

Abstract
Lysyl oxidase (LOX) is a secreted copper-dependent amine oxidase that cross-links collagens and elastin in the extracellular matrix and is a critical mediator of tumor growth and metastatic spread. LOX is a target for cancer therapy, and thus the search for therapeutic agents against LOX has been widely sought. We report herein the medicinal chemistry discovery of a series of LOX inhibitors bearing an aminomethylenethiophene (AMT) scaffold. High-throughput screening provided the initial hits. Structure–activity relationship (SAR) studies led to the discovery of AMT inhibitors with sub-micromolar half-maximal inhibitory concentrations (IC50) in a LOX enzyme activity assay. Further SAR optimization yielded the orally bioavailable LOX inhibitor CCT365623 with good anti-LOX potency, selectivity, pharmacokinetic properties, as well as anti-metastatic efficacy.
Introduction
Lysyl oxidase (LOX) and its family members LOX-like (LOX-L) 1–4 are copper-dependent amine oxidases that covalently cross-link collagens and elastin in the tumor extracellular matrix.1−4 LOX is secreted as a catalytically inactive 50 kDa pro-protein, which is cleaved to an active 32 kDa enzyme by proteases such as procollagen C-proteinase. LOX and LOXL1–4 have variable N-termini, and they share a highly conserved C-terminus, where the catalytic domain is located. The catalytic site comprises a copper binding motif and a covalently bound lysine tyrosylquinone (LTQ) cofactor, where peptidyl lysine residues (H2NCH2R) are converted to the corresponding α-aminoadipic-δ-semialdehyde (O=CHR) in an oxidative deamination reaction.3 The newly formed aldehyde residues undergo spontaneous cross-linking with adjacent nucleophilic functionalities, leading to the insoluble extracellular protein matrices.
LOX and LOXL2 also have important roles in promoting tumor growth in many types of cancer.5−12 In particular, LOX has been demonstrated to be a critical mediator of cancer metastasis.13 Therapeutic agents targeting the activity of LOX are thus proposed as cancer treatments, especially against metastasis where no effective therapeutic methods are currently available.
Until recently, no druglike small molecule inhibitors of LOX itself have been reported. Noticeably, the irreversible inhibitor β-aminopropionitrile14,15 (BAPN) has found widespread applications in LOX-family-related biological studies (Figure 1), although the lack of amenable sites for chemical modification has prevented its development into a clinically optimal drug. More recently, haloallylamine-based inhibitors PXS-S1A and PXS-S2A (full structures not disclosed)16 and trifluoromethyl (CF3)-substituted aminomethylene-pyridine 1 were reported to be potent selective inhibitors of one of the family members, LOXL2; the latter also showed weak inhibition against LOX.17,18 Intriguingly, analogues of pyridine 1 without the CF3 functionality were less selective toward LOXL2, with low micromolar IC50s against LOX.
Figure 1.

Small molecule inhibitors of LOX-family enzymes.
We have recently reported the elucidation of a mechanism by which LOX drives tumor progression in breast cancer19 and that treatment with the aminomethylenethiophene (AMT) inhibitor CCT365623 (9f) led to significant reduction in tumor growth and, importantly, in metastatic burden too, in a LOX-dependent breast tumor transgenic mouse model. In our current study, we present the medicinal chemistry development leading to the discovery of the orally efficacious AMT inhibitor 9f.
Results and Discussion
LOX Inhibition, Initial SAR
We ran a high-throughput screen (HTS) at Evotec, of 267 000 diverse compounds and 5000 fragments, on LOX, which yielded a hit rate of 0.4%. (5-(Piperidin-1-ylsulfonyl)thiophen-2-yl)methanamine 2a was identified as a positive hit with a mean IC50 of 19 μM. Since no crystal structure of LOX is available, the design of inhibitors could not be aided by crystallographic or in silico methods. Therefore, the SAR of enzyme inhibition is largely elucidated by introducing systematic modifications to different regions of the hit molecule.
Substitutions at the 5-Sulfonyl Linker, Sulfonamides
SAR exploration commenced with the investigation of sulfonamide substitutions on LOX inhibition (Table 1). Acyclic sulfonamides show no improvement (2b and 2c vs 2a), whereas 2-amido- and 2-hydroxymethylpyrrolidine substitutions exhibit comparable or better LOX potencies (2d and 2e vs 2a). 2-Phenylpyrrolidine 2f is also effective against LOX, as is the bicyclic indoline 2g, which is ∼10-fold more potent than the piperidine hit 2a. Similarly, tetrahydroquinoline 2h is equipotent to indoline 2g. Replacement of the piperidine ring with morpholine does not improve LOX inhibition (2i vs 2a), whereas homopiperazine (2j) substitution leads to ∼2-fold improvement in IC50. Functionalization of the free homopiperazine nitrogen with small groups leads to gains in potency compared with the initial hit, as exemplified in N-methyl analogue 2k, ethyl urea 2l and, in particular, sulfonamide 2m.
Table 1. Effects of Sulfonamide Substitution on LOX Potency.
![]()

Reported IC50 values were determined in at least two separate experiments (n ≥ 2). When n = 2, individual IC50 values are shown. When n > 2, the values are reported as the geometric mean with the error in square brackets expressed as the 95% confidence interval of the geometric mean.
Substitution at the 5-Sulfonyl Linker, Sulfones
The effect of alkyl and aryl substitutions at the 5-sulfonyl linker on LOX inhibition was investigated next (Table 2). Replacements of the piperidine moiety on HTS hit 2a with cyclohexyl (3a) and phenyl groups (3b) are beneficial, as are pyridine (3c) and thiophene (3d). Biphenylsulfone is a weaker inhibitor than the phenyl analogue (3e vs 3b), whereas 2-naphthalylsulfone 3g is more potent than the 1-regiosiomer 3f. Methanesulfonamido-phenyl analogue 3h moderately inhibits LOX, whereas methanesulfonylphenyl sulfone 3i is an excellent LOX inhibitor with an IC50 of 0.26 μM, ∼70-fold more potent than the HTS hit 2a. Replacement of the phenyl moiety of inhibitor 3i with an alkyl group (3j) leads to a reduction in potency. The SAR data illustrate that the attachment of cyclic alkyl or aryl groups to the sulfonyl linker greatly improves LOX potency and the inhibitory effect is further enhanced by the addition of a second sulfonyl group.
Table 2. Effects of Sulfonyl-Alkyl and -Aryl Substitutions on LOX Potency.
![]()

Reported IC50 values were determined in at least two separate experiments (n ≥ 2). When n = 2, individual IC50 values are shown. When n > 2, the values are reported as the geometric mean with the error in square brackets expressed as the 95% confidence interval of the geometric mean.
Modification of the 5-Sulfonyl Linker
The impact of the sulfonyl linker was subsequently examined (Table 3). A noticeable correlation can be observed between the electron-withdrawing ability of the linker and the LOX IC50, with the most electron-withdrawing sulfonyl group achieving the most potent inhibition (LOX IC50: 3b < 4a < 4b). Carboxamide substitution does not improve LOX inhibition (4d vs 4c). Finally, exchanging the sulfonyl linker and the phenyl ring leads to reduction in potency (4e vs 3b). It is thus apparent that the sulfonyl moiety is the optimum linker for the AMT core and that the electron-withdrawing effect of the linker is likely to play an important role in the mechanism of inhibition even though the possibility of H-bond and dipole–dipole interactions cannot be ruled out.
Table 3. Effects of Sulfonyl Linker (X) Modification on LOX Potency.
![]()

Reported IC50 values were determined in at least two separate experiments (n ≥ 2). When n = 2, individual IC50 values are shown. When n > 2, the values are reported as the geometric mean with the error in square brackets expressed as the 95% confidence interval of the geometric mean.
Modification of the Thiophene Ring
Aminomethylene-pyridine 5a is ∼10-fold weaker than the aminomethylenethiophene counterpart (Table 4; 5a vs 5b). 1,4-Thiazole 5c is considerably less potent as an inhibitor than the 1,3-regiomer 5d, whereas 1,3-thiazole 5d demonstrates potency that is similar to the thiophene 3g. Furan replacement does not improve the effectiveness of the inhibitors (5e vs 4d). Additional substitutions on the thiophene ring can potentially be a useful handle for the development of the series, but the introduction of a small methyl group is highly unfavorable (5f vs 2g). Overall, although the replacements of the thiophene core with some unsubstituted 5-membered heterocycles are tolerated for LOX inhibition, they are not superior to thiophene itself.
Table 4. Effects of Thiophene Modifications on LOX Potency.

Reported IC50 values were determined in at least two separate experiments (n ≥ 2). When n = 2, individual IC50 values are shown. When n > 2, the values are reported as the geometric mean with the error in square brackets expressed as the 95% confidence interval of the geometric mean.
Modification of the Aminomethylene Moiety
All inhibitors contain the aminomethylene moiety (H2NCH2), which forms a part of the core AMT scaffold. We therefore investigate a series of modifications to this group where the replacement moieties are sufficiently diverse for probing noncovalent interactions, such as H-bond, electrostatic, and dipolar interactions, while small enough to minimize unfavorable steric clashes. The SAR data reveals that all substitutions or modifications at this site result in total loss of activity (Table 5, 6a–6f). It is thus apparent that the aminomethylene moiety has a unique role in LOX inhibition; it is likely to be involved in the formation of a Schiff base similar to that of the natural lysyl substrates.
Table 5. Effects of Aminomethylene Modifications (R1) on LOX Potency.
![]()

Reported IC50 values were determined in at least two separate experiments (n ≥ 2). When n = 2, individual IC50 values are shown. When n > 2, the values are reported as the geometric mean with the error in square brackets expressed as the 95% confidence interval of the geometric mean.
Next, regiosiomers on the thiophene ring were investigated (Table 6). Both 2-aminomethyl-3-sulfonyl-thiophene 7a and 3-aminomethyl-4-sulfonylthiophene 7b show no inhibitory activity against LOX, whereas 2-aminomethyl-4-sulfonylthiophene 7c is a weak inhibitor. Therefore, from these modifications, the thiophene ring is the optimal ring type and the aminomethylene and sulfonyl groups are the most effective substituents when placed on the 2- and 5-positions of the ring, respectively.
Table 6. Effects of Regioisomers on the Thiophene Ring on LOX Potency.

Reported IC50 values were determined in at least two separate experiments (n ≥ 2). When n = 2, individual IC50 values are shown. When n > 2, the values are reported as the geometric mean with the error in square brackets expressed as the 95% confidence interval of the geometric mean.
Although the exact mode of binding of these AMT inhibitors remains unclear due to the absence of a cocrystal structure or a homology model, the observed SAR suggests that a stable Schiff base formed from the inhibitor and the LTQ cofactor (Figure 2) is likely to be involved. Although the formation of the Schiff base is reversible, its stabilization by the sulfonyl substituent on the thiophene ring by resonance stabilization and/or direct binding to the enzyme by noncovalent processes, such as H-bonding, electrostatic, dipolar, or van der Waals interactions, can lead to a tightly bound enzyme–inhibitor complex. This can potentially rationalize the improvement in potency observed in Table 3. The SAR also suggests an additional noncovalent interaction between the methanesulfonylphenyl moiety and the enzyme, which further enhances the potency of bis-sulfonyl inhibitors such as compound 9f.
Figure 2.

