

Abstract
Bone morphogenetic protein 10 (BMP10) is a cardiac peptide growth factor belonging to the transforming growth factor β superfamily that critically controls cardiovascular development, growth, and maturation. It has been shown that BMP10 elicits its intracellular signaling through a receptor complex of activin receptor-like kinase 1 with morphogenetic protein receptor type II or activin receptor type 2A. Previously, we generated and characterized a transgenic mouse line expressing BMP10 from the α-myosin heavy chain gene promoter and found that these mice have normal cardiac hypertrophic responses to both physiological and pathological stimuli. In this study, we report that these transgenic mice exhibit significantly reduced levels of cardiomyocyte apoptosis and cardiac fibrosis in response to a prolonged administration of the β-adrenoreceptor agonist isoproterenol. We further confirmed this cardioprotective function with a newly generated conditional Bmp10 transgenic mouse line, in which Bmp10 was activated in adult hearts by tamoxifen. Moreover, the intraperitoneal administration of recombinant human BMP10 was found to effectively protect hearts from injury, suggesting potential therapeutic utility of using BMP10 to prevent heart failure. Gene profiling and biochemical analyses indicated that BMP10 activates the SMAD-mediated canonical pathway and, unexpectedly, also the signal transducer and activator of transcription 3 (STAT3)–mediated signaling pathway both in vivo and in vitro. Additional findings further supported the notion that BMP10's cardioprotective function likely is due to its dual activation of SMAD- and STAT3-regulated signaling pathways, promoting cardiomyocyte survival and suppressing cardiac fibrosis.
Keywords: bone morphogenetic protein (BMP), SMAD transcription factor, STAT transcription factor, heart failure, cell death, fibrosis
Introduction
Heart failure is a leading cause of death globally. It can result from a variety of pathogenic events, including inheritable cardiomyopathies, structural anomalies from congenital heart defects, ischemia/reperfusion injury, viral myocarditis, and exposure to cardiotoxic pharmacological agents. Despite the broad range of triggers, the progression of heart failure after the initial injury often follows a common pathway: cardiac injury or stress gives rise to cardiomyocyte death, which in turn gives rise to adverse cardiac remodeling (e.g. the recruitment and activation of fibroblasts and excessive deposition of extracellular matrix) and ultimately leads to the impairment of cardiac contractile function (1–3). The critical contribution of cardiomyocyte death to heart failure progression has been well-documented (1–3). Taking measures to slow or prevent cardiomyocytes from undergoing apoptosis and cardiac fibroblasts from depositing excessive extracellular matrix are considered key steps in preventing the progression of heart failure (1–3).
BMP10 is a peptide growth factor belonging to the TGFβ5 superfamily (6, 7). BMP family members mediate a diverse spectrum of developmental events throughout evolution in species ranging from insects to mammals (8, 9). It has been shown that BMP10, as well as BMP9, elicits intracellular signaling through the receptor complex of ALK1 and morphogenetic protein receptor type II or activin receptor type 2A (10, 11). Cardiac expression of BMP10 is observed in chick, mouse, and human (6, 7, 12–16). In normal developing hearts, BMP10 is more enriched in trabecular myocardium (17) and then becomes restricted to the right atrium in postnatal hearts (17). Interestingly, BMP10 transcription is reported to be elevated in adult ventricular cardiomyocytes in response to hypertension, suggesting the involvement of BMP10 in postnatal cardiac physiology (14, 16). However, BMP10 expression is not altered in response to β-adrenergic agonist isoproterenol from previous published work (18, 19) and their deposited data of transcriptomic analyses of β-adrenergic agonist isoproterenol (ISO)–regulated genes (GSE102612 and GSE18801). Previously, we utilized the mouse α-myosin heavy chain (αMHC) promoter to express mouse Bmp10 specifically in postnatal cardiomyocytes (20, 21). Although the transgenic mice exhibited normal embryonic cardiac development, there was a dramatic inhibition of perinatal-to-postnatal cardiac hypertrophic growth (20), which resulted in a significant reduction in cardiomyocyte size, as well as overall heart size, in adult αMHC–Bmp10 mice. In the current study, we tested whether αMHC–Bmp10 transgenic hearts had a normal response to β-adrenergic stimulation using ISO treatment. Despite a marked and proportionally similar increase in cardiac mass in ISO-treated αMHC–Bmp10 mice and nontransgenic littermates (20), cardiomyocyte apoptosis and myocardial fibrosis were markedly blunted in the αMHC–Bmp10 hearts, suggesting that BMP10 was able to promote cardiomyocyte survival and prevent cardiac adverse remodeling. Additional experiments with the de novo activation of BMP10 in the postnatal heart and the treatment of recombinant human BMP10 (rhBMP10) further confirmed this observation. More importantly, transcriptomic and biochemical analyses suggested a novel activity of BMP10 in activating both SMAD- and STAT3-mediated signaling pathways, which likely contributed to BMP10's cardioprotective activity.
Results
Assessment of cardiac function in αMHC–Bmp10 mice in response to prolonged isoproterenol treatment
In an attempt to determine whether elevated levels of Bmp10 expression impacted pathological hypertrophic growth induced by the β-adrenergic agonist ISO, we performed a series of morphological and histological analyses on the ISO-treated αMHC–Bmp10 (TG) and nontransgenic (NTG) littermate hearts and compared them with age-matched saline-treated αMHC–Bmp10 and nontransgenic littermate control hearts. ISO treatment (Alzet osmotic mini-pumps, model 2001, flow rate of 1 μl/h, 0.028 g/ml ISO dissolved in saline) for 7 days in mice normally increases cardiac size 30–40% and reduces cardiac systolic function, accompanied by prominent cardiomyocyte apoptosis and cardiac fibrosis. Previously, we demonstrated that the elevated Bmp10 level did not prevent pathological hypertrophic growth (20). Our current data further demonstrated that cardiac Bmp10 overexpression dramatically reduced the level of cardiac fibrosis as shown by Masson's trichrome staining and qRT-PCR analyses of collagen (e.g. Col1a1 and Col3a1) expression (Fig. 1A).
Figure 1.

Preserved cardiac function in αMHC–Bmp10 mice in response to prolonged isoproterenol stimulation. A, Masson's trichrome staining showing a significantly reduced level of collagen deposition in ISO-treated TG heart compared with ISO-treated littermate NTG control heart (left panel); quantification of fibrosis area using ImageJ software (middle panel); qRT-PCR analysis to determine the mRNA expression level of Col1a1 and Col3a1 (right two panels). B, echocardiographic analysis of cardiac systolic function of ISO-treated TG mice and ISO-treated littermate NTG control mice (left panel); compared with the reduced level of fraction shortening (FS%) and ejection fraction (EF%) in ISO-treated littermate NTG mice, ISO-treated TG mice preserved a normal cardiac function. C, the number of apoptotic cells was significantly less in the ISO-treated TG hearts compared with NTG control hearts; shown is a representative image of activated caspase 3 immunohistochemical staining, the dark brown signal with a long, rough shape indicated an activated caspase 3–positive cardiomyocyte (inset). D, Western blotting analysis of the level of cleaved PARP, confirming the reduced level of cell death in ISO-treated TG hearts. E, Western blotting analysis of the level of cell survival factors, confirming the up-regulation of Bcl-xL in ISO-treated TG hearts. The quantification of the Western blotting is shown in Fig. S1.
