitution into permeabilized CMP (Fig. 4).
Our chemical cross-linking results suggest that the C terminus of cTnT is in proximity (∼1 nm) to the N-helix of cTnC. It is therefore not surprising that the cTnC I4M variant impacted the binding affinity for both full-length and C-terminal peptide of cTnT. Recent molecular dynamics simulations performed on cTn in the Ca2+-saturated state have suggested that the C terminus of cTnT most frequently samples a conformation that is situated in a cleft between the two lobes of cTnC, simultaneously interacting with the inhibitory peptide and N-terminal regions of cTnI (59). In this conformation, it would appear that the cTnT C terminus could influence intrasubunit cTnI contacts (28). Interestingly, it has been shown that mutation of conserved acidic residues to alanine in the regulatory N-domain of skeletal muscle TnC simultaneously reduced TnT binding and disrupted the Ca2+-induced structural rearrangements of troponin I (60). Considering the abundance of basic residues at the C terminus of cTnT, this region is expected to electrostatically interact with this patch of acidic residues in cTnC. Although it has been recognized that the cardiac thin filament represents an extensive allosteric system, the molecular mechanisms are not fully understood (61–63). Our findings in this report raise an intriguing possibility that the C terminus of cTnT allosterically controls thin filament activation via modulation of the cTnI–cTnC switching mechanism.
It is reasonable to expect that the C terminus of cTnT would have a different binding partner at low (diastolic) levels of Ca2+. The conformational plasticity of intrinsically disordered regions allows for interactions with a diverse set of binding partners in the cell (64, 65). We could find no evidence for cTnC–cTnT binding in the absence of Ca2+ bound to the regulatory site of cTnC.3 This is consistent with previous findings from binding studies with the skeletal muscle isoforms of TnT and TnC, which were shown to be Ca2+-dependent (23, 66, 67). Moreover, hydrogen–deuterium exchange experiments performed on cTn revealed that the C terminus of cTnT is stabilized by Ca2+, suggestive of the formation of inter- or intrasubunit unit contacts in cTn (68). One possibility is that the C terminus of cTnT binds to actin or tropomyosin in the absence of Ca2+ to stabilize the nonforce-generating B-state of the thin filament (25). Whereas much of this discussion is merely speculation, one certainty is that further investigations are needed to unravel the complexity of myofilament protein–protein interactions underlying modulation of contractile regulation in the myocardium.
Time-dependent fluctuations in atomic coordinates of proteins (i.e. protein dynamics) have emerged as a critical determinant of function (69). The observed alterations in cTnT–cTnC binding led us to ask the question of whether this cardiomyopathic variant impacted the structural dynamics of cTnC. Our group and others have previously reported on the importance of cTnC dynamics for its regulatory function (37, 70–72). The NMR relaxation results revealed remote molecular dynamics changes in the cTnC molecule caused by the I4M substitution, because the residues distant from the mutation site showed different NMR relaxation rates when compared with the same residues in WT cTnC (Fig. 10). Our bis-ANS fluorescence studies revealed similar hydrophobic exposure profiles in the apo (minus Ca2+) form. However, upon titration with Ca2+, hydrophobic exposure was markedly blunted for cTnC–I4M (Fig. 8), suggesting that this variant might stabilize the closed conformation or destabilize the open conformation of cTnC. One possibility is that introduction of a methionine at a solvent-exposed position in the N-helix of cTnC could render the cTnC molecule vulnerable to oxidation-induced conformational destabilization (73). Our CSP analysis by NMR suggested that key acidic residues in cTnC–I4M experienced a different chemical environment compared with cTnC–WT. We found that the regulatory N-domain of cTnC, the expected region of cTnC C-terminal binding, experienced relatively slower backbone dynamics as a direct result of this N-helix variant. These results offer a potential atomic-level explanation for the enhanced binding affinity between cTnC–I4M and the C terminus of cTnT.
Although we addressed this study mechanistically from a biophysical perspective, it should be acknowledged that DCM is a complex, heterogeneous disorder that is influenced by additional factors (e.g. modifier genes, epigenetics, and environment); myofilament dysregulation represents just one conceivable explanation for the genotype–phenotype relation (4, 74). In some instances, sarcomeric gene variants associated with cardiomyopathy have been suggested to render a host susceptible to bacterial or viral infections and further exacerbate cardiac dysfunction (75). Intriguingly, immunofluorescence studies have indicated nuclear localization of cTnC, raising the possibility that pathogenic cTnC variants might also disrupt cellular processes beyond contractile regulation (76). With this complexity in mind, it is not surprising that unequivocally establishing a genotype–phenotype relation for cardiomyopathic variants remains an enormous challenge, particularly in the case of de novo variants associated with DCM. While this study focused on an isolated rare variant, we surmise that other cardiomyopathic variants in the contractile apparatus might alter cTnC–cTnT-based regulation of myocardial contractility.
Experimental procedures
Clinical and molecular genetics
Physicians obtained family history and performed physical examination of the proband. Trio Whole-Exome Sequencing was performed at Centogene (Centogene AG, Germany). Written informed consent was obtained from the parents of the proband.
Molecular visualization and protein sequence alignment
The cTnC–I4M variant was visualized by UCSF Chimera using Protein Data Bank code 1J1E (22, 77). Multiple protein sequence alignments were performed with Clustal Omega, 1.2.4 (EMBL-EMI).
Mutagenesis, cloning, protein expression, and purification
QuikChange (Agilent) PCR site-directed mutagenesis was used to generate the cTnC mutants from pET-3d/11d constructs. Plasmid DNA sequences were verified by sequencing. Recombinant human cTnC, cTnT, and cTnI were cloned, expressed, and purified as described previously (41, 78). Both cTnT and cTnI were stored in aliquots at −80 °C, and cTnC–WT and -I4M were lyophilized and stored at 4 °C. For NMR spectroscopy experiments, the cTnC–I4M construct was transformed into competent Escherichia coli cells (strain BL21-DE3) and inoculated for growth in [15N]ammonium chloride (NLM-467-PK, Cambridge Isotope Laboratories, Inc.)-enriched minimal media as described previously (37). Native porcine cardiac tropomyosin was purified from left ventricular tissue, and filamentous actin was prepared from rabbit skeletal muscle acetone powder as described previously (41). SDS-PAGE was used to assess protein purity and potential degradation.
Protein quantification
NanoDrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA) was used to quantify protein concentrations (absorbance at 280 nm) for peptide-binding experiments. Extinction coefficients used for cTnC and a 14-amino acid C-terminal cTnT peptide were 4080 and 5960 cm−1 m−1, respectively. Pierce Coomassie Plus/Bradford assay kit (Thermo Fisher ScientificTM, catalogue no. 23236) was used for all other experiments with BSA as the standard.