Proposed binding mode of AMT inhibitors.
Optimization toward In Vivo-Compatible Inhibitors
As our aim is to discover LOX inhibitors that can be administered orally, metabolic stability and pharmacokinetic (PK) studies were performed on the most promising AMT inhibitors described above. The highly potent bis-sulfonylphenyl analogue 3i has good stability against mouse microsomal (MLM) degradation (Table 9), but its poor detectability by mass spectrometry renders it unsuitable for in vivo studies. Bis-sulfonylhomopiperazine 2m also exhibits good anti-LOX potency, but it cannot be progressed further due to poor stability against microsomal metabolism. Naphthalenesulfone 3g demonstrates good MLM stability but only moderate plasma exposure (AUC = 4.2 μM h) when administered orally in mice at 50 mg/kg. It was apparent that further medicinal chemistry development was necessary to achieve both potent LOX inhibition and oral plasma drug exposure compatible with in vivo studies.
Table 9. In Vitro Mouse Liver Microsome (MLM) Stability and in Vivo Pharmacokinetic (PK) Properties of AMT Inhibitors.
| compound | MLM stability (%)a | Cmax(PO) (μM)b | AUC(PO) (μM h)c | CL(IV) (mL/(min kg))d | t1/2(IV) (h)e | F (%)f |
|---|---|---|---|---|---|---|
| 2m | 37 | |||||
| 3i | 67 | ND | ND | |||
| 8j | 91 | 0.38 | 0.36 | |||
| 8d | 59 | 0.83 | 0.22 | |||
| 8e | 63 | 1.5 | 0.46 | |||
| 9k | 41 | 1.6 | 1.4 | |||
| 9b | 67 | 6.3 | 2.3 | |||
| 9j | 90 | 9.6 | 2.8 | |||
| 3g | 100 | 15 | 4.2 | |||
| 9a | 68 | 16 | 6.5 | |||
| 9g | 60 | 6.7 | 11 | 67 | 1.0 | 39 |
| 9l | 95 | 9.4 | 12 | 106 | 0.4 | 74 |
| 9f | 65 | 17 | 15 | 49 | 1.2 | 45 |
Mouse liver microsome (MLM) stability values represent the percentage of compound remaining after 30 min; mouse plasma PK parameters were determined following a single dose by oral gavage (PO) or intravenous injection (IV) at 50 or 10 mg/kg, respectively.
Cmax: maximum concentration.
AUC: area under curve.
CL: clearance.
t1/2: half-life.
F: bioavailability; ND: could not be detected by MS.
Due to its superior LOX potency and ease of synthesis, inhibitor 3i was chosen as the platform for the next phase of discovery. Initial SAR established that modifications of the aminomethylene group, the thiophene ring, and the sulfonyl linker were unfavorable to target inhibition. Therefore, optimization to improve PK properties focused on the aryl ring and its side-chain substituents. The aim of the subsequent studies therefore targeted improvement to oral in vivo PK exposure whilst maintaining/improving LOX potency.
Side-Chain Substitutions
Further SAR studies began with the investigation of the sulfonyl side-chain substitutions (Table 7). The attachment of the sulfonyl side chain through the 3-position (with respect to the thiophene sulfone) affords inhibitors with similar LOX IC50s as the 4-regioisomers (8a vs 3i, 8d vs 8c), whilst 2-substitution is disfavored (8b vs 3i). Sulfonylpyrrolidines exhibit similar potencies as the methyl sulfone counterparts (8c–8e vs 3i and 8a), but phenyl sulfone 8f is a weaker inhibitor. Small alkyl substituents on the sulfonyl group are generally well tolerated (8g–8k). Inhibitors 8d, 8e, and 8j show good anti-LOX potency and MLM stability (Table 9) and were thus selected for in vivo PK evaluation. Unfortunately, they have poor plasma exposure in mice when dosed orally.
Table 7. Effects of Side-Chain Substitutions on LOX Potency of Phenylsulfonyl-AMT Analogues.
![]()

Reported IC50 values were determined in at least two separate experiments (n ≥ 2). When n = 2, individual IC50 values are shown. When n > 2, the values are reported as the geometric mean with the error in square brackets expressed as the 95% confidence interval.
Phenyl Ring Substitution
1,3- and 1,4-Bis-sulfonylphenyl-AMT inhibitors containing an additional substituent on the 5-position of the phenyl ring were evaluated against LOX inhibition (Table 8). All of tert-butyl, tert-butoxy, trimethylsilylethynyl, N-methylpyrazolyl, and pyridinyl substitutions on the 1,3-bisulfonylphenyl motif lead to moderate reduction in LOX potency (9a–9e vs 8a), whereas a phenyl substituent is well tolerated (9f). Replacement of the phenyl group with p-tolyl affords an equipotent LOX inhibitor (9g vs 9f), but m-xylyl (9h) and o-ethylphenyl (9i) substitutions are disfavored. For the 1,4-bis-sulfonylphenyl-AMT motif, all of the N-methylpyrazolyl (9j), phenyl (9k), and p-tolyl (9l) analogues exhibit sub-micromolar LOX IC50 values.
Table 8. Effects of 5-Substitutions on LOX Potency of Bis-sulfonylphenyl-AMT Analogues.
![]()

In vivo mouse PK studies were then conducted for selected 5-substituted compounds. Both tert-butoxy (9b) and N-methylpyrazolyl (9j) analogues exhibit low AUCs of 2.3 and 2.8 μM h, respectively (Table 9), although this improves to 6.5 μM h when the oxygen atom of the tert-butoxy group is removed from the parent compound (9a vs 9b). Pleasingly, the 5-p-tolyl-substituted bis-sulfones have greatly improved and therapeutically relevant plasma exposures (11 and 12 μM h for 9g and 9l respectively) are obtained. Although the exposure for 5-phenyl-1,4-bis-sulfone 9k is disappointing (AUC = 1.4 μM h), 5-phenyl-1,3-bis-sulfonyl-AMT 9f achieves a desirable in vivo PK profile, with the highest AUC, Cmax, longest half-life, lowest clearance of the series (albeit this is still moderate), and a respectable intermediate oral bioavailability (F) of 45%.
Profile of Orally Available AMT Inhibitors
We have discovered three AMT inhibitors 9g, 9l, and 9f that are highly effective in inhibiting LOX activity as well as possessing therapeutically relevant PK profiles. These compounds are equally potent inhibitors of LOXL2 (Table 10), which is as expected due to the highly conserved catalytic site across the LOX-family members. Pleasingly, all three inhibitors are inactive against common amine oxidases, including the copper-containing diamine oxidase (DAO), semicarbazide-sensitive amine oxidase (SSAO), and the flavin-containing monoamine oxidases (MAO) A and B. It is noteworthy that AMT 9f is a substrate of SSAO; thus, SSAO-catalyzed metabolism could be a potential mechanism of degradation in vivo.
Table 10. Potency, Selectivity, and Permeability of Optimized AMT Inhibitors.
| Caco-2 Pappb (10–6 cm/s) |
||||||||
|---|---|---|---|---|---|---|---|---|
| inhibitor | LOX IC50 (μM)a | LOXL2 IC50 (μM)a | DAO IC50 (μM)a | SSAO IC50 (μM)a | MAO-A MAO-B IC50 (μM)a | hERG IC50 (μM) | A → B | B → A |
| 9g | 1.1 [0.75, 1.62] | 2.0 [1.3, 3.0] | >100, >100 | 49, 59 | A: >100, >100 | 4.6 | 8.0 | 17 |
| B: 87, 89 | ||||||||
| 9l | 0.76, 0.86 | 2.2 [1.4, 3.7] | >100, >100 | >100, 100 | A: 33, 33 | 68 | 18 | 25 |
| B: >100, >100 | ||||||||
| 9f | 0.90 [0.55, 1.48] | 1.5 [0.28, 8.1] | >100, >100 | 48, 90 | A: >100, >100 | 25 | 8.5 | 35 |
| B: >100, >100 | ||||||||
Reported IC50 values were determined in at least two separate experiments (n ≥ 2). When n = 2, individual IC50 values are shown. When n > 2, the values are reported as the geometric mean with the error in square brackets expressed as the 95% confidence interval of the geometric mean.
Papp: permeability coefficient.
AMT 9g is a weak inhibitor of the human potassium-ion channel hERG, whereas inhibitors 9l and 9f are highly selective. All three inhibitors exhibit high permeability through colon Caco-2 cells, which is reflected in their good oral availabilities in mice (Table 9). AMT inhibitor 9f achieves the most favorable overall profile and was therefore chosen for in vivo efficacy studies.
Evaluation of Anti-metastatic Efficacy
Compound 9f was assessed in a LOX-driven genetically engineered mouse model (GEMM) of breast cancer that metastasizes to the lungs.19 Mice were dosed daily by oral gavage (70 mg/kg) from day 60 when primary tumors start to be palpable (Figure 3). The metastatic nodules in the lungs are measured when the primary tumors reach an ethical size limit. Pleasingly, compound 9f reduces lung metastasis significantly, as measured by the total surface area (Figure 3B).
Figure 3.
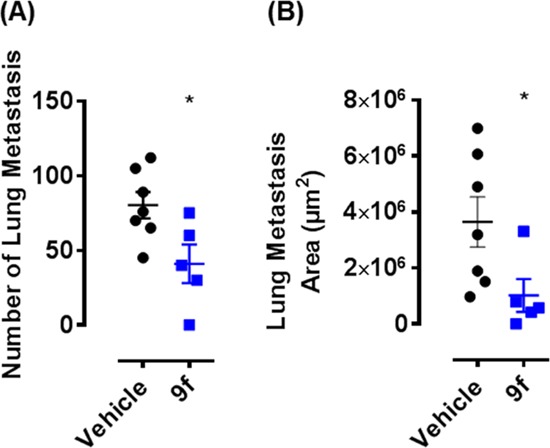
Anti-metastatic efficacy of compound 9f in LOX-driven GEMM model. Animals treated with vehicle (black) or compound 9f at 70 mg/kg qd (blue). All values are reported as the arithmetic mean with the error expressed as the standard error of the means. (A) Number of lung metastasis, vehicle, n = 7; 9f treated, n = 5; (B) Lung metastasis area in μm2, vehicle, n = 7; 9f treated, n = 5.
Synthetic Chemistry
AMT-Sulfonamides
All sulfonamide analogues were synthesized from the sulfonyl chloride intermediates 11a and 11b by condensation with the corresponding amines in dichloromethane (DCM) (Scheme 1). Subsequent trifluoroacetamide hydrolysis or methanolysis using aqueous NaOH or 7 N NH3 in methanol furnishes the desired amines 2a–2i. 3-Methylthiophene sulfonamide 5f (Table 4) was also synthesized by this method. Sulfonyl chlorides 11a and 11b were derived from the commercially available thiophen-2-ylmethanamines 10a/10b in straightforward steps. Access to sulfonylhomopiperazine analogues with additional functionalization at the free amino group could be achieved via the amine hydrochloride intermediate 12, where N-substituted sulfonylhomopiperazines 2k, 2l, and 2m were obtained in three steps. The unsubstituted sulfonylhomopiperazine 2j was synthesized from sulfonyl chloride 11a by condensation with N-Boc-homopiperazine and trifluoroacetamide hydrolysis, followed by Boc removal.
Scheme 1. General Synthetic Routes to AMT-Sulfonamide Analogues 2a–n and 5f.

Reagents and conditions: (a) TFAA, Et3N, DCM, room temperature (rt); (b) ClSO3H, DCM, −78 °C to rt, then H2O; (c) oxalyl chloride, dimethylformamide (DMF), DCM, rt; (d) R2NH, Et3N, DCM, rt; (e) aq. NaOH, MeOH, rt or 7 N NH3 in MeOH, rt; (f) N-Boc-homopiperazine, Et3N, DCM, rt; (g) 2 M HCl in Et2O, rt; (h) R′-Cl (EtNCO for 2l), Et3N, DCM, rt.
AMT-Sulfones
Synthesis of 2-pyridinesulfonyl 3c proceeded by the lithiation of 2-methylthiophene 14 followed by condensation with aldrithiol-2 to afford the corresponding sulfide, which underwent S-oxidation with mCPBA to afford sulfone 15 (Scheme 2). Subsequent methyl bromination was followed by the displacement of the bromide by sodium azide to give the resulting alkyl azide, which was catalytically hydrogenated to afford the desired 2-pyridinesulfonyl-AMT 3c. Similarly, ring opening of 1,2-dithiane with lithiated 2-methylthiophene followed by condensation with iodomethane afforded the corresponding bis-sulfide, which underwent subsequent S-oxidation to afford bis-sulfone 16. This intermediate was subsequently converted to bis-sulfonylbutyl-AMT 3j by the method described above.
Scheme 2. Synthetic Routes to AMT-Sulfone Analogues 3c and 3j.