To assess cardiac function, noninvasive echocardiography was performed to determine the impact of ISO infusion on systolic function in αMHC–Bmp10 mice. As demonstrated in Fig. 1B, cardiac overexpression of Bmp10 significantly preserved cardiac systolic function when compared with ISO treatment in nontransgenic mice. Importantly, using immunohistochemical staining for activated caspase 3, we found a significant reduction in apoptotic cardiomyocytes in ISO-treated αMHC–Bmp10 hearts (Fig. 1C, left panel, inset, shows a representative image of a cardiomyocyte positive for activated caspase-3 immune reactivity). Poly(ADP-ribose) polymerase (PARP) is a nuclear polymerase, and its substrate caspase 3 is commonly used as a molecular marker for apoptosis (as inferred by the detection of a PARP cleavage product) (22–24). Western blotting analysis of the level of cleaved PARP showed a great reduction in Bmp10 transgenic hearts compared with nontransgenic hearts (Fig. 1D and Fig. S1A). In addition, consistent with the above findings, the cell survival factor Bcl-xL was up-regulated in Bmp10 transgenic hearts compared with nontransgenic control hearts (Fig. 1E and Fig. S1B). These observations suggested that Bmp10 was able to promote cardiomyocyte survival, reduce cardiac fibrosis, and preserve cardiac function in response to cardiac pathological stimulation.
De novo activation of BMP10 in postnatal hearts
Although the above data suggested that BMP10 was an important cardioprotective molecule, interpreting the data was complicated because of the reduced cardiomyocyte sizes in αMHC–Bmp10 transgenic hearts. It was possible that the apparent cardioprotective phenotype could simply be a secondary phenomenon resulting from smaller cardiomyocytes (20). To circumvent this problem, we generated an inducible Bmp10 transgenic mouse model (αMHC–eGFPf–Bmp10) (Fig. 2A). This model used a floxed-eGFP–stop cassette (eGFPf) that was placed between the mouse αMHC promoter and a cDNA fragment encoding the full-length mouse Bmp10. In the absence of Cre recombinase, Bmp10 expression was silent because of the eGFPf cassette. Following exposure to Cre or inducible Cre recombinase (e.g. MerCreMer), Bmp10 expression was activated because of the removal of the eGFPf cassette (Fig. 2A). αMHC–eGFPf–Bmp10 mice exhibited a normal life span and growth and normal cardiac histology (Fig. 2B) and function. We confirmed the activation of Bmp10 expression in αMHC–eGFPf–Bmp10 by crossing αMHC–MerCreMer mice (25), in which the MerCreMer transgenic line expressed a Cre recombinase protein fused to two mutant estrogen-receptor (Mer) ligand-binding domains. Cre recombinase activity depended upon the presence of the estrogen analog tamoxifen. Tamoxifen treatment resulted in the activation of Bmp10 expression in αMHC–eGFPf–Bmp10:αMHC–MerCreMer hearts, as evidenced by qRT-PCR (Fig. 2C).
Figure 2.
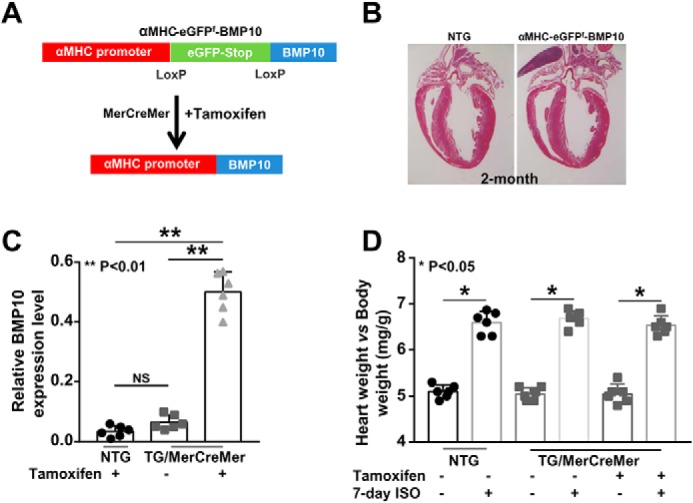
De novo activation of Bmp10 in mouse adult heart. A, a schematic diagram of the inducible Bmp10 transgenic mouse model and the de novo activation of Bmp10 in mouse adult hearts using an inducible Cre-LoxP transgenic strategy. B, hematoxylin and eosin staining of αMHC–eGFPf–Bmp10 and NTG control hearts to show normal cardiac histology in transgenic mice. C, validation of the successful activation of Bmp10 expression in adult ventricle by tamoxifen. D, normal cardiac hypertrophic response in tamoxifen-induced αMHC–eGFPf–Bmp10 mice in response to ISO treatment. NS, not significant.
De novo Bmp10 induction improves cardiac function in response to isoproterenol infusion
To determine whether de novo Bmp10 expression in postnatal hearts exerted cardioprotective activity, we subjected 2-month-old αMHC–eGFPf–Bmp10:αMHC–MerCreMer mice (with or without tamoxifen induction), as well as nontransgenic mice, to ISO infusion for 7 days as previously described (20). We compared the degree of cardiac hypertrophy (Fig. 2D). All mice, regardless of genotype or tamoxifen treatment, exhibited a comparable increase in heart weight/body weight ratio, indicating that de novo Bmp10 expression did not alter the hypertrophic response to isoproterenol treatment, which was consistent with our previous finding in the constitutive αMHC–Bmp10 model (20). Importantly, by Sirius red/fast green staining, hearts from the tamoxifen-induced αMHC–eGFPf–Bmp10:αMHC–MerCreMer mice had a significantly reduced level of collagen deposition following 7-day ISO treatment compared with the hearts from αMHC–eGFPf–Bmp10:αMHC–MerCreMer mice treated with vehicle (Fig. 3A). By immunohistological staining, there was a significant reduction in cardiomyocyte-activated caspase-3 immune reactivity in hearts from ISO-treated tamoxifen-induced αMHC–eGFPf–Bmp10:αMHC–MerCreMer transgenic mice compared with mice lacking tamoxifen induction (Fig. 3B; inset: a representative image of a cardiomyocyte positive for activated caspase-3 immune reactivity). The levels of cleaved PARP were also greatly reduced in hearts from ISO-treated tamoxifen-induced αMHC–eGFPf–Bmp10:αMHC–MerCreMer mice compared with the control hearts without tamoxifen induction (Fig. 3C and Fig. S2).
Figure 3.