Formation of troponin complexes
Recombinant human cardiac troponin complexes were assembled following standard procedures in our laboratory (15, 79). Briefly, troponin complexes were formed by first dialyzing the individual subunits (≥1.0 mg/ml) for 4 h in 4 liters of troponin complex buffer (3 m urea, 1 m KCl, 10 mm MOPS, 1 mm DTT, pH 7.0) and then twice against 4 liters of troponin complex buffer without urea for 4 h each. After a 1-h incubation on ice, recombinant cTnT, cTnI, and cTnC–WT or -I4M subunits were combined at 1.3:1.3:1 ratio into dialysis tubing and sequentially dialyzed against 4 liters of troponin complex buffer containing 1 mm MgCl2 with decreasing salt concentrations (0.7, 0.5, 0.3, and 0.1 m KCl) under constant stirring at 4 °C for 4 h each (15, 79). Precipitants were removed by centrifugation at 19,000 rpm in a JA-20 (Beckman Coulter) rotor for 15 min at 4 °C. Complexation was confirmed by native PAGE (4% stacking, 8% resolving).
Peptide synthesis
The C-terminal peptide (acetyl–Ser-Lys-Thr-Arg-Gly-Lys-Ala-Lys-Val-Thr-Gly-Arg-Trp-Lys–amide), corresponding to the last 14 amino acid residues in human cTnT (Uniprot ID P45379–6), was purchased from Genscript USA (Piscataway, NJ) and supplied in lyophilized form at 99.1% purity and stored at −20 °C. Peptide sequence and purity were assessed by MS and analytical HPLC.
Preparation of buffered Ca2+ solutions for CMP experiments
Calculations for buffered pCa (−log[Ca2+]free) solutions were carried out using the computer program, pCa calculator (80). Composition of the pCa solutions is as follows: 20 mm MOPS, 7 mm EGTA, 15 mm phosphocreatine, 15 units ml−1 creatine phosphokinase, 2.5 mm MgATP2−, 1 mm free Mg2+, constant ionic strength of 150 mm (anion = propionate), varying [Ca2+], pH 7.0. The solutions were prepared at room temperature (∼21 °C), as described previously (48). Experiments performed at different temperatures (as indicated in the figure legends) and involving dilutions (CaCl2 titrations) were accounted for in the calculations.
Cardiac muscle preparations
Fresh porcine hearts were procured from a local abattoir and transported in an ice-cold, O2-sparged buffer containing the following: 140 mm NaCl, 4 mm KCl, 1.8 mm CaCl2, 1 mm MgCl2, 1.8 mm NaH2PO4, 5.5 mm d-glucose, 50 mm HEPES, pH 7.4. Strips of papillary muscle were dissected from the myocardium on ice and permeabilized for 4 h in pCa 8.0 relaxing solution containing 1% (v/v) Triton X-100 at 4 °C. The CMP were further dissected to an average length of ∼1 mm, clipped on each end with aluminum T-clips, and stored in pCa 8.0 relaxing solution containing 52% (v/v) glycerol at −20 °C until used for experiments, as described previously (11).
Muscle mechanics
Extraction–reconstitution experiments on CMP were performed as described previously (11, 13). Briefly, CMP were mounted between a force transducer (model 403A, Aurora Scientific Inc.) and a high-speed servomotor (model 322C, Aurora Scientific Inc.) in pCa 8.0. Sarcomere length (SL) was set to 2.1 μm by He-Ne laser diffraction. Endogenous cTnC was extracted by incubation of a CMP with extraction buffer (5.0 mm CDTA, 25 mm Tris, pH 8.4). Drops of 55 μm cTnC–WT or -I4M were applied directly to the cTnC-extracted CMP. Measurements and analysis of myofilament Ca2+ sensitivity of steady-state isometric tension generation and kTR were performed as described previously with minor modifications (48). The pCa-force relation was fitted with a two-parameter, sigmoid Hill equation (48). The ends of the CMP were fixed with 1% glutaraldehyde (w/v) to minimize end compliance (81). In conditions where force was below 15% of the maximal force, the CMP were excluded for kTR analysis. Extraction and reconstitution of cTnC, as well as recording of initial maximal tension, were carried out at 21 °C to prevent CMP rundown. However, the temperature of the chambers was switched to 30 °C immediately prior to Ca2+-dependent activation (71). Mathematical modeling of tension kinetics was carried out in the MatLab environment as described previously (48).
bis-ANS fluorescence
Equimolar concentrations (2 μm) of bis-ANS (Sigma D4162) and cTnC–WT or -I4M were combined in fluorescence buffer (120 mm MOPS, 1.25 mm MgCl2, 90 mm KCl, 2 mm EGTA, 1 mm fresh DTT, pH 7.0) to a final volume of 2 ml in quartz cuvettes. Incremental (μl) amounts of 1 m CaCl2 were added to achieve the desired pCa based on the calculations described above. bis-ANS was excited at 380 nm, and emission maxima were monitored at ∼440–600 nm. Spectra were acquired on a four-cell Jasco FP-8300 spectrofluorometer connected to a compact recirculating cooler (Julabo) set to 21 °C. Acquisition parameters were set to 5.0-nm slit widths, a constant photomultiplier (PMT) voltage of 340 V, and automated stirring (600 rpm).
Intrinsic tryptophan fluorescence
The 14-amino acid C-terminal cTnT peptide, cTnC–WT and cTnC–I4M, was suspended and dialyzed in 25 mm HEPES, 60 mm NaCl, 3 mm MgCl2, 0.5 mm CaCl2, 2 mm β-mercaptoethanol (βME), pH 7.0, prior to titrations. Incremental (μl) amounts, from 0 to 50 μm, of cTnC–WT- or -I4M were titrated into 5 μm peptide. To account for potential contribution of fluorescence intensity changes upon titration of cTnC, cTnC–WT or -I4M was titrated into buffer without peptide and subtracted from the fluorescence intensity values for each condition. Intrinsic tryptophan was excited at 295 nm, and the wavelength emission maximum was monitored within the range 300–400 nm. Spectra were acquired on a four-cell Jasco FP-8300 spectrofluorometer connected to a compact recirculating cooler (Julabo) set to 21 °C. Acquisition parameters were set to 5.0 nm slit widths, a constant PMT voltage of 300 V, and automated stirring (600 rpm). Results were fitted using OriginPro software (Northampton, MA) with a modified Hill equation that describes one class of binding site (Equation 1) where START and END are the fluorescence values at the beginning and end of the curve, respectively; n is the Hill coefficient, and k represents the binding constant. Experiments are shown as average ± S.E. of five independent experiments.
| (Eq. 1) |
MST
Fluorescent labeling of cTnC–WT or -I4M was carried out using the Red-NHS 2nd Generation labeling kit (catalogue no. MO-L011, NanoTemper Technologies) following the manufacturer's protocol. The fluorescently-labeled proteins were exchanged into an assay buffer containing 200 mm MOPS, 150 mm KCl, 1.25 mm MgCl2, 100 μm CaCl2, 1 mm fresh DTT, 0.05% Tween 20, pH 7.0. Full-length human recombinant cTnT in a stock solution containing 0.35 m KCl, 0.1 m MOPS, 2 mm EGTA, 4 mm nitrilotriacetic acid, 1.25 mm MgCl2, pH 7.2, was titrated into fluorescently-labeled targets (cTnC–WT or -I4M). The final concentrations of fluorescently-labeled targets were kept constant at 20 nm, and the ligand (cTnT) varied from 1.2 nm to 38 μm. MST experiments were performed on a Monolith NT.115 (NanoTemper Technologies) at ambient temperature (21 °C) using premium-treated capillaries (catalogue no. MO-K025). The instrument parameters were set to 20% LED power and medium MST power. The equilibrium dissociation constants were derived from the MST software (MO Affinity Analysis software, version 3.2, NanoTemper Technologies) using the Kd model based on four independently titrated experiments per condition.
