

Abstract
The inhibitory co-receptor programmed cell death 1 (PD-1, Pdcd1) plays critical roles in the regulation of autoimmunity, anticancer immunity, and immunity against infections. Immunotherapies targeting PD-1 have revolutionized cancer management and instigated various trials of improved cancer immunotherapies. Moreover, extensive trials are underway to potentiate PD-1 function to suppress harmful immune responses. Here we found that both natural and synthetic glucocorticoids (GCs) up-regulate PD-1 on T cells without altering the expression levels of other co-receptors and cell surface molecules. GC-induced up-regulation of PD-1 depended on transactivation of PD-1 transcription mediated through the glucocorticoid receptor. We further found that a GC response element 2525 bp upstream of the transcription start site of Pdcd1 is responsible for GC-mediated transactivation. We also observed that in vivo administration of GCs significantly up-regulates PD-1 expression on tumor-infiltrating T cells. By analyzing T cells differing in PD-1 expression, we directly demonstrated that the amount of PD-1 on the cell surface correlates with its inhibitory effect. Accordingly, GCs potentiated the capacity of PD-1 to inhibit T cell activation, suggesting that this PD-1-mediated inhibition contributes, at least in part, to the anti-inflammatory and immunosuppressive effects of GCs. In light of the critical roles of PD-1 in the regulation of autoimmunity, we expect that the potentiation of PD-1 activity may offer a promising therapeutic strategy for managing inflammatory and autoimmune diseases. Our current findings provide a rationale for strategies seeking to enhance the inhibitory effect of PD-1 by increasing its expression level.
Keywords: gene expression, glucocorticoid, immunosuppression, immunology, T cell, T cell receptor (TCR), cytokine induction, coreceptor, immune checkpoint, PD-1
Introduction
The inhibitory co-receptors programmed cell death 1 (PD-1) is inducibly expressed on T cells upon activation and inhibits T cell receptor (TCR)4 signaling by recruiting SHP-2, a protein tyrosine phosphatase, in a manner dependent on engagement by either of its two ligands, PD-L1 or PD-L2 (1, 2). PD-1–dependent regulation of the TCR signal is required for establishment and maintenance of immune self-tolerance and suppression of excess immune responses to pathogens. Mice deficient in PD-1 spontaneously develop tissue-specific autoimmune diseases and die of severe inflammatory tissue damage upon infection with pathogens that normally establish chronic infection in PD-1–sufficient mice (3, 4). On the other hand, this immunoregulatory function of PD-1 is often hijacked by tumors to escape from cancer immune surveillance. Cancer immunotherapies targeting PD-1 successfully eradicate various types of tumors by restoring the tumoricidal activities of tumor-specific T cells (5, 6). As anticipated from the autoimmune phenotypes of PD-1–deficient mice, targeted blockade of PD-1 activates not only tumor-specific T cells but also self-reactive T cells to provoke inflammatory tissue damage called immune-related adverse events (irAEs) (7, 8).
Various kinds of immunosuppressants have been developed and are widely used for treatment of autoimmune diseases, allergic diseases, transplant rejection, and so on (9). However, most of these drugs have a low therapeutic index and can cause various side effects that are dependent on time and dose, requiring special caution regarding their use. Although the molecular mechanisms of action of immunosuppressants have been extensively analyzed, the actual effects of immunosuppressants on the expression and function of immune-related molecules, including immune checkpoint molecules, are not fully understood, which makes rational design of regimens with higher efficacy and safety difficult.
Among various immunosuppressants, glucocorticoids (GCs) have been mainstay drugs for the treatment of numerous inflammatory diseases, including irAEs of cancer immunotherapies. GCs, a class of steroid hormones playing critical roles in diverse physiological processes, have profound anti-inflammatory and immunosuppressive activities (10). The pharmacological effects of GCs are predominantly mediated through the glucocorticoid receptor (GR), a member of the nuclear receptor superfamily of ligand-dependent transcription factors (TFs). Upon interacting with GC in the cytoplasm, GR translocates into the nucleus, where it functions either as a transcriptional activator or repressor (11). GCs attenuate the expression of pro-inflammatory cytokines, induce apoptotic cell death, and impede recruitment of immune cells by inhibiting the expression of adhesion molecules and chemokines in suppression of inflammation (10). In T cells, GCs suppress T cell activation by abrogating TCR-induced gene expression or inhibiting dendritic cell maturation (12–15). Despite their potent anti-inflammatory effects, recent studies revealed that GCs might also augment immune responses by up-regulating genes involved in innate immunity (16, 17). In addition to genomic effects, which involve the induction/suppression of genes, GCs have also been reported to function in a nongenomic manner (18). Thus, GCs have extremely diverse and complicated effects on immune and non-immune cells that are not fully understood.
In this study, we found that natural and synthetic GCs up-regulate PD-1 on T cells by augmenting PD-1 transcription without changing the expression levels of other co-receptors. We also observed that in vivo administration of GCs significantly up-regulated PD-1 expression on tumor-infiltrating T cells. We identified a GC response element (GRE) responsible for GC-mediated transactivation in the promoter region of the PD-1 gene. By analyzing T cells expressing PD-1 to a variable degree, we directly demonstrated that the amount of PD-1 on the cell surface correlated with its inhibitory effect. Accordingly, GCs potentiated the inhibitory effect of PD-1 on antigen-dependent functional T cell activation. These results provide new insights into the mechanisms underlying the immunosuppressive effects of GCs and provide a rationale for the strategy to enhance the inhibitory effect of PD-1 by augmenting its expression level.
Results
Dexamethasone strongly enhances PD-1 expression on T hybridoma cells
A variety of drugs have been developed as immunosuppressants with different mechanisms of action. First we assessed the effects of immunosuppressants on PD-1 expression using DO11.10 T hybridoma cells that endogenously express PD-1 on their surface and up-regulate PD-1 expression upon TCR-dependent activation. Treatment with cyclophosphamide monohydrate (an alkylating agent inhibiting DNA synthesis) and mizoribine (an imidazole nucleoside inhibiting the de novo synthesis of guanosine) did not affect PD-1 expression levels at all (Fig. 1, A and B). The expression levels of PD-1 were slightly enhanced by everolimus (a selective inhibitor of the serine–threonine kinase mTOR) (Fig. 1, A and B), consistent with a previous report that mTOR induces T-box transcription factor (T-bet), which represses transcription of PD-1 mRNA (18, 19). High-dose cyclosporin A (a selective inhibitor of calcineurin) reduced PD-1 expression (Fig. 1, A and B), which suggests that endogenous expression of PD-1 on DO11.10 T hybridoma cells depends on Ca2+–calcineurin–nuclear factor of activated T cells (NFAT) signaling, as is the case with PD-1 expression upon T cell activation (20). Notably, we found that PD-1 expression was strongly augmented by the treatment with dexamethasone (a synthetic GC with potent anti-inflammatory activities) in a dose- and time-dependent manner (Fig. 1, A–C). Although dexamethasone also induced apoptotic cell death of DO11.10 T hybridoma cells in a dose- and time-dependent manner (Fig. 1, D and E, and Fig. S1), augmentation of PD-1 expression was observed in live cells and preceded cell death, indicating that PD-1 up-regulation was not due to nonspecific effects caused by cell death. Consistently, cell death, but not PD-1 up-regulation by dexamethasone, was canceled by benzyloxycarbonyl-VAD-fluoromethyl ketone, an inhibitor of apoptotic cell death (Fig. S1). In addition, no statistically significant correlation was detected between changes in PD-1 expression level and viability of cells treated with immunosuppressants at various concentrations (Fig. S2). We also confirmed that apoptosis inducers do not necessarily up-regulate PD-1 expression (Fig. S3). Collectively, PD-1 up-regulation by dexamethasone is likely mediated by its specific pharmacological action.