De novo activation of Bmp10 preserves cardiac function. A, Sirus red/fast green staining showing a significantly reduced level of collagen deposition (red staining signal) in tamoxifen-induced transgenic heart in response to ISO treatment. B, significantly reduced number of activated caspase 3–positive cardiomyocytes in tamoxifen-induced transgenic hearts with ISO treatment. C, significantly reduced level of cleaved PARP in tamoxifen-induced transgenic hearts with ISO treatment. The quantification of the Western blotting is in Fig. S2. D, well-preserved cardiac systolic function in tamoxifen-induced transgenic mice with ISO treatment. E, i.p. injection of rhBMP10 into WT mice (DBA/2J) reduced cardiac fibrosis in response to ISO treatment compared with saline-injected control mice in response to ISO treatment. F, i.p. injection of rhBMP10 into WT mice (DBA/2J) significantly preserved cardiac systolic function in response to ISO treatment. EF%, ejection fraction; FS%, fractional shortening.
To assess cardiac function, echocardiography was performed to assess the impact of isoproterenol infusion on systolic function in αMHC–eGFPf–Bmp10:αMHC–MerCreMer mice in the presence and absence of tamoxifen induction. As demonstrated in Fig. 3D, the induction of myocardial Bmp10 expression preserved cardiac systolic function well. Collectively, these data confirmed that Bmp10 had a strong cardioprotective activity.
To further test whether this BMP10-mediated cardioprotection could be achieved by intraperitoneal delivery of exogenous BMP10, we injected rhBMP10 (500 ng/day/mouse, R&D Systems) daily into mice during 7-day ISO administration, which was followed by echocardiographic and histological analyses. As demonstrated in Fig. 3 (E and F), the administration of rhBMP10 effectively preserved cardiac function and significantly reduced the cardiac fibrosis induced by prolonged ISO treatment.
Altered SMAD- and STAT3-mediated signaling pathways in BMP10 transgenic hearts
Previously, we used an Affymetrix mouse exon array to compare the transcriptomes between αMHC–Bmp10 transgenic and nontransgenic littermate hearts (20, 21). Based on the chosen false discovery rate (<0.05) and the fold change (over 1.5), we re-evaluated the data set and found a total of 402 genes to be differentially expressed between αMHC–Bmp10 transgenic hearts and NTG control hearts (Fig. 4A). Among these 402 genes, 219 were up-regulated, and 183 were down-regulated (Fig. 4A). Gene Ontology analysis of these differentially expressed genes indicated that cell adhesion and angiogenesis were among the top affected biological processes, extracellular region and extracellular matrix were among the top affected cellular components, and heparin binding (key regulator of BMP signaling) and other cell membrane-mediated signaling pathways were the top affected molecular functions (Fig. 4B). These findings were further confirmed by Kyoto Encyclopedia of Gene and Genomes analysis, which showed that TGFβ and its mediated pathways were the top affected signaling pathways in Bmp10 transgenic hearts (Fig. 4C). We also used ingenuity pathway analysis (IPA) software to evaluate potential signaling cascades that were most altered in Bmp10 transgenic hearts. As expected, based on the p value and z score, TGFβ/BMP was confirmed as the top affected signaling pathway (Fig. 4, D–F). Interestingly, STAT3 was also among the top affected signaling pathways (Fig. 4, D, G, and H). This series of in silico analyses suggested that in addition to the commonly known TGFβ/BMP–Smad pathway (known as the canonical pathway), BMP10 was able to activate the STAT3-mediated pathway, which was likely an important noncanonical mediator of Bmp10 signaling and a key contributor to the cardioprotective phenotype observed in Bmp10 transgenic hearts, especially considering that STAT3 is a critical cell survival regulator.
Figure 4.

In silico analysis of altered signaling pathways in BMP10 transgenic hearts. A, volcano plots of gene expression profiles using a previously published gene array data set comparing 4-week-old TG hearts versus NTG hearts. B, functional categorization of all differentially expressed genes based on Gene Ontology annotations. C, pathway analysis of all differentially expressed genes by the Kyoto Encyclopedia of Genes and Genomes (KEGG). D, top 10 upstream regulators predicted by IPA. The predicted regulators were sorted by overlapping p value, predicted activation state, and activation Z score. E, regulatory network of the BMP/TGFβ signaling pathway in TG hearts. The color intensity of the nodes indicates the expression level of the genes, with red representing up-regulation and green representing down-regulation. Connecting lines show the predicted relationships to BMP/TGFβ signaling, with orange representing predicted activation, blue representing inhibition, yellow representing inconsistency, and gray representing “to be determined.” The direct interaction is represented as a solid line, whereas indirect interaction is indicated by a dotted line. F, heat map of BMP/TGFβ regulated differentially expressed genes in TG versus NTG hearts. G, regulatory network of STAT3 signal pathway in TG hearts. H, heat map of STAT3-regulated differentially expressed genes in TG versus NTG hearts. ECM, extracellular matrix; PI3K, phosphatidylinositol 3-kinase.
Confirmation of the role of BMP10 in activating both SMAD and STAT3 signaling
In general, through binding to type I and II serine/threonine kinase receptors, BMPs exert their biological activity by phosphorylating Smad1/5/8. In addition, numerous studies have also suggested that several Smad-independent noncanonical signaling pathways are involved in regulating TGFβ/BMP-mediated cellular function, such as TAK1/MEKK1, Ras, Rho, and PP2A pathways (26, 27). Previously, we have shown Smad1/5/8 was persistently up-regulated in αMHC–Bmp10 (20). To analyze the potential activation of STAT3, as well as other noncanonical signaling pathways downstream of TGFβ/BMPs, as shown in Fig. 5A, Western blotting analysis demonstrated that pY-STAT3 was up-regulated in BMP10 transgenic hearts, which was consistent with IPA analysis. In contrast, the TAK1–MEKK1–mediated pathway appeared to be unaffected by overexpression of Bmp10, because the levels of phospho-p38 and pAkt were similar between αMHC–Bmp10 and NTG control hearts (Fig. 5A and Fig. S3A). Immunohistochemical staining was used to confirm that STAT3 was indeed activated in αMHC–Bmp10 transgenic cardiomyocytes (Fig. 5B, red arrows indicate positive nuclear staining). To determine whether the activation of pY-STAT3 was a specific function of BMP10, we treated P19 cells with rhBMP10 in vitro. P19 is a well-characterized mouse embryo-derived teratocarcinoma cell line that can be differentiated into cardiomyocytes and is widely used to study BMP signaling (28–30). We examined pSmad1/5/8 or pY-STAT3 levels in a dose-response assay by incubating these cells with rhBMP10 or rhBMP4 for 60 min, respectively (Fig. 5C and Fig. S3B). As expected, Smad1/5/8 was activated by rhBMP10. Interestingly, STAT3 activation by rhBMP10 exhibited a bell-shaped dose-response curve, with the maximal effect observed at a concentration of 50 ng/ml (Fig. 5D and Fig. S3C). Interestingly, rhBMP4 did not effectively activate STAT3 in P19 cells, although Smad1/5/8 was efficiently activated (Fig. 5C; note that the blot for analyzing pY-STAT3 and STAT3 protein levels was the one used for pSmad1/5/8 and anti-Smad1 analysis (20)). This finding suggested that the activation of STAT3 was a direct Bmp10-dependent event and not a Smad-mediated secondary event. Consistent with this notion, the time course for STAT3 activation indicated that pY-STAT3 was activated within 15 min of the rhBMP10 treatment (Fig. 5D and Fig. S3C). It has been shown that JAK1 interacts with ALK5 (TGFβR1) and is involved in STAT3 activation by TGFβ. To determine whether JAK1 is also involved in BMP10-mediated STAT3 activation, we applied JAK1 inhibitor (AG490, 5 μm) to rhBMP10 (50 ng/ml)–stimulated cells. As shown in Fig. 5E, AG490 was able to inhibit BMP10-mediated STAT3 activation, suggesting that JAK1 was involved in BMP10-mediated STAT3 activation.