Solution-state NMR spectroscopy
Solution-state NMR experiments on 15N-labeled cTnC–I4M (at 396 μm) were carried out at 25 °C in the following buffer: 200 mm MOPS, 50 mm KCl, 2 mm EGTA, 2 mm MgCl2 (free), 10 mm DTT, 10% D2O, 4,4-dimethyl-4-silapentane-1-sulfonic acid, pH 7.0. Heteronuclear 1H–15N HSQC NMR spectra were acquired on a Bruker Avance III 900-MHz spectrometer equipped with a 5-mm trip resonance TXI probe at the National Center of Nuclear Magnetic Resonance, Jiri Jonas (Rio de Janeiro, Brazil) or Bruker Avance III 700 MHz equipped with a cryoprobe at the Department of Chemistry and Biochemistry, Florida State University (Tallahassee, FL). We performed the 1H–15N assignment of cTnC–I4M using the following procedures: (i) cross-peak transferring from our previous cTnC–WT assignment (37) to the -I4M 1H–15N HSQC spectrum, and (ii) peak–by–peak evaluation to decide whether transferred cross-peaks belong to its closest neighbor peak. The inclusion criteria to assign was naked eye inspection of the surrounding area and clear evidence that a selected cross-peak exclusively belongs to the closest neighbor. We did not assign few peaks at crowded areas because dualities arose (37). CSP analysis was performed as described previously using Equation 2,
| (Eq. 2) |
where ΔδH and ΔδN represent the change in chemical shifts for 1H and 15N, respectively, between cTnC–WT and -I4M. All spectra were processed using Topspin 3.2, and chemical-shift values were measured using the software CCPN Analysis 2.4.1. The diffusion properties (R1, R2, and NOE) of I4M were obtained as described previously (37).15N R1 and 15N R2 relaxation rates were measured from spectra with different relaxation delays: T1 = 0.05, 0.1, 0.2, 0.3, 0.4, 0.5, 0.7, 0.9, and 1.1 s for R1 and T2 = 0.01696, 0.03392, 0.05088, 0.06784, 0.0848, 0.10176, 0.13568, and 0.1696 s for R2. The errors in the peak intensities were calculated from the standard deviation of background noise levels in relation to each residue peak intensities (82). The heteronuclear NOEs were determined from the ratio of peak intensities with and without the saturation of the amide protons. NOE spectra was acquired using recycle relaxation delays of 6 s between the scans. Errors in HetNOE values were calculated from the peak intensities and noise levels in the reference and saturated spectra. Rotation correlation times, τc, of the N- and C-terminal domains were calculated separately from R2/R1 rates assuming isotropic tumbling and using Equation 3,
| (Eq. 3) |
where ν15N is the Larmor frequency of 15N. For these calculations, all residues showing a higher contribution of local dynamics on picosecond–nanosecond timescales, characterized by HetNOE rates lower than 0.6, and those showing contribution of conformational exchange, characterized by R2/R1 ratio values above the average value plus the standard deviation, were excluded. Relaxation rates R1, R2, and HetNOE were also analyzed with the Lipari-Szabo formalism (83, 84) using the software Tensor 2.0 (85). For this analysis, the N- and the C-terminal domains were analyzed separately because they showed very different values of rotational correlational times.
The maximum theoretical value (HetNOEmax) for cTnC was estimated using the software HYDRONMR (86). HYDRONMR simulates NMR relaxation data by calculating anisotropic rotational diffusion and by modeling protein structure (PDB code 1J1E) with a bead model. The following parameters used in the calculation were: temperature = 298 K; solvent viscosity = 0.01 poise; magnetic field = 21.15 tesla; gyromagnetic ratio of 1H = 2.675 × 104 rad s−1 G−1; gyromagnetic ratio of 15N = −2.713 × 103 rad s−1 G−1; internuclear H–N distance = 1.02 Å; and 15N chemical-shift anisotropy = −160 ppm.
Co-sedimentation assay
Co-sedimentation assays were performed following previously described methods with minor modifications (67). Prior to running the assay, troponin subunits were centrifuged to remove precipitates, and the supernatants were run on a gel to confirm solubility. Tropomyosin (2.86 μm), actin (20 μm), and cTnT (2.86 μm) were combined in a final volume of 100 μl of reaction buffer containing 25 mm HEPES, 60 mm NaCl, 3 mm MgCl2, 0.5 mm CaCl2, 2 mm βME, pH 7.0. Next, cTnC–WT or cTnC–I4M was added to a final concentration of 40 μm. The samples were then gently mixed, incubated on ice for 30 min, and centrifuged at 54,000 rpm in a fixed-angle TLA-100 rotor (Beckman Coulter) for 10 min at 4 °C. The protein-containing pellets were washed twice with reaction buffer and subsequently resuspended in 100 μl of 1× Laemmli buffer, boiled, and resolved by SDS-PAGE on a 15% gel. As a control, the experiment was repeated exactly as described but with omission of cTnT from the reactions. Proteins were visualized by Coomassie staining and imaged on an Odyssey IR system (LI-COR Biosciences). Image Studio Lite (LI-COR Biosciences) was used for densitometric quantification.
Chemical cross-linking-MS
WT cTn was subjected to BS3 chemical cross-linking followed by MS analysis using a recently published protocol (87). A 25 mm stock of equimolar BS3-d0 and BS3-d4 (bis(sulfosuccinimidyl) 2,2,7,7-suberate, Thermo Fisher Scientific) was freshly prepared in double-distilled water. A series of incubation times and varying concentrations of BS3-d0/d4 were tested to optimize cross-linking conditions. In a final reaction volume of 20 μl, cross-linking was initiated by addition of equimolar BS3-d0/d4 (0.25 mm, final concentration) to 1 mg/ml cTn in 25 mm HEPES, 60 mm NaCl, 3 mm MgCl2, 0.5 mm CaCl2, 2 mm βME, pH 7.0. The reaction was incubated in a MixMate (Eppendorf) at 350 rpm for 1 h at room temperature and quenched by addition of glycine, pH 8.0 (100 mm, final concentration). The uncross-linked and cross-linked samples were prepared for SDS-PAGE and run on a 4–15% Mini-PROTEAN TGX gel (Bio-Rad).
