

Abstract
Expression of human leukocyte antigen (HLA)-B27 is strongly associated with predisposition toward ankylosing spondylitis (AS) and other spondyloarthropathies. However, the exact involvement of HLA-B27 in disease initiation and progression remains unclear. The homodimer theory, which proposes that HLA-B27 heavy chains aberrantly form homodimers, is a central hypothesis that attempts to explain the role of HLA-B27 in disease pathogenesis. Here, we examined the ability of the eight most prevalent HLA-B27 allotypes (HLA-B*27:02 to HLA-B*27:09) to form homodimers. We observed that HLA-B*27:03, a disease-associated HLA-B27 subtype, showed a significantly reduced ability to form homodimers compared with all other allotypes, including the non-disease-associated/protective allotypes HLA-B*27:06 and HLA-B*27:09. We used X-ray crystallography and site-directed mutagenesis to unravel the molecular and structural mechanisms in HLA-B*27:03 that are responsible for its compromised ability to form homodimers. We show that polymorphism at position 59, which differentiates HLA-B*27:03 from all other allotypes, is responsible for its compromised ability to form homodimers. Indeed, histidine 59 in HLA-B*27:03 leads to a series of local conformational changes that act in concert to reduce the accessibility of the nearby cysteine 67, an essential amino acid residue for the formation of HLA-B27 homodimers. Considered together, the ability of both protective and disease-associated HLA-B27 allotypes to form homodimers and the failure of HLA-B*27:03 to form homodimers challenge the role of HLA-B27 homodimers in AS pathoetiology. Rather, this work implicates other features, such as peptide binding and antigen presentation, as pivotal mechanisms for disease pathogenesis.
Keywords: autoimmune disease, major histocompatibility complex (MHC), crystallography, arthritis, single-nucleotide polymorphism (SNP), Ankylosing spondylitis, HLA heavy chain homodimers, HLA-B*27:03, HLA-B27, X-ray crystallography
Introduction
Ankylosing spondylitis (AS)6 is an immune-mediated inflammatory disease, which affects primarily the spine and belongs to the family of rheumatoid diseases called spondyloarthropathies (1, 2). For more than 40 years, the human leukocyte antigen (HLA)-B27 gene has been known to be linked with AS pathology (3, 4). This is one of the strongest genetic associations that has ever been described between an autoimmune disease and an HLA gene, with an odds ratio greater than 100. HLA-B27 is expressed by 90–95% of all AS patients, and studies on HLA-B27+ twins showed a concordance rate for developing AS of 63 and 23% for monozygotic and dizygotic twin pairs, respectively (5, 6). Moreover, HLA-B27+ individuals have a 6–16 times greater risk of developing AS if they have a first-degree relative with AS (3). Although recent genome- and phenome-wide association studies suggested that additional loci are associated with AS, such as ERAP1 (endoplasmic reticulum aminopeptidase 1), HLA-B27 has been repeatedly confirmed as the most relevant genetic predisposition for the development and onset of AS (7–10).
Despite extensive studies during the last few decades, the exact role of HLA-B27 in disease initiation and progression remains unknown, but several theories have been suggested to explain its apparent role in AS (11). One of the central hypotheses is the “HLA-B27 homodimer theory,” which proposes that the formation and subsequent cell-surface presentation of HLA-B27 heavy-chain homodimers triggers an aberrant immune response leading to autoimmunity. In support of this theory, HLA-B27 molecules exhibit an atypically high propensity to form homodimers, and stably expressed HLA-B27 homodimers have been observed both in vitro and in vivo (12–16). In addition, HLA-B27 homodimers have been shown to interact with immunoreceptors, such as killer-cell immunoglobulin-like receptors and leukocyte immunoglobulin-like receptors, although it is unknown whether these interactions are responsible for the onset of autoimmunity (17–20). At a molecular level, HLA-B27 homodimers are formed by two HLA-B27 heavy chains, which are covalently linked through a disulfide bridge involving a conserved, unpaired cysteine residue (Cys-67) (12, 13, 21), although other cysteine residues may be involved as well (15, 21). β2-Microglobulin (β2m), the auxiliary subunit of all canonical trimeric HLA class I complexes, is almost certainly not retained upon homodimer formation (12, 13). Moreover, the addition of oxidizing or apoptosis-inducing agents has been shown to increase the levels of HLA-B*27:05 homodimers on both the EBV-transformed Jesthom B-cell and the leukemic CEM T-cell lines, suggesting that alterations in the cellular redox environment can induce the formation of HLA-B27 homodimers (15).
To date, more than 160 different HLA-B27 allotypes have been described (22–24), but not all allotypes appear to be associated with AS, and two alleles in particular—HLA-B*27:06 (common in the southeast Asian population) and HLA-B*27:09 (found primarily in an ethnic Sardinian population)—seem to protect against or have little association with AS (25, 26). In contrast, the most prevalent allotypes, including HLA-B*27:02, :03, :04, :05, :07, and :08, have been linked to the disease (25, 27). All previous studies that focused on HLA-B27 homodimer formation have predominantly analyzed the most prevalent HLA-B*27:05 allotype and, to a lesser extent, HLA-B*27:04 and HLA-B*27:09 (13, 14, 28–30). In addition, a recent report used comparative flow cytometric analyses to show that the arthritis-associated allotype HLA-B*27:05 forms more cell-surface homodimers than the non-disease-associated subtype HLA-B*27:09 (18), which suggests that expression levels of HLA-B27 homodimer might correlate with disease pathogenesis.
Here, we examined the ability of the eight most prevalent HLA-B27 allotypes (HLA-B*27:02 to HLA-B*27:09) to form homodimers, which included the non-disease-associated allotypes HLA-B*27:06 and HLA-B*27:09. We showed that the disease-associated allotype HLA-B*27:03 (31–34) shows a very weak propensity to form homodimers compared with other HLA-B27 allomorphs, independently of their disease association (protection or progression). Using X-ray crystallography in combination with site-directed mutagenesis, we provide the molecular bases for the compromised ability of HLA-B*27:03 homodimer formation. The observation that a disease-associated HLA-B27 subtype is barely able to form homodimers may suggest that HLA-B27 homodimers are not as important in disease initiation and progression as previously anticipated.
Results
HLA-B*27:03 shows a reduced propensity to form homodimers
The addition of hydrogen peroxide (H2O2) to the human Epstein–Barr virus–transformed, HLA-B*27:05–positive Jesthom B-cell and the leukemic CEM T-cell lines expressing HLA-B*27:05 can increase the formation of HLA-B*27:05 homodimers in cellulo (15). To confirm and extend this observation to other HLA-B27 allotypes in a different cell type, we incubated human B lymphocyte C1R cells expressing matched levels of the eight most common allotypes (HLA-B*27:02 to :09 (35); Fig. S1A) with 250 μm H2O2 for 8 h and analyzed the formation of HLA-B27 homodimers by Western blotting using the HC10 antibody (36, 37), which recognizes HLA class I heavy chains (Fig. 1 (A and B) and Fig. S2). Of note, any HC-10 reactivity from those C1R transfectants can be solely attributed to HLA-B27, as WT C1R cells do not form homodimers because they only express very low levels of endogenous HLA-B*35:03 and HLA-C*04:01 (38–41). As expected, a significant increase in homodimer formation was observed for the HLA-B*27:05 allotype expressed on C1R cells upon treatment with H2O2, indicating that HLA-B*27:05 is able to robustly form homodimers in a cell line–independent manner. In addition, following exposure to H2O2, we also observed significantly elevated levels of homodimers for the disease-associated subtypes HLA-B*27:02, HLA-B*27:04, HLA-B*27:07, and HLA-B*27:08 as well as for the non-disease-associated subtypes HLA-B*27:06 and HLA-B*27:09, with no major differences between these various allotypes (Fig. S2). In agreement with previous studies (12, 13, 21), the addition of the reducing reagent DTT resulted in a complete loss of the homodimers, confirming that HLA-B27 homodimers are covalently linked via disulfide bridges (data not shown). Moreover, we demonstrated that the unpaired Cys-67 plays a major role in homodimer formation, as a C67S point mutation in HLA-B*27:05 abolishes homodimer formation (Fig. S3).
Figure 1.