Reagents and conditions: (a) nBuLi, tetrahydrofuran (THF), −40 °C, then aldrithiol-2, −40 °C to rt; (b) mCPBA, DCM, rt; (c) NBS, Bz2O2, 1,2-dichloroethane (DCE), 70 °C; (d) NaN3, DMF, rt; (e) H2, Pd/C, THF, rt; (f) nBuLi, THF, −40 °C, then 1,2-dithiane, −40 °C to rt, then MeI, rt.
The commercially available 5-bromo-2-thiophenecarbonitrile 17a (interchangeable with 5-chloro-2-thiophenecarbonitrile) and 2-bromothiazole-5-carbonitrile 17b served as valuable building blocks for the AMT-sulfone inhibitors (Scheme 3). Nucleophilic aromatic substitution with a range of thiols afforded the corresponding sulfides, which were oxidized with mCPBA to afford sulfone intermediates 18. Subsequent nitrile reduction with borane–tetrahydrofuran complex afforded AMT-sulfones 3a, 3b, 3d, 3f, 3g, 3i, 5b, 8a, 8b, and 9b. For sulfonylaniline 3h, the product from the initial condensation 19 underwent an additional sulfonamide formation step. Subsequent mCPBA-mediated oxidation afforded the usual sulfone intermediate 18, which was converted to the desired target. Analogues 3e and 5c were synthesized from carbamates 20, which were derived from nitriles 17a and 17b, respectively, by condensation with the corresponding thiols, followed by nitrile reduction and subsequent Boc protection.
Scheme 3. Synthetic Routes to AMT-sulfone Analogues 3a, 3b, 3d–3i, 5b, 5c, 8a, 8b, and 9b.

Reagents and conditions: (a) RSH, K2CO3, DMF, heat; (b) mCPBA, DCM, rt; (c) BH3·THF, THF, rt; (d) MsCl, Et3N, DCM, rt; (e) Boc2O, Et3N, DCM, rt; (f) 4 M HCl in dioxane, rt.
Sulfonamide-substituted phenylsulfonyl-AMT analogues 8c–8e were synthesized from the corresponding thiols 22 (Scheme 4). Hence, condensation of 3- or 4-fluorophenylsulfonyl chloride 21 with the desired amines (HNR2) afforded the corresponding sulfonamides, which were thiolated by treatment with sodium thiomethoxide to afford intermediate thiols 22. The thiols were condensed with 5-chloro-2-thiophene-carbonitrile to yield sulfides 23. The nitrile group of sulfides 23 was then converted to the corresponding trifluoroacetamide by nitrile reduction and amide formation. S-oxidation of sulfides 24 and subsequent amine deprotection affords the desired AMT targets 8c–8e.
Scheme 4. Synthetic Route to Sulfonamide-Substituted Phenylsulfonyl-AMTs 8c–8e.

Reagents and conditions: (a) HNR2, DCM; (b) NaSMe, DMF, 165 °C; (c) 5-chloro-2-thiophene-carbonitrile, K2CO3, DMF, 120–130 °C; (d) BH3·THF, THF, rt; (e) TFAA, Et3N, DCM; (f) mCPBA, DCM, rt; (g) 7 N NH3 in MeOH, rt.
The sulfonyl side chains of AMT analogues 8g–8k were introduced through the nucleophilic substitution of the chlorine atom on intermediate 26 (Scheme 5), which could be obtained in straightforward steps using methods previously described in Scheme 3. Condensation of intermediate 26 with 2-mercaptoethanol or 3-mercaptopropanol followed by sulfide oxidation led to alkanol intermediates 27. After Boc removal, these intermediates afforded AMT targets 8g and 8h. Alternatively, treatment of hydroxyethylsulfone 27 (n = 1) with methanesulfonyl chloride led to concomitant sulfonylation and elimination, furnishing vinyl sulfone 28. Addition of pyrrolidine to vinyl sulfone 28 followed by Boc removal gives AMT target 8i. Ammonia addition to intermediate 28 provided the corresponding aminoethyl sulfone, which was treated with acetic anhydride to afford the corresponding AMT-acetamide 8j after Boc removal. Methoxyethylsulfone 8k was also obtained by from vinyl sulfone 28 by methoxide addition followed by Boc removal.
Scheme 5. Synthetic Routes to Phenylsulfonyl-AMTs with Sulfonyl Side-Chain Modifications (8g–8k).

Reagents and conditions: (a) 2-mercaptoethanol or 3-mercaptopropanol, K2CO3, DMF, 50 °C; (b) mCPBA, DCM, rt (45–61% over two steps); (c) 4 M HCl in dioxane, rt; (d) MsCl, Et3N, DCM, rt (quant.); (e) pyrrolidine, DCM, rt (85% for steps e + c); (f) 7 N NH3 in MeOH, rt; (g) Ac2O, Et3N, DCM, rt; (h) K2CO3, MeOH, rt.
Phenyl sulfide 29, obtained from methods previously described in Scheme 3, underwent a Friedel–Crafts sulfonylation to afford the corresponding phenylsulfonyl (Scheme 6), which was subsequently oxidized to bis-sulfone 30 using mCPBA. Reduction of the nitrile group with borane–tetrahydrofuran complex afforded the bis-sulfonylphenyl-AMT analogue 8f.
Scheme 6. Synthetic Route to Bis-sulfonylphenyl-AMT 8f.

Reagents and conditions: (a) PhSO2Cl, FeCl3, neat, 100 °C; (b) mCPBA, DCM, rt (c) BH3·THF, THF, rt.
tert-Butyl-bis-sulfonylphenyl-AMT 9a was synthesized from 1,3-dibromo-5-(tert-butyl)benzene 31 (Scheme 7). tert-Butyllithium-mediated lithium-bromine exchange of dibromide 31 and treatment of the resultant lithium phenylate with dimethyl disulfide afforded the corresponding methyl sulfide, which was oxidized to sulfone 32 using mCPBA. This underwent Pd-catalyzed cross coupling with thiophene-2-thiol to yield the corresponding thiophene sulfide, which was formylated under Vilsmeier–Haack conditions to give aldehyde 33. After reduction of the aldehyde with sodium borohydride and subsequent sulfide oxidation, the hydroxyl group was substituted with azide using diphenyl phosphoryl azide (DPPA) before it was subsequently reduced to the desired AMT target 9a.
Scheme 7. Synthetic Route to tert-Butyl-bis-sulfonylphenyl-AMT 9a.

Reagents and conditions: (a) tBuLi, THF, −78 °C then MeSSMe, −78 °C to rt; (b) mCPBA, DCM, rt; (c) thiophene-2-thiol, Pd2(dba)3, xantphos, K2CO3, p-xylene; 140 °C; (d) POCl3, DMF, 50 °C; (e) NaBH4, THF, rt; (f) DPPA, PPh3, diisopropyl azodicarboxylate, THF, rt; (g) H2, Pd/C, EtOH, rt.
5-Substituted 1,3-bis-sulfonylphenyl-AMTs 9c–9i were synthesized from aryl bromide 34.19 Hence, Pd-catalyzed cross coupling of aryl bromide 34 with trimethylsilylacetylene under Sonogashira conditions afforded the corresponding aryl-alkyne (Scheme 8), which was subjected to HCl-mediated Boc deprotection to afford AMT analogue 9c. The Suzuki–Miyaura coupling with boronic acids gave the corresponding biaryl products, which underwent Boc removal to furnish the desired AMT inhibitors 9d–9i.
Scheme 8. Synthetic Routes to Substituted Bis-sulfonylphenyl-AMTs 9c–9i.

Reagents and conditions: (a) Pd(PPh3)4, CuI, Et3N, trimethylsilylacetylene, dioxane, rt; (b) 4 M HCl in dioxane, DCM, rt; (c) Pd(PPh3)4, RB(OR′)2, Cs2CO3, dioxane, 100 °C.
5-Substituted 1,4-bis-sulfonylphenyl-AMTs 9j–9l were synthesized from bromofluorobenzene intermediate 36 (Scheme 9), which could be obtained from the commercially available 5-bromothiophene-2-carbonitrile 17a and 3-bromo-4-fluorobenzenethiol 35 by methods previously described in Scheme 3. Selective substitution of the fluorine atom of intermediate 36 with thiomethoxide afforded the corresponding sulfide, which was oxidized to sulfone 37 using mCPBA. The Suzuki–Miyaura coupling with boronic acids and subsequent Boc removal furnished the desired AMT inhibitors 9j–9l.
Scheme 9. Synthetic Routes to Substituted Bis-sulfonylphenyl-AMTs 9j–9l.

Reagents and conditions: (a) NaSMe, DMF, rt; (b) mCPBA, DCM, rt; (c) Pd(PPh3)4, RB(OR′)2, Cs2CO3, dioxane, 100 °C; (d) 4 M HCl in dioxane, DCM, rt.
AMT-Sulfide, AMT-Sulfoxide, and Aminomethylene Modifications
AMT-sulfide 4b and AMT-sulfoxide 4a (Scheme 10) were derived from intermediate sulfide 29 (see Scheme 3; R = Ph). Mono S-oxidation of sulfide 29 with 1 equivalent of mCPBA and subsequent nitrile reduction leads to AMT-sulfoxide 4a. Reduction of intermediate 29 with borane–tetrahydrofuran complex, followed by Boc protection affords carbamate 38. Removal of Boc from carbamate 38 led to AMT-sulfide 4b, whereas N-methylation using sodium hydride/iodomethane and subsequent oxidation with mCPBA affords intermediate 39, which was deprotected to afford the N-methyl AMT analogue 6a. Alternatively, sulfone 6c can be obtained from the condensation of bromothiophene 17a and sodium benzenesulfinate in DMF. This intermediate was converted to the corresponding thiopheneacetimidamide 6d and thiophenecarboxamide 6e by treatment with indium trichloride (InCl3) and lithium hexamethyldisilazide (LiHMDS), respectively.
Scheme 10. Synthetic Routes to Compounds 4a, 4b, 6a, 6c–6e.

Reagents and conditions: (a) mCPBA (2 equiv), DCM, rt; (b) mCPBA (1 equiv), DCM, rt; (c) BH3·THF, THF, rt; (d) InCl3, acetaldoxime, toluene, reflux; (e) LiHMDS, Et2O, rt, then 2 M HCl, rt; (f) Boc2O, Et3N, DCM, rt; (g) NaH, MeI, THF, rt; (h) 4 M HCl in dioxane, DCM, rt; (i) sodium benzenesulfinate, DMF, 135 °C.
Condensation of 1-(5-chlorothiophen-2-yl)ethan-1-one 40 with thiophenol afforded the corresponding sulfide (Scheme 11), which was oxidized to sulfone 41 using mCPBA. Subsequent oxime formation followed by reduction using zinc powder in trifluoroacetic acid furnished C-methylated AMT 6b. Sulfide 42 was obtained in straightforward steps by methods previously illustrated in Scheme 3. Oxidation of sulfide 42 with mCPBA afforded the corresponding sulfone 43. This was reduced to the corresponding aldehyde with DIBAL-H before conversion to oxime 6f by treatment with hydroxylamine hydrochloride.
Scheme 11. Synthetic Routes to Compounds 6b and 6f.