In-gel protein digestion
The band corresponding to the cross-linked troponin complex band was cut from gel and digested with trypsin. Briefly, the gel band was de-stained with a de-staining buffer (1:1 H2O/acetonitrile with 50 mm ammonium bicarbonate), cut into ∼2-mm pieces, shrunk with acetonitrile, and dried in SpeedVac. Then, the gel pieces were rehydrated with a digestion buffer (10% aqueous acetonitrile with 50 mm ammonium bicarbonate) and reduced with 0.5 mm tris(2-carboxyethyl)phosphine (Sigma, catalogue no. C4706) at 37 °C for 10 min. After cooling to room temperature, 1 mm iodoacetamide (Sigma, catalogue no. I1149) was added to the mixture and vortexed for 10 min. Then 0.005 μg/μl trypsin (Thermo Fisher Scientific, catalogue no. 90058) was added, and the mixture was incubated overnight at 37 °C. The supernatant was collected. 0.5% formic acid was added to the residual gel pieces; the mixture was incubated at 37 °C for 15 min, and the supernatant was collected. The residual gel pieces were dried after adding acetonitrile, and incubation was at 37 °C for 10 min. The combined supernatant was dried in SpeedVac (Thermo Fisher Scientific).
Nano-LC nLC/MS2
The dried tryptic peptide mixture was reconstituted in 0.1% aqueous formic acid and separated by nano-LC (nLC). An Easy Nano LC II system (Thermo Fisher Scientific) equipped with a 75 μm × 10-cm C18AQ analytical column (catalogue no. SC003, Thermo Fisher Scientific) and a 100 μm × 2-cm trap column (easy column, catalogue no. SC001, Thermo Fisher Scientific) was used. A 3-h linear gradient from 1 to 35% B was performed with a flow rate of 300 nl/min (mobile phase A: 99.9% H2O and 0.1% formic acid; mobile phase B: 99.9% acetonitrile and 0.1% formic acid). Eluate was ionized online with 2 kV spray voltage and detected by a Velos LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific). The precursor ions were detected with a mass resolution of 60 K (at m/z of 800 Da) and Automatic Gain Control (AGC) of 1e6 in the Orbitrap. Centroid data-dependent MS2 was carried out on the top 10 most abundant precursor ions with AGC of 5e4 with collision-induced dissociation (2.0 m/z isolation window and 35 normalized collision energy) in the LTQ.
Data analysis
An open-access software ProteoWizard MSConvert (ProteoWizard version 3.0.10158) (88) was used to convert the original .raw file to .mzXML file. The converted file was then analyzed by another open-access software StavroX (StavroX version 3.6.6) to identify cross-linked peptides (89). The following parameters were used in StavroX: protein sequence FASTA database containing the three human cardiac troponin complex proteins (cTnC, cTnI, and cTnT); up to three trypsin missed cleavages; static modification of cysteine to carbamidomethyl cysteine; variable modification of methionine by oxidation; precursor precision of <3 ppm; fragment ion precision of <0.8 Da; signal/noise ratio above 2; false discovery rate cutoff of <5.0%. Software-generated results were then manually checked for correct assignments of those cross-linked peptide MS2 spectra. For every confirmed cross-linked peptide, the extracted ion chromatogram and MS of the corresponding precursor ions were checked for presence of peptide doublets, peptides cross-linked by BS3-d0/d4. The cross-linked peptide doublets should have a separation of 4.0256 Da (the mass difference with 4 H and 4 D). A list of selected peptide sequences identified, precursor charge, m/z for each assignment, peptide identification scores, and LC retention time can be found in Table S3. All cross-linked peptides identified and annotated MS/MS spectra can be found in Fig. S6.
Statistical analyses
Statistical analyses were performed using SigmaPlot version 12.0 or OriginPro software (Northampton, MA). Experimental results are reported as mean ± S.D. as indicated in each figure legend. Student's t-test (paired or unpaired, indicated in figure legends) was used to determine statistical significance (p < 0.05) between control and experimental groups. Sample size (N) for a given condition is reported in each respective figure legend.
Author contributions
J. R. J., G. A. P. d. O., and J. R. P. conceptualization; J. R. J., M. L.-V., M. A. M., D. G.-M., H. H., A. I., Y. W., E. B., and N. Z. data curation; J. R. J., M. L.-V., G. A. P. d. O., D. G.-M., A. H. M., H. H., A. I., Y. W., E. B., N. Z., and P. B. C. formal analysis; J. R. J., J. L. S., P. B. C., and J. R. P. supervision; J. R. J., G. A. P. d. O., A. H. M., H. H., and P. B. C. validation; J. R. J., M. L.-V., M. A. M., G. A. P. d. O., H. H., A. I., E. B., N. Z., and P. B. C. investigation; J. R. J., M. L.-V., M. A. M., G. A. P. d. O., H. H., E. B., and P. B. C. visualization; J. R. J., M. L.-V., M. A. M., G. A. P. d. O., A. H. M., H. H., and P. B. C. methodology; J. R. J. and G. A. P. d. O. writing-original draft; J. R. J., P. B. C., and J. R. P. project administration; J. R. J., P. B. C., and J. R. P. writing-review and editing; A. H. M. and H. H. software; E. B., N. Z., J. L. S., and J. R. P. resources; J. R. P. funding acquisition.
Supplementary Material
Acknowledgments
We thank the family members for participating in this study, as well as Cheryl Pye, Dr. Brian Washburn (Molecular Cloning Facility, Florida State University), Dr. Steve Miller (DNA Sequencing Facility, Florida State University), and Dr. Banghao Chen (NMR Facility, Florida State University) for their assistance and technical expertise.
This work was supported by American Heart Association Grant 19PRE34380628 (to J. R. J.) and National Institutes of Health Grant HL128683 from NHLBI (to J. R. P.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S6 and Tables S1–S3.
The raw mass spectrometry files are available through Zenodo accession no. 3523344.
J. R. Pinto, J. R. Johnston, and P. B. Chase, unpublished observations.
- DCM
- dilated cardiomyopathy
- HCM
- hypertrophic cardiomyopathy
- cTn
- cardiac troponin complex
- TNNC1
- slow-skeletal/cardiac troponin C
- cTnC
- cardiac troponin C
- cTnT
- cardiac troponin T
- cTnI
- cardiac troponin I
- CMP
- cardiac muscle preparation
- CDTA
- trans-1,2-diaminocyclohexane-N,N,N′,N′-tetraacetic acid
- bis-ANS
- 4,4′-dianilino-1,1′-binaphthyl-5,5′-disulfonic acid dipotassium salt
- HSQC
- heteronuclear single-quantum coherence
- CSP
- chemical-shift perturbation
- R2/R1
- transverse/longitudinal relaxation rate
- HetNOE
- heteronuclear nuclear Overhauser effect
- MST
- microscale thermophoresis
- βME
- β-mercaptoethanol
- PDB
- Protein Data Bank
- PMT
- photomultiplier
- N
- newton
- AGC
- Automatic Gain Control
- kTR
- rate of isometric tension redevelopment.