Figure 5.

Biochemical analysis of BMP10-mediated activation of STAT3. A, Western blotting analysis of key signaling molecules in TG and NTG hearts showing STAT3 was significantly activated. B, immunohistochemical analysis of the subcellular localization of STAT3 in TG and NTG hearts, with positive nuclear STAT3 staining (red arrows) indicating the activation of STAT3 and its mediated signaling pathway. C, Western blotting analysis of STAT3 activation in P19 cells in response to rhBMP10 and rhBMP4 at different concentrations compared with Smad activation. D, Western blotting analysis of the time course of STAT3 activation by BMP10. E, Western blotting analysis of STAT3 activation in response to BMP10 and JAK1 inhibitor AG490. After overnight starvation, the cells are treated with rhBMP10 (50 ng/ml) with either DMSO or AG490 (5 μm in DMSO). The quantification of the Western blotting is shown in Fig. S3.
Taken together, these data suggested that STAT3 activation was a direct, specific, and immediate response to BMP10 stimulation and was likely an important effector in the BMP10 signaling pathway. Collectively, the above data also suggest that BMP10 can simultaneously activate both the Smad-mediated pathway (canonical pathway) and STAT3-mediated signaling pathway (noncanonical pathway).
STAT3 deficiency reduces Bmp10-mediated cardioprotection
As shown in Fig. 3E, using the prolonged ISO treatment model, rhBMP10 could well-protect the hearts from ISO-induced loss of cardiac systolic function. Previously, we showed that STAT3 was an important cardioprotective factor in response to ISO treatment (31). To test whether STAT3 was essential to BMP10-mediated cardioprotection, we administrated rhBMP10 (500 ng/daily) to STAT3 cardiomyocyte-specific knockout (STAT3cKO) mice. As demonstrated in Fig. 6A, rhBMP10 failed to protect cardiac function in STAT3cKO hearts in response to 7-day ISO treatment as measured by echocardiographic analysis (Fig. 6A). Consistent with this observation, the activated caspase 3 index was comparable between ISO-treated STAT3cKO hearts with rhBMP10 treatment and ISO-treated STAT3cKO hearts without rhBMP10 treatment (Fig. 6B), suggesting that myocardial STAT3 was essential for BMP10-mediated cardiomyocyte survival. However, when we assessed the state of cardiac fibrosis using Sirus red/fast green staining, a significantly reduced level of extracellular matrix deposition was found in ISO-treated STAT3cKO hearts with rhBMP10 treatment compared with ISO-treated STAT3cKO hearts without rhBMP10 treatment (Fig. 6C), suggesting that the reduced level of collagen deposition in BMP10-treated hearts was likely independent of myocardial STAT3. Taken together, this finding confirmed that STAT3 was essential for BMP10-mediated cardioprotection by preventing cardiomyocyte death.
Figure 6.

The loss of BMP10-mediated cardiac protection in STAT3 cardiomyocyte-restrictive knockout mice (STAT3cko). A, echocardiographic measurement to show a similarly reduced level of fraction shortening and ejection fraction in ISO-treated STAT3cKO mice with or without rhBMP10 treatment. B, comparable number of activated caspase 3–positive cardiomyocytes in ISO-treated STAT3cKO heart with or without rhBMP10 treatment. C, representative images of Sirus red/fast green staining (left panel). The level of cardiac fibrosis was significantly reduced in ISO-treated STAT3cKO mice with rhBMP10 treatment compared with ISO-treated STAT3cKO mice without rhBMP10 treatment. EF%, ejection fraction; FS%, fractional shortening.
BMP10-mediated inhibition of excessive extracellular matrix deposition
We were intrigued by the possibility that the BMP10 pathway was also involved in the inhibition of cardiac fibroblast activity. The major extracellular matrix proteins produced by fibroblasts in the heart are type I and type III collagens (e.g. Col1a1 and Col3a1). As shown in our gene array analysis, both type I and type III collagens were found down-regulated in Bmp10 transgenic hearts. To test whether BMP10 had a direct function on adult cardiac fibroblasts (ACFs), we isolated ACFs from adult mouse hearts to perform a [3H]proline incorporation assay as described under “Experimental procedures” in which we used TGFβ1 (1 ng/ml) as an inducer of collagen production. As shown in Fig. 7A, rhBMP10 (50 ng/ml) was able to effectively inhibit the production of collagen. Furthermore, qRT-PCR confirmed that both Col1a1 and Col3a1 transcripts were significantly reduced by rhBMP10 treatment (Fig. 7B). As shown in Fig. 7C, TGFβ1 and BMP10 effectively activated pSamd2/3 and pSmad1/5/8 in ACFs, respectively, confirming the involvement of Smad-mediated signaling in ACFs.
Figure 7.

BMP10-mediated inhibition of excessive extracellular matrix deposition. A, [3H]proline assay as described under “Experimental procedures” to determine the specific function of BMP10 in inhibiting collagen production in adult cardiac fibroblasts. B, qRT-PCR confirmation of both Col1a1 and Col3a1 mRNA levels in cultured cardiac fibroblasts treated with TGFβ1 (1 ng/ml) or combined treatment with TGFβ1 (1 ng/ml) and rhBMP10 (50 ng/ml). C, Western blotting confirmation of the activation of pSmad2/3 and pSmad1/5/8 by TGFβ1 and BMP10, respectively, to validate the cardiac fibroblast culture system. D, a representative data set of a qRT-PCR-based miRNA profiling of cardiac fibroblasts isolated from αMHC–Bmp10 hearts (4 weeks old) and littermate NTG control hearts. E, the relationship between cardiac fibrosis and the differentially expressed miRNAs analyzed by IPA, in which blue lines represent inhibition and yellow lines represent inconsistency. The color intensity of the nodes indicates the expression level of the genes, with red representing up-regulation and green representing down-regulation. F, the use of anti-miR29 anti-miRs to validate its role in Col1a1 and Col3a1 expression; anti-miR29 reduced BMP10's inhibition of Col1a1 and Col3a1 mRNA expression.