Y59H polymorphism affects homodimer formation. A, C, and E, C1R-B*27:03, C1R-B*27:05, C1R-B*27:09, C1R-B*27:07, C1R-B*27:07-Y59H, C1R-B*27:08, and C1R-B*27:08-Y59H cell lysates were separated by 12% SDS-PAGE and analyzed by immunoblotting using the HC10 antibody and an antibody recognizing GAPDH as loading control. Representative blots are shown. The addition of H2O2 to the cells is indicated above the lanes. Red arrow, HLA-B27 homodimers; blue arrow, monomeric forms. The addition of H2O2 to the cells as well as the presence of DTT in the SDS-PAGE loading buffer is indicated above the lanes. B, D, F, background-corrected densitometric analyses showing the ratio of HLA-B27 homodimer to monomer. n = 4; mean ± S.E. (error bars). p value was calculated using the Welch's t test (*, p < 0.05; **, p < 0.001; NS, nonsignificant).
However, the disease-associated subtype HLA-B*27:03 showed severely reduced levels of homodimer formation after the addition of H2O2 (Fig. 1 (A and B) and Fig. S2). This discrepancy was evident at steady-state conditions, under which HLA-B*27:03 homodimers were barely visible compared with other allotypes, suggesting that HLA-B*27:03 exhibits a severely reduced propensity to form homodimers, which is an intriguing observation, given that HLA-B*27:03 is associated with AS (6, 31–34, 42, 43).
Polymorphism at position 59 is responsible for the lack of HLA-B*27:03 homodimers
To further investigate this finding, we compared the amino acid sequence of HLA-B*27:03 against the other HLA-B27 allotypes (Table 1), which revealed that HLA-B*27:03 differs from other HLA-B27 allotypes at position 59, whereby the conserved tyrosine residue is replaced by a histidine residue (Y59H) (Table 1). Of note, Y59H is the only polymorphism that differentiates HLA-B*27:03 from the archetypal HLA-B*27:05 allomorph, suggesting that this single-amino acid polymorphism is sufficient to impact the formation of HLA-B27 homodimers. Nevertheless, to further test this notion, we introduced the Y59H polymorphism into the HLA-B*27:07 and HLA-B*27:08 allotypes (Fig. S4A) and expressed matching levels of these mutant versions in C1R cells (C1R-HLA-B*27:07-Y59H and C1R-HLA-B*27:08-Y59H; Fig. S1B). We have chosen these particular allomorphs, as they differ from HLA-B*27:05 by several polymorphisms (Table 1) and are more diverged from this archetypal allotype than the other allomorphs. In agreement with our postulate, the presence of this single point mutation in HLA-B*27:07 and HLA-B*27:08 severely reduced the formation of homodimers under both steady-state and H2O2-inducing conditions (Fig. 1, C–F). These results demonstrate that the presence of a histidine residue at position 59 is sufficient to impact the ability of HLA-B27 allotypes to form homodimers in a dominant-negative fashion.
Table 1.
Amino acid differences among the eight most common HLA-B27 allotypes
| Amino acid position |
|||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 47a | 59 | 60a | 67b | 77 | 80 | 81 | 82 | 83 | 97 | 113 | 114 | 116 | 131 | 152 | 164 | 211 | |
| B*27:05 | P | Y | W | C | D | T | L | L | R | N | Y | H | D | S | V | C | A |
| B*27:02 | N | I | A | ||||||||||||||
| B*27:03 | H | ||||||||||||||||
| B*27:04 | S | E | G | ||||||||||||||
| B*27:06 | S | D | Y | E | G | ||||||||||||
| B*27:07 | S | H | N | Y | R | ||||||||||||
| B*27:08 | S | N | R | G | |||||||||||||
| B*27:09 | H | ||||||||||||||||
a Positions 47 and 60 are located in the loop surrounding Cys-67. Mutations at these positions are expected to affect homodimer formation.
b Key cysteine residue involved in the formation of homodimers.
Short-range conformational rearrangements govern the accessibility of Cys-67
Due to the three-dimensional proximity of the Y59H polymorphism to Cys-67, which has been shown to be the key cysteine residue involved in the formation of homodimers through disulfide bonds (12, 13), we hypothesized that the presence of His-59 causes conformational changes that may impact the environment of Cys-67. To unravel the effects of this polymorphic position on HLA-B*27:03 and HLA-B*27:05, we analyzed the structures of these HLA molecules in complex with two different peptide ligands. We selected the endogenous self-peptide LRNQSVFNF (LRN), which has been identified in our comprehensive HLA-B27 peptide database as a ligand for both HLA-B*27:03 and HLA-B*27:05 allotypes (35), as well as the well-characterized KK10 peptide (KRWIILGLNK) derived from HIV. The structure of HLA-B*27:05 in complex with KK10 was published previously (44, 45), and we determined the structures of the remaining three complexes (of HLA-B*27:03 in complex with both LRN and KK10 peptide and of HLA-B*27:05 in complex with the LRN peptide).
All structures were solved at high resolution and in the same space group, independently of the bound peptide or the allomorph crystallized (Table 2). The HLA-B*27:05KK10 and HLA-B*27:03KK10 structures overlaid well, with a root mean square deviation (r.m.s.d.) of 0.19 Å on the antigen-binding cleft and of 0.42 Å for the peptide itself (Fig. 2A). As expected, the KK10 peptide was observed to bind in a similar extended conformation in both molecules with the characteristic anchor residues P2-Arg and PΩ-Lys binding the B and F pockets, respectively. The central part of the KK10 peptide, P6-Leu and P7-Gly, was poorly defined in the electron density map, indicating flexibility in both complexes. The structures of the HLA-B*27:03 and HLA-B*27:05 in complex with the self-derived LRN peptide also showed a similar conformation of the HLA antigen binding cleft (r.m.s.d. of 0.14 Å) and of the peptide (r.m.s.d. of 0.06 Å) (Fig. 2B). However, in contrast to the KK10 peptide, the electron density for 9-mer LRN peptide was well-defined with all residues clearly observed, indicating a more rigid structure than the KK10 peptide (Fig. S5). Similar thermal stability was observed for four peptide-HLA (pHLA) complexes (Table 3) with a Tm ranging from 60 to 63 °C, indicating that the polymorphism at position 59 has no impact on the stability of these pHLAs.
Table 2.
Data collection and refinement statistics
| Data collection | HLA-B*27:03KK10 | HLA-B*27:03LRN | HLA-B*27:05LRN | HLA-B*27:03-P47GLRN | HLA-B*27:05-W60ALRN |
|---|---|---|---|---|---|
| Space group | P212121 | P212121 | P212121 | P212121 | P212121 |
| Cell dimensions: a, b, c (Å) | 50.85, 85.65, 104.75 | 51.05, 82.51, 109.62 | 50.78, 82.70, 104.98 | 50.98, 82.79, 109.90 | 50.96, 82.49, 110.02 |
| Resolution (Å) | 44.24–1.52 (1.55–1.52) | 46.28–1.53 (1.56–1.53) | 45.71–1.44 (1.46–1.44) | 28.00–1.45 (1.47–1.45) | 36.67–1.38 (1.40–1.38) |
| Total no. of observations | 500,083 (24,151) | 499,886 (19,314) | 586,220 (27,692) | 499,862 (24,458) | 770,543 (32,004) |
| No. of unique observations | 68,733 (3336) | 70,002 (3190) | 80,761 (3937) | 82,939 (4047) | 95,569 (4403) |
| Multiplicity | 7.3 (7.2) | 7.1 (6.1) | 7.3 (7.0) | 6.0 (6.0) | 8.1 (7.3) |
| Data completeness (%) | 100 (100) | 99.6 (92.0) | 100 (99.9) | 99.8 (99.9) | 99.5 (93.9) |
| I/σI | 14.5 (2.1) | 13.0 (2.1) | 16.8 (2.1) | 18.3 (2.1) | 20.7 (2.4) |
| Rpima (%) | 4.0 (42.3) | 3.9 (33.9) | 3.6 (42.0) | 2.5 (36.2) | 2.0 (31.9) |
| Refinement | |||||
| Non-hydrogen atoms | |||||
| Protein | 3213 | 3220 | 3238 | 3223 | 3233 |
| Water | 424 | 517 | 425 | 626 | 533 |
| Rfactorb (%) | 20.14 | 19.35 | 20.20 | 18.94 | 20.36 |
| Rfreeb (%) | 23.02 | 22.34 | 22.03 | 23.13 | 22.02 |
| r.m.s.d. from ideality | |||||
| Bond lengths (Å) | 0.010 | 0.010 | 0.010 | 0.010 | 0.010 |
| Bond angles (degrees) | 1.03 | 1.01 | 1.04 | 1.01 | 1.08 |
| Ramachandran plot (%) | |||||
| Favored region | 97.9 | 97.9 | 97.7 | 97.7 | 98.2 |
| Disallowed region | 0 | 0 | 0.25 | 0 | 0 |
aRp.i.m. = Σhkl(1/(N − 1))½ Σi|Ihkl,i − 〈Ihkl〉|/Σhkl 〈Ihkl〉.
b Rfactor = Σhkl‖Fo| − |Fc‖/Σhkl|Fo| for all data except ∼5%, which were used for Rfree calculation.