References
- 1. Khush K. K., Cherikh W. S., Chambers D. C., Goldfarb S., Hayes D. Jr., Kucheryavaya A. Y., Levvey B. J., Meiser B., Rossano J. W., Stehlik J., and International Society for Heart and Lung Transplantation. (2018) The International Thoracic Organ Transplant Registry of the International Society for Heart and Lung Transplantation: 35th adult heart transplantation report–2018; focus theme: multiorgan transplantation. J. Heart Lung Transplant. 37, 1155–1168 10.1016/j.healun.2018.07.022 [DOI] [PubMed] [Google Scholar]
- 2. Rossano J. W., Cherikh W. S., Chambers D. C., Goldfarb S., Hayes D. Jr., Khush K. K., Kucheryavaya A. Y., Toll A. E., Levvey B. J., Meiser B., and Stehlik J. (2018) The International Thoracic Organ Transplant Registry of the International Society for Heart and Lung Transplantation: 21st Pediatric Heart Transplantation Report–2018; Focus Theme: Multiorgan Transplantation. J. Heart Lung Transplant. 37, 1184–1195 10.1016/j.healun.2018.07.018 [DOI] [PubMed] [Google Scholar]
- 3. Benjamin E. J., Muntner P., Alonso A., Bittencourt M. S., Callaway C. W., Carson A. P., Chamberlain A. M., Chang A. R., Cheng S., Das S. R., Delling F. N., Djousse L., Elkind M. S. V., Ferguson J. F., Fornage M., et al. (2019) Heart Disease and Stroke Statistics–2019 Update: A Report From the American Heart Association. Circulation 139, e56–e528 10.1161/CIR.0000000000000659 [DOI] [PubMed] [Google Scholar]
- 4. McNally E. M., and Mestroni L. (2017) Dilated cardiomyopathy: genetic determinants and mechanisms. Circ. Res. 121, 731–748 10.1161/CIRCRESAHA.116.309396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. McNally E. M., Golbus J. R., and Puckelwartz M. J. (2013) Genetic mutations and mechanisms in dilated cardiomyopathy. J. Clin. Invest. 123, 19–26 10.1172/JCI62862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Schultheiss H. P., Fairweather D., Caforio A. L. P., Escher F., Hershberger R. E., Lipshultz S. E., Liu P. P., Matsumori A., Mazzanti A., McMurray J., and Priori S. G. (2019) Dilated cardiomyopathy. Nat. Rev. Dis. Primers 5, 32 10.1038/s41572-019-0084-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Burke M. A., Cook S. A., Seidman J. G., and Seidman C. E. (2016) Clinical and mechanistic insights into the genetics of cardiomyopathy. J. Am. Coll. Cardiol. 68, 2871–2886 10.1016/j.jacc.2016.08.079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van der Velden J., and Stienen G. J. M. (2019) Cardiac disorders and pathophysiology of sarcomeric proteins. Physiol. Rev. 99, 381–426 10.1152/physrev.00040.2017 [DOI] [PubMed] [Google Scholar]
- 9. Lipshultz S. E., Law Y. M., Asante-Korang A., Austin E. D., Dipchand A. I., Everitt M. D., Hsu D. T., Lin K. Y., Price J. F., Wilkinson J. D., and Colan S. D. (2019) Cardiomyopathy in children: classification and diagnosis: a scientific statement from the American Heart Association. Circulation 140, e9–e68 10.1161/CIR.0000000000000682 [DOI] [PubMed] [Google Scholar]
- 10. Walsh R., Thomson K. L., Ware J. S., Funke B. H., Woodley J., McGuire K. J., Mazzarotto F., Blair E., Seller A., Taylor J. C., Minikel E. V., Exome Aggregation Consortium, MacArthur D. G., Farrall M., Cook S. A., and Watkins H. (2017) Reassessment of Mendelian gene pathogenicity using 7,855 cardiomyopathy cases and 60,706 reference samples. Genet. Med. 19, 192–203 10.1038/gim.2016.90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pinto J. R., Siegfried J. D., Parvatiyar M. S., Li D., Norton N., Jones M. A., Liang J., Potter J. D., and Hershberger R. E. (2011) Functional characterization of TNNC1 rare variants identified in dilated cardiomyopathy. J. Biol. Chem. 286, 34404–34412 10.1074/jbc.M111.267211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Norton N., Robertson P. D., Rieder M. J., Züchner S., Rampersaud E., Martin E., Li D., Nickerson D. A., Hershberger R. E., and National Heart, Lung and Blood Institute GO Exome Sequencing Project. (2012) Evaluating pathogenicity of rare variants from dilated cardiomyopathy in the exome era. Circ. Cardiovasc. Genet. 5, 167–174 10.1161/CIRCGENETICS.111.961805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dweck D., Reynaldo D. P., Pinto J. R., and Potter J. D. (2010) A dilated cardiomyopathy troponin C mutation lowers contractile force by reducing strong myosin-actin binding. J. Biol. Chem. 285, 17371–17379 10.1074/jbc.M109.064105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Willott R. H., Gomes A. V., Chang A. N., Parvatiyar M. S., Pinto J. R., and Potter J. D. (2010) Mutations in troponin that cause HCM, DCM, and RCM: what can we learn about thin filament function? J. Mol. Cell. Cardiol. 48, 882–892 10.1016/j.yjmcc.2009.10.031 [DOI] [PubMed] [Google Scholar]
- 15. Parvatiyar M. S., Landstrom A. P., Figueiredo-Freitas C., Potter J. D., Ackerman M. J., and Pinto J. R. (2012) A mutation in TNNC1-encoded cardiac troponin C, TNNC1-A31S, predisposes to hypertrophic cardiomyopathy and ventricular fibrillation. J. Biol. Chem. 287, 31845–31855 10.1074/jbc.M112.377713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Repetti G. G., Toepfer C. N., Seidman J. G., and Seidman C. E. (2019) Novel therapies for prevention and early treatment of cardiomyopathies. Circ. Res. 124, 1536–1550 10.1161/CIRCRESAHA.119.313569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Filatov V. L., Katrukha A. G., Bulargina T. V., and Gusev N. B. (1999) Troponin: structure, properties, and mechanism of functioning. Biochemistry 64, 969–985 [PubMed] [Google Scholar]
- 18. Kobayashi T., and Solaro R. J. (2005) Calcium, thin filaments, and the integrative biology of cardiac contractility. Annu. Rev. Physiol. 67, 39–67 10.1146/annurev.physiol.67.040403.114025 [DOI] [PubMed] [Google Scholar]
- 19. Holroyde M. J., Robertson S. P., Johnson J. D., Solaro R. J., and Potter J. D. (1980) The calcium and magnesium binding sites on cardiac troponin and their role in the regulation of myofibrillar adenosine triphosphatase. J. Biol. Chem. 255, 11688–11693 [PubMed] [Google Scholar]
- 20. Wei B., and Jin J. P. (2016) TNNT1, TNNT2, and TNNT3: isoform genes, regulation, and structure-function relationships. Gene 582, 1–13 10.1016/j.gene.2016.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Heller W. T., Finley N. L., Dong W. J., Timmins P., Cheung H. C., Rosevear P. R., and Trewhella J. (2003) Small-angle neutron scattering with contrast variation reveals spatial relationships between the three subunits in the ternary cardiac troponin complex and the effects of troponin I phosphorylation. Biochemistry 42, 7790–7800 10.1021/bi0341509 [DOI] [PubMed] [Google Scholar]