Reagents and conditions: (a) thiophenol, K2CO3, DMF, 120 °C; (b) mCPBA, DCM, rt; (c) H2NOH·HCl, N,N-diisopropylethylamine (DIPEA), EtOH, rt; (d) Zn, TFA, rt; (e) DIBAL-H, DCM, 45 °C; (f) H2NOH·HCl, K2CO3, EtOH, 70 °C.
AMT- and Aminomethylenefuran-amides
Synthesis of AMT-amide 4d and aminomethylenefuran-amide 5f were accomplished via methyl esters 44 (Scheme 12; A = S or O).20,21 Esters 44 first underwent saponification with aqueous hydroxide, and the resultant carboxylic acids were then converted to acid chlorides 45 using oxalyl chloride. Condensation with benzylamine in dichloromethane followed by catalytic hydrogenation using 10% Pd/C in tetrahydrofuran furnished the desired amides 4d and 5e.
Scheme 12. Synthetic Routes to Amide Analogues 4d and 5e.

Reagents and conditions: (a) 1 M NaOH, MeOH, rt; (b) (COCl)2, DMF, DCM, rt; (c) benzylamine, DCM, rt; (d) H2, Pd/C, THF, rt.
Aminomethylene-pyridine, Aminomethylene-1,3-Thiazole, and Aminomethylene-Imidazole
Aminomethylene-pyridine 5a was obtained from the commercially available tert-butyl ((6-chloropyridin-3-yl)methyl)carbamate 46. Condensation with sodium benzylthiolate afforded the corresponding sulfide 47 (Scheme 13). Sulfide oxidation with mCPBA and subsequent HCl-mediated Boc removal furnished the desired target 5a. 2-(Aminomethylene)-1,3-thiazole 5d was obtained from sulfide intermediate 49 in a similar manner. Sulfide 49 was synthesized from tert-butyl ((5-bromothiazol-2-yl)methyl)carbamate2248 and naphthalene-2-thiol by a Pd-catalyzed cross-coupling reaction.
Scheme 13. Synthetic Routes to Aminomethylene-Pyridine, Aminomethylene-Thiazole, and Aminomethylene-Imidazole 5a and 5d.

Reagents and conditions: (a) NaH, benzylmercaptan, DMF, 70 °C; (b) mCPBA, DCM, rt; (c) 2 M HCl in Et2O or 4 M HCl in dioxane, rt; (d) Pd2(dba)3, xantphos, naphthalene-2-thiol, NaOtBu, tBuOH/toluene, 100 °C.
AMT-Sulfone Regiosiomers
The AMT regioisomers 7a–7c were synthesized by two different methods (Scheme 14). The synthesis of 2,3-regioisomer 7a began with nucleophilic aromatic substitution between 3-bromothiophene-2-carbonitrile 51 and naphthalene-2-thiol. The resultant sulfide underwent subsequent sulfide oxidation followed by nitrile reduction to afford the desired target 7a. The initial nucleophilic aromatic substitution step was replaced with a Pd-catalyzed cross coupling for the synthesis of the 3,4- and 2,4-regioisomers 7b and 7c, starting from bromothiophenes 53 and 55. The resultant sulfides were oxidized to sulfones 54 and 56, which were subsequently converted to the corresponding AMT targets 7b and 7c by nitrile reduction.
Scheme 14. Synthetic Routes to AMT Regioisomers 7a–7c.

Reagents and conditions: (a) naphthalene-2-thiol, K2CO3, DMF, 120 °C; (b) mCPBA, DCM, rt; (c) BH3·THF, THF, rt; (d) Pd2dba3, xantphos, naphthalene-2-thiol, NaOtBu, toluene, 110 °C.
Finally, the Suzuki–Miyaura coupling between 5-bromothiophene-2-carbonitrile 17a and (4-(methylthio)phenyl)boronic acid 57 affords phenyl thiophene 58 (Scheme 15). This was converted to carbamate 59 by nitrile reduction, Boc protection, and sulfide oxidation. Subsequent Boc removal furnished the phenyl-linked AMT analogue 4e.
Scheme 15. Synthetic Route to Phenyl-Linked AMT 4e.