Figure 2.

Molecular structures of pHLA-B*27:03 and pHLA-B*27:05. A–F, overlays or zoomed-in views of HLA-B*27:05 (pink) and HLA-B*27:03 (orange) in complex with the HIV peptide KK10 in pale colors (A and C) or with the self-peptide LRN in bright colors (B, D, E, and F). The HLA clefts and peptides are represented as a cartoon. The polymorphic residue at position 59 as well as key residues that are impacting on the HLA homodimer formation are represented in stick form. The third loop, which spans from Ser-38 to Pro-50, is colored in yellow in E and F. G, superposition of E and F: HLA-B*27:05 (pink) and HLA-B*27:03 (orange) presenting the LRN peptide. H and I, the relative B factor of residues from the HLA cleft's third loop (residues 37–46) has been calculated for each pHLA complex. For each residue, the B factor of the main chain (Bresidue) has been used to calculate the mobility relative to the cleft of each corresponding molecule (Bα1α2) using the formula, 100 × (Bresidue − Bα1α2)/Bα1α2. The higher the relative B factor, the more mobile the residue compared with the HLA cleft. The two graphs represent comparisons of the relative B factors of the third loops of the pHLA complexes. H, average relative B factor of HLA-B*27:05KK10 = 23.8; average relative B factor of HLA-B*27:03KK10 = 5.5; significance: p = 0.1933 (ns), unpaired t test. I, average relative B factor of HLA-B*27:05LRN = 64.5; average relative B factor of HLA-B*27:03LRN = 20.8; significance: p = 0.0309 (*), unpaired t test.
Table 3.
Thermal stability of pHLA complexes
Tm is the temperature required to unfold half of the protein sample. Results are mean ± S.E. (n = 2).
| pHLA complex | Tm |
|---|---|
| °C | |
| HLA-B*27:05KK10 | 62.3 ± 0.6 |
| HLA-B*27:03KK10 | 60.5 ± 0.5 |
| HLA-B*27:05LRN | 63.5 ± 0.7 |
| HLA-B*27:03LRN | 60.7 ± 0.7 |
The polymorphic residue at position 59 is located at the beginning of the HLA α1-helix in close proximity with the P1 residue of the peptide. This residue is a Tyr in HLA-B*27:05 and a His in the HLA-B*27:03 molecule. The side chains of both Tyr-59 and His-59 are pointing down in the antigen-binding cleft. In HLA-B*27:05, the Tyr-59 hydroxyl group is interacting directly or via a water-mediated bond with the Tyr-171 and Tyr-7, respectively, as well as interacting with the hydrophobic residues Ile-52, Phe-33, and Val-34. The aromatic ring of Tyr-59 sits above the Trp-60 and is sandwiched by two negatively charged residues, Glu-55 and Glu-63 (Fig. S6A). In HLA-B*27:03, the His-59 has an environment similar to that of Tyr-59, with some localized movement. The lack of a hydroxyl group allows for the presence of an additional water molecule bonding His-59 and Tyr-7 in HLA-B*27:03 structures (Fig. S6B). The smaller side chain of the His-59, compared with Tyr-59, allows the Trp-60 side chain to be closer to residue 59 in HLA-B*27:03 (Fig. 2, C and D). The repositioning of the Trp-60 side chain was more obvious in the KK10-bound structures than in the LRN-bound ones, with a maximum displacement of 1.8 and 0.6 Å, respectively. Interestingly, the Trp-60 sits above the Pro-47, which is part of the HLA molecule's third loop (Fig. S7). This loop is located beneath the Cys-67 residue and spans from Ser-38 (at the end of the third β-strand) to Pro-50 (at the start of the α1-helix; Fig. 2, E–G). The Cys-67 residue is located within the α1-helix and shielded by Phe-36, which is located upstream of the third loop. The third loop itself is stabilized by hydrophobic side chain stacking of Pro-47, Trp-60, and the polymorphic residue Tyr/His-59.
Whereas the overall structures of the HLA-B*27:03 and HLA-B*27:05 molecules were similar irrespective of the bound peptide, we observed major differences in the flexibility of the third loop of the antigen-binding cleft based on the B factor analysis, which is the “temperature factor” representing the dynamic mobility of each atom. Namely, we compared the B factor of each residue in the loop located between Phe-36 and Pro-47 with the overall B factor of the antigen-binding cleft (residues 1–180 (46)). The relative B factors are higher for the loop in HLA-B*27:05 compared with HLA-B*27:03 (Fig. 2, H and I), which revealed a higher mobility of the third loop in HLA-B*27:05. The relative B factor of this loop was nearly 5 times higher for HLA-B*27:05KK10 (average of 23.8) than HLA-B*27:03KK10 (average of 5.5; p = 0.1933 (ns), unpaired t test) and more than 3 times higher for HLA-B*27:05LRN than HLA-B*27:03LRN (average of 64.5 compared with 20.8, respectively; p = 0.0309; *, unpaired t test).
Based on these structural rearrangements observed for Tyr/His-59, Trp-60, and Pro-47 in combination with the apparent differences in the B factor of the third loops, we concluded that the lower flexibility of the third loop in HLA-B*27:03 may limit the accessibility of the buried Cys-67 residue compared with HLA-B*27:05 molecule, which reduces the ability of HLA-B*27:03 to form homodimers.
Proximal loop mobility dictates Cys-67 accessibility and homodimer formation
To test whether the position and mobility of the proximal third loop plays an important role in the formation of homodimers, we introduced specific point mutations into the HLA-B*27:03 and HLA-B*27:05 heavy chains and analyzed their ability to form homodimers using Western blotting with the HC10 antibody (36, 37). We first mutated the Pro-47 in HLA-B*27:03 to a glycine residue (P47G). We hypothesized that the P47G mutation would increase the flexibility of the third loop, which should increase the formation of homodimers in HLA-B*27:03. Indeed, HLA-B*27:03-P47G showed a significant increase in homodimer levels compared with WT HLA-B*27:03 (Fig. 3). Second, we mutated the bulky Trp-60 to a small alanine residue (W60A) in HLA-B*27:05. Due to the interaction interplay between His–Tyr-59/Trp-60/Pro-47, the hypothesis was that the lack of side chain at position 60 would help the third loop of HLA-B*27:05 to be more rigid and adopt an “HLA-B*27:03-like” position resulting in reduced levels of homodimers. Consistent with this hypothesis, we were unable to observe any homodimers in HLA-B*27:05-W60A–expressing cells in contrast to WT HLA-B*27:05 (Fig. 3) despite comparable cell-surface expression levels (Fig. 1C). However, by mutating the tryptophan residue to alanine, we destroyed one of the key residues of the HC10 epitope (57PXXWDR62 (36, 37)), which results in suboptimal antibody binding and explains the very faint signals for HLA-B*27:05-W60A. Nonetheless, these observations provide direct evidence for our hypothesis that naturally occurring and intentional substitutions in this loop dictate Cys-67 accessibility and heavy-chain homodimer formation.
Figure 3.