Figure 1.

Effects of immunosuppressants on PD-1 expression. A, surface PD-1 expression levels on DO11.10 T hybridoma cells after treatment with immunosuppressants. Representative histogram plots are shown for cells treated with the indicated immunosuppressants (100 nm). Gray-shaded histograms represent isotype control Ig staining. B–E, dose- and time-dependent effects of immunosuppressants on PD-1 expression (B and C) and cell viability (D and E). DO11.10 T hybridoma cells were cultured in the presence of the indicated immunosuppressants at the indicated doses (B and D) for 24 h or dexamethasone (100 nm, C and E) for the indicated hours and analyzed by flow cytometry. -Fold changes in PD-1 expression relative to DMSO-treated cells (the relative geometric mean fluorescence intensity (geoMFI) of PD-1, B and C) and the percentages of live (propidium iodide (PI)–negative) cells are shown (D and E). Representative plots of three independent experiments (A) or the mean ± S.D. of three independent experiments (B–E) are shown. Two-way ANOVA with Dunnett's multiple comparisons test (compared with DMSO-treated cells, B and D) or with Bonferroni's multiple comparisons test (compared with DMSO-treated cells, E) and one-way ANOVA with Dunnett's multiple comparisons test (compared with before treatment, C) were used. *, p < 0.05; ***, p < 0.001. CyA, cyclosporin A; CPA, cyclophosphamide monohydrate; MZR, mizoribine; ERL, everolimus; Dex, dexamethasone.
GCs selectively enhance PD-1 expression on T hybridoma cells
In addition to dexamethasone, various synthetic analogues of GC have been developed for treatment of inflammatory diseases. When we examined natural and synthetic GCs, all GCs tested enhanced PD-1 expression on DO11.10 T hybridoma cells to a similar extent as dexamethasone. Intriguingly, the expression levels of other cell surface molecules tested were not affected by dexamethasone or hydrocortisone (HC) (Fig. 2, A and B, and Fig. S4, A–C).
Figure 2.

Selective up-regulation of PD-1 expression on T hybridoma cells by GCs. A, surface PD-1 expression levels on DO11.10 T hybridoma cells after treatments with GCs. Cells were treated with the indicated GCs (100 nm) for 24 h, and their PD-1 expression level was analyzed by flow cytometry. B, expression levels of cell surface molecules on DO11.10 T hybridoma cells after dexamethasone treatment. Cells were treated with dexamethasone (100 nm) for 24 h, and the expression levels of the indicated molecules were analyzed by flow cytometry. C, expression of cell surface molecules on DO11.10 T hybridoma cells after antigenic stimulation in the presence of dexamethasone. DO11.10 T hybridoma cells were stimulated by co-culturing with pOVA323–339–pulsed IIA1.6 cells for 24 h in the presence dexamethasone (100 nm) and expression of the indicated molecules on live DO11.10 T hybridoma cells (PI−B220−TCR-β+) cells were analyzed by flow cytometry. Representative histogram plots and the mean ± S.D. of three independent experiments are shown (A–C). Gray-shaded histograms represent isotype control Ig staining (A–C). One-way ANOVA with Dunnett's multiple comparisons test (compared with DMSO-treated cells, A) and with Tukey's multiple comparisons test (B and C) was used. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
DO11.10 T hybridoma cells recognize the 323–339 segment of chicken ovalbumin (pOVA323–339) in the context of I-Ad. When we stimulated DO11.10 T hybridoma cells by co-culturing with pOVA323–339-pulsed IIA1.6 B lymphoma cells expressing I-Ad, DO11.10 T hybridoma cells were activated to express or up-regulate activation markers such as CD69, CD28, 4-1BB, and receptor activator of nuclear factor kappa-B ligand (RANKL) as well as PD-1 to a variable degree (Fig. 2C). Addition of dexamethasone and HC to the co-culture attenuated activation-induced expression/up-regulation of most activation markers, which likely reflects the reduced levels of T cell activation by GC treatment. On the other hand, the expression level of PD-1 was further augmented by GCs, indicating that the magnitude of PD-1 up-regulation by GCs outweighs the magnitude of PD-1 down-regulation because of the reduced T cell activation by GCs (Fig. 2C and Fig. S4D). Therefore, the augmentation of expression by treatment with GCs was highly specific to PD-1.
GCs selectively enhance PD-1 expression on primary T cells
We next tested whether GCs also augment PD-1 expression on primary T cells. Because naïve T cells do not express PD-1, we induced PD-1 expression by stimulating naïve T cells with anti-CD3ϵ and anti-CD28 Abs for 16 h. When we added GCs during stimulation, the expression levels of PD-1 were augmented both on CD4+ and CD8+ T cells with a clear dependence on the dose of GCs (Fig. 3, A and B, and Fig. S5, A and B). In contrast, induction of activation markers such as CD25, CD44, and 4-1BB was attenuated by GCs (Fig. 3C). The levels of their down-regulation by GCs were smaller compared with those in stimulated DO11.10 T hybridoma cells, probably because anti-CD3ϵ and anti-CD28 Abs activated primary T cells so strongly that GCs could only partially inhibit T cell activation.
Figure 3.