It has been shown that miRNAs play an important role in cardiac fibroblast proliferation and collagen production (32). We used a qRT-PCR-based miRNA panel to profile the expression of miRNAs in ACFs isolated from αMHC–Bmp10 transgenic and control littermate hearts (Fig. 7D). miR29, miR30, miR1, miR23, and miR199 were significantly up-regulated, and miR34 was significantly down-regulated. Among these, miR29c was the most elevated miRNA (over a 3-fold increase, p < 0.001). Interestingly, these miRNAs were previously known for their roles in regulating cardiac fibrosis either by inhibiting cardiac fibroblast proliferation (e.g. miR1) (33) or collagen production (e.g. miR29) (34) or by regulating cardiomyocyte survival (e.g. miR34) (35, 36). As shown in Fig. 7E, IPA disease and biological function enrichment analysis demonstrated a largely consistent function of these differentially expressed miRNAs in the reduced level of cardiac fibrosis in Bmp10 transgenic hearts.
Previously, miR29 was shown down-regulated in hearts in response to myocardial infarction (34) and ISO treatment (43). TGFβ also potently down-regulated miR29 in fibroblasts (34). Along with the known function of miR29 in inhibiting collagen production in cardiac fibroblasts (34), we thought it an important possibility that Bmp10, in addition to preventing cardiomyocyte death, was likely to act on the cardiac fibroblasts and to inhibit the deposition of excessive amounts of extracellular matrix. To determine whether the down-regulated levels of Col1a1 and Col3a1 in BMP10-treated ACFs were miR29-dependent, AFC cultures were supplemented with anti-miR29s. As shown in Fig. 7F, these anti-miRs significantly reduced BMP10-mediated activity, further supporting the role of miR29 in Bmp10-mediated inhibition of ECM deposition.
Discussion
BMPs belong to the TGFβ superfamily and mediate a diverse spectrum of morphogenetic events and physiological functions throughout evolution in species ranging from insects to mammals. Although all members of the BMP family share similar protein structures, each member has a unique protein sequence and displays different patterns of tissue distribution and unique biological functions (26, 37). There are approximately six BMP members found in the heart, including BMP2, BMP4, BMP5, BMP6, BMP7, and BMP10 (38). Among them, BMP10 has a rather specific and enriched expression pattern in the heart (17). In adult hearts, BMP10 mRNA is highly expressed in the right atria, whereas BMP10 protein can be detected throughout the heart. The biological function of BMP10 in adult hearts is largely elusive. Our current study suggests that BMP10 is likely an important cardiac cytokine that protects the heart from potential injuries caused by various stresses and cardiotoxicities. Future studies of Bmp10 cardiac-restrictive knockout will help to test this hypothesis and will determine whether this cardioprotective phenomenon reflects the actual physiological function of BMP10 or simply the gain-of-function effect of BMP10. Nevertheless, this study suggests a potential therapeutic use of BMP10 in combating the pathogenesis of heart failure.
Our analyses also demonstrate that BMP10 activated two important intracellular pathways (Fig. 8), the Smad-mediated canonical pathway and the STAT3-mediated noncanonical pathway. Based on our phenotypic analysis of STAT3cKO hearts, the STAT3-mediated pathway played an essential role in BMP10 cardioprotection most like via its regulation of cardiomyocyte survival. This BMP10-mediated immediate/direct STAT3 activation was supported by the data from transcriptomic and biochemical analyses. The cross-talk between STAT3-mediated and Smad-mediated pathways is well-recognized (39, 40). However, this direct activation of STAT3 by BMP10 was initially unexpected. A recent study by Tang et al. (41) provided a potential clue. They demonstrated that JAK1 constitutively interacted with TGFβR1 (also known as ALK5) and that TGFβ was able to directly activate the JAK1–STAT3 pathway independent of Smad3 activation in hepatic cell lines (41). More interestingly, this immediate and Smad3-independent activation of STAT3 was hypersensitive to TGFβ stimulation, which was followed by a second phase of STAT3 activation, a Smad3-dependent process (41). This prior study was consistent with our findings, suggesting that the immediate activation of STAT3 by BMP10 was not dependent upon Smad activation. Intriguingly, receptor ALK1 was known to serve as the type 1 receptor for BMP10, BMP9, and TGFβ (11, 42). This unique characteristic of ALK1 raised the possibility that ALK1 was similar to ALK5 in its direct interaction with JAK1. Additional biochemical analyses in the future will further verify this potential underlying mechanism.
Figure 8.
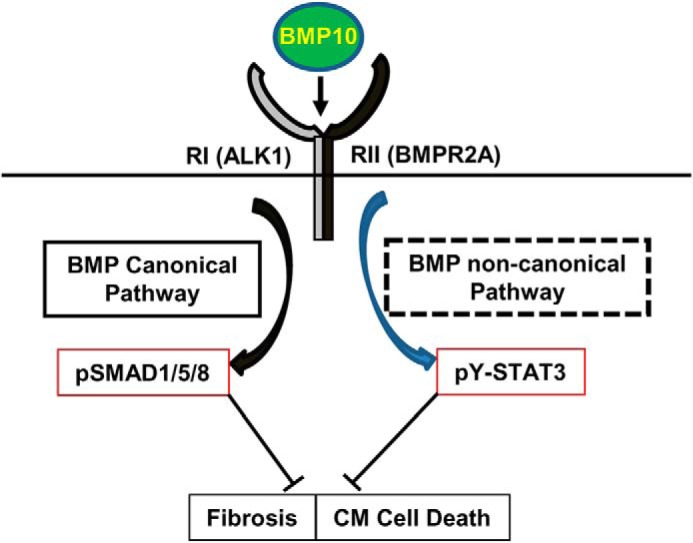
Schematic diagram of BMP10-mediated dual activation of Smad and STAT3 pathways. BMP10 is able to activate Smad-mediated canonical pathway and potentially STAT3-mediated noncanonical pathway, both of which likely contribute to preserving the normal cardiac function via the inhibition of cardiomyocyte death and cardiac fibrosis. Future work will determine the underlying mechanism for BMP10-mediated STAT3 activation.
Another important finding of our study was that BMP10 was able to regulate miRNA expression in AFCs. Interestingly, many of the miRNAs had an important role in regulating cardiomyocyte survival and cardiac fibrosis. In the current study, we showed that the up-regulation of miR29 in BMP10-treated AFCs reduced collagen production. miR29 has been shown down-regulated in hearts in response to myocardial infarction, β-adrenergic agonist phenylephrine treatment (34), and ISO treatment (43). TGFβ also potently down-regulates miR29 in fibroblasts (34). It will be interesting to determine the underlying molecular mechanism by which BMP10 regulates the expression of these miRNAs in cardiomyocyte survival/death and cardiac fibrosis, especially whether the up-regulation of miR29 depends on BMP10-Smad canonical pathway or BMP10–STAT3 noncanonical pathways.
In conclusion, our study revealed a novel function of BMP10 in preserving heart function in response to pathogenic stimulation by preventing cardiomyocyte death and inhibiting the excessive deposition of extracellular matrix. Our findings suggest a potential therapeutic use of BMP10 in preventing or slowing the pathogenesis of heart failure.