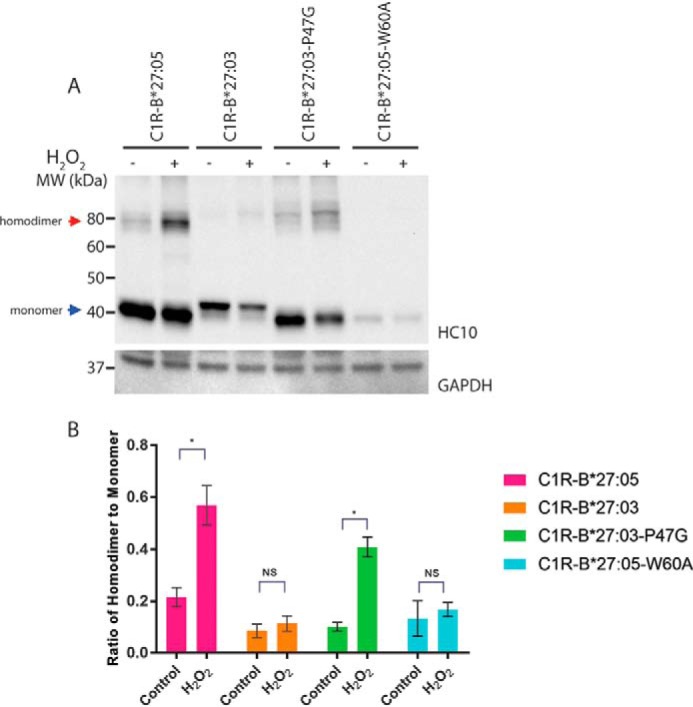
P47G promotes and W60A abrogates homodimer formation. A, C1R-B*27:05, C1R-B*27:03, C1R-B*27:03-P47G, and C1R-B*27:05-W60A cell lysates were separated by 12% SDS-PAGE and analyzed by immunoblotting using the HC10 antibody and an antibody recognizing GAPDH as loading control. Representative blots are shown. The addition of H2O2 to the cells is indicated above the lanes. Red arrow, HLA-B27 homodimers; blue arrow, monomeric forms. B, background-corrected densitometric analyses showing the ratio of HLA-B27 homodimer to monomer. n = 3, mean ± S.E. (error bars). p value was calculated using the Welch's t test (*, p < 0.05; **, p < 0.001; NS, nonsignificant).
To gain molecular insight into the impact of these loop mutations, we solved the structures of the HLA-B*27:03-P47GLRN and HLA-B*27:05-W60ALRN complex and compared them with their WT counterparts (Table 2). The overall conformation of HLA-B*27:03 was not disturbed by the P47G mutation, with an r.m.s.d. of 0.10 Å on the overall pHLA complex. In addition, the P47G mutation did not change the conformation of the third loop but had a minor effect on the Trp-60 side chain, which was slightly shifted (0.5 Å of the Cζ2 atom) (Fig. 4A). However, in agreement with our hypothesis, the overall relative B factor of the third loop in the HLA-B*27:03-P47GLRN mutant was almost twice as high (average of 35.3) compared with WT HLA-B*27:03LRN (average of 20.8; p = 0.439 (ns), unpaired t test; Fig. 4B). This suggests that even a minor increase in the third loop mobility may have an impact on the homodimer formation.
Figure 4.

Structural impact of P47G and W60A. A, C, and D, overlays of HLA-B*27:03 (orange), HLA-B*27:03-P47G (green), HLA-B*27:05 (pink), and HLA-B*27:05-W60A (cyan) presenting the LRN peptide. The residues at positions 47, 59, and 60 are represented in stick form, and the Gly-47 Cα atom in A is represented as a sphere. B and E, see legend to Fig. 2 (H and I). B, average relative B factor of HLA-B*27:03-P47GLRN = 35.3; average relative B factor of HLA-B*27:03LRN = 20.8; significance: p = 0.439 (ns), unpaired t test. E, average relative B factor of HLA-B*27:05-W60ALRN = 16.7; average relative B factor of HLA-B*27:05LRN = 64.5; significance: p = 0.0111 (*), unpaired t test.
Similar to the P47G mutation in HLA-B*27:03, the W60A mutation did not impact the overall conformation of the HLA-B*27:05 antigen cleft or the peptide (r.m.s.d. of 0.47 Å on overall pHLA). However, the W60A substitution had an impact on the conformation of the third loop as well as on the beginning of the α1-helix (residues 50–57) (Fig. 4, C and D). A section of the third loop (residues 40–43) changed conformation in HLA-B*27:05-W60A when compared with WT HLA-B*27:05, with a maximum displacement of 1.5 Å for the Ala-41 Cα atom (Fig. 4C). The conformation of HLA-B*27:05-W60ALRN was more similar to the HLA-B*27:03LRN complex (r.m.s.d. of 0.20 Å on overall pHLA) than HLA-B*27:05LRN complex (r.m.s.d. of 0.47 Å) (Fig. 4D). This confirms our hypothesis that HLA-B*27:05-W60A would adopt a “HLA-B*27:03-like” configuration. In addition, the third loop in HLA-B*27:05-W60ALRN showed an almost 4-fold decrease in relative B factor (average of 16.7) when compared with the HLA-B*27:05LRN complex (average of 64.5) (Fig. 4E; p = 0.0111 (*), unpaired t test). The structural rearrangements observed in HLA-B*27:05-W60ALRN decreased third loop flexibility significantly, as well as the ability of HLA-B*27:05-W60A to form homodimers (Fig. 3). Our data also show that the Trp-60, which is directly contacted by the polymorphic residue 59, has a greater impact on the third loop mobility than observed for Pro-47, which sits underneath Trp-60.
To validate biochemically that the Cys-67 residue in HLA-B*27:03 is less accessible compared with HLA-B*27:05, we performed Ellman's assays (47) with HLA-B*27:03 and HLA-B*27:05 by adding 5,5′-dithiobis-2-nitrobenzoic acid (DTNB; Ellman's reagent) to the KK10-refolded HLA molecules. In brief, DTNB reacts with free thiol groups and stoichiometrically produces a 2-nitro-5-thiobenzoate dianion (TNB2−) of yellow color that can be colorimetrically monitored over time. Given that HLA-B*27:03 and HLA-B*27:05 are identical apart from a single polymorphism at position 59 (Table 1), and the only free thiol group in both molecules is the cysteine residue at position 67, any difference in the reaction rate with DTNB can be directly attributed to variations in the accessibility of the Cys-67 residue. In agreement with our hypothesis, significant differences were observed in the absorbance at 412 nm between HLA-B*27:03 and HLA-B*27:05 (Fig. 5), suggesting that the reduced flexibility of the third loop in HLA-B*27:03 causes the bulky DTNB compound to react slower with its free Cys-67 residue compared with HLA-B*27:05.
Figure 5.
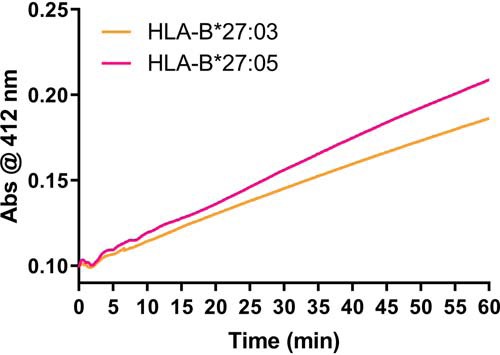
Ellman's assay. The diagram shows the reaction rate of KK10-refolded HLA-B*27:03 (orange) and HLA-B*27:05 (pink) with DTNB (Ellman's reagent). The production of the 2-nitro-5-thiobenzoate dianion was measured colorimetrically for 1 h at an absorbance of 412 nm. n = 3; a representative result is shown.
Taken together, we demonstrate that the flexibility and conformation of the third loop of the HLA-B27 molecule has a major impact on its ability to form homodimers. The Tyr-59 in HLA-B*27:05—and presumably in all other HLA-B27 allotypes carrying the conserved Tyr-59 at this position—impacts the third loop conformation through its interaction with Trp-60 and Pro-47. These changes impact the mobility of the third loop, which in turn might modulate the accessibility of Cys-67 and homodimer formation. The presence of the smaller His-59 residue in HLA-B*27:03 allows the third loop to adopt a more rigid conformation, which reduces homodimer formation probably by shielding the underlying Cys-67 residue.