- 22. Takeda S., Yamashita A., Maeda K., and Maéda Y. (2003) Structure of the core domain of human cardiac troponin in the Ca2+-saturated form. Nature 424, 35–41 10.1038/nature01780 [DOI] [PubMed] [Google Scholar]
- 23. Potter J. D., Sheng Z., Pan B. S., and Zhao J. (1995) A direct regulatory role for troponin T and a dual role for troponin C in the Ca2+ regulation of muscle contraction. J. Biol. Chem. 270, 2557–2562 10.1074/jbc.270.6.2557 [DOI] [PubMed] [Google Scholar]
- 24. Thierfelder L., Watkins H., MacRae C., Lamas R., McKenna W., Vosberg H. P., Seidman J. G., and Seidman C. E. (1994) α-Tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy: a disease of the sarcomere. Cell 77, 701–712 10.1016/0092-8674(94)90054-X [DOI] [PubMed] [Google Scholar]
- 25. Franklin A. J., Baxley T., Kobayashi T., and Chalovich J. M. (2012) The C terminus of troponin T is essential for maintaining the inactive state of regulated actin. Biophys. J. 102, 2536–2544 10.1016/j.bpj.2012.04.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Gafurov B., Fredricksen S., Cai A., Brenner B., Chase P. B., and Chalovich J. M. (2004) The Δ14 mutation of human cardiac troponin T enhances ATPase activity and alters the cooperative binding of S1-ADP to regulated actin. Biochemistry 43, 15276–15285 10.1021/bi048646h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Johnson D., Angus C. W., and Chalovich J. M. (2018) Stepwise C-terminal truncation of cardiac troponin T alters function at low and saturating Ca2. Biophys. J. 115, 702–712 10.1016/j.bpj.2018.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brunet N. M., Chase P. B., Mihajlović G., and Schoffstall B. (2014) Ca2+-regulatory function of the inhibitory peptide region of cardiac troponin I is aided by the C-terminus of cardiac troponin T: effects of familial hypertrophic cardiomyopathy mutations cTnI R145G and cTnT R278C, alone and in combination, on filament sliding. Arch. Biochem. Biophys. 552, 11–20 10.1016/j.abb.2013.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., Grody W. W., Hegde M., Lyon E., Spector E., Voelkerding K., Rehm H. L., and ACMG Laboratory Quality Assurance Committee. (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17, 405–424 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Karczewski K. J., Francioli L. C., Tiao G., Cummings B. B., Alföldi J., Wang Q., Collins R. L., Laricchia K. M., Ganna A., Birnbaum D. P., Gauthier L. D., Brand H., Solomonson M., Watts N. A., Rhodes D., et al. (2019) Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv 10.1101/531210 [DOI] [Google Scholar]
- 31. Landrum M. J., Lee J. M., Riley G. R., Jang W., Rubinstein W. S., Church D. M., and Maglott D. R. (2014) ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 42, D980–D985 10.1093/nar/gkt1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schwarz J. M., Cooper D. N., Schuelke M., and Seelow D. (2014) MutationTaster2: mutation prediction for the deep-sequencing age. Nat. Methods 11, 361–362 10.1038/nmeth.2890 [DOI] [PubMed] [Google Scholar]
- 33. Pejaver V., Urresti J., Lugo-Martinez J., Pagel K. A., Lin G. N., Nam H.-J., Mort M., Cooper D. N., Sebat J., Iakoucheva L. M., Mooney S. D., and Radivojac P. (2017) MutPred2: inferring the molecular and phenotypic impact of amino acid variants. bioRxiv 10.1101/134981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kumar P., Henikoff S., and Ng P. C. (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4, 1073–1081 10.1038/nprot.2009.86 [DOI] [PubMed] [Google Scholar]
- 35. Adzhubei I. A., Schmidt S., Peshkin L., Ramensky V. E., Gerasimova A., Bork P., Kondrashov A. S., and Sunyaev S. R. (2010) A method and server for predicting damaging missense mutations. Nat. Methods 7, 248–249 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Landesberg A., and Sideman S. (1994) Coupling calcium binding to troponin C and cross-bridge cycling in skinned cardiac cells. Am. J. Physiol. 266, H1260–H1271 10.1152/ajpheart.1994.266.3.H1260 [DOI] [PubMed] [Google Scholar]
- 37. Marques M. A., Pinto J. R., Moraes A. H., Iqbal A., de Magalhães M. T., Monteiro J., Pedrote M. M., Sorenson M. M., Silva J. L., and de Oliveira G. A. (2017) Allosteric transmission along a loosely structured backbone allows a cardiac troponin C mutant to function with only one Ca2+ ion. J. Biol. Chem. 292, 2379–2394 10.1074/jbc.M116.765362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mogensen J., Murphy R. T., Shaw T., Bahl A., Redwood C., Watkins H., Burke M., Elliott P. M., and McKenna W. J. (2004) Severe disease expression of cardiac troponin C and T mutations in patients with idiopathic dilated cardiomyopathy. J. Am. Coll. Cardiol. 44, 2033–2040 10.1016/j.jacc.2004.08.027 [DOI] [PubMed] [Google Scholar]
- 39. Li M. X., and Hwang P. M. (2015) Structure and function of cardiac troponin C (TNNC1): implications for heart failure, cardiomyopathies, and troponin modulating drugs. Gene 571, 153–166 10.1016/j.gene.2015.07.074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Acuna-Hidalgo R., Veltman J. A., and Hoischen A. (2016) New insights into the generation and role of de novo mutations in health and disease. Genome Biol. 17, 241 10.1186/s13059-016-1110-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dweck D., Hus N., and Potter J. D. (2008) Challenging current paradigms related to cardiomyopathies. Are changes in the Ca2+ sensitivity of myofilaments containing cardiac troponin C mutations (G159D and L29Q) good predictors of the phenotypic outcomes? J. Biol. Chem. 283, 33119–33128 10.1074/jbc.M804070200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bers D. M. (2002) Cardiac excitation-contraction coupling. Nature 415, 198–205 10.1038/415198a [DOI] [PubMed] [Google Scholar]