Experimental procedures
Antibodies and reagents
The antibodies used in the Western blots were as follows: anti-PARP1 (Abcam catalog no. ab191217), anti-STAT3 (Cell Signaling catalog no. 9139), anti-pSTAT3 (Cell Signaling catalog no. 9145), anti-Smad1 (Cell Signaling catalog no. 9743), anti-pSmad1/5/8 (Cell Signaling catalog no. 13820), anti-Akt (Cell Signaling catalog no. 4691), anti-pAkt (Cell Signaling catalog no. 4060), anti-p38 (Cell Signaling catalog no. 9212), anti-p-p38 (Cell Signaling catalog no. 4511), and anti-GAPDH (Proteintech catalog no. 60004-Ig). The anti-pSTAT3 (Cell Signaling catalog no. 9145) antibody was also used for immunohistochemical analysis.
Animals
The studies were approved by the Laboratory Animal Welfare & Ethics Committee of Jiangnan University (protocol no. 20180530c1851010) and Indiana University School of Medicine Institutional Animal Care and Research Advisory Committee. αMHC–BMP10 transgenic mice were generated and maintained as previously described (20). αMHC–eGFPf–Bmp10 mice were generated as previously described (20). A floxed-eGFP–stop cassette (eGFPf) was placed between the mouse αMHC promoter and a cDNA encoding the full-length mouse Bmp10, followed by SV40 early-region transcription termination and polyadenylation sites. The transgenic mice were generated at the mouse transgenic core of the Indiana University Cancer Center as previously described. In brief, the transgene insert (αMHC–eGFPf–Bmp10) was purified and microinjected into inbred C3HeB/FeJ zygotes (Jackson Laboratories), which were then implanted into the oviducts of pseudopregnant Swiss Webster mice. The resulting pups were screened by diagnostic PCR.
Isoproterenol infusion
Two-month-old male transgenic mice and their nontransgenic siblings were implanted with isoproterenol-filled Alzet osmotic mini-pumps (7-day infusion, model 2001, flow rate of 1 μl/h, 0.028 g/ml isoproterenol dissolved in saline) as previously described (20). Infusion of saline was used as a vehicle control in the assay.
Echocardiographic analysis
Echocardiographic analysis was performed with a high-resolution micro-ultrasound system (Vevo770 or Vevo 2100, VisualSonics Inc., Toronto, Canada) equipped with a 40-MHz mechanical scan probe as previously described (4). Briefly, the mice were anesthetized using 2.5% isoflurane and placed on a temperature-controllable surgical table. M-mode images at the midpapillary level were derived from a short-axis view. The cardiac parameter measurements included the left ventricular internal diameter at diastole, left ventricular internal diameter at systole, left ventricular volume at diastole, and left ventricular volume at systole. Calculated ejection fraction and fractional shortening were used to compare the cardiac systolic function between experimental groups.
Histological examination
Heart tissues were harvested for cryosectioning or paraffin embedding/sectioning following the standard procedure. Masson's trichrome staining and Sirus red/fast green were performed according to a standard protocol (4). The terminal deoxynucleotidyl transferase dUTP nick end labeling assay (Promega catalog no. G3250) was performed according to the manufacturer's instructions. Activated caspase-3 immune reactivity (antibody category G7481; Promega) was performed on postfixed sections as described previously (4).
Western blotting analysis
For Western blotting, 4–20% Mini-PROTEAN® TGX Stain-FreeTM precast gels (Bio-Rad catalog no. 4568096) were used. The samples were mixed with 2× Laemmli sample loading buffer (Bio-Rad catalog no. 1610737), boiled at 95 °C for 5 min before cooling at room temperature, and electrophoresed in Tris/glycine/SDS running buffer (Bio-Rad catalog no. 1610771) at 200 V followed by semidry transfer at 15 V for 30 min. The blots were incubated with the respective primary antibodies overnight at 4 °C, followed by secondary antibody incubation for 1 h at room temperature and ECL (PerkinElmer) for 1 min. The signals were visualized either on X-ray film or by a ChemiDoc (Bio-Rad).
Reverse transcription and qRT-PCR
Total RNA was extracted from dissected hearts using TRIzol reagent (Invitrogen catalog no. 15596018). The first-strand cDNA was synthesized by iScriptTM reverse transcription Supermix for qRT-PCR (Bio-Rad catalog no. 1708841). Quantitative PCR was performed using PowerUpTM SYBRTM Green Master Mix (Applied Biosystems catalog no. A25780). The relative expression level was normalized to the housekeeping gene RPL7 (ribosomal protein L7). The mRNA expression levels of each gene were calculated according to the 2−ΔΔCT method. The primers used in the assay are listed in Table S1.
ACF isolation and [3H]proline incorporation assay
Adult mouse cardiac fibroblasts were collected from 3-month-old mouse hearts by perfusion with Liberase (19 μg/ml, Roche)–containing buffer, and cultured in Dulbecco's modified Eagle's medium supplemented with 15% fetal bovine serum (Fisher catalog no. SV3001403HI) and seeded in laminin-coated 60-mm dishes (BD, catalog no. 354405). Collagen synthesis was measured using the [3H]proline incorporation assay similar to a previous method (5). ACFs were serum-starved with 0.1% fetal bovine serum for 48 h, followed by rhBMP10 (50 ng/ml, R&D Systems) pretreatment for 30 min, followed by rhTGFβ1 (1 ng/ml, R&D Systems) treatment for 48 h. Then 1 μCi/ml [3H]proline (PerkinElmer catalog no. NET483001MC) was added to the culture 8 h after the addition of TGFβ1. The cultured ACFs were washed with ice-cold PBS, and 10% TCA (Sigma catalog no. T0699) was used to fix the cells at 4 °C for 30 min. The cell pellets were then further washed with cold PBS and solubilized in 0.2 n NaOH at room temperature for 1 h before liquid scintillation counting by a liquid scintillation spectrometry (Beckman, model LS6000-SC). [3H]Proline incorporation was measured as radioactive cpm, which was normalized by total protein content.
Statistical analysis
All values are presented as means ± S.E. Statistical significance (p < 0.05 or 0.01) was determined by Student's t test (for groups of two).
Author contributions
X. Q., Y. L., D. C., J. C., P. Z., X. L., W. S., and H. C. data curation; X. Q., Y. L., D. C., W. S., and H. C. formal analysis; X. Q., Y. L., D. C., J. C., Z. L., H. J., Y. C., W. Z., P. Z., D.X., X. L., W. S., and H. C. investigation; X. Q., Y. L., D. C., D.X., X. L., W. S., and H. C. methodology; X. Q., W. S., and H. C. writing-original draft; Y. L., D. C., W. S., and H. C. validation; Y. L., X. L., and W. S. project administration; D. C. and X. L. software; D. C., X. L., and W. S. visualization; X. L., W. S., and H. C. conceptualization; X. L., W. S., and H. C. supervision; W. S. and H. C. funding acquisition; W. S. resources; W. S. and H. C. writing-review and editing.
Supplementary Material
Acknowledgments
We thank Indiana University's Transgenic & Knock-Out Mouse Core for generating the transgenic mouse lines. We also thank Dr. Bartholomew A. Pederson and Dr. Mark Soonpaa for discussion and proofreading.