Discussion
In this study, we analyzed the ability of the eight most prevalent HLA-B27 allotypes (HLA-B*27:02 to HLA-B*27:09) to form homodimers under oxidizing conditions. With the exception of HLA-B*27:04, HLA-B*27:05, and HLA-B*27:09, none of these HLA-B27 allotypes have been tested previously for their ability to form homodimers, and, to our knowledge, this is the first report that compared homodimer formation of multiple HLA-B27 allotypes in the one parental cell line. In line with previous results (15, 18), we observed that HLA-B*27:05 and HLA-B*27:09 are able to form homodimers that can be effectively induced by the addition of H2O2, and Cys-67 appears to be essential for the formation of such redox-induced dimers (Fig. S3), although Cys-325 has been reported to be involved as well (15). Importantly, we did not observe any major differences in the level of homodimer formation between this disease-associated and the non-disease-associated HLA-B27 allotypes. This observation is in contrast to a previous report (18), which shows that the arthritis-associated HLA-B*27:05 allele forms more cell-surface homodimers than the nonassociated HLA-B*27:09 allele. HLA-B27 expression levels, cell lines, and/or sensitivity of reporter assays might account for these discrepancies, in addition to conflicting behaviors between cell surface–localized and intracellular HLA-B27 homodimers.
All other tested HLA-B27 allotypes—apart from HLA-B*27:03—were observed to form inducible homodimers (Fig. S2), which are sensitive to the addition of the reducing agent DTT, confirming that they are covalently linked by disulfide bonds. Surprisingly, we observed that HLA-B*27:03, an arthritis-associated HLA-B27 subtype that has been found to be present in many AS patients (6, 31–34), is barely able to form homodimers and shows significantly reduced homodimer levels under both steady-state and oxidizing conditions (Fig. 1 (A and B) and Fig. S2). These apparent differences are clearly evident compared with other disease-associated subtypes as well as with both non-disease-associated subtypes HLA-B*27:06 and HLA-B*27:09, and it is difficult to reconcile our observations with the “HLA-B27 homodimer theory.” A comparison of the HLA-B*27:03 amino acid sequence against the other HLA-B27 alleles suggested that the polymorphism at position 59 (Y59H) is accountable for its compromised ability to form homodimers. This notion was further confirmed by the observation (i) that Y59H is the only polymorphism differentiating HLA-B*27:03 and HLA-B*27:05 and (ii) that the HLA-B*27:07 and HLA-B*27:08 allotypes, which are able to readily form homodimers, lose this ability after the substitution of the Tyr-59 by His-59. These findings prompted us to use X-ray crystallography to understand the consequences of this polymorphism at the molecular level. As expected—given that HLA-B*27:03 and HLA-B*27:05 differ by only one polymorphism—the antigen-binding cleft conformations are highly similar, but some notable local conformational differences were observed surrounding the polymorphic residue 59. The bulky Tyr-59 in HLA-B*27:05 exerts an effect on the position of the third loop of the antigen-binding cleft (residues 38–50) via hydrophobic interactions with Trp-60 and Pro-47. In contrast, the presence of the smaller His-59 in HLA-B*27:03 allows the loop to sit closer to the α1-helix. Although these conformational changes were modest, the resulting mobility changes of the third loops were significant. In HLA-B*27:03, the third loop was found to be relatively rigid, which in turn reduces the accessibility of the buried Cys-67 residue and leads to decreased levels of homodimer. In contrast, the third loop in HLA-B*27:05 was found to be relatively flexible, such that the underlying Cys-67 residue becomes more accessible to form homodimers. Somewhat reminiscent changes in mobility have been described for a hinged unit containing the conserved 310 helix in MHC class I molecules that shows differential mobility, depending on whether a peptide is bound or not (48). Although highly speculative, the third loop in HLA-B27 might also reflect a peptide-dependent region that may be stabilized or destabilized in the context of appropriate peptides. In addition, the protonation level of the His-59 residue might be different under physiological conditions than in the crystals at pH 5.6, but those changes in charge states will have presumably only marginal impact on the structure.
To confirm our observations, we performed biochemical assays (Ellman's assays) with HLA-B*27:03 and HLA-B*27:05, which showed that the Cys-67 residue in HLA-B*27:03 is less accessible compared with HLA-B*27:05. In addition, we introduced specific, site-directed point mutations (W60A and P47G) into the interaction cascade linking the polymorphic residue at position 59. We succeeded in changing homodimer levels by interfering with the cascade, which directly altered the flexibility of the third loop. When analyzing HLA-B*27:05-W60A by Western blotting using the HC10 antibody, we observed very faint signals even for the monomeric bands (Fig. 3), which was not due to decreased expression levels of HLA-B*27:05-W60A, as shown by flow cytometry with the HLA-B27–specific antibody ME1 (Fig. S1C). However, by mutating the tryptophan residue to alanine, we destroyed one of the key residues of the HC10 epitope (57PXXWDR62 (36, 37)), resulting in suboptimal antibody binding.
Taken together, we provide compelling evidence that HLA-B*27:03—a disease-associated subtype—is barely able to form homodimers. These observations are difficult to reconcile with the “HLA-B27 homodimer theory” and strongly suggest that HLA-B27 homodimers are not as important in disease initiation and progression as previously anticipated. Although we cannot entirely rule out the involvement of HLA-B27 homodimers in AS, the polymorphisms underpinning disease association implicate peptide-binding features and classical antigen presentation in disease pathogenesis.
Experimental procedures
HLA constructs
To introduce the Y59H polymorphism into HLA-B*27:07 and HLA-B*27:08, the BglII section from the pcDNA3.1(+) Hygro-B*27:03 construct (35) was swapped with the corresponding region of the constructs pcDNA3.1(+)Hygro-B*27:07 and pcDNA3.1(+)Hygro-B*27:08 (35), resulting in pcDNA3.1(+) Hygro-B27:07-Y59H and pcDNA3.1(+)Hygro-B27:08-Y59H (Fig. S4A).
To introduce the P47G and/or W60A polymorphisms into HLA-B*27:03 and HLA-B*27:05, commercially synthesized HLA-B27 DNA fragments were introduced into the pcDNA3.1(+)Hygro vector using the restriction sites BamHI and XhoI (Fig. S4B).
To generate HLA-B27 constructs for bacterial expression, commercially synthesized and Escherichia coli codon-optimized HLA-B*27:03 and HLA-B*27:05 DNA fragments with and without the P47G and/or W60A mutations were introduced into the pET30a vector using NdeI and HindIII (Fig. S4C). Only the extracellular domains of the HLA-B27 alleles have been used for bacterial expression. All DNA constructs were sequenced to confirm their integrity.
Cell lines
The generation of the C1R cell lines expressing HLA-B*27:02 to HLA-B*27:09 has been described previously (35).
All pcDNA3.1(+)Hygro constructs (Fig. S4, A and B) were stably transfected into C1R cells by electroporation using a Gene Pulser XcellTM electroporation system (Bio-Rad). Stable transfectants were cultured under hygromycin B selection (0.3 mg/ml; Gibco®, Invitrogen) in RPMI 1640 medium (Life TechnologiesTM, Invitrogen) supplemented with 10% fetal calf serum, 1% penicillin/streptomycin (Gibco®, Invitrogen), MEM nonessential amino acids (Gibco®, Invitrogen), 1 mm β-mercaptoethanol, 100 mm HEPES (Sigma), and 40 mm l-glutamine (MP Biomedicals).
Genomic DNA of all established cell lines was extracted at various stages throughout the study. The HLA-B27 locus was PCR-amplified and sequenced to confirm allotype and mutant identities.
H2O2 treatment of cells
2.5 × 106 cells were plated in 5 ml of RPMI 1640 medium at least 2 h prior to H2O2 treatment. H2O2 (Sigma) was added to the culture medium to a final concentration of 250 μm, and the cells were incubated for 8 h at 37 °C. The cell viability was monitored using trypan blue exclusion.
Cell lysis, SDS-PAGE, and immunoblotting
Cells were washed twice with cold PBS, resuspended in lysis buffer (1% (v/v) IGEPAL, 50 mm Tris (pH 8.0), 150 mm NaCl, 1 mm phenylmethylsulfonyl fluoride, 10 mm iodoacetamide, and protease inhibitor mixture (Roche Applied Science)), and incubated for 30 min on ice. Insoluble materials were removed by centrifugation (16,000 × g for 15 min at 4 °C), and the protein concentration of the supernatant was determined using a Direct Detect® spectrometer (Millipore).