Selective up-regulation of PD-1 expression on primary T cells by GCs. A–C, expression levels of PD-1 (A and B) and cell surface molecules (C) on primary T cells stimulated with anti-CD3ϵ/CD28 Abs in the presence of dexamethasone. Splenocytes from C57BL/6J mice were cultured in the presence of soluble anti-CD3ϵ Ab, anti-CD28 Ab, and dexamethasone for 16 h and analyzed by flow cytometry. D–F, effects of dexamethasone on the expression of PD-1 (D and E) and cell surface molecules (F) of preactivated primary T cells. Splenocytes from C57BL/6J mice were preactivated with soluble anti-CD3ϵ and anti-CD28 Abs for 48 h. The preactivated cells were cultured in the presence of dexamethasone for 24 h and analyzed by flow cytometry. Representative histogram plots of cells treated with dexamethasone (1 μm) and changes in the expression levels of the indicated molecules are shown for CD4+ (gated on PI−TCR-β+CD4+ cells, A), CD8+ (gated on PI−TCR-β+CD8+ cells, B and C), and preactivated CD4+ (D) and preactivated CD8+ (E and F) T cells. Gray-shaded histograms represent isotype control Ig staining (A–F). Data indicate the mean ± S.D. of biological triplicates in one representative experiment (A–F). Data are representative of at least two independent experiments (A–F). One-way ANOVA with Dunnett's multiple comparisons test (compared with DMSO-treated cells, A–F). *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Addition of GCs for 24 h after stimulation also resulted in augmentation of PD-1 expression on CD4+ and CD8+ T cells (Fig. 3, D and E, and Fig. S5, C and D). As is the case with unstimulated DO11.10 T hybridoma cells, dexamethasone and HC did not substantially affect the expression levels of most cell surface molecules on preactivated CD8+ T cells (Fig. 3F and Fig. S5E). These results indicate that GCs selectively up-regulate PD-1 on primary T cells as well.
In vivo treatment with GC up-regulates PD-1 expression on tumor-infiltrating CD8+ T cells
GCs are commonly used to mitigate irAEs in cancer patients receiving immunotherapy targeting PD-1 and CTLA-4 (8). To investigate the effect of GCs on PD-1 expression in the tumor setting, we analyzed PD-1 expression on T cells infiltrating into tumors of CT26 mouse colon carcinoma cells. As is well known, tumor-infiltrating CD4+ and CD8+ T cells expressed PD-1 at substantial levels (Fig. 4A). Notably, administration of dexamethasone significantly increased the expression levels of PD-1 as well as the proportion of PD-1–expressing cells among tumor-infiltrating CD8+ T cells compared with control mice treated with the solvent DMSO (Fig. 4, A–C). Although not statistically significant, tumor-infiltrating CD4+ T cells also exhibited a tendency toward increased levels of PD-1 expression upon dexamethasone treatment (Fig. 4, A–C). These results indicate that GCs have the potential to up-regulate PD-1 expression in vivo.
Figure 4.

Up-regulation of PD-1 on tumor-infiltrating CD8+ T cells by GCs. A–C, surface PD-1 expression levels on T cells infiltrating tumors of CT26 colon carcinoma cells. BALB/c mice were subcutaneously administered DMSO or dexamethasone (400 μg) 10 and 12 days after subcutaneous inoculation of CT26 cells. On day 13, the percentages of tumor-infiltrating CD4+ and CD8+ T cells expressing PD-1 (gated on PI−CD45+CD4+ and PI−CD45+ CD8+, respectively; A and B) and the expression levels of PD-1 on PD-1–expressing CD4+ and CD8+ T cells (geoMFI, C) were analyzed by flow cytometry. Representative histogram plots of three independent experiments are shown (A). Each symbol represents an individual mouse (n = 10 each), and horizontal lines denote the mean ± S.E. of 10 biological replicates pooled from three independent experiments. Two-tailed Student's t test. *, p < 0.05; **, p < 0.01.
The GC–GR complex up-regulates PD-1 mRNA by binding to a GRE in the promoter region
We tested the involvement of the GR in up-regulation of PD-1 and cell death by GCs using RU486, a GR antagonist. Up-regulation of PD-1 as well as cell death by dexamethasone were canceled by addition of RU486, indicating that up-regulation of PD-1 and cell death by GCs are mediated through the GR (Fig. 5A). Then we examined the effects of GCs on PD-1 expression at the mRNA level. The amount of PD-1 mRNA was increased about 2-fold by 1 h after addition of GCs and further augmented over time, which was canceled by RU486 (Fig. 5, B and C). These results indicate that GCs augment PD-1 expression at the mRNA level through binding to the GR.
Figure 5.
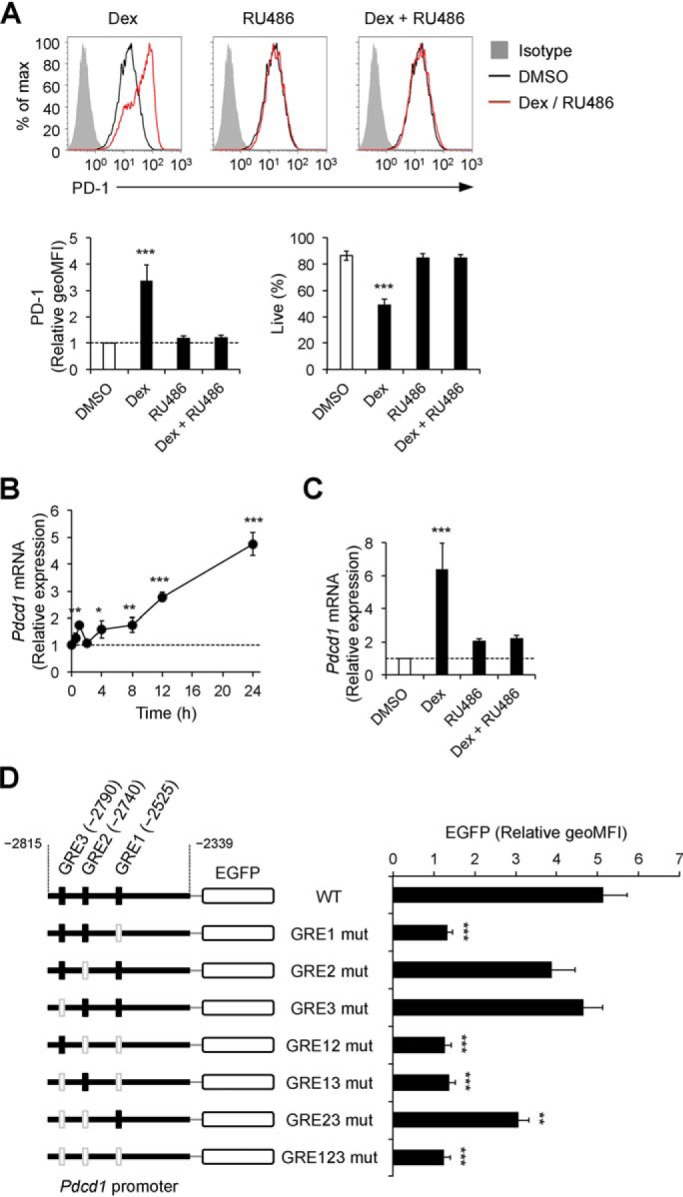
GR-dependent augmentation of PD-1 transcription by GC treatment. A, abrogation of GC-mediated PD-1 up-regulation and cell death by the glucocorticoid receptor antagonist RU486. DO11.10 T hybridoma cells were cultured in the presence of dexamethasone (100 nm) and RU486 (10 μm) for 24 h and analyzed by flow cytometry. -Fold changes in PD-1 expression relative to DMSO-treated cells and the percentages of live cells are shown. Gray-shaded histograms represent isotype control Ig staining. B, the time course of PD-1 mRNA expression in DO11.10 T hybridoma cells after treatment with dexamethasone. Expression levels of PD-1 mRNA were determined by real-time quantitative PCR and normalized to those of GAPDH mRNA. -Fold changes relative to DMSO-treated cells are shown. C, abrogation of GC-mediated PD-1 mRNA up-regulation by RU486. DO11.10 T hybridoma cells were cultured in the presence of dexamethasone (100 nm) and RU486 (10 μm) for 24 h, and -fold changes of PD-1 mRNA expression relative to DMSO-treated cells were determined as in B. D, promoter activity of DNA sequences containing three putative GREs in the promoter region of the PD-1 gene upon GC treatment. Shown are schematics of reporter constructs encoding EGFP cDNA and the promoter region of the PD-1 gene (2790–2339 bp upstream of the transcription start site) with or without nucleotide mutations in the putative GREs (left). DO11.10 T hybridoma cells transduced with mouse GR and the indicated reporter constructs were cultured in the presence of dexamethasone (100 nm), phorbol 12-myristate 13-acetate (50 ng/ml), and ionomycin (500 ng/ml) for 24 h and analyzed by flow cytometry. Relative geoMFIs of EGFP compared with DMSO-treated cells are shown. The mean ± S.D. (A and C) or S.E. (D) of three independent experiments or the mean ± S.D. of technical triplicates of one of three independent experiments (B) are shown. One-way ANOVA with Dunnett's multiple comparisons test compared with DMSO-treated cells (A–C) or cells with the WT reporter construct (D) was used. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Then we investigated whether the GR–GC complex directly increases transcription of the PD-1 gene. By using the TF binding site prediction database, we identified three putative GREs 2525, 2740, and 2790 bp upstream of the transcription start site of PD-1 mRNA and termed them as GRE1, GRE2, and GRE3, respectively. To examine whether augmentation of PD-1 expression by GCs was mediated through these GREs, we generated a series of reporter constructs containing intact or mutated GREs. Dexamethasone treatment strongly augmented promoter activity in the presence of GRE1, suggesting that GRE1 is responsible for augmentation of PD-1 expression by GCs (Fig. 5D).
GCs potentiate the inhibitory effect of PD-1 by increasing the cell surface amount of PD-1
Upon antigen stimulation, DO11.10 T hybridoma cells secrete IL-2 in a manner dependent on the amount of antigen. Thus, we can evaluate the magnitude of functional T cell activation based on the amount of secreted IL-2. As reported previously, IL-2 production from DO11.10 T hybridoma cells upon antigen stimulation was strongly inhibited when IIA1.6 cells overexpressing PD-L1 (IIA1.6–PD-L1 cells) were used as antigen-presenting cells, and the inhibitory effect of PD-1 was completely canceled by anti-PD-L1 blocking Abs (21, 22). PD-L1–mediated inhibition was abolished by targeted deletion of the PD-1 gene in DO11.10 T hybridoma cells and restored by retroviral reconstitution of PD-1 (Fig. 6, A and B). By using this system, we examined the correlation between the cell surface amount of PD-1 and its inhibitory effect. We overexpressed PD-1 at various levels using five different promoters (EF1α, cytomegalovirus, cytomegalovirus enhancer/chicken β-actin, MC1, and SV40) with or without a polyadenylation signal, which plays essential roles in the stabilization of mRNA (Fig. 6, C and D). These cells were stimulated with pOVA323–339–pulsed IIA1.6–PD-L1 cells, and PD-1–mediated inhibitory effects were calculated by comparing the amount of secreted IL-2 in the presence or absence of anti-PD-L1–blocking Ab. We observed a strong positive correlation of the cell surface expression level of PD-1 with its inhibitory effect (Fig. 6E).
Figure 6.

Potentiation of the inhibitory effect of PD-1 by GCs. A, surface PD-1 expression levels on DO11.10 T hybridoma cells with or without deletion and overexpression of PD-1. B, restoration of PD-1–mediated inhibitory effects by overexpression of PD-1 in DO11.10 T hybridoma cells with targeted deletion of the PD-1 gene. IL-2 concentration in the culture supernatant is shown for the indicated DO11.10 T hybridoma cells stimulated with pOVA323–339–pulsed IIA1.6–PD-L1 cells in the presence or absence of anti-PD-L1–blocking Ab for 24 h. C, schematics of retroviral expression vectors. Mouse PD-1 cDNA was overexpressed in DO11.10 T hybridoma cells with targeted deletion of the PD-1 gene using the indicated promoters with or without a poly(A) signal (pA). D, flow cytometric analysis of DO11.10 T hybridoma cells overexpressing PD-1 at various levels by using the retroviral expression vectors shown in C. E, the correlation between the percent inhibition of IL-2 production by PD-1 and the expression level of PD-1 (MFI). DO11.10 T hybridoma cells with different PD-1 expression levels were stimulated as in B. F and G, augmentation of PD-1–mediated inhibitory effects by dexamethasone. DO11.10 T hybridoma cells were treated with dexamethasone (100 nm) or DMSO for 24 h and rested for 6 h. The same numbers of dexamethasone- or DMSO-treated live cells were stimulated for 24 h as in B. IL-2 concentration in the culture supernatant (F) and percent inhibition of IL-2 production by PD-1 (G) are shown. Data are the mean ± S.D. of technical duplicates of one representative experiment (B and F) or the mean ± S.D. of four independent experiments (G). Data are representative of at least three independent experiments (A, B, and D–F). Two-tailed Student's t test (G) was used. CMV, cytomegalovirus; CAG, cytomegalovirus enhancer/chicken β-actin. *, p < 0.05.
Then we evaluated the functional consequence of PD-1 up-regulation by GCs. DO11.10 T hybridoma cells were treated with dexamethasone, and live cells were stimulated with pOVA323–339. As anticipated from the immunosuppressive activity of GCs, pretreatment with dexamethasone substantially reduced the amount of IL-2 secreted from DO11.10 T hybridoma cells upon antigen stimulation. PD-1 engagement further reduced IL-2 production from activated DO11.10 T hybridoma cells. Intriguingly, when we compared the levels of PD-1-dependent inhibition with or without dexamethasone pretreatment, PD-1 inhibited IL-2 production more efficiently when DO11.10 T hybridoma cells were pretreated with dexamethasone (Fig. 6, F and G). Thus, GCs can potentiate the inhibitory effect of PD-1 by increasing the cell surface amount of PD-1.
No substantial effect of GCs on ectopic PD-1
To confirm the specificity of PD-1 up-regulation by GCs, we evaluated the effect of GCs on ectopic PD-1. We treated DO11.10 T hybridoma cells that express exogenous PD-1 under the retroviral long terminal repeat promoter but lack endogenous PD-1 expression with GCs. As expected, GCs failed to augment the cell surface amount of ectopic PD-1 (Fig. S6A). Accordingly, GCs failed to potentiate the inhibitory effect of ectopic PD-1 (Fig. S6, B and C). These results strongly suggest that the GC–GR complex transactivates PD-1 transcription by directly binding to GRE1 located in the promoter region of the PD-1 gene.