Reagents and conditions: (a) Pd(PPh3)4, Cs2CO3, 1,4-dioxane, 100 °C; (b) BH3·THF, THF, rt; (c) Boc2O, Et3N, DCM, rt; (d) mCPBA, DCM, rt; (e) 4 M HCl in dioxane, DCM, rt.
Conclusions
We described herein a series of LOX inhibitors containing a 2-aminomethylene-5-sulfonyl-thiophene core. The attachment of a sulfonylphenyl side chain to the core scaffold via the 5-sulfonyl linker furnishes inhibitors with sub-micromolar LOX IC50 values. Further SAR optimization leads to the discovery of inhibitor 9f with potent anti-LOX activity as well as desirable selectivity and PK profile, making it a valuable asset for LOX research. More importantly, we have described its ability to reduce the growth of spontaneous breast tumor lung metastasis in a GEMM,19 thus demonstrating the promise of 9f as a drug candidate.
Experimental Section
Synthesis of Inhibitors
Commercial building blocks, reagents, and solvents for reactions were reagent grade and used as purchased. Flash chromatography was performed on a Biotage Isolera flash purification system using prepacked silica gel cartridges (Biotage) with HPLC grade solvents. Thin-layer chromatography analysis was performed using silica gel 60 F-254 thin-layer plates. Liquid chromatography mass spectrometry (LCMS) and high-resolution mass spectrometry (HRMS) analyses of chemical compounds were performed on an Agilent 1200 series HPLC and a diode array detector coupled to a 6210 time-of-flight mass spectrometer with a multimode ESI source or a Waters Acquity UPLC and diode array detector coupled to a Waters G2 QToF mass spectrometer fitted with a multimode ESI/APCI source. 1H and 13C NMR spectra were recorded on a Bruker Avance 500 MHz or a 300 MHz spectrometer using an internal deuterium lock. NMR data is given as follows: chemical shift (δ) in ppm, multiplicity, coupling constants (J) given in hertz and integration. All final inhibitors submitted for biological evaluation were at least 95% pure by HPLC, apart from compound 2g, which has a purity of 94%.
General Procedures GP1
Alkylamine was added to a solution of sulfonyl chloride 11a in DCM, and the reaction mixture was stirred at rt for 1–16 h. One molar HCl was added, and the aqueous phase was extracted with DCM (3×). The combined organic layer was dried over MgSO4 and filtered, and the solvent was removed under reduced pressure. MeOH and aq. NaOH were then added, and the mixture was stirred at rt for 16 h (MeOH and aq. NaOH can be replaced by 7 N NH3 in MeOH). H2O was added, and the aqueous phase was extracted with DCM (3×). The combined organic phase was dried over MgSO4 and filtered, and the solvent was removed under reduced pressure to afford the desired AMT-sulfonamide, which could be further purified if necessary.
General Procedures GP2
Four molar HCl in dioxane or 2 M HCl in Et2O was added to tert-butyl carbamate (neat or as a solution in DCM), and the mixture was stirred at rt for 1–16 h. EtOAc was added to precipitate the solids if necessary. The solids were collected by filtration or centrifugation, washed with EtOAc, and dried under vacuum to afford the desired amine hydrochloride. If necessary, this can be further purified by chromatography in its free amine form, which can be obtained by treatment with 7 N NH3 in MeOH.
General Procedures GP3
BH3·THF was added to a solution of heteroaryl nitrile in THF, and the mixture was stirred at rt for 1–5 h. EtOH (equal volume to reaction mixture) was then carefully added to quench the reaction. The solution was subsequently heated at 70 °C for 1 h to aid borane decomplexation. The solvent was removed under reduced pressure to afford the desired amine, which could be purified if necessary.
General Procedures GP4
A mixture of 5-bromothiophene-2-carbonitrile 17a (interchangeable with 5-chorothiophene-2-carbonitrile) or 2-bromothiazole-5-carbonitrile 17b, alkyl or aryl thiol, K2CO3, and DMF was stirred at 50–140 °C. After cooling to rt, the mixture was diluted with EtOAc. The organic phase was washed with 1:1 H2O/brine (3×), dried over MgSO4, and filtered. The solvent was removed under reduced pressure to afford the desired sulfide product, which could be further purified if necessary.
General Procedures GP5
mCPBA (>2 equiv for sulfones, 1.0 equiv for sulfoxides) was added in small portions to a solution of sulfide in DCM at 0 °C, and the mixture was stirred at rt (0 °C for sulfoxides) for 3–16 h. When complete conversion was achieved, EtOAc was added. The organic phase was washed with sat. NaHCO3 (3×) and sat. Na2S2O3 (until no color was detected on starch iodide paper), dried over MgSO4, and filtered. The solvent was removed under reduced pressure to afford the desired sulfone/sulfoxide, which could be further purified if necessary.
General Procedures GP6
The alkylamine was dissolved in DCM. Et3N followed by Boc2O were added, and the mixture was stirred at rt for 16 h. When complete conversion was achieved, DCM was added. The organic phase was washed with H2O and brine, dried over MgSO4, and filtered. The solvent was removed under reduced pressure to afford the desired tert-butyl-carbamate, which could be further purified if necessary.
General Procedures GP7
A mixture of aryl bromide, Pd(PPh3)4, ArB(OR′)2 (boronic acid or pinacolatoboronate), Cs2CO3, and 1,4-dioxane was degassed with argon and then stirred at 100 °C for 16 h. After cooling to rt, the mixture was filtered through celite and washed with EtOAc. The solvent was removed under reduced pressure to afford the desired biaryl, which could be further purified if necessary.
General Procedures GP8
A mixture of aryl bromide, Pd2(dba)3, Xantphos, alkyl or aryl thiol, NaOtBu, and tBuOH/toluene (1:4) was degassed with argon and then stirred at 100 °C for 16 h. After cooling to rt, the suspension was filtered through celite and washed with DCM. The solvent was removed under reduced pressure to afford the desired sulfide, which could be further purified if necessary.
(5-(Piperidin-1-ylsulfonyl)thiophen-2-yl)methanamine (2a)
The titled compound was synthesized according to general procedure GP1, from (i) sulfonyl chloride 11a (150 mg, 0.487 mmol), piperidine (96.3 μL, 0.975 mmol), and DCM (2.4 mL), 16 h, rt and (ii) 1 M NaOH (2 mL) and MeOH (2 mL), rt, 16 h. Compound 2a was obtained as a white crystalline solid (109 mg, 86%) and did not require further purification. 1H NMR (500 MHz, chloroform-d) δ 7.37 (d, J = 3.6 Hz, 1H), 6.92 (d, J = 3.6 Hz, 1H), 4.20 (br, 2H), 3.11–2.96 (m, 4H), 1.82–1.12 (m, 8H). 13C NMR (126 MHz, chloroform-d) δ 134.51, 132.45, 123.57, 47.10, 25.23, 23.58. HRMS (ESI) for C10H17N2O2S2 ([M + H]+): calculated 261.0726; observed 261.0743; error = 4.8 ppm.
5-(Aminomethyl)-N,N-dimethylthiophene-2-sulfonamide (2b)
The titled compound was synthesized according to general procedure GP1, from (i) sulfonyl chloride 11a (100 mg, 0.325 mmol), dimethylamine (40% in H2O; 0.206 mL, 1.62 mmol), and DCM (1.63 mL), 16 h, rt and (ii) 30% NaOH (0.5 mL) and MeOH (3 mL), rt, 16 h. Compound 2b was obtained as a white solid (38 mg, 53%) and did not require further purification. 1H NMR (500 MHz, chloroform-d) δ 7.41 (d, J = 3.7 Hz, 1H), 6.95 (dt, J = 3.7, 1.0 Hz, 1H), 4.12 (d, J = 0.6 Hz, 2H), 2.76 (s, 6H), 1.61 (s, 2H). 13C NMR (126 MHz, chloroform-d) δ 155.65, 133.60, 132.76, 123.60, 41.66, 38.18. HRMS (ESI) for C7H13N2O2S2 ([M + H]+): calculated 221.0413; observed 221.0409; error = 1.8 ppm.
5-(Aminomethyl)-N-ethylthiophene-2-sulfonamide (2c)
The titled compound was synthesized according to general procedure GP1, from (i) sulfonyl chloride 11a (100 mg, 0.325 mmol), ethylamine (2.0 M in MeOH; 0.812 mL, 1.62 mmol), and DCM (1.63 mL), 16 h, rt and (ii) 30% NaOH (0.5 mL) and MeOH (3 mL), rt, 16 h. Compound 2c was obtained as a colorless oil (23 mg, 32%) and did not require further purification. 1H NMR (500 MHz, chloroform-d) δ 7.46 (d, J = 3.7 Hz, 1H), 6.88 (d, J = 3.7 Hz, 1H), 4.68 (br, 1H), 4.09 (s, 2H), 3.09 (q, J = 7.2 Hz, 2H), 1.76 (br, 2H), 1.15 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, chloroform-d) δ 155.38, 138.81, 132.43, 123.47, 41.60, 38.62, 15.11. HRMS (ESI) for C7H10NO2S2 ([M – NH2]+): calculated 204.1480; observed 204.01460; error = 0.98 ppm.
(S)-1-(5-(Aminomethyl)thiophen-2-ylsulfonyl)pyrrolidine-2-carboxamide (2d)
The titled compound was synthesized according to general procedure GP1, from (i) sulfonyl chloride 11a (75 mg, 0.244 mmol), prolinamide hydrochloride (30.6 mg, 0.268 mmol), Et3N (74.7 μL, 0.536 mmol), and DCM (1.2 mL), 16 h, rt and (ii) 1 M NaOH (1.2 mL) and MeOH (1.2 mL), rt, 16 h. The crude was purified by chromatography (1 N NH3 in MeOH/DCM 0 → 20%) to afford an orange oil (10 mg, 14%). [α]D21 −134.2 (c 0.16, MeOH/CHCl3). 1H NMR (500 MHz, methanol-d4) δ 7.58 (d, J = 3.8 Hz, 1H), 7.15 (d, J = 3.8 Hz, 1H), 4.10 (s, 2H), 4.06 (dd, J = 8.3, 4.0 Hz, 1H), 3.61 (ddd, J = 10.2, 6.1, 4.6 Hz, 1H), 3.32 (m, 1H), 1.99 (m, 1H), 1.93–1.82 (m, 2H), 1.67 (m, 1H). 13C NMR (126 MHz, methanol-d4) δ 177.35, 154.70, 135.83, 134.45, 126.75, 63.79, 50.93, 41.12, 32.07, 25.51. HRMS (ESI) for C10H13N2O3S2 ([M – NH2]+): calculated 273.0362; observed 273.0334; error = 10 ppm.
(S)-(1-(5-(Aminomethyl)thiophen-2-ylsulfonyl)pyrrolidin-2-yl)methanol (2e)
The titled compound was synthesized according to general procedure GP1, from (i) sulfonyl chloride 11a (100 mg, 0.325 mmol), (S)-pyrrolidin-2-ylmethanol (35.3 μL, 0.358 mmol), Et3N (100 μL, 0.715 mmol), and DCM (1.6 mL), 2 h, rt and (ii) 7 N NH3 in MeOH (5 mL), rt, 20 h. The crude was purified by chromatography (1 N NH3 in MeOH/DCM 0 → 15%) to afford a yellow oil (47 mg, 52%). [α]D21 −33.6 (c 2.76, MeOH/CHCl3). 1H NMR (500 MHz, chloroform-d) δ 7.47 (d, J = 3.8 Hz, 1H), 6.94 (dt, J = 3.7, 1.0 Hz, 1H), 4.11 (d, J = 0.8 Hz, 2H), 3.80–3.57 (m, 3H), 3.53–3.45 (m, 1H), 3.30 (dt, J = 10.5, 7.1 Hz, 1H), 2.18–1.95 (m, 3H), 1.90–1.69 (m, 3H), 1.60–1.50 (m, 1H). 13C NMR (126 MHz, chloroform-d) δ 155.82, 134.49, 133.01, 123.57, 65.67, 62.29, 50.22, 41.55, 28.90, 24.41. HRMS (ESI) for C10H17N2O3S2 ([M + H]+): calculated 277.0675; observed 277.0629; error = 17 ppm.