Proteins were separated by SDS-PAGE using 12% Mini-PROTEAN® TGX precast gels (Bio-Rad), and 0.1 m DTT was added where necessary. The resolved proteins were transferred onto nitrocellulose membranes by Western blotting at 110 mA for 1 h, and the membranes were blocked with 2.5% milk powder dissolved in TBS-T. The blocked membranes were probed with the HC10 antibody (which binds HLA class I heavy chains (36, 37)), which was harvested from hybridoma supernatants, followed by an anti-mouse horseradish peroxidase–conjugated secondary antibody (Abcam). For loading control, the membranes were probed with an anti-GAPDH antibody (Abcam) followed by an anti-rat horseradish peroxidase–conjugated secondary antibody (Abcam). Membranes were scanned with a ChemiDoc Touch imaging system (Bio-Rad), using Western Lightning Plus-ECL enhanced chemiluminescence reagent (PerkinElmer Life Sciences). Densitometry analyses were performed using the Bio-Rad ImageLab software (version 5.2.1) in combination with ImageJ (version 1.5i), Microsoft Excel 2016, and GraphPad Prism 7.
Flow cytometry
Cells were washed with ice-cold PBS, resuspended in flow buffer (1% fetal calf serum in PBS), and incubated on ice with the primary antibody ME1 (which binds to HLA-B27 (49)), which was harvested from hybridoma supernatants). Anti-mouse FITC or PE antibodies were used as secondary antibodies (Abcam). Propidium iodide was added to monitor cell viability, and 1% paraformaldehyde (in PBS) was used as fixative prior to antibody staining when required. Flow cytometric analysis was performed on a BD LSR II flow cytometer (BD Biosciences), and data were analyzed using FlowJo version 10.
Protein expression and purification
Soluble HLA class I-β2m heterodimers of WT and mutated HLA-B*27:03 and HLA-B*27:05 in complex with the KK10 (KRWIILGLNK) or LRN (LRNQSVFNF) peptide were prepared as described previously (44). Briefly, β2m and the extracellular section of HLA class I were expressed as insoluble inclusion bodies in E. coli cells and purified to be refolded with the peptide. The pHLA complexes were further purified via anion-exchange chromatography, and their conformational integrity was confirmed by binding to the mAb W6/32 (50).
Crystallization, data collection, and structure determination
Crystals of each pHLA complex were grown by the hanging-drop, vapor-diffusion method at 20 °C with a protein/reservoir drop ratio of 1:1, at a concentration of 10 mg/ml in 10 mm Tris-HCl, pH 8, 150 mm NaCl using 20–30% PEG 4000, 0.2 m sodium acetate, and 0.1 m sodium citrate, pH 5.6. The pHLA crystals were soaked in mother liquor solution with an increased PEG 4000 concentration to 30% (w/v) prior to being flash-frozen in liquid nitrogen. All data were collected on the MX1 (51) and MX2 beamlines at the Australian Synchrotron, Clayton, using the ADSC-Quantum or Eiger detectors (at 100 K). Data were processed using XDS (52) and scaled with the Aimless software from the CCP4 suite (57). The structures were determined by molecular replacement using the PHASER (53) program with the HLA-B*27:05KK10 for the MHC model without the peptide (Protein Data Bank accession number 4G9G (44)). Manual model building was conducted using the Coot software (54), followed by maximum-likelihood refinement with the Buster program (55). The final models have been validated using the Protein Data Base validation web site, and the final refinement statistics are summarized in Table 2. The Protein Data Bank accession numbers are as follows: HLA-B*27:03KK10 (6PYL); HLA-B*27:03LRN (6PZ5); HLA-B*27:05LRN (6PYJ); HLA-B*27:03-P47GLRN (6PYV); HLA-B*27:05-W60ALRN (6PYW). All molecular graphics representations were created using PyMOL version 1.7.6.4 (56).
Thermal stability assay
To assess the stability of the allomorphs and the impact of the various polymorphisms, a thermal shift assay was performed. The fluorescent dye Sypro orange was used to monitor the protein unfolding. The assay was performed in a real-time detection system (Corbett RotorGene 3000) originally designed for PCR. Each pHLA complex was measured twice in duplicate at two concentrations (0.5 and 1 mg/ml) in 10 mm Tris-HCl, pH 8, 150 mm NaCl and heated from 30 to 95 °C with a heating rate of 1 °C/min. The fluorescence intensity was measured with excitation at 530 nm and emission at 555 nm. The Tm, or thermal melt point, represents the temperature for which 50% of the protein is unfolded (Table 3).
Ellman's assay
500 μg of KK10-refolded HLA-B*27:03 or HLA-B*27:05 was resuspended in 500 μl of buffer containing 20 mm Tris-HCl, 300 mm NaCl, and 1 mm EDTA. Ellman's reagent (DTNB, Sigma) was added to a final concentration of 100 μm. The reaction was monitored at 412 nm for 1 h using a Jasco V-750 spectrophotometer.
Author contributions
T. C. L. K. S., S. I., P. B., S. G., A. W. P., and R. B. S. conceptualization; T. C. L. K. S., P. B., J. R., S. G., A. W. P., and R. B. S. data curation; T. C. L. K. S., S. I., H. H., A. G., M. J. C., P. B., J. R., S. G., A. W. P., and R. B. S. formal analysis; T. C. L. K. S., S. I., P. B., S. G., A. W. P., and R. B. S. supervision; T. C. L. K. S., H. H., M. J. C., P. B., J. R., S. G., A. W. P., and R. B. S. validation; T. C. L. K. S., S. I., H. H., A. G., M. J. C., P. B., A. W. P., and R. B. S. investigation; T. C. L. K. S. and H. H. visualization; T. C. L. K. S., H. H., P. B., A. W. P., and R. B. S. writing-original draft; T. C. L. K. S., S. I., P. B., S. G., A. W. P., and R. B. S. project administration; T. C. L. K. S., S. I., P. B., J. R., S. G., A. W. P., and R. B. S. writing-review and editing; S. I., M. J. C., P. B., J. R., S. G., A. W. P., and R. B. S. resources; S. I., P. B., J. R., S. G., A. W. P., and R. B. S. funding acquisition.
Supplementary Material
Acknowledgments
We thank the Macromolecular Crystallization Facility staff and the staff at the Australian synchrotron for technical assistance. We would also like to thank Dr. Nicole Mifsud for providing expertise on extracting and sequencing genomic DNA.
This work was supported by the Australian Research Council (ARC) and the National Health and Medical Research Council (NHMRC) Project 1085017. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S7.
- AS
- ankylosing spondylitis
- HLA
- human leukocyte antigen
- β2m
- β2-microglobulin
- r.m.s.d.
- root mean square deviation
- DTNB
- 5,5′-dithio-bis-2-nitrobenzoic acid
- MHC
- major histocompatibility complex
- GAPDH
- glyceraldehyde-3-phosphate dehydrogenase
- pHLA
- peptide-HLA.