This work was supported in part by National Institutes of Health Grants R01HL145060, R01HL81092, P01HL134599, and P01HL85098 (to W. S.), a grant from the Indiana University Showalter Research Trust Fund (to H. C.), and National Science Foundation China Projects of International Cooperation and Exchanges Grant 81720102004 and National Science Foundation China Grants 81570279 and 81974019 (to P. Z.) to support P. Z. as a visiting Scholar in Shou Lab. The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Table S1 and Figs. S1–S4.
- TGF
- transforming growth factor
- ACF
- adult cardiac fibroblast
- ALK1
- activin receptor-like kinase 1
- MHC
- myosin heavy chain
- ISO
- isoproterenol
- PARP
- poly(ADP-ribose) polymerase
- STAT
- signal transducer and activator of transcription
- rh
- recombinant human
- TG
- transgenic
- NTG
- nontransgenic
- qRT-PCR
- quantitative RT-PCR
- IPA
- ingenuity pathway analysis.
References
- 1. Foo R. S., Mani K., and Kitsis R. N. (2005) Death begets failure in the heart. J. Clin. Invest. 115, 565–571 10.1172/JCI24569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kung G., Konstantinidis K., and Kitsis R. N. (2011) Programmed necrosis, not apoptosis, in the heart. Circ. Res. 108, 1017–1036 10.1161/CIRCRESAHA.110.225730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Whelan R. S., Kaplinskiy V., and Kitsis R. N. (2010) Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu. Rev. Physiol. 72, 19–44 10.1146/annurev.physiol.010908.163111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Zhu W., Soonpaa M. H., Chen H., Shen W., Payne R. M., Liechty E. A., Caldwell R. L., Shou W., and Field L. J. (2009) Acute doxorubicin cardiotoxicity is associated with p53-induced inhibition of the mammalian target of rapamycin pathway. Circulation 119, 99–106 10.1161/CIRCULATIONAHA.108.799700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Teunissen B. E., Smeets P. J., Willemsen P. H., De Windt L. J., Van der Vusse G. J., and Van Bilsen M. (2007) Activation of PPARδ inhibits cardiac fibroblast proliferation and the transdifferentiation into myofibroblasts. Cardiovasc. Res. 75, 519–529 10.1016/j.cardiores.2007.04.026 [DOI] [PubMed] [Google Scholar]
- 6. Neuhaus H., Rosen V., and Thies R. S. (1999) Heart specific expression of mouse BMP-10 a novel member of the TGF-β superfamily. Mech. Dev. 80, 181–184 10.1016/S0925-4773(98)00221-4 [DOI] [PubMed] [Google Scholar]
- 7. Pashmforoush M., Lu J. T., Chen H., Amand T. S., Kondo R., Pradervand S., Evans S. M., Clark B., Feramisco J. R., Giles W., Ho S. Y., Benson D. W., Silberbach M., Shou W., and Chien K. R. (2004) Nkx2–5 pathways and congenital heart disease; loss of ventricular myocyte lineage specification leads to progressive cardiomyopathy and complete heart block. Cell 117, 373–386 10.1016/S0092-8674(04)00405-2 [DOI] [PubMed] [Google Scholar]
- 8. Ducy P., and Karsenty G. (2000) The family of bone morphogenetic proteins. Kidney Int. 57, 2207–2214 10.1046/j.1523-1755.2000.00081.x [DOI] [PubMed] [Google Scholar]
- 9. Nakayama T., Cui Y., and Christian J. L. (2000) Regulation of BMP/Dpp signaling during embryonic development. Cell Mol. Life Sci. 57, 943–956 10.1007/PL00000736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chen H., Brady Ridgway J., Sai T., Lai J., Warming S., Chen H., Roose-Girma M., Zhang G., Shou W., and Yan M. (2013) Context-dependent signaling defines roles of BMP9 and BMP10 in embryonic and postnatal development. Proc. Natl. Acad. Sci. U.S.A. 110, 11887–11892 10.1073/pnas.1306074110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. David L., Mallet C., Mazerbourg S., Feige J. J., and Bailly S. (2007) Identification of BMP9 and BMP10 as functional activators of the orphan activin receptor-like kinase 1 (ALK1) in endothelial cells. Blood 109, 1953–1961 10.1182/blood-2006-07-034124 [DOI] [PubMed] [Google Scholar]
- 12. Teichmann U., and Kessel M. (2004) Highly restricted BMP10 expression in the trabeculating myocardium of the chick embryo. Dev. Genes Evol. 214, 96–98 10.1007/s00427-003-0380-2 [DOI] [PubMed] [Google Scholar]
- 13. Somi S., Buffing A. A., Moorman A. F., and Van Den Hoff M. J. (2004) Expression of bone morphogenetic protein-10 mRNA during chicken heart development. Anat. Rec. A Discov. Mol. Cell Evol. Biol. 279, 579–582 [DOI] [PubMed] [Google Scholar]
- 14. Nakano N., Hori H., Abe M., Shibata H., Arimura T., Sasaoka T., Sawabe M., Chida K., Arai T., Nakahara K., Kubo T., Sugimoto K., Katsuya T., Ogihara T., Doi Y., et al. (2007) Interaction of BMP10 with Tcap may modulate the course of hypertensive cardiac hypertrophy. Am. J. Physiol. Heart Circ. Physiol. 293, H3396–H3403 10.1152/ajpheart.00311.2007 [DOI] [PubMed] [Google Scholar]
- 15. Kahr P. C., Piccini I., Fabritz L., Greber B., Schöler H., Scheld H. H., Hoffmeier A., Brown N. A., and Kirchhof P. (2011) Systematic analysis of gene expression differences between left and right atria in different mouse strains and in human atrial tissue. PLoS One 6, e26389 10.1371/journal.pone.0026389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Piotrowska I. (2007) Functional Implication of Bone Morphogenetic Protein 10 (BMP10) Expression in Pathological Hearts., Ph.D. Thesis, University of Giessen, Giessen, Germany [Google Scholar]
- 17. Chen H., Shi S., Acosta L., Li W., Lu J., Bao S., Chen Z., Yang Z., Schneider M. D., Chien K. R., Conway S. J., Yoder M. C., Haneline L. S., Franco D., and Shou W. (2004) BMP10 is essential for maintaining cardiac growth during murine cardiogenesis. Development 131, 2219–2231 10.1242/dev.01094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Maruyama S., Wu C. L., Yoshida S., Zhang D., Li P. H., Wu F., Parker Duffen J., Yao R., Jardin B., Adham I. M., Law R., Berger J., Di Marchi R., and Walsh K. (2018) Relaxin family member insulin-like peptide 6 ameliorates cardiac fibrosis and prevents cardiac remodeling in murine heart failure models. J. Am. Heart Assoc. 7, e008441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Galindo C. L., Skinner M. A., Errami M., Olson L. D., Watson D. A., Li J., McCormick J. F., McIver L. J., Kumar N. M., Pham T. Q., and Garner H. R. (2009) Transcriptional profile of isoproterenol-induced cardiomyopathy and comparison to exercise-induced cardiac hypertrophy and human cardiac failure. BMC Physiol. 9, 23 10.1186/1472-6793-9-23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen H., Yong W., Ren S., Shen W., He Y., Cox K. A., Zhu W., Li W., Soonpaa M., Payne R. M., Franco D., Field L. J., Rosen V., Wang Y., and Shou W. (2006) Overexpression of bone morphogenetic protein 10 in myocardium disrupts cardiac postnatal hypertrophic growth. J. Biol. Chem. 281, 27481–27491 10.1074/jbc.M604818200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhang W., Chen H., Wang Y., Yong W., Zhu W., Liu Y., Wagner G. R., Payne R. M., Field L. J., Xin H., Cai C. L., and Shou W. (2011) Tbx20 transcription factor is a downstream mediator for bone morphogenetic protein-10 in regulating cardiac ventricular wall development and function. J. Biol. Chem. 286, 36820–36829 10.1074/jbc.M111.279679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Satoh M. S., and Lindahl T. (1992) Role of poly(ADP-ribose) formation in DNA repair. Nature 356, 356–358 10.1038/356356a0 [DOI] [PubMed] [Google Scholar]