(5-(2-Phenylpyrrolidin-1-ylsulfonyl)thiophen-2-yl)methanamine (2f)
The titled compound was synthesized according to general procedure GP1, from (i) sulfonyl chloride 11a (100 mg, 0.325 mmol), 2-phenylpyrrolidine (40.7 mg, 0.357 mmol), Et3N (100 μL, 0.715 mmol), and DCM (1.6 mL), 2 h, rt and (ii) 7 N NH3 in MeOH (4 mL), rt, 16 h. The crude was purified by chromatography (1 N NH3 in MeOH/DCM 0 → 20%) to afford a brown oil (40 mg, 38%). 1H NMR (500 MHz, chloroform-d) δ 7.42 (d, J = 3.7 Hz, 1H), 7.38–7.21 (m, 5H), 6.90 (dt, J = 3.7, 1.0 Hz, 1H), 4.80 (dd, J = 8.0, 3.6 Hz, 1H), 4.10 (d, J = 0.9 Hz, 2H), 3.68 (m, 1H), 3.45 (dt, J = 10.5, 7.3 Hz, 1H), 2.04 (m, 1H), 1.96–1.81 (m, 2H), 1.70 (m, 1H), 1.53 (s, 2H). 13C NMR (126 MHz, chloroform-d) δ 155.35, 143.01, 135.67, 132.68, 128.44, 127.18, 126.22, 123.38, 63.71, 49.77, 41.57, 35.82, 24.08. HRMS (ESI) for C15H19N2O2S2 ([M + H]+): calculated 323.0883; observed 323.0873; error = 3.1 ppm.
(5-(Indolin-1-ylsulfonyl)thiophen-2-yl)methanamine (2g)
The titled compound was synthesized according to general procedure GP1, from (i) sulfonyl chloride 11a (100 mg, 0.325 mmol), indoline (142 mg, 0.358 mmol), Et3N (100 μL, 0.715 mmol), and DCM (1.6 mL), 2 h, rt and (ii) 7 N NH3 in MeOH (5 mL) at rt, for 20 h. The crude was purified by chromatography (1 N NH3 in MeOH/DCM 0 → 15%) to afford a brown solid (73 mg, 76%). 1H NMR (500 MHz, chloroform-d) δ 7.58 (d, J = 8.1 Hz, 1H), 7.42 (d, J = 3.8 Hz, 1H), 7.24–7.16 (m, 1H), 7.12 (d, J = 7.4 Hz, 1H), 7.00 (td, J = 7.4, 0.9 Hz, 1H), 6.88–6.78 (m, 1H), 4.01 (s, 2H), 3.95 (d, J = 8.4 Hz, 2H), 2.98 (t, J = 8.4 Hz, 2H), 1.56 (s, 2H). 13C NMR (126 MHz, chloroform-d) δ 156.23, 141.59, 133.90, 132.97, 131.84, 127.81, 125.24, 124.07, 123.43, 115.10, 50.30, 50.25, 41.51, 27.91. HRMS (ESI) for C13H15N2O2S2 ([M + H]+): calculated 295.0569; observed 295.0577; error = 2.7 ppm.
(5-(3,4-Dihydroquinolin-1(2H)-ylsulfonyl)thiophen-2-yl)methanamine (2h)
The titled compound was synthesized according to general procedure GP1, from (i) sulfonyl chloride 11a (100 mg, 0.325 mmol), tetrahydroquinoline (44.9 μL, 0.357 mmol), Et3N (100 μL, 0.715 mmol), and DCM (1.6 mL), 2 h, rt and (ii) 7 N NH3 in MeOH (5 mL), rt, 20 h. The crude was purified by chromatography (1 N NH3 in MeOH/DCM 0 → 10%) to afford a brown foam (27 mg, 27%). 1H NMR (500 MHz, chloroform-d) δ 7.78 (d, J = 8.3 Hz, 1H), 7.25–7.16 (m, 2H), 7.13–7.02 (m, 2H), 6.84 (d, J = 3.7 Hz, 1H), 4.04 (s, 2H), 3.87–3.81 (m, 2H), 2.54 (t, J = 6.7 Hz, 2H), 2.37–1.82 (m, 2H), 1.80–1.72 (br, 2H). 13C NMR (126 MHz, chloroform-d) δ 137.52, 136.43, 132.42, 130.96, 129.27, 126.69, 125.28, 125.03, 123.78, 46.98, 41.34, 26.93, 21.78. HRMS (ESI) for C14H17N2O2S2 ([M + H]+): calculated 309.0726; observed 309.0732; error = 1.9 ppm.
(5-(Morpholinosulfonyl)thiophen-2-yl)methanamine (2i)
The titled compound was synthesized according to general procedure GP1, from (i) sulfonyl chloride 11a (75 mg, 0.244 mmol), morpholine (42.7 μL, 0.487 mmol), and DCM (1.2 mL), 16 h, rt and (ii) 1 M NaOH (1 mL) and MeOH (1 mL), rt, 16 h. The crude was dissolved in MeOH, passed through an SCX ion exchange (sulfonic acid) column, and washed with MeOH. The amine was released by the addition of 1 N NH3 in MeOH to afford a crystalline solid (47 mg, 73%). 1H NMR (500 MHz, chloroform-d) δ 7.40 (d, J = 3.7 Hz, 1H), 6.95 (d, J = 3.6 Hz, 1H), 4.08 (br, 2H), 3.90–3.65 (m, 4H), 3.15–2.89 (m, 4H), 1.64 (br, 2H). 13C NMR (126 MHz, chloroform-d) δ 133.16, 133.01, 123.70, 66.13, 46.12. HRMS (ESI) for C9H15N2O3S2 ([M + H]+): calculated 263.0519; observed 263.0533; error = 5.3 ppm.
(5-(1,4-Diazepan-1-ylsulfonyl)thiophen-2-yl)methanamine Dihydrochloride (2j)
tert-Butyl 4-((5-(aminomethyl)thiophen-2-yl)sulfonyl)-1,4-diazepane-1-carboxylate was synthesized according to general procedure GP1, from (i) sulfonyl chloride 11a (75 mg, 0.244 mmol), N-Boc-homopiperazine (53.7 mg, 0.268 mmol), Et3N (74.7 μL, 0.536 mmol), and DCM (1.2 mL), rt, 16 h and (ii) 1 M NaOH (5 mL), MeOH (5 mL), rt, 16 h. The crude was used in the subsequent transformation immediately.
Compound 2j was synthesized according to general procedure GP2, from tert-butyl 4-((5-(aminomethyl)thiophen-2-yl)sulfonyl)-1,4-diazepane-1-carboxylate (crude) and 2 M HCl in Et2O (10 mL), rt, 16. Compound 2j was obtained as a pink solid (43 mg, 63%). 1H NMR (500 MHz, D2O) δ 7.77 (br, 1H), 7.44 (br, 1H), 4.55 (s, 2H), 3.75 (br, 2H), 3.60–3.45 (m, 6H), 2.23 (br, 2H). 13C NMR (126 MHz, D2O) δ 141.90, 137.80, 133.32, 130.36, 47.20, 46.84, 44.85, 44.38, 37.31, 25.22. HRMS (ESI) for C10H18N3O2S2 ([M + H]+): calculated 276.0835; observed 276.0771; error = 23 ppm.
(5-(4-Methyl-1,4-diazepan-1-ylsulfonyl)thiophen-2-yl)methanamine (2k)
A mixture of sulfonyl chloride 11a (645 mg, 2.10 mmol), N-Boc-homopiperazine (449 μL, 2.31 mmol), Et3N (643 μL, 4.61 mmol), and DCM (10 mL) was stirred at rt for 2 h. One molar HCl (30 mL) was added. The aqueous phase was extracted with DCM (3 × 15 mL). The combined organic phase was dried over MgSO4 and filtered, and the solvent was removed under reduced pressure. The intermediate was dissolved in 2 M HCl in Et2O (10 mL), and the mixture was stirred at rt for 16 h. The solids were collected, washed with Et2O, and dried under vacuum to afford N-((5-((1,4-diazepan-1-yl)sulfonyl)thiophen-2-yl)methyl)-2,2,2-trifluoroacetamide hydrochloride as a beige solid (580 mg, 68%). 1H NMR (500 MHz, D2O) δ 7.70 (m, 1H), 7.27 (m, 1H), 3.74–3.70 (m, 2H), 3.56–3.42 (m, 6H), 2.27–2.13 (m, 2H). HRMS (ESI) for C12H17F3N3O3S2 ([M + H]+): calculated 372.0658.
A mixture of N-((5-((1,4-diazepan-1-yl)sulfonyl)thiophen-2-yl)methyl)-2,2,2-trifluoroacetamide hydrochloride (80 mg, 0.196 mmol), MeI (18.0 μL, 0.294 mmol), Et3N (68.0 μL, 0.490 mmol), and DCM (1.0 mL) was stirred at rt for 48 h. H2O (10 mL) was added. The aqueous phase was extracted with DCM (3 × 10 mL). The combined organic phase was removed under reduced pressure. The intermediate was dissolved in 7 N NH3 in MeOH (3 mL), and the mixture was stirred at rt for 24 h. The solvent was removed under reduced pressure, and the crude was purified by chromatography (1 N NH3 in MeOH/DCM 0 → 20%) to afford compound 2k as a brown oil (12 mg, 21%). 1H NMR (500 MHz, chloroform-d) δ 7.40 (d, J = 3.7 Hz, 1H), 6.89 (dt, J = 3.7, 0.9 Hz, 1H), 4.10 (d, J = 0.8 Hz, 2H), 3.45–3.38 (m, 4H), 2.69–2.61 (m, 4H), 2.36 (s, 2H), 1.88 (dt, J = 11.7, 6.2 Hz, 2H), 1.69 (s, 3H). 13C NMR (126 MHz, chloroform-d) δ 155.05, 137.16, 131.90, 123.37, 58.55, 56.84, 48.04, 47.38, 46.58, 41.66, 27.55. HRMS (ESI) for C11H20N3O2S2 ([M + H]+): calculated 290.0992; observed 290.0861; error = 45 ppm.
4-(5-(Aminomethyl)thiophen-2-ylsulfonyl)-N-ethyl-1,4-diazepane-1-carboxamide (2l)
A mixture of N-((5-((1,4-diazepan-1-yl)sulfonyl)thiophen-2-yl)methyl)-2,2,2-trifluoroacetamide hydrochloride (80 mg, 0.196 mmol), EtNCO (17.0 μL, 0.216 mmol), Et3N (54.7 μL, 0.392 mmol), and DCM (1.0 mL) was stirred at rt for 2 h. DCM (10 mL) was added. The organic phase was washed with H2O and brine (10 mL each), dried over MgSO4, and filtered, and the solvent was removed under reduced pressure. The intermediate was dissolved in 7 N NH3 in MeOH (3 mL), and the mixture was stirred at rt for 16 h. The solvent was removed under reduced pressure, and the crude was purified by chromatography (MeOH/DCM 0 → 20%) to afford compound 2l as a colorless oil (60 mg, 88%). 1H NMR (500 MHz, chloroform-d) δ 7.38 (d, J = 3.7 Hz, 1H), 6.88 (dt, J = 3.7, 0.9 Hz, 1H), 4.39 (s, 1H), 4.08 (s, 2H), 3.62–3.57 (m, 2H), 3.49 (t, J = 6.4 Hz, 2H), 3.36–3.32 (m, 2H), 3.29–3.20 (m, 4H), 2.02–1.94 (m, 2H), 1.69 (s, 2H), 1.11 (t, J = 7.2 Hz, 3H). 13C NMR (126 MHz, chloroform-d) δ 157.38, 155.48, 136.96, 132.01, 123.40, 50.69, 48.51, 48.13, 45.28, 41.58, 35.79, 28.24, 15.70. HRMS (ESI) for C13H23N4O3S2 ([M + H]+): calculated 347.1206; observed 347.1196; error = 2.9 ppm.
(5-(4-(Methylsulfonyl)-1,4-diazepan-1-ylsulfonyl)thiophen-2-yl)methanamine (2m)
A mixture of N-((5-((1,4-diazepan-1-yl)sulfonyl)thiophen-2-yl)methyl)-2,2,2-trifluoroacetamide hydrochloride (80 mg, 0.196 mmol), MsCl (16.7 μL, 0.216 mmol), Et3N (68.0 μL, 0.490 mmol), and DCM (1.0 mL) was stirred at rt for 48 h·H2O (10 mL) was added. The aqueous phase was extracted with DCM (3 × 10 mL). The combined organic phase was dried over MgSO4 and filtered, and the solvent was removed under reduced pressure. The intermediate was dissolved in 7 N NH3 in MeOH (3 mL), and the mixture was stirred at rt for 24 h. The solvent was removed under reduced pressure, and the crude was purified by chromatography (1 N NH3 in MeOH/DCM 0 → 20%) to afford compound 2m as a white solid (44 mg, 63%). 1H NMR (500 MHz, chloroform-d) δ 7.39 (d, J = 3.8 Hz, 1H), 6.88 (dt, J = 3.7, 1.0 Hz, 1H), 4.08 (d, J = 0.9 Hz, 2H), 3.54–3.34 (m, 8H), 2.85 (s, 3H), 2.00 (pentet, J = 6.3 Hz, 2H), 1.62 (s, 2H). 13C NMR (126 MHz, chloroform-d) δ 155.81, 136.76, 132.10, 123.37, 51.25, 50.49, 47.76, 47.27, 41.56, 38.13, 29.33. HRMS (ESI) for C11H20N3O4S3 ([M + H]+): calculated 354.0611; observed 354.0530; error = 23 ppm.
(5-(Cyclohexylsulfonyl)thiophen-2-yl)methanamine (3a)