- 43. Alves M. L., Warren C. M., Simon J. N., Gaffin R. D., Montminy E. M., Wieczorek D. F., Solaro R. J., and Wolska B. M. (2017) Early sensitization of myofilaments to Ca2+ prevents genetically linked dilated cardiomyopathy in mice. Cardiovasc. Res. 113, 915–925 10.1093/cvr/cvx068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shettigar V., Zhang B., Little S. C., Salhi H. E., Hansen B. J., Li N., Zhang J., Roof S. R., Ho H. T., Brunello L., Lerch J. K., Weisleder N., Fedorov V. V., Accornero F., Rafael-Fortney J. A., et al. (2016) Rationally engineered Troponin C modulates in vivo cardiac function and performance in health and disease. Nat. Commun. 7, 10794 10.1038/ncomms10794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Chung J. H., Biesiadecki B. J., Ziolo M. T., Davis J. P., and Janssen P. M. (2016) Myofilament calcium sensitivity: role in regulation of in vivo cardiac contraction and relaxation. Front. Physiol. 7, 562 10.3389/fphys.2016.00562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stehle R., and Iorga B. (2010) Kinetics of cardiac sarcomeric processes and rate-limiting steps in contraction and relaxation. J. Mol. Cell. Cardiol. 48, 843–850 10.1016/j.yjmcc.2009.12.020 [DOI] [PubMed] [Google Scholar]
- 47. Wolff M. R., McDonald K. S., and Moss R. L. (1995) Rate of tension development in cardiac muscle varies with level of activator calcium. Circ. Res. 76, 154–160 10.1161/01.RES.76.1.154 [DOI] [PubMed] [Google Scholar]
- 48. Gonzalez-Martinez D., Johnston J. R., Landim-Vieira M., Ma W., Antipova O., Awan O., Irving T. C., Chase P. B., and Pinto J. R. (2018) Structural and functional impact of troponin C-mediated Ca2+ sensitization on myofilament lattice spacing and cross-bridge mechanics in mouse cardiac muscle. J. Mol. Cell. Cardiol. 123, 26–37 10.1016/j.yjmcc.2018.08.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Baker A. J., Figueredo V. M., Keung E. C., and Camacho S. A. (1998) Ca2+ regulates the kinetics of tension development in intact cardiac muscle. Am. J. Physiol. 275, H744–H750 10.1152/ajpheart.1998.275.3.H744 [DOI] [PubMed] [Google Scholar]
- 50. Loong C. K., Takeda A. K., Badr M. A., Rogers J. S., and Chase P. B. (2013) Slowed dynamics of thin filament regulatory units reduces Ca2+-sensitivity of cardiac biomechanical function. Cell. Mol. Bioeng. 6, 183–198 10.1007/s12195-013-0269-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Brenner B. (1988) Effect of Ca2+ on cross-bridge turnover kinetics in skinned single rabbit psoas fibers: implications for regulation of muscle contraction. Proc. Natl. Acad. Sci. U.S.A. 85, 3265–3269 10.1073/pnas.85.9.3265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Brenner B., and Eisenberg E. (1986) Rate of force generation in muscle: correlation with actomyosin ATPase activity in solution. Proc. Natl. Acad. Sci. U.S.A. 83, 3542–3546 10.1073/pnas.83.10.3542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Regnier M., Rivera A. J., Chase P. B., Smillie L. B., and Sorenson M. M. (1999) Regulation of skeletal muscle tension redevelopment by troponin C constructs with different Ca2+ affinities. Biophys. J. 76, 2664–2672 10.1016/S0006-3495(99)77418-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kawai M., Johnston J. R., Karam T., Wang L., Singh R. K., and Pinto J. R. (2017) Myosin rod hypophosphorylation and CB kinetics in papillary muscles from a TnC-A8V KI mouse model. Biophys. J. 112, 1726–1736 10.1016/j.bpj.2017.02.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bing W., Fraser I. D., and Marston S. B. (1997) Troponin I and troponin T interact with troponin C to produce different Ca2+-dependent effects on actin-tropomyosin filament motility. Biochem. J. 327, 335–340 10.1042/bj3270335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. McKillop D. F., and Geeves M. A. (1993) Regulation of the interaction between actin and myosin subfragment 1: evidence for three states of the thin filament. Biophys. J. 65, 693–701 10.1016/S0006-3495(93)81110-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Risi C., Eisner J., Belknap B., Heeley D. H., White H. D., Schröder G. F., and Galkin V. E. (2017) Ca2+-induced movement of tropomyosin on native cardiac thin filaments revealed by cryoelectron microscopy. Proc. Natl. Acad. Sci. U.S.A. 114, 6782–6787 10.1073/pnas.1700868114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ripoll-Vera T., Gámez J. M., Góvea N., Gomez Y., Núñez J., Socías L., Escandell Á., and Rosell J. (2016) Clinical and prognostic profiles of cardiomyopathies caused by mutations in the troponin T gene. Rev. Esp. Cardiol. 69, 149–158 10.1016/j.recesp.2015.06.028 [DOI] [PubMed] [Google Scholar]
- 59. Zamora J. E., Papadaki M., Messer A. E., Marston S. B., and Gould I. R. (2016) Troponin structure: its modulation by Ca2+ and phosphorylation studied by molecular dynamics simulations. Phys. Chem. Chem. Phys. 18, 20691–20707 10.1039/C6CP02610A [DOI] [PubMed] [Google Scholar]
- 60. Kobayashi T., Zhao X., Wade R., and Collins J. H. (1999) Involvement of conserved, acidic residues in the N-terminal domain of troponin C in calcium-dependent regulation. Biochemistry 38, 5386–5391 10.1021/bi981320m [DOI] [PubMed] [Google Scholar]
- 61. Solís C., Kim G. H., Moutsoglou M. E., and Robinson J. M. (2018) Ca2+ and myosin cycle states work as allosteric effectors of troponin activation. Biophys. J. 115, 1762–1769 10.1016/j.bpj.2018.08.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Lehrer S. S., and Geeves M. A. (1998) The muscle thin filament as a classical cooperative/allosteric regulatory system. J. Mol. Biol. 277, 1081–1089 10.1006/jmbi.1998.1654 [DOI] [PubMed] [Google Scholar]
- 63. Williams M. R., Lehman S. J., Tardiff J. C., and Schwartz S. D. (2016) Atomic resolution probe for allostery in the regulatory thin filament. Proc. Natl. Acad. Sci. U.S.A. 113, 3257–3262 10.1073/pnas.1519541113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dyson H. J., and Wright P. E. (2005) Intrinsically unstructured proteins and their functions. Nat. Rev. Mol. Cell Biol. 6, 197–208 10.1038/nrm1589 [DOI] [PubMed] [Google Scholar]
- 65. Na I., Kong M. J., Straight S., Pinto J. R., and Uversky V. N. (2016) Troponins, intrinsic disorder, and cardiomyopathy. Biol. Chem. 397, 731–751 10.1515/hsz-2015-0303 [DOI] [PubMed] [Google Scholar]
- 66. Blumenschein T. M., Tripet B. P., Hodges R. S., and Sykes B. D. (2001) Mapping the interacting regions between troponins T and C. Binding of TnT and TnI peptides to TnC and NMR mapping of the TnT-binding site on TnC. J. Biol. Chem. 276, 36606–36612 10.1074/jbc.M105130200 [DOI] [PubMed] [Google Scholar]