Discussion
GCs have been widely used as anti-inflammatory and immunosuppressive agents for treatment of a variety of inflammatory and autoimmune diseases. However, despite extensive clinical and experimental studies, the extremely diverse and complicated effects of GCs on immune and non-immune cells have not been fully understood. In this study, we found that GCs augment the expression level of PD-1 on T cells but not other cell surface molecules, including CD25, CD44, CD69, 4-1BB, RANKL, and LAG-3. Dexamethasone has been reported to enhance expression of PD-1 and CTLA-4 on T cells, but the underlying mechanism and the functional consequence of up-regulation were largely unknown (23–25). We revealed that treatment with GCs leads to transactivation of PD-1 expression in a manner dependent on GR and GRE in the promoter region of the PD-1 gene. We directly demonstrated that the amount of PD-1 on the cell surface strongly correlated with its inhibitory effect. Accordingly, augmented expression of PD-1 by GCs led to proportional enhancement of the inhibitory effects of PD-1 against antigen-dependent functional T cell activation. These findings suggest that PD-1–mediated inhibition contributes, at least in part, to the anti-inflammatory and immunosuppressive effects of GCs.
Naïve T cells do not express PD-1 but rapidly express PD-1 upon antigen stimulation. This initial induction of PD-1 is driven by multiple TFs, including nuclear factor of activated T cells 1 (NFATc1) and c-Fos/activator protein-1 (AP-1), which are activated by signaling through the TCR or Notch (20, 26, 27). In the setting of chronic viral infection and tumors, chronic antigen exposure induces high and sustained expression of PD-1 on CD8+ T cells, resulting in their functional exhaustion (4, 28). Forkhead box protein O1 (FoxO1), NFATc1, and nuclear receptor subfamily 4 group A (NR4A) have been reported to mediate the expression of PD-1 on exhausted CD8+ T cells (29–32). Although these TFs function as transcriptional activators in PD-1 expression, T-bet and Blimp-1 have been reported to function as transcriptional repressors in PD-1 expression (19, 33). These TFs cooperatively or competitively regulate PD-1 expression by directly binding to the promoter region of the PD-1 gene. GRs are known to exert diverse functions by binding to promoter sequences together with other TFs or by physically interacting with other TFs (11). In this study, we identified a GRE in the promoter region of the PD-1 gene that is responsible for transactivation by GC treatment. Therefore, it is likely that GR directly binds to the promoter region of the PD-1 gene in response to GC treatment and acts as a transcriptional activator to augment PD-1 transcription. Further studies are expected to reveal a possible positive or negative cooperation of the GC–GR complex with other TFs in regulation of PD-1 expression.
Endogenous GCs (cortisol in humans and corticosterone in mice) play critical roles in the regulation of various physiological and developmental processes. It was recently reported that endogenous GCs produced upon infection with mouse cytomegalovirus induced PD-1 expression on natural killer (NK) cells to restrain IFN-γ production from NK cells, leading to prevention of immunopathology (34). Another recent study showed that deletion of the GR in regulatory T cells (Tregs) resulted in a reduction of PD-1-expressing Tregs in the spleen (35). These findings suggest that PD-1 expression is positively modulated by endogenous GCs. It is possible that the GRE found in the current study also mediates the expression of PD-1 on NK cells and Tregs by endogenous GCs under physiological and pathological conditions.
Because of the recent success of cancer immunotherapy targeting PD-1 and CTLA-4, many other inhibitory co-receptors are extensively investigated with the aim of developing new cancer immunotherapies with higher efficacy. Revisiting the significance of inhibitory co-receptors in maintaining immunotolerance to self and preventing excess immune responses, potentiation of their activities is expected to be a promising therapeutic strategy for inflammatory and autoimmune diseases. Our current findings clearly demonstrate that the expression level of PD-1 strongly correlates with its inhibitory function, just like LAG-3, another inhibitory co-receptor (36). These results provide a rationale for the strategy to treat inflammatory and autoimmune diseases by augmenting the expression levels of inhibitory co-receptors.
Experimental procedures
Reagents
The immunosuppressants, natural and synthetic glucocorticoids, inhibitors of cell death, and apoptosis inducers used in this study are as follows: cyclosporin A (chemical abstracts service number (CAS no.) 1202.635, Tokyo Chemical Industry), cyclophosphamide monohydrate (CAS no. 6055-19-2, Tokyo Chemical Industry), mizoribine (CAS no. 50924-49-7, Tokyo Chemical Industry), everolimus (CAS no. 159351-69-6, Selleck Chemicals), dexamethasone (CAS no. 50-02-2, Cayman Chemical), HC (CAS no. 50-23-7, Sigma-Aldrich), prednisolone (CAS no. 50-24-8, Tokyo Chemical Industry), bethamethasone valerate (CAS no. 2152-44-5, LKT Laboratories), fluocinolone acetonide (CAS no. 67-73-2, Tokyo Chemical Industry), hydrocortisone 17-butyrate (CAS no. 25122-46-7, Tokyo Chemical Industry), budesonide (CAS no. 51333-22-3, Tokyo Chemical Industry), benzyloxycarbonyl-VAD-fluoromethyl ketone (CAS no. 187389-52-2, AdooQ Bioscience), necrostatin-1 (CAS no. 4311-88-0, Selleck Chemicals), IM-54 (CAS no. 861891-50-1, Cayman Chemical), necrosulfonamide (CAS no. 1360614-48-7, Cayman Chemical), cisplatin (CAS no. 15663-27-1, Tokyo Chemical Industry), camptothecin (CAS no. 7689-03-4, Tokyo Chemical Industry), etoposide (CAS no. 33419-42-0, Tokyo Chemical Industry), and nitidine chloride (CAS no. 13063-04-2, Sigma-Aldrich). The glucocorticoid receptor antagonist RU486 (mifepristone, CAS no. 84371-65-3) was purchased from Cayman Chemical. These reagents were dissolved in DMSO (Wako).
Cell culture
DO11.10 mouse hybridoma T cells and CT26 mouse colon carcinoma cells were kindly provided by Tasuku Honjo (Kyoto University). IIA1.6 mouse B lymphoma cells were kindly provided by Tomohiro Kurosaki (Osaka University). PD-1 knockout DO11.10 T hybridoma cells were generated using the CRISPR/Cas9 system (37). These cell lines were maintained in RMPI 1640 medium (Gibco) supplemented with 10% (v/v) FBS (Biowest), 0.5 mm monothioglycerol (Wako), 2 mm l-alanyl-l-glutamine dipeptide (Gibco), 100 units/ml penicillin (Nacalai Tesque), and 100 μg/ml streptomycin (Nacalai Tesque). Plat-E cells, which were kindly provided by Toshio Kitamura (University of Tokyo), were maintained in DMEM (Gibco) supplemented with 10% (v/v) FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin.