NaH (60% in mineral oil; 39.6 mg, 0.985 mmol) was added to a solution of 5-bromothiophene-2-carbonitrile 17a (100 μL, 0.901 mmol) and cyclohexylmercaptan (121 μL, 0.991 mmol) in DMF (3.0 mL), and the mixture was stirred at 130 °C for 16 h. After cooling to rt, EtOAc (15 mL) was added. The organic phase was washed with 1:1 H2O/brine (3 × 15 mL), dried over MgSO4, and filtered. The solvent was removed under reduced pressure, and the intermediate was dissolved in DCM (3.0 mL). mCPBA (77%; 489 mg, 2.18 mmol) was added in small portions, and the mixture was stirred at rt for 3 h. DCM (15 mL) was added. The organic phase was washed with 1 M NaOH (3 × 15 mL) and brine (15 mL), dried over MgSO4, and filtered. The solvent was removed under reduced pressure, and the crude was purified by chromatography (EtOAc/cyclohexane 0 → 20%) to afford 5-(cyclohexylsulfonyl)thiophene-2-carbonitrile as a white crystalline solid (50 mg, 22%). 1H NMR (500 MHz, chloroform-d) δ 7.65 (d, J = 4.0 Hz, 1H), 7.61 (d, J = 4.0 Hz, 1H), 3.03 (tt, J = 12.1, 3.4 Hz, 1H), 2.19–2.11 (m, 2H), 1.97–1.88 (m, 2H), 1.72 (m, 1H), 1.65–1.10 (m, 5H). LCMS (ESI) m/z 278 [M + Na]+.
Compound 3a was synthesized according to general procedure GP3, from BH3 (1.0 M in THF; 0.58 mL, 0.58 mmol), 5-(cyclohexylsulfonyl)thiophene-2-carbonitrile (49 mg, 0.192 mmol), and THF (1.9 mL), rt, 1 h. The crude was purified by chromatography (MeOH/DCM 0 → 15%) to afford a white solid (12 mg, 24%). 1H NMR (500 MHz, chloroform-d) δ 7.50 (d, J = 3.8 Hz, 1H), 6.96 (m, 1H), 4.12 (s, 2H), 2.95 (tt, J = 12.1, 3.4 Hz, 1H), 2.20–2.12 (m, 2H), 1.92–1.84 (m, 2H), 1.68 (d, J = 21.7 Hz, 3H), 1.51–1.38 (m, 2H), 1.33–1.08 (m, 3H). 13C NMR (126 MHz, chloroform-d) δ 158.07, 135.80, 135.11, 123.83, 64.77, 41.72, 25.97, 25.27, 25.19. HRMS (ESI) for C11H17 NO2S2 ([M + H]+): calculated 260.0773; observed 260.0785; error = 4.6 ppm.
(5-(Phenylsulfonyl)thiophen-2-yl)methanamine (3b)
A mixture of nitrile 6c (39 mg, 0.156 mmol), LiAlH4 (1.0 M in THF; 160 μL, 0.160 mmol), and THF (1.6 mL) was stirred at 0 °C for 1 h. H2O (5 mL) was slowly added, and the aqueous phase was extracted with DCM (3 × 8 mL). The combined organic phase was washed with brine (10 mL), dried over MgSO4, and filtered. The solvent was removed under reduced pressure, and the crude was purified by chromatography (7 N NH3 in MeOH/DCM 0 → 60%) to afford compound 3b as a light brown crystalline solid (15 mg, 38%). 1H NMR (500 MHz, chloroform-d) δ 8.01–7.95 (m, 2H), 7.63–7.48 (m, 4H), 6.88 (dt, J = 3.8, 1.0 Hz, 1H), 4.07 (s, 2H), 1.58 (s, 2H). 13C NMR (126 MHz, chloroform-d) δ 158.19, 142.50, 140.63, 133.70, 133.26, 129.39, 127.41, 123.80, 41.71. HRMS (ESI) for C11H12NO2S2 ([M + H]+): calculated 254.0304; observed 254.0309; error = 2.0 ppm.
(5-(Pyridin-2-ylsulfonyl)thiophen-2-yl)methanamine (3c)
A mixture of 2-((5-methylthiophen-2-yl)sulfonyl)pyridine 15 (310 mg, 1.30 mmol), Bz2O2 (75%; 20.9 mg, 0.0648 mmol), NBS (253 mg, 1.42 mmol), and DCE (6.5 mL) was stirred at 80 °C for 16 h. The solvent was removed under reduced pressure, and the crude was purified by chromatography (EtOAc/cyclohexane 20 → 40%) to afford 2-((5-(bromomethyl)thiophen-2-yl)sulfonyl)pyridine as a white solid (294 mg, 71%). 1H NMR (500 MHz, chloroform-d) δ 8.74 (s, 1H), 8.19 (s, 1H), 7.97 (s, 1H), 7.71 (s, 1H), 7.53 (s, 1H), 7.13 (d, J = 3.9 Hz, 1H), 4.66 (s, 2H). LCMS (ESI) m/z 318/320 [M + H]+.
A mixture of 2-((5-(bromomethyl)thiophen-2-yl)sulfonyl)pyridine (133 mg, 0.418 mmol), NaN3 (32.6 mg, 0.502 mmol), and DMF (2.1 mL) was stirred at 70 °C for 16 h. After cooling to rt, EtOAc (20 mL) was added. The organic phase was washed with 1:1 H2O/brine (2 × 20 mL), dried over MgSO4, and filtered. The solvent was removed under reduced pressure, and the intermediate was dissolved in THF (2.1 mL), and Pd/C (10%; 44 mg, 0.0418 mmol) was then added. The mixture was stirred at rt under a H2 atmosphere (balloon) for 16 h and subsequently filtered through celite. The solvent was removed under reduced pressure, and the crude was purified by chromatography (MeOH/DCM 0 → 20%) to afford compound 3c as a white solid (53 mg, 50%). 1H NMR (500 MHz, chloroform-d) δ 8.69 (ddd, J = 4.7, 1.7, 0.9 Hz, 1H), 8.15 (dt, J = 7.9, 1.0 Hz, 1H), 7.92 (td, J = 7.8, 1.7 Hz, 1H), 7.70 (d, J = 3.8 Hz, 1H), 7.46 (ddd, J = 7.6, 4.7, 1.1 Hz, 1H), 6.92 (dt, J = 3.9, 1.0 Hz, 1H), 4.07 (d, J = 0.9 Hz, 2H), 1.62 (s, 2H). 13C NMR (126 MHz, chloroform-d) δ 159.60, 159.12, 150.48, 138.27, 136.95, 135.65, 127.01, 123.94, 121.78, 41.71. HRMS (ESI) for C10H11N2O2S2 ([M + H]+): calculated 255.0257; observed 255.0276; error = 7.5 ppm.
(5-(Thiophen-2-ylsulfonyl)thiophen-2-yl)methanamine Hydrochloride (3d)
The titled compound was synthesized according to general procedures GP4, GP5, and GP3, from (i) 5-bromothiophene-2-carbonitrile 17a (1.0 g, 5.32 mmol), thiophene-2-thiol (586 mg, 5.05 mmol), K2CO3 (881 mg, 6.38 mmol), and DMF (17 mL), 80 °C, 16 h; (ii) mCPBA (77%; 1.30 g, 11.2 mmol) and DCM (36 mL), rt, 3 h; and (iii) BH3 (1.0 M in THF; 10.6 mL, 10.6 mmol) and THF (10.6 mL), rt, 1 h. Chromatography (EtOH/cyclohexane 0 → 100%) afforded a brown gum (323 mg, 25%). 1H NMR (500 MHz, chloroform-d) δ 7.64 (dd, J = 5.0, 1.3 Hz, 1H), 7.52 (dd, J = 3.7, 1.3 Hz, 1H), 7.43 (d, J = 3.7 Hz, 1H), 7.11 (dd, J = 5.0, 3.7 Hz, 1H), 6.90 (dt, J = 3.7, 0.9 Hz, 1H), 3.93 (s, 2H), 1.63 (br, 2H). 13C NMR (126 MHz, chloroform-d) δ 155.85, 147.91, 145.56, 131.64, 130.91, 129.86, 127.48, 123.47, 41.84. HRMS (ESI) for C9H7O2S3 ([M – NH2]+): calculated 242.9608; observed 242.9608; error = 0 ppm.
(5-(Biphenyl-3-ylsulfonyl)thiophen-2-yl)methanamine Hydrochloride (3e)
5-([1,1′-Biphenyl]-3-ylthio)thiophene-2-carbonitrile was synthesized according to general procedures GP4, from 5-bromothiophene-2-carbonitrile 17a (160 mg, 1.11 mmol), [1,1′-biphenyl]-3-thiol (223 mg, 1.22 mmol), K2CO3 (308 mg, 2.23 mmol), and DMF (4.6 mL), 120 °C, 16 h. Chromatography (EtOAc/cyclohexane 0 → 20%) afforded a brown oil (327 mg, 92%). 1H NMR (500 MHz, methanol-d4) δ 7.13–7.80 (m, 11H). LCMS (ESI) m/z 294 [M + H]+.
tert-Butyl ((5-([1,1′-biphenyl]-3-ylthio)thiophen-2-yl)methyl)carbamate was synthesized according to general procedures GP3 and GP6, from (i) 5-([1,1′-biphenyl]-3-ylthio)thiophene-2-carbonitrile (264 mg, 0.910 mmol), BH3 (1.0 M in THF; 2.7 mL, 2.70 mmol), THF (2.7 mL), rt, 1 h and (ii) Boc2O (590 mg, 2.70 mmol), Et3N (250 μL, 1.80 mmol), and DCM (3.5 mL), rt, 16 h. Chromatography (EtOAc/cyclohexane 0 → 30%) afforded a yellow oil (64 mg, 18%). 1H NMR (500 MHz, chloroform-d) δ 7.56 (m, 1H), 7.55 (m, 1H), 7.49 (m, 1H), 7.46 (m, 2H), 7.43–7.33 (m, 3H), 7.20 (m, 2H), 6.93 (d, J = 3.3 Hz, 1H), 5.04 (s, 1H), 4.49 (s, 2H), 1.50 (s, 9H). LCMS (ESI) m/z 281 [M – BocNH]+.
Compound 3e was synthesized according to general procedures GP5 and GP2, from (i) tert-butyl ((5-([1,1′-biphenyl]-3-ylthio)thiophen-2-yl)methyl)carbamate (64 mg, 0.161 mmol), mCPBA (77%; 69 mg, 0.402 mmol), and DCM (2.0 mL), 45 °C, 1.5 h and (ii) 4 M HCl in dioxane (8.0 mL), rt, 16 h. The white precipitate was filtered and washed with excess EtOAc to afford a white solid (25 mg, 42% over two steps). 1H NMR (500 MHz, methanol-d4) δ 8.19 (m, 1H), 7.99 (ddd, J = 7.8, 1.8, 1.0 Hz, 1H), 7.96 (ddd, J = 7.8, 1.8, 1.0 Hz, 1H), 7.81 (d, J = 3.9 Hz, 1H), 7.70 (t, J = 7.7 Hz, 1H), 7.65 (m, 2H), 7.52 (m, 2H), 7.44 (m, 1H), 7.32 (d, J = 3.8 Hz, 1H), 4.38 (s, 2H). 13C NMR (126 MHz, chloroform-d) δ 144.2, 143.8, 142.8, 142.4, 138.8, 133.8, 131.9, 130.3, 130.1, 128.9, 128.2, 126.7, 125.7, 125.1, 37.1. HRMS (ESI) for C17H13O2S2 ([M – NH2]+): calculated 313.0357; observed 313.0359; error = 0.64 ppm.
(5-(Naphthalen-1-ylsulfonyl)thiophen-2-yl)methanamine (3f)
5-(Naphthalen-1-ylsulfonyl)thiophene-2-carbonitrile was synthesized according to general procedures GP4 and GP5, from (i) 5-chlorothiophene-2-carbonitrile 17a (95 μL, 0.901 mmol), naphthalene-1-thiol (137 μL, 0.991 mmol), K2CO3 (250 mg, 1.80 mmol), and DMF (3.0 mL), 120 °C, 16 h and (ii) mCPBA (77%; 504 mg, 2.25 mmol) and DCM (15 mL), rt, 16 h. Chromatography (EtOAc/cyclohexane 0 → 15%) afforded a colorless oil (170 mg, 63%). 1H NMR (500 MHz, chloroform-d) δ 8.78 (m, 1H), 8.51 (dd, J = 7.4, 1.2 Hz, 1H), 8.17 (d, J = 8.2 Hz, 1H), 7.96 (d, J = 8.2 Hz, 1H), 7.75–7.67 (m, 2H), 7.67–7.59 (m, 2H), 7.49 (d, J = 4.1 Hz, 1H). LCMS (ESI) m/z 300 [M + H]+.
Compound 3f was synthesized according to general procedure GP3, from BH3 (1.0 M in THF; 1.10 mL, 1.10 mmol), 5-(naphthalen-1-ylsulfonyl)thiophene-2-carbonitrile (110 mg, 0.367 mmol), and THF (1.8 mL), rt, 2 h. Chromatography (MeOH/DCM 0 → 20%) afforded a white crystalline solid (25 mg, 23%). 1H NMR (500 MHz, chloroform-d) δ 8.86 (dd, J = 8.7, 0.8 Hz, 1H), 8.47 (dd, J = 7.4, 1.2 Hz, 1H), 8.09 (d, J = 8.2 Hz, 1H), 7.95–7.87 (m, 1H), 7.70–7.64 (m, 2H), 7.63–7.54 (m, 2H), 6.84 (dt, J = 3.9, 1.0 Hz, 1H), 4.01 (d, J = 0.9 Hz, 2H), 1.58 (s, 3H). 13C NMR (126 MHz, chloroform-d) δ 157.61, 141.08, 137.18, 135.25, 134.40, 133.68, 129.61, 129.23, 128.54, 127.07, 124.65, 124.63, 123.50, 41.67. HRMS (ESI) for C15H11O2S2 ([M – NH2]+): calculated 287.0195; observed 287.0226; error = 11 ppm.
(5-(Naphthalen-2-ylsulfonyl)thiophen-2-yl)methanamine (3g)
Compound 3g was synthesized according to general procedure GP3, from BH3 (1.0 M in THF; 5.10 mL, 5.10 mmol), 5-(naphthalen-2-ylsulfonyl)thiophene-2-carbonitrile 43 (510 mg, 1.71 mmol) and THF (10 mL), rt, 3 h. Chromatography (EtOH/cyclohexane 30 → 100%) afforded a white solid (273 mg, 54%). 1H NMR (500 MHz, chloroform-d) δ 8.58 (s, 1H), 8.01–7.87 (m, 4H), 7.68–7.57 (m, 3H), 6.88 (m, 1H), 4.05 (s, 2H), 1.56 (s, 2H). 13C NMR (126 MHz, chloroform-d) δ 158.24, 140.73, 139.30, 135.17, 133.73, 132.40, 129.75, 129.59, 129.25, 128.71, 128.06, 127.75, 123.82, 122.58, 41.68. HRMS (ESI) for C15H11O2S2 ([M – NH2]+): calculated 287.0195; observed 287.0207; error = 4.2 ppm.