- 67. Malnic B., Farah C. S., and Reinach F. C. (1998) Regulatory properties of the NH2- and COOH-terminal domains of troponin T. ATPase activation and binding to troponin I and troponin C. J. Biol. Chem. 273, 10594–10601 10.1074/jbc.273.17.10594 [DOI] [PubMed] [Google Scholar]
- 68. Kowlessur D., and Tobacman L. S. (2012) Significance of troponin dynamics for Ca2+-mediated regulation of contraction and inherited cardiomyopathy. J. Biol. Chem. 287, 42299–42311 10.1074/jbc.M112.423459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Henzler-Wildman K., and Kern D. (2007) Dynamic personalities of proteins. Nature 450, 964–972 10.1038/nature06522 [DOI] [PubMed] [Google Scholar]
- 70. Marques M. A., Parvatiyar M. S., Yang W., de Oliveira G. A. P., and Pinto J. R. (2019) The missing links within troponin. Arch. Biochem. Biophys. 663, 95–100 10.1016/j.abb.2018.12.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Veltri T., de Oliveira G. A. P., Bienkiewicz E. A., Palhano F. L., Marques M. A., Moraes A. H., Silva J. L., Sorenson M. M., and Pinto J. R. (2017) Amide hydrogens reveal a temperature-dependent structural transition that enhances site-II Ca2+-binding affinity in a C-domain mutant of cardiac troponin C. Sci. Rep. 7, 691 10.1038/s41598-017-00777-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Stevens C. M., Rayani K., Singh G., Lotfalisalmasi B., Tieleman D. P., and Tibbits G. F. (2017) Changes in the dynamics of the cardiac troponin C molecule explain the effects of Ca2+-sensitizing mutations. J. Biol. Chem. 292, 11915–11926 10.1074/jbc.M116.770776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kim G., Weiss S. J., and Levine R. L. (2014) Methionine oxidation and reduction in proteins. Biochim. Biophys. Acta 1840, 901–905 10.1016/j.bbagen.2013.04.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Deranek A. E., Klass M. M., and Tardiff J. C. (2019) Moving beyond simple answers to complex disorders in sarcomeric cardiomyopathies: the role of integrated systems. Pflugers Arch. 471, 661–671 10.1007/s00424-019-02269-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Cannatá A., Artico J., Gentile P., Merlo M., and Sinagra G. (2019) Myocarditis evolving in cardiomyopathy: when genetics and offending causes work together. Eur. Heart J. Suppl. 21, B90–B95 10.1093/eurheartj/suz033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Johnston J. R., Chase P. B., and Pinto J. R. (2018) Troponin through the looking-glass: emerging roles beyond regulation of striated muscle contraction. Oncotarget 9, 1461–1482 10.18632/oncotarget.22879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., and Ferrin T. E. (2004) UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 10.1002/jcc.20084 [DOI] [PubMed] [Google Scholar]
- 78. Landstrom A. P., Parvatiyar M. S., Pinto J. R., Marquardt M. L., Bos J. M., Tester D. J., Ommen S. R., Potter J. D., and Ackerman M. J. (2008) Molecular and functional characterization of novel hypertrophic cardiomyopathy susceptibility mutations in TNNC1-encoded troponin C. J. Mol. Cell. Cardiol. 45, 281–288 10.1016/j.yjmcc.2008.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Pinto J. R., Parvatiyar M. S., Jones M. A., Liang J., and Potter J. D. (2008) A troponin T mutation that causes infantile restrictive cardiomyopathy increases Ca2+ sensitivity of force development and impairs the inhibitory properties of troponin. J. Biol. Chem. 283, 2156–2166 10.1074/jbc.M707066200 [DOI] [PubMed] [Google Scholar]
- 80. Dweck D., Reyes-Alfonso A. Jr, and Potter J. D. (2005) Expanding the range of free calcium regulation in biological solutions. Anal. Biochem. 347, 303–315 10.1016/j.ab.2005.09.025 [DOI] [PubMed] [Google Scholar]
- 81. Chase P. B., and Kushmerick M. J. (1988) Effects of pH on contraction of rabbit fast and slow skeletal muscle fibers. Biophys. J. 53, 935–946 10.1016/S0006-3495(88)83174-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Farrow N. A., Muhandiram R., Singer A. U., Pascal S. M., Kay C. M., Gish G., Shoelson S. E., Pawson T., Forman-Kay J. D., and Kay L. E. (1994) Backbone dynamics of a free and phosphopeptide-complexed Src homology 2 domain studied by 15N NMR relaxation. Biochemistry 33, 5984–6003 10.1021/bi00185a040 [DOI] [PubMed] [Google Scholar]
- 83. Lipari G., Szabo A., and Levy R. M. (1982) Protein dynamics and NMR relaxation: comparison of simulations with experiment. Nature 300, 197–198 10.1038/300197a0 [DOI] [Google Scholar]
- 84. Morin S. (2011) A practical guide to protein dynamics from 15N spin relaxation in solution. Prog. Nucl. Magn. Reson. Spectrosc. 59, 245–262 10.1016/j.pnmrs.2010.12.003 [DOI] [PubMed] [Google Scholar]
- 85. d'Auvergne E. J., and Gooley P. R. (2008) Optimisation of NMR dynamic models II. A new methodology for the dual optimisation of the model-free parameters and the Brownian rotational diffusion tensor. J. Biomol. NMR 40, 121–133 10.1007/s10858-007-9213-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. García de la Torre J., Huertas M. L., and Carrasco B. (2000) HYDRONMR: prediction of NMR relaxation of globular proteins from atomic-level structures and hydrodynamic calculations. J. Magn. Reson. 147, 138–146 10.1006/jmre.2000.2170 [DOI] [PubMed] [Google Scholar]
- 87. Schmidt C., and Robinson C. V. (2014) A comparative cross-linking strategy to probe conformational changes in protein complexes. Nat. Protoc. 9, 2224–2236 10.1038/nprot.2014.144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Chambers M. C., Maclean B., Burke R., Amodei D., Ruderman D. L., Neumann S., Gatto L., Fischer B., Pratt B., Egertson J., Hoff K., Kessner D., Tasman N., Shulman N., Frewen B., Baker T. A., et al. (2012) A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 30, 918–920 10.1038/nbt.2377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Götze M., Pettelkau J., Schaks S., Bosse K., Ihling C. H., Krauth F., Fritzsche R., Kühn U., and Sinz A. (2012) StavroX–a software for analyzing crosslinked products in protein interaction studies. J. Am. Soc. Mass. Spectrom. 23, 76–87 10.1007/s13361-011-0261-2 [DOI] [PubMed] [Google Scholar]
- 90. Pinto J. R., Reynaldo D. P., Parvatiyar M. S., Dweck D., Liang J., Jones M. A., Sorenson M. M., and Potter J. D. (2011) Strong cross-bridges potentiate the Ca2+ affinity changes produced by hypertrophic cardiomyopathy cardiac troponin C mutants in myofilaments: a fast kinetic approach. J. Biol. Chem. 286, 1005–1013 10.1074/jbc.M110.168583 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.