- 23. Oliver F. J., de la Rubia G., Rolli V., Ruiz-Ruiz M. C., de Murcia G., and Murcia J. M. (1998) Importance of poly(ADP-ribose) polymerase and its cleavage in apoptosis. Lesson from an uncleavable mutant. J. Biol. Chem. 273, 33533–33539 10.1074/jbc.273.50.33533 [DOI] [PubMed] [Google Scholar]
- 24. Trucco C., Oliver F. J., de Murcia G., and Ménissier-de Murcia J. (1998) DNA repair defect in poly(ADP-ribose) polymerase-deficient cell lines. Nucleic Acids Res. 26, 2644–2649 10.1093/nar/26.11.2644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sohal D. S., Nghiem M., Crackower M. A., Witt S. A., Kimball T. R., Tymitz K. M., Penninger J. M., and Molkentin J. D. (2001) Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ. Res. 89, 20–25 10.1161/hh1301.092687 [DOI] [PubMed] [Google Scholar]
- 26. Derynck R., and Zhang Y. E. (2003) Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 425, 577–584 10.1038/nature02006 [DOI] [PubMed] [Google Scholar]
- 27. von Bubnoff A., and Cho K. W. (2001) Intracellular BMP signaling regulation in vertebrates: pathway or network? Dev. Biol. 239, 1–14 10.1006/dbio.2001.0388 [DOI] [PubMed] [Google Scholar]
- 28. Gianakopoulos P. J., and Skerjanc I. S. (2009) Cross talk between hedgehog and bone morphogenetic proteins occurs during cardiomyogenesis in P19 cells. In Vitro Cell Dev. Biol. Anim. 45, 566–572 10.1007/s11626-009-9228-z [DOI] [PubMed] [Google Scholar]
- 29. Angello J. C., Kaestner S., Welikson R. E., Buskin J. N., and Hauschka S. D. (2006) BMP induction of cardiogenesis in P19 cells requires prior cell-cell interaction (s). Dev. Dyn. 235, 2122–2133 10.1002/dvdy.20863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Harada K., Ogai A., Takahashi T., Kitakaze M., Matsubara H., and Oh H. (2008) Crossveinless-2 controls bone morphogenetic protein signaling during early cardiomyocyte differentiation in P19 cells. J. Biol. Chem. 283, 26705–26713 10.1074/jbc.M801485200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang W., Qu X., Chen B., Snyder M., Wang M., Li B., Tang Y., Chen H., Zhu W., Zhan L., Yin N., Li D., Xie L., Liu Y., Zhang J. J., et al. (2016) Critical roles of STAT3 in β-adrenergic functions in the heart. Circulation 133, 48–61 10.1161/CIRCULATIONAHA.115.017472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang J., Liew O. W., Richards A. M., and Chen Y. T. (2016) Overview of microRNAs in cardiac hypertrophy, fibrosis, and apoptosis. Int. J. Mol. Sci. 17, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Valkov N., King M. E., Moeller J., Liu H., Li X., and Zhang P. (2019) MicroRNA-1–mediated inhibition of cardiac fibroblast proliferation through targeting cyclin D2 and CDK6. Front. Cardiovasc. Med. 6, 65 10.3389/fcvm.2019.00065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. van Rooij E., Sutherland L. B., Thatcher J. E., DiMaio J. M., Naseem R. H., Marshall W. S., Hill J. A., and Olson E. N. (2008) Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. U.S.A. 105, 13027–13032 10.1073/pnas.0805038105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ooi J. Y. Y., Bernardo B. C., Singla S., Patterson N. L., Lin R. C. Y., and McMullen J. R. (2017) Identification of miR-34 regulatory networks in settings of disease and antimiR-therapy: Implications for treating cardiac pathology and other diseases. RNA Biol. 14, 500–513 10.1080/15476286.2016.1181251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bernardo B. C., Gao X. M., Winbanks C. E., Boey E. J., Tham Y. K., Kiriazis H., Gregorevic P., Obad S., Kauppinen S., Du X. J., Lin R. C., and McMullen J. R. (2012) Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodeling and improves heart function. Proc. Natl. Acad. Sci. U.S.A. 109, 17615–17620 10.1073/pnas.1206432109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Massagué J., Seoane J., and Wotton D. (2005) Smad transcription factors. Genes Dev. 19, 2783–2810 10.1101/gad.1350705 [DOI] [PubMed] [Google Scholar]
- 38. Schneider M. D., Gaussin V., and Lyons K. M. (2003) Tempting fate: BMP signals for cardiac morphogenesis. Cytokine Growth Factor Rev. 14, 1–4 10.1016/S1359-6101(02)00053-9 [DOI] [PubMed] [Google Scholar]
- 39. Bryson B. L., Junk D. J., Cipriano R., and Jackson M. W. (2017) STAT3-mediated SMAD3 activation underlies oncostatin M–induced senescence. Cell Cycle 16, 319–334 10.1080/15384101.2016.1259037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rajan P., Panchision D. M., Newell L. F., and McKay R. D. (2003) BMPs signal alternately through a SMAD or FRAP–STAT pathway to regulate fate choice in CNS stem cells. J. Cell Biol. 161, 911–921 10.1083/jcb.200211021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Tang L. Y., Heller M., Meng Z., Yu L. R., Tang Y., Zhou M., and Zhang Y. E. (2017) Transforming growth factor-β (TGF-β) directly activates the JAK1–STAT3 axis to induce hepatic fibrosis in coordination with the SMAD pathway. J. Biol. Chem. 292, 4302–4312 10.1074/jbc.M116.773085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cunha S. I., and Pietras K. (2011) ALK1 as an emerging target for antiangiogenic therapy of cancer. Blood 117, 6999–7006 10.1182/blood-2011-01-330142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ning B. B., Zhang Y., Wu D. D., Cui J. G., Liu L., Wang P. W., Wang W. J., Zhu W. L., Chen Y., and Zhang T. (2017) Luteolin-7-diglucuronide attenuates isoproterenol-induced myocardial injury and fibrosis in mice. Acta Pharmacol. Sin. 38, 331–341 10.1038/aps.2016.142 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.