N-(4-(5-(Aminomethyl)thiophen-2-ylsulfonyl)phenyl)methanesulfonamide (3h)
A mixture of 5-((4-aminophenyl)thio)thiophene-2-carbonitrile 19 (120 mg, 0.517 mmol), MsCl (44.0 μL, 0.568 mmol), and pyridine (1.7 mL) was stirred at rt for 16 h. Two molar HCl (30 mL) was added. The aqueous phase was extracted with DCM (3 × 20 mL). The combined organic phase was dried over MgSO4 and filtered. The solvent was removed under reduced pressure, and the crude was dissolved in DCM (2.6 mL). mCPBA (77%; 254 mg, 1.14 mmol) was added, and the mixture was stirred at rt for 5 h. sat. NaHCO3 (40 mL) was added, and the aqueous phase was extracted with EtOAc (3 × 40 mL). The combined organic phase was dried over MgSO4 and filtered. The solvent was removed under reduced pressure, and the crude was purified by chromatography (MeOH/DCM 0 → 15%) to afford N-(4-((5-cyanothiophen-2-yl)sulfonyl)phenyl)methanesulfonamide as a white solid (69 mg, 39%). 1H NMR (500 MHz, chloroform-d) δ 7.50 (d, J = 3.9 Hz, 1H), 7.41–7.37 (m, 2H), 7.26–7.19 (m, 3H), 7.09 (d, J = 3.9 Hz, 1H), 3.05 (s, 3H). LCMS (ESI) m/z 365 [M + Na]+.
Compound 3h was synthesized according to general procedure GP3, from BH3 (1.0 M in THF; 0.61 mL, 0.61 mmol), N-(4-((5-cyanothiophen-2-yl)sulfonyl)phenyl)methanesulfonamide (69 mg, 0.202 mmol), and THF (0.6 mL), rt, 1 h. Chromatography (MeOH/DCM 5 → 25%) afforded a yellow solid (2 mg, 3%). 1H NMR (500 MHz, methanol-d4) δ 7.90–7.84 (m, 2H), 7.58 (d, J = 3.8 Hz, 1H), 7.38–7.32 (m, 2H), 7.04 (d, J = 3.8 Hz, 1H), 4.02 (s, 2H), 3.03 (s, 3H). 13C NMR (126 MHz, methanol-d4) δ 156.79, 146.06, 142.74, 136.92, 134.52, 130.00, 126.62, 119.69, 41.34, 40.06. HRMS (ESI) for C12H15N2O4S3 ([M + H]+): calculated 347.0189; observed 347.0190; error = 0.29 ppm.
(5-(4-(Methylsulfonyl)phenylsulfonyl)thiophen-2-yl)methanamine (3i)
5-((4-(Methylsulfonyl)phenyl)sulfonyl)thiophene-2-carbonitrile was synthesized according to general procedures GP4 and GP5, from (i) 5-chlorothiophene-2-carbonitrile (500 mg, 3.48 mmol), 4-(methylthio)benzenethiol (599 mg, 3.83 mmol), K2CO3 (960 mg, 7.00 mmol), and DMF (11.6 mL), 120 °C, 16 h and (ii) mCPBA (77%; 3.90 g, 17.4 mmol) and DCM (23 mL), rt, 2 h. A white solid was obtained, which did not require further purification (770 mg, 68%). 1H NMR (500 MHz, DMSO-d6) δ 8.35–8.29 (m, 2H), 8.24–8.18 (m, 2H), 8.13–8.08 (m, 2H), 3.31 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 147.29, 145.83, 144.19, 140.29, 134.79, 128.78, 128.56, 116.87, 112.57, 42.92.
Compound 3i was synthesized according to general procedure GP3, from BH3 (1.0 M in THF; 8.40 mL, 8.40 mmol), 5-((4-(methylsulfonyl)phenyl)sulfonyl)thiophene-2-carbonitrile (921 mg, 2.81 mmol), and THF (18 mL), 50 °C, 3 h. Chromatography (MeOH/DCM 0 → 20%) afforded a white solid (310 mg, 34%). 1H NMR (500 MHz, chloroform-d) δ 8.19–8.15 (m, 2H), 8.11–8.06 (m, 2H), 7.63 (d, J = 3.9 Hz, 1H), 6.93 (dt, J = 3.8, 1.0 Hz, 1H), 4.09 (d, J = 0.8 Hz, 2H), 3.08 (s, 3H), 1.61 (s, 2H). 13C NMR (126 MHz, chloroform-d) δ 160.12, 147.61, 144.80, 138.67, 134.95, 128.64, 128.46, 124.12, 44.41, 41.70. HRMS (ESI) for C12H14NO4S3 ([M + H]+): calculated 332.0080; observed 332.0070; error = 3.0 ppm.
(5-(4-(Methylsulfonyl)butylsulfonyl)thiophen-2-yl)methanamine (3j)
A mixture of 2-methyl-5-((4-(methylsulfonyl)butyl)sulfonyl)thiophene 16 (860 mg, 2.90 mmol), Bz2O2 (75%; 46.8 mg, 0.145 mmol), NBS (568 mg, 3.19 mmol), and DCE (14.5 mL) was stirred at 70 °C for 16 h. The solvent was removed under reduced pressure, and the crude was dissolved in DMF (14.5 mL). NaN3 (226 mg, 3.40 mmol) was added and the mixture was stirred at 70 °C for 5 h. After cooling to rt, EtOAc (30 mL) was added. The organic phase was washed with 1:1 H2O/brine (3 × 30 mL), dried over MgSO4, and filtered. The solvent was removed under reduced pressure, and the crude was purified by chromatography (EtOAc/DCM 5 → 30%) to afford 2-(azidomethyl)-5-((4-(methylsulfonyl)butyl)sulfonyl)thiophene as a colorless oil (414 mg, 42%). 1H NMR (500 MHz, chloroform-d) δ 7.61 (d, J = 3.9 Hz, 1H), 7.09 (m, 1H), 4.59 (s, 2H), 3.33–3.22 (m, 2H), 3.13–3.01 (m, 2H), 2.92 (s, 3H), 2.13–1.92 (m, 4H). LCMS (ESI) m/z 360 [M + Na]+.
A mixture of (azidomethyl)-5-((4-(methylsulfonyl)butyl)sulfonyl)-thiophene (410 mg, 1.22 mmol) and Pd/C (10%; 129 mg, 0.122 mmol) in THF (6.1 mL) was stirred at rt under H2 atmosphere (balloon) for 16 h and subsequently filtered through celite. The solvent was removed under reduced pressure, and the crude was purified by chromatography (MeOH/DCM 0 → 20%) to afford compound 3j as a white solid (27 mg, 6%). 1H NMR (500 MHz, chloroform-d) δ 7.54 (d, J = 3.8 Hz, 1H), 6.95 (mm, 1H), 4.11 (s, 2H), 3.25–3.19 (m, 2H), 3.05–3.00 (m, 2H), 2.89 (s, 3H), 2.03–1.91 (m, 4H), 1.71 (s, 2H). 13C NMR (126 MHz, chloroform-d) δ 158.73, 137.14, 134.65, 123.97, 56.81, 53.92, 41.63, 40.85, 22.16, 21.11. HRMS (ESI) for C10H18NO4S3 ([M + H]+): calculated 312.0393; observed 312.0417; error = 7.7 ppm.
(5-(Phenylsulfinyl)thiophen-2-yl)methanamine (4a)
5-(Phenylsulfinyl)thiophene-2-carbonitrile was synthesized according to general procedures GP5, from mCPBA (77%; 143 mg, 0.636 mmol) and 5-(phenylthio)thiophene-2-carbonitrile 29 (138 mg, 0.636 mmol) in DCM (3 mL), rt, 16 h. Chromatography (EtOAc/cyclohexane 0 → 20%) afforded a colorless oil (127 mg, 86%). 1H NMR (500 MHz, chloroform-d) δ 7.76–7.70 (m, 2H), 7.60–7.50 (m, 4H), 7.44 (d, J = 4.0 Hz, 1H). LCMS (ESI) m/z 233 [M + H]+.
Compound 4a was synthesized according to general procedure GP3, from BH3 (1.0 M in THF; 1.70 mL, 1.70 mmol), 5-(naphthalen-2-ylsulfonyl)thiophene-2-carbonitrile (132 mg, 0.568 mmol), and THF (2.8 mL), rt, 2 h. Chromatography (MeOH/DCM 0 → 20%) afforded a yellow oil (53 mg, 40%). 1H NMR (500 MHz, chloroform-d) δ 7.72–7.65 (m, 2H), 7.54–7.43 (m, 4H), 6.86 (m, 1H), 4.00 (s, 2H), 1.80 (s, 2H). 13C NMR (126 MHz, chloroform-d) δ 156.03, 145.85, 145.18, 132.03, 131.13, 129.26, 124.42, 123.46, 41.74. HRMS (ESI) for C11H12NOS2 ([M + H]+): calculated 238.0355; observed 238.0378; error = 9.7 ppm.
(5-(Phenylthio)thiophen-2-yl)methanamine Hydrochloride (4b)
The titled compound was synthesized according to general procedure GP2, from 4 M HCl in dioxane (2.5 mL) and tert-butyl ((5-(phenylthio)thiophen-2-yl)methyl)carbamate (60.1 mg, 0.189 mmol), rt, 3 h. A white solid was obtained, which did not require further purification (29 mg, 60%). 1H NMR (500 MHz, methanol-d4) δ 7.36–7.14 (m, 7H), 4.32 (s, 2H). 13C NMR (126 MHz, methanol-d4) δ 141.06, 138.75, 136.99, 135.89, 131.40, 130.24, 129.20, 127.91, 38.79. HRMS (ESI) for C11H9S2 ([M – NH2]+): calculated 205.0146; observed 205.1050; error = 2.0 ppm.
5-(Aminomethyl)-N-benzylthiophene-2-sulfonamide (4c)
The titled compound was synthesized according to general procedure GP1, from (i) sulfonyl chloride 11a (100 mg, 0.325 mmol), benzylamine (88.7 μL, 0.812 mmol), and DCM (1.63 mL), 16 h, rt and (ii) 30% NaOH (0.5 mL) and MeOH (3 mL), rt, 16 h. A colorless oil (59 mg, 64%) was obtained, which did not require further purification. 1H NMR (500 MHz, chloroform-d) δ 7.44 (d, J = 3.7 Hz, 1H), 7.32–7.20 (m, 5H), 6.84 (d, J = 3.6 Hz, 1H), 5.24 (br, 1H), 4.19 (s, 2H), 4.02 (s, 2H), 1.87 (br, 2H). 13C NMR (126 MHz, chloroform-d) δ 132.60, 128.77, 128.02, 127.98, 123.55, 47.50, 41.43. HRMS (ESI) for C12H15N2O2S2 ([M + H]+): calculated 283.0570; observed 283.0573; error = 1.1 ppm.
5-(Aminomethyl)-N-benzylthiophene-2-carboxamide (4d)
A mixture of methyl 5-(azidomethyl)thiophene-2-carboxylate 44 (A = S) (1.77 g, 8.98 mmol), 1 M NaOH (30 mL), and MeOH (30 mL) was stirred at rt for 16 h. The pH was subsequently adjusted to <2 with 2 M HCl. The aqueous phase was extracted with DCM (3 × 30 mL). The combined organic phase was dried over MgSO4 and filtered, and the solvent was removed under reduced pressure. The crude was dissolved in DCM (21.8 mL), and oxalyl chloride (0.61 mL, 7.20 mmol) was added, followed by DMF (two drops). The mixture was stirred at rt for 3 h, and the solvent was removed under reduced pressure to afford acyl chloride 45 (A = S) as an orange oil (1.25 g, 75%). Acyl chloride 45 was used in the subsequent transformation without further purification
Benzylamine (120 μL, 1.09 mmol) was added to a solution of acyl chloride 45 (100 mg, 0.496 mmol) and DCM (2.5 mL), and the mixture was stirred at rt for 3 h. Two molar HCl (10 mL) was added, and the aqueous phase was extracted with DCM (3 × 10 mL). The combined organic phase was dried over MgSO4, filtered, and the solvent was removed under reduced pressure. The crude was dissolved in THF (2.5 mL), and 10% Pd/C (53 mg) was added. The mixture was stirred under a H2 atmosphere (balloon) at rt for 24 h and was subsequently filtered through celite. The solvent was removed under reduced pressure, and the crude was purified by chromatography (1 N NH3 in MeOH/DCM 0 → 20%) to afford compound 4d as a white solid (55 mg, 45%). 1H NMR (500 MHz, methanol-d4) δ 7.36–7.19 (m, 5H), 7.04 (d, J = 3.4 Hz, 1H), 6.37 (d, J = 3.3 Hz, 1H), 4.52 (s, 2H), 3.81 (s, 2H). 13C NMR (126 MHz, methanol-d4) δ 160.84, 159.70, 147.96, 140.02, 129.53, 128.59, 128.25, 116.26, 109.12, 43.74, 39.37. HRMS (ESI) for C13H15N2OS ([M + H]+): calculated 247.0905; observed 247.0907; error = 0.81 ppm.
(5-(4-(Methylsulfonyl)phenyl)thiophen-2-yl)methanamine Hydrochloride (4e)
The titled compound was synthesized according to general procedures GP2, from tert-butyl ((5-(4-(methylsulfonyl)phenyl)thiophen-2-yl)methyl)carbamate 59 (267 mg, 0.797 mmol), 4 M HCl in dioxane (4 mL), and DCM (4 mL), rt, 16 h. A light yellow solid was