References
- 1. Colbert R. (2004) The immunobiology of HLA-B27: variations on a theme. Curr. Mol. Med. 4, 21–30 10.2174/1566524043479293 [DOI] [PubMed] [Google Scholar]
- 2. Reveille J. D. (2011) Epidemiology of spondyloarthritis in North America. Am. J. Med. Sci. 341, 284–286 10.1097/MAJ.0b013e31820f8c99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. van der Linden S. M., Valkenburg H. A., de Jongh B. M., and Cats A. (1984) The risk of developing ankylosing spondylitis in HLA-B27 positive individuals. Arthritis Rheum. 27, 241–249 10.1002/art.1780270301 [DOI] [PubMed] [Google Scholar]
- 4. Schlosstein L., Terasaki P. I., Bluestone R., and Pearson C. M. (1973) High association of an HL-A antigen, W27, with ankylosing spondylitis. N. Engl. J. Med. 288, 704–706 10.1056/NEJM197304052881403 [DOI] [PubMed] [Google Scholar]
- 5. Braun J., and Sieper J. (2007) Ankylosing spondylitis. Lancet 369, 1379–1390 10.1016/S0140-6736(07)60635-7 [DOI] [PubMed] [Google Scholar]
- 6. Brown M. A., Kennedy L. G., and MacGregor A. J., Darke C., Duncan E., Shatford J. L., Taylor A., Calin A., and Wordsworth P. (1997) Susceptibility to ankylosing spondylitis in twins the role of genes, HLA, and the environment. Arthritis Rheum. 40, 1823–1828 10.1002/art.1780401015 [DOI] [PubMed] [Google Scholar]
- 7. Brown M. A. (2011) Progress in the genetics of ankylosing spondylitis. Brief. Funct. Genomics 10, 249–257 10.1093/bfgp/elr023 [DOI] [PubMed] [Google Scholar]
- 8. Cortes A., Pulit S. L., Leo P. J., Pointon J. J., Robinson P. C., Weisman M. H., Ward M., Gensler L. S., Zhou X., Garchon H. J., Chiocchia G., Nossent J., Lie B. A., Førre Ø., Tuomilehto J., et al. (2015) Major histocompatibility complex associations of ankylosing spondylitis are complex and involve further epistasis with ERAP1. Nat. Commun. 6, 7146 10.1038/ncomms8146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Karnes J. H., Bastarache L., Shaffer C. M., Gaudieri S., Xu Y., Glazer A. M., Mosley J. D., Zhao S., Raychaudhuri S., Mallal S., Ye Z., Mayer J. G., Brilliant M. H., Hebbring S. J., Roden D. M., et al. (2017) Phenome-wide scanning identifies multiple diseases and disease severity phenotypes associated with HLA variants. Sci. Transl. Med. 9, eaai8708 10.1126/scitranslmed.aai8708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lin H., and Gong Y.-Z. (2017) Association of HLA-B27 with ankylosing spondylitis and clinical features of the HLA-B27-associated ankylosing spondylitis: a meta-analysis. Rheumatol. Int. 37, 1267–1280 10.1007/s00296-017-3741-2 [DOI] [PubMed] [Google Scholar]
- 11. Bowness P. (2015) HLA-B27. Annu. Rev. Immunol. 33, 29–48 10.1146/annurev-immunol-032414-112110 [DOI] [PubMed] [Google Scholar]
- 12. Allen R. L., O'Callaghan C. A., McMichael A. J., and Bowness P. (1999) Cutting edge: HLA-B27 can form a novel β2-microglobulin-free heavy chain homodimer structure. J. Immunol. 162, 5045–5048 [PubMed] [Google Scholar]
- 13. Bird L. A., Peh C. A., Kollnberger S., Elliott T., McMichael A. J., and Bowness P. (2003) Lymphoblastoid cells express HLA-B27 homodimers both intracellularly and at the cell surface following endosomal recycling. Eur. J. Immunol. 33, 748–759 10.1002/eji.200323678 [DOI] [PubMed] [Google Scholar]
- 14. Kollnberger S., Bird L., Sun M. Y., Retiere C., Braud V. M., McMichael A., and Bowness P. (2002) Cell-surface expression and immune receptor recognition of HLA–B27 homodimers. Arthritis Rheum. 46, 2972–2982 10.1002/art.10605 [DOI] [PubMed] [Google Scholar]
- 15. Makhadiyeva D., Lam L., Moatari M., Vallance J., Zheng Y., Campbell E. C., and Powis S. J. (2012) MHC class I dimer formation by alteration of the cellular redox environment and induction of apoptosis. Immunology 135, 133–139 10.1111/j.1365-2567.2011.03518.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Santos S. G., Lynch S., Campbell E. C., Antoniou A. N., and Powis S. J. (2008) Induction of HLA-B27 heavy chain homodimer formation after activation in dendritic cells. Arthritis Res. Ther. 10, R100 10.1186/ar2492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bowness P., Ridley A., Shaw J., Chan A. T., Wong-Baeza I., Fleming M., Cummings F., McMichael A., and Kollnberger S. (2011) Th17 cells expressing KIR3DL2+ and responsive to HLA-B27 homodimers are increased in ankylosing spondylitis. J. Immunol. 186, 2672–2680 10.4049/jimmunol.1002653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cauli A., Shaw J., Giles J., Hatano H., Rysnik O., Payeli S., McHugh K., Dessole G., Porru G., Desogus E., Fiedler S., Hölper S., Carette A., Blanco-Gelaz M. A., Vacca A., et al. (2013) The arthritis-associated HLA-B*27:05 allele forms more cell surface B27 dimer and free heavy chain ligands for KIR3DL2 than HLA-B*27:09. Rheumatology 52, 1952–1962 10.1093/rheumatology/ket219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chan A. T., Kollnberger S. D., Wedderburn L. R., and Bowness P. (2005) Expansion and enhanced survival of natural killer cells expressing the killer immunoglobulin-like receptor KIR3DL2 in spondylarthritis. Arthritis Rheum. 52, 3586–3595 10.1002/art.21395 [DOI] [PubMed] [Google Scholar]
- 20. Wong-Baeza I., Ridley A., Shaw J., Hatano H., Rysnik O., McHugh K., Piper C., Brackenbridge S., Fernandes R., Chan A., Bowness P., and Kollnberger S. (2013) KIR3DL2 binds to HLA-B27 dimers and free H chains more strongly than other HLA class I and promotes the expansion of T cells in ankylosing spondylitis. J. Immunol. 190, 3216–3224 10.4049/jimmunol.1202926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Antoniou A. N., Ford S., Taurog J. D., Butcher G. W., Powis S. J. (2004) Formation of HLA-B27 homodimers and their relationship to assembly kinetics. J. Biol. Chem. 279, 8895–8902 10.1074/jbc.M311757200 [DOI] [PubMed] [Google Scholar]
- 22. Gonzalez-Galarza F. F., Christmas S., Middleton D., and Jones A. R. (2011) Allele frequency net: a database and online repository for immune gene frequencies in worldwide populations. Nucleic Acids Res. 39, D913–D919 10.1093/nar/gkq1128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Robinson J., Halliwell J. A., Hayhurst J. D., Flicek P., Parham P., and Marsh S. G. (2015) The IPD and IMGT/HLA database: allele variant databases. Nucleic Acids Res. 43, D423–D431 10.1093/nar/gku1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Robinson J., Soormally A. R., Hayhurst J. D., and Marsh S. (2016) The IPD-IMGT/HLA Database—new developments in reporting HLA variation. Hum. Immunol. 77, 233–237 10.1016/j.humimm.2016.01.020 [DOI] [PubMed] [Google Scholar]
- 25. Khan M. A. (2010) Remarkable polymorphism of HLA-B27: an ongoing saga. Curr. Rheumatol. Rep. 12, 337–341 10.1007/s11926-010-0126-x [DOI] [PubMed] [Google Scholar]
- 26. Reveille J. D., and Maganti R. M. (2009) Subtypes of HLA-B27: history and implications in the pathogenesis of ankylosing spondylitis. Adv. Exp. Med. Biol. 649, 159–176 10.1007/978-1-4419-0298-6_12 [DOI] [PubMed] [Google Scholar]
- 27. Khan M. A. (2000) Update: the twenty subtypes of HLA-B27. Curr. Opin. Rheumatol. 12, 235–238 10.1097/00002281-200007000-00001 [DOI] [PubMed] [Google Scholar]
- 28. Giquel B., Carmouse S., Denais C., Cherfa A., Chimenti M. S., Fert I., Hacquard-Bouder C., Breban M., and André C. (2007) Two HLA–B27 alleles differently associated with spondylarthritis, B*2709 and B*2705, display similar intracellular trafficking and oligomer formation. Arthritis Rheum. 56, 2232–2242 10.1002/art.22725 [DOI] [PubMed] [Google Scholar]