Plasmid and retroviral gene transduction
Fragments of cDNA were amplified by PCR and cloned into retroviral expression plasmid vectors modified from pFB-ires-neo (Agilent). To control the expression levels of PD-1, fragments of cDNA were cloned into retroviral expression plasmid vectors modified from pSUPER.retro.puro (Oligoengine), whose promoter region was replaced with promoters of EF-1α (human elongation factor-1α), cytomegalovirus, cytomegalovirus enhancer/chicken β-actin), MC1 (polyoma virus enhancer/herpes simplex virus thymidine kinase), and SV40 (simian virus 40) coupled with or without a poly(A) signal. Plasmids were transfected using FuGENE HD (Promega) into Plat-E cells cultured in high-glucose DMEM (Gibco) supplemented with 20% (v/v) FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin, and supernatants containing viruses were used to transduce genes into target cells. Infected cells were selected with G418 (Wako), puromycin (Sigma-Aldrich), or blasticidin (InvivoGen).
Mice
C57BL/6J and BALB/c mice were obtained from Charles River Laboratories Japan and Japan SLC, respectively, and housed under specific pathogen-free conditions in environmentally controlled clean rooms. All experimental procedures were planned and conducted according to institutional regulations complying with the Act on Welfare and Management of Animals and the related guidelines in Japan. All mouse protocols were approved by the Animal Experimentation Committee of Tokushima University.
Stimulation of DO11.10 T hybridoma cells and primary T cells
DO11.10 T hybridoma cells (5 × 104 cells/well) were stimulated with IIA1.6 cells (1 × 104 cells/well) in the presence of antigenic peptide (323–339 segment of chicken ovalbumin, pOVA323–339, ISQAVHAAHAEINEAGR, >95% purity, Eurofins) in 96-well round-bottom plate (BD Biosciences) for 24 h. Where indicated, 1 μg/ml of anti-PD-L1–blocking Ab (1–111A) or rat IgG2a isotype control Ig (2A3, Bio X Cell) was added. Activated primary T cells were prepared by stimulating splenocytes from C57BL/6J mice with soluble anti-CD3ϵ (0.5–1 μg/ml, 145-2C-11, BioLegend) and anti-CD28 (0.5–1 μg/ml, 37.51, BioLegend) Abs. The concentration of IL-2 in the culture supernatant was determined by ELISA (BioLegend). The percentage of PD-1–mediated inhibition of IL-2 production was calculated as the ratio of IL-2 concentration in the presence or absence of anti-PD-L1–blocking Ab (1–111A). IL-2 concentrations with different amounts of antigens were summed for calculation of the percentage of PD-1–mediated inhibition.
Antibody and flowcytometric analysis
Cultured cell lines and primary cells were stained with the indicated Abs. Data were obtained with Gallios (Beckman Coulter) and analyzed using FlowJo (Tree Star). CF633 dye–labeled anti-mouse LAG-3 Ab (TKB58) was prepared as described previously (38). All other Abs used in this study were purchased from BioLegend: anti-mouse PD-1 (RMP1–30), TCR-β (H57-597), CD3ϵ (145-2C-11), CD44 (IM7), CD45 (30-F11), Thy1.2 (53-2.1), CD69 (H1.2F3), CD28 (37.51), 4-1BB (17B5), RANKL (IL22/5), CD25 (7D4), CD4 (RM4-5), and CD8a (53-6.7). Apoptotic cells were detected by using Annexin V (BioLegend).
Real-time quantitative PCR
Total RNA was extracted from cells using TRIzol reagent (Ambion) and then subjected to reverse transcription using a high-capacity cDNA reverse transcription kit (Applied Biosystems). Gene expression was analyzed by quantitative PCR using Power SYBR Green PCR Master Mix (Applied Biosystems) on a 7900HT fast real-time PCR system (Applied Biosystems). Values were normalized to the expression of Gapdh. The following specific primer sets were used: Gapdh forward (5′-TTCACCACCATGGAGAAGGC-3′) and reverse (5′-GGCATGGACTGTGGTCATGA-3′) and Pdcd1 forward (5′-ACCCTGGTCATTCACTTGGG-3′) and reverse (5′-CATTTGCTCCCTCTGACACTG-3′).
Reporter assay
Putative GREs in the promoter region of the PD-1 gene were predicted using the JASPAR database (39) and Genomatix Genome Analyzer (Genomatix). A DNA fragment containing GRE1, GRE2, and GRE3 (2790–2339 bp upstream of the transcription start of PD-1 mRNA) was amplified from a C57BL/6N mouse BRC clone (B6Ng01-240G08, RIKEN BioResource Research Center) by PCR and cloned into a retroviral expression plasmid vector modified from pSUPER.retro.puro. together with a synthetic minimal promoter sequence and EGFP cDNA. Where indicated, GRE sequences (AGAACAnnnTGTTCT) were mutated by overhang PCR to AGGTCAnnnTGACCT. Reporter constructs were retrovirally introduced into DO11.10 T hybridoma cells overexpressing the mouse GR. After selection with puromycin, cells were cultured in the presence of dexamethasone (100 nm), phorbol 12-myristate 13-acetate (50 ng/ml, Sigma-Aldrich), and ionomycin (500 ng/ml, Sigma-Aldrich) for 24 h and analyzed by flow cytometry.
Preparation of tumor-infiltrating T cells
Male and female BALB/c mice (8–10 weeks old) were inoculated subcutaneously with 1 × 106 CT26 murine colon carcinoma cells on a shaved back. On days 10 and 12, dexamethasone (400 μg) in 50% DMSO/PBS was subcutaneously injected near the site of tumor inoculation. On day 13, tumor tissues were dissected and mechanically dissociated using a gentleMACS Octo Dissociator with Heaters (Miltenyi Biotec) in RPMI 1640 medium containing collagenase (1 mg/ml, Wako), hyaluronidase (50 μg/ml, Sigma-Aldrich), and DNase I (10 μg/ml, Sigma-Aldrich). Single-cell suspensions prepared from tumor tissues were used for flow cytometric analysis.
Statistical analysis
Two-way ANOVA with Dunnett's and Bonferroni's multiple comparisons test, one-way ANOVA with Dunnett's multiple comparisons test, Pearson's correlation test, and two-tailed Student's t test were used to evaluate statistical significance. p < 0.05 was considered statistically significant. These statistical analyses were performed using GraphPad Prism 8 (GraphPad Software).
Author contributions
N. M., T. M., and T. O. conceptualization; N. M. and T. M. data curation; N. M. and T. M. formal analysis; N. M., T. M., D. S., K. S., and I.-m. O. investigation; N. M., T. M., and T. O. visualization; N. M. and T. M. writing-original draft; N. M., T. M., D. S., K. S., I.-m. O., and T. O. writing-review and editing; T. M. and T. O. funding acquisition; T. M. validation; T. M., D. S., K. S., I.-m. O., and T. O. methodology; D. S., K. S., I.-m. O., and T. O. resources; I.-m. O. and T. O. project administration; T. O. supervision.
Supplementary Material
Acknowledgments
We thank Drs. T. Honjo, T. Kurosaki, and T. Kitamura for kindly providing cell lines; Y. Okamoto, M. Aoki, A. Otsuka, H. Tsuduki, and R. Matsumura for technical and secretarial assistance; and all other members of our laboratory for helpful discussions.
This work was supported in part by the Core Research for Evolutional Science and Technology Program of the Japan Science and Technology Agency, Basic Science and Platform Technology Program for Innovative Biological Medicine of the Japan Agency for Medical Research and Development (JP18am0301007) and Grants-in-Aid by the Japan Society for the Promotion of Science JP18H05417, JP19H01029, and 19K16694. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S6.
- TCR
- T cell receptor
- irAE
- immune-related adverse event
- GC
- glucocorticoid
- GR
- glucocorticoid receptor
- TF
- transcription factor
- GRE
- glucocorticoid response element
- HC
- hydrocortisone
- Ab
- antibody
- NK
- natural killer
- Treg
- regulatory T cell
- cDNA
- complementary DNA
- EGFP
- enhanced GFP
- ANOVA
- analysis of variance
- geoMFI
- geometric mean fluorescence intensity
- PI
- propidium iodide.
References
- 1. Okazaki T., Chikuma S., Iwai Y., Fagarasan S., and Honjo T. (2013) A rheostat for immune responses: the unique properties of PD-1 and their advantages for clinical application. Nat. Immunol. 14, 1212–1218 10.1038/ni.2762 [DOI] [PubMed] [Google Scholar]
- 2. Sharpe A. H., and Pauken K. E. (2018) The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 18, 153–167 10.1038/nri.2017.108 [DOI] [PubMed] [Google Scholar]
- 3. Barber D. L., Mayer-Barber K. D., Feng C. G., Sharpe A. H., and Sher A. (2011) CD4 T cells promote rather than control tuberculosis in the absence of PD-1-mediated inhibition. J. Immunol. 186, 1598–1607 10.4049/jimmunol.1003304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Barber D. L., Wherry E. J., Masopust D., Zhu B., Allison J. P., Sharpe A. H., Freeman G. J., and Ahmed R. (2006) Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439, 682–687 10.1038/nature04444 [DOI] [PubMed] [Google Scholar]
- 5. Ribas A., and Wolchok J. D. (2018) Cancer immunotherapy using checkpoint blockade. Science 359, 1350–1355 10.1126/science.aar4060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sun C., Mezzadra R., and Schumacher T. N. (2018) Regulation and function of the PD-L1 checkpoint. Immunity 48, 434–452 10.1016/j.immuni.2018.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Martins F., Sofiya L., Sykiotis G. P., Lamine F., Maillard M., Fraga M., Shabafrouz K., Ribi C., Cairoli A., Guex-Crosier Y., Kuntzer T., Michielin O., Peters S., Coukos G., Spertini F., et al. (2019) Adverse effects of immune-checkpoint inhibitors: epidemiology, management and surveillance. Nat. Rev. Clin. Oncol. 16, 563–580 10.1038/s41571-019-0218-0 [DOI] [PubMed] [Google Scholar]
- 8. Postow M. A., Sidlow R., and Hellmann M. D. (2018) Immune-related adverse events associated with immune checkpoint blockade. N. Engl. J. Med. 378, 158–168 10.1056/NEJMra1703481 [DOI] [PubMed] [Google Scholar]
- 9. Halloran P. F. (2004) Immunosuppressive drugs for kidney transplantation. N. Engl. J. Med. 351, 2715–2729 10.1056/NEJMra033540 [DOI] [PubMed] [Google Scholar]
- 10. Cain D. W., and Cidlowski J. A. (2017) Immune regulation by glucocorticoids. Nat. Rev. Immunol 17, 233–247 10.1038/nri.2017.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Weikum E. R., Knuesel M. T., Ortlund E. A., and Yamamoto K. R. (2017) Glucocorticoid receptor control of transcription: precision and plasticity via allostery. Nat. Rev. Mol. Cell Biol. 18, 159–174 10.1038/nrm.2016.152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Moser M., De Smedt T., Sornasse T., Tielemans F., Chentoufi A. A., Muraille E., Van Mechelen M., Urbain J., and Leo O. (1995) Glucocorticoids down-regulate dendritic cell function in vitro and in vivo. Eur. J. Immunol. 25, 2818–2824 10.1002/eji.1830251016 [DOI] [PubMed] [Google Scholar]
- 13. Petrillo M. G., Fettucciari K., Montuschi P., Ronchetti S., Cari L., Migliorati G., Mazzon E., Bereshchenko O., Bruscoli S., Nocentini G., and Riccardi C. (2014) Transcriptional regulation of kinases downstream of the T cell receptor: another immunomodulatory mechanism of glucocorticoids. BMC Pharmacol. Toxicol. 15, 35 10.1186/2050-6511-15-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Piemonti L., Monti P., Allavena P., Sironi M., Soldini L., Leone B. E., Socci C., and Di Carlo V. (1999) Glucocorticoids affect human dendritic cell differentiation and maturation. J. Immunol. 162, 6473–6481 [PubMed] [Google Scholar]
- 15. Tsitoura D. C., and Rothman P. B. (2004) Enhancement of MEK/ERK signaling promotes glucocorticoid resistance in CD4+ T cells. J. Clin. Invest. 113, 619–627 10.1172/JCI200418975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Boldizsar F., Talaber G., Szabo M., Bartis D., Palinkas L., Nemeth P., and Berki T. (2010) Emerging pathways of non-genomic glucocorticoid (GC) signalling in T cells. Immunobiology 215, 521–526 10.1016/j.imbio.2009.10.003 [DOI] [PubMed] [Google Scholar]
- 17. van de Garde M. D., Martinez F. O., Melgert B. N., Hylkema M. N., Jonkers R. E., and Hamann J. (2014) Chronic exposure to glucocorticoids shapes gene expression and modulates innate and adaptive activation pathways in macrophages with distinct changes in leukocyte attraction. J. Immunol. 192, 1196–1208 10.4049/jimmunol.1302138 [DOI] [PubMed] [Google Scholar]