- 29. McHugh K., Rysnik O., Kollnberger S., Shaw J., Utriainen L., Al-Mossawi M. H., Payeli S., Belaunzaran O. M., Milling S., Renner C., and Bowness P. (2014) Expression of aberrant HLA-B27 molecules is dependent on B27 dosage and peptide supply. Ann. Rheum. Dis. 73, 763–770 10.1136/annrheumdis-2012-203080 [DOI] [PubMed] [Google Scholar]
- 30. Yu H.-C., Huang K.-Y., Lu M.-C., Huang H.-L., Liu S.-Q., Lai N.-S., and Huang H.-B. (2017) Targeted delivery of the HLA-B*27-binding peptide into the endoplasmic reticulum suppresses the IL-23/IL-17 axis of immune cells in spondylarthritis. Mediators Inflamm. 2017, 4016802 10.1155/2017/4016802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Armas J. B., Gonzalez S., Martinez-Borra J., Laranjeira F., Ribeiro E., Correia J., Ferreira M. L., Toste M., López-Vazquez A., and López-Larrea C. (1999) Susceptibility to ankylosing spondylitis is independent of the Bw4 and Bw6 epitopes of HLA-B27 alleles. Tissue Antigens 53, 237–243 10.1034/j.1399-0039.1999.530303.x [DOI] [PubMed] [Google Scholar]
- 32. Gonzalez-Roces S., Alvarez M. V., Gonzalez S., Dieye A., Makni H., Woodfield D. G., Housan L., Konenkov V., Abbadi M. C., Grunnet N., Coto E., and López-Larrea C. (1997) HLA-B27 polymorphism and worldwide susceptibility to ankylosing spondylitis. Tissue Antigens 49, 116–123 10.1111/j.1399-0039.1997.tb02724.x [DOI] [PubMed] [Google Scholar]
- 33. Liu X., Hu L. H., Li Y. R., Chen F. H., Ning Y., and Yao Q. F. (2010) The association of HLA-B*27 subtypes with ankylosing spondylitis in Wuhan population of China. Rheumatol. Int. 30, 587–590 10.1007/s00296-009-1018-0 [DOI] [PubMed] [Google Scholar]
- 34. Ben Radhia K., Ayed-Jendoubi S., Sfar I., Ben Romdhane T., Makhlouf M., Gorgi Y., and Ayed K. (2008) Distribution of HLA-B*27 subtypes in Tunisians and their association with ankylosing spondylitis. Joint Bone Spine 75, 172–175 10.1016/j.jbspin.2007.05.020 [DOI] [PubMed] [Google Scholar]
- 35. Schittenhelm R. B., Lim Kam Sian T. C. C., Wilmann P. G., Dudek N. L., and Purcell A. W. (2015) Revisiting the arthritogenic peptide theory: quantitative not qualitative changes in the peptide repertoire of HLA–B27 allotypes. Arthritis Rheumatol. 67, 702–713 10.1002/art.38963 [DOI] [PubMed] [Google Scholar]
- 36. Perosa F., Luccarelli G., Prete M., Favoino E., Ferrone S., and Dammacco F. (2003) β2-Microglobulin-free HLA class I heavy chain epitope mimicry by monoclonal antibody HC-10-specific peptide. J. Immunol. 171, 1918–1926 10.4049/jimmunol.171.4.1918 [DOI] [PubMed] [Google Scholar]
- 37. Stam N. J., Vroom T. M., Peters P. J., Pastoors E. B., and Ploegh H. L. (1990) HLA-A- and HLA-B-specific monoclonal antibodies reactive with free heavy chains in Western blots, in formalin-fixed, paraffin-embedded tissue sections and in cryo-immuno-electron microscopy. Int. Immunol. 2, 113–125 10.1093/intimm/2.2.113 [DOI] [PubMed] [Google Scholar]
- 38. Edwards P. A. W., Smith C. M., Neville M. A., and O'Hare M. J. (1982) A human-human hybridoma system based on a fast-growing mutant of the ARH-77 plasma cell leukemia-derived line. Eur. J. Immunol. 12, 641–648 10.1002/eji.1830120804 [DOI] [PubMed] [Google Scholar]
- 39. Schittenhelm R. B., Dudek N. L., Croft N. P., Ramarathinam S. H., and Purcell A. W. (2014) A comprehensive analysis of constitutive naturally processed and presented HLA-C*04:01 (Cw4)–specific peptides. Tissue Antigens 83, 174–179 10.1111/tan.12282 [DOI] [PubMed] [Google Scholar]
- 40. Storkus W. J., Howell D. N., Salter R. D., Dawson J. R., and Cresswell P. (1987) NK susceptibility varies inversely with target cell class I HLA antigen expression. J. Immunol. 138, 1657–1659 [PubMed] [Google Scholar]
- 41. Zemmour J., Little A. M., Schendel D. J., and Parham P. (1992) The HLA-A, B “negative” mutant cell line C1R expresses a novel HLA-B35 allele, which also has a point mutation in the translation initiation codon. J. Immunol. 148, 1941–1948 [PubMed] [Google Scholar]
- 42. Khan M. A., Mathieu A., Sorrentino R., and Akkoc N. (2007) The pathogenetic role of HLA-B27 and its subtypes. Autoimmun. Rev. 6, 183–189 10.1016/j.autrev.2006.11.003 [DOI] [PubMed] [Google Scholar]
- 43. Ziegler A., Loll B., Misselwitz R., and Uchanska-Ziegler B. (2009) Implications of structural and thermodynamic studies of HLA-B27 subtypes exhibiting differential association with ankylosing spondylitis. Adv. Exp. Med. Biol. 649, 177–195 10.1007/978-1-4419-0298-6_13 [DOI] [PubMed] [Google Scholar]
- 44. Ladell K., Hashimoto M., Iglesias M. C., Wilmann P. G., McLaren J. E., Gras S., Chikata T., Kuse N., Fastenackels S., Gostick E., Bridgeman J. S., Venturi V., Arkoub Z. A., Agut H., van Bockel D. J., et al. (2013) A molecular basis for the control of preimmune escape variants by HIV-specific CD8+ T cells. Immunity 38, 425–436 10.1016/j.immuni.2012.11.021 [DOI] [PubMed] [Google Scholar]
- 45. Stewart-Jones G. B. E., di Gleria K., Kollnberger S., McMichael A. J., Jones E. Y., and Bowness P. (2005) Crystal structures and KIR3DL1 recognition of three immunodominant viral peptides complexed to HLA-B*2705. Eur. J. Immunol. 35, 341–351 10.1002/eji.200425724 [DOI] [PubMed] [Google Scholar]
- 46. Gras S., Burrows S. R., Kjer-Nielsen L., Clements C. S., Liu Y. C., Sullivan L. C., Bell M. J., Brooks A. G., Purcell A. W., McCluskey J., and Rossjohn J. (2009) The shaping of T cell receptor recognition by self-tolerance. Immunity 30, 193–203 10.1016/j.immuni.2008.11.011 [DOI] [PubMed] [Google Scholar]
- 47. Ellman G. L. (1959) Tissue sulfhydryl groups. Arch. Biochem. Biophys. 82, 70–77 10.1016/0003-9861(59)90090-6 [DOI] [PubMed] [Google Scholar]
- 48. Mage M. G., Dolan M. A., Wang R., Boyd L. F., Revilleza M. J., Robinson H., Natarajan K., Myers N. B., Hansen T. H., and Margulies D. H. (2012) The peptide-receptive transition state of MHC class I molecules: insight from structure and molecular dynamics. J. Immunol. 189, 1391–1399 10.4049/jimmunol.1200831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ellis S. A., Taylor C., and McMichael A. (1982) Recognition of HLA-B27 and related antigen by a monoclonal antibody. Hum. Immunol. 5, 49–59 10.1016/0198-8859(82)90030-1 [DOI] [PubMed] [Google Scholar]
- 50. Barnstable C. J., Bodmer W. F., Brown G., Galfre G., Milstein C., Williams A. F., and Ziegler A. (1978) Production of monoclonal antibodies to group A erythrocytes, HLA and other human cell surface antigens—new tools for genetic analysis. Cell 14, 9–20 10.1016/0092-8674(78)90296-9 [DOI] [PubMed] [Google Scholar]
- 51. Cowieson N. P., Aragao D., Clift M., Ericsson D. J., Gee C., Harrop S. J., Mudie N., Panjikar S., Price J. R., Riboldi-Tunnicliffe A., Williamson R., and Caradoc-Davies T. (2015) MX1: a bending-magnet crystallography beamline serving both chemical and macromolecular crystallography communities at the Australian Synchrotron. J. Synchrotron Radiat. 22, 187–190 10.1107/S1600577514021717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Read R. J. (2001) Pushing the boundaries of molecular replacement with maximum likelihood. Acta Crystallogr. D Biol. Crystallogr. 57, 1373–1382 10.1107/S0907444901012471 [DOI] [PubMed] [Google Scholar]
- 54. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bricogne G., Blanc E., Brandl M., Flensburg C., Keller P., Paciorek W., Roversi P., Sharff A., Smart O. S., Vonrhein C., and Womack T. O. (2017) BUSTER version 1.7.6.3., Global Phasing Ltd., Cambridge, UK [Google Scholar]
- 56. DeLano W. L. (2015) The PyMOL Molecular Graphics System, version 1.7.6.4, Schroedinger, LLC, New York [Google Scholar]
- 57. Collaborative Computational Project Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol Crystallogr. 760–763 10.1107/S0907444994003112 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.