- 18. Rao R. R., Li Q., Odunsi K., and Shrikant P. A. (2010) The mTOR kinase determines effector versus memory CD8+ T cell fate by regulating the expression of transcription factors T-bet and Eomesodermin. Immunity 32, 67–78 10.1016/j.immuni.2009.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kao C., Oestreich K. J., Paley M. A., Crawford A., Angelosanto J. M., Ali M. A., Intlekofer A. M., Boss J. M., Reiner S. L., Weinmann A. S., and Wherry E. J. (2011) Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nat. Immunol. 12, 663–671 10.1038/ni.2046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Oestreich K. J., Yoon H., Ahmed R., and Boss J. M. (2008) NFATc1 regulates PD-1 expression upon T cell activation. J. Immunol. 181, 4832–4839 10.4049/jimmunol.181.7.4832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mizuno R., Maruhashi T., Sugiura D., Shimizu K., Watada M., Okazaki I. M., and Okazaki T. (2019) PD-1 efficiently inhibits T cell activation even in the presence of co-stimulation through CD27 and GITR. Biochem. Biophys. Res. Commun. 511, 491–497 10.1016/j.bbrc.2019.02.004 [DOI] [PubMed] [Google Scholar]
- 22. Mizuno R., Sugiura D., Shimizu K., Maruhashi T., Watada M., Okazaki I. M., and Okazaki T. (2019) PD-1 primarily targets TCR signal in the inhibition of functional T cell activation. Front. Immunol. 10, 630 10.3389/fimmu.2019.00630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Giles A. J., Hutchinson M. N. D., Sonnemann H. M., Jung J., Fecci P. E., Ratnam N. M., Zhang W., Song H., Bailey R., Davis D., Reid C. M., Park D. M., and Gilbert M. R. (2018) Dexamethasone-induced immunosuppression: mechanisms and implications for immunotherapy. J. Immunother. Cancer 6, 51 10.1186/s40425-018-0371-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xia M., Gasser J., and Feige U. (1999) Dexamethasone enhances CTLA-4 expression during T cell activation. Cell Mol. Life Sci. 55, 1649–1656 10.1007/s000180050403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Xing K., Gu B., Zhang P., and Wu X. (2015) Dexamethasone enhances programmed cell death 1 (PD-1) expression during T cell activation: an insight into the optimum application of glucocorticoids in anti-cancer therapy. BMC Immunol. 16, 39 10.1186/s12865-015-0103-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mathieu M., Cotta-Grand N., Daudelin J. F., Thébault P., and Labrecque N. (2013) Notch signaling regulates PD-1 expression during CD8+ T-cell activation. Immunol. Cell Biol. 91, 82–88 10.1038/icb.2012.53 [DOI] [PubMed] [Google Scholar]
- 27. Xiao G., Deng A., Liu H., Ge G., and Liu X. (2012) Activator protein 1 suppresses antitumor T-cell function via the induction of programmed death 1. Proc. Natl. Acad. Sci. U.S.A. 109, 15419–15424 10.1073/pnas.1206370109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Blackburn S. D., Shin H., Haining W. N., Zou T., Workman C. J., Polley A., Betts M. R., Freeman G. J., Vignali D. A., and Wherry E. J. (2009) Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 10, 29–37 10.1038/ni.1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chen J., López-Moyado I. F., Seo H., Lio C. J., Hempleman L. J., Sekiya T., Yoshimura A., Scott-Browne J. P., and Rao A. (2019) NR4A transcription factors limit CAR T cell function in solid tumours. Nature 567, 530–534 10.1038/s41586-019-0985-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liu X., Wang Y., Lu H., Li J., Yan X., Xiao M., Hao J., Alekseev A., Khong H., Chen T., Huang R., Wu J., Zhao Q., Wu Q., Xu S., et al. (2019) Genome-wide analysis identifies NR4A1 as a key mediator of T cell dysfunction. Nature 567, 525–529 10.1038/s41586-019-0979-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Martinez G. J., Pereira R. M., Äijö T., Kim E. Y., Marangoni F., Pipkin M. E., Togher S., Heissmeyer V., Zhang Y. C., Crotty S., Lamperti E. D., Ansel K. M., Mempel T. R., Lähdesmäki H., Hogan P. G., and Rao A. (2015) The transcription factor NFAT promotes exhaustion of activated CD8+ T cells. Immunity 42, 265–278 10.1016/j.immuni.2015.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Staron M. M., Gray S. M., Marshall H. D., Parish I. A., Chen J. H., Perry C. J., Cui G., Li M. O., and Kaech S. M. (2014) The transcription factor FoxO1 sustains expression of the inhibitory receptor PD-1 and survival of antiviral CD8+ T cells during chronic infection. Immunity 41, 802–814 10.1016/j.immuni.2014.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lu P., Youngblood B. A., Austin J. W., Mohammed A. U., Butler R., Ahmed R., and Boss J. M. (2014) Blimp-1 represses CD8 T cell expression of PD-1 using a feed-forward transcriptional circuit during acute viral infection. J. Exp. Med. 211, 515–527 10.1084/jem.20130208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Quatrini L., Wieduwild E., Escaliere B., Filtjens J., Chasson L., Laprie C., Vivier E., and Ugolini S. (2018) Endogenous glucocorticoids control host resistance to viral infection through the tissue-specific regulation of PD-1 expression on NK cells. Nat. Immunol. 19, 954–962 10.1038/s41590-018-0185-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rocamora-Reverte L., Tuzlak S., von Raffay L., Tisch M., Fiegl H., Drach M., Reichardt H. M., Villunger A., Tischner D., and Wiegers G. J. (2019) Glucocorticoid receptor-deficient Foxp3+ regulatory T cells fail to control experimental inflammatory bowel disease. Front. Immunol. 10, 472 10.3389/fimmu.2019.00472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Maeda T. K., Sugiura D., Okazaki I. M., Maruhashi T., and Okazaki T. (2019) Atypical motifs in the cytoplasmic region of the inhibitory immune co-receptor LAG-3 inhibit T cell activation. J. Biol. Chem. 294, 6017–6026 10.1074/jbc.RA119.007455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sugiura D., Maruhashi T., Okazaki I. M., Shimizu K., Maeda T. K., Takemoto T., and Okazaki T. (2019) Restriction of PD-1 function by cis-PD-L1/CD80 interactions is required for optimal T cell responses. Science 364, 558–566 10.1126/science.aav7062 [DOI] [PubMed] [Google Scholar]
- 38. Maruhashi T., Okazaki I. M., Sugiura D., Takahashi S., Maeda T. K., Shimizu K., and Okazaki T. (2018) LAG-3 inhibits the activation of CD4+ T cells that recognize stable pMHCII through its conformation-dependent recognition of pMHCII. Nat. Immunol. 19, 1415–1426 10.1038/s41590-018-0217-9 [DOI] [PubMed] [Google Scholar]
- 39. Mathelier A., Zhao X., Zhang A. W., Parcy F., Worsley-Hunt R., Arenillas D. J., Buchman S., Chen C. Y., Chou A., Ienasescu H., Lim J., Shyr C., Tan G., Zhou M., Lenhard B., et al. (2014) JASPAR 2014: an extensively expanded and updated open-access database of transcription factor binding profiles. Nucleic Acids Res. 42, D142–D147 10.1093/nar/gkt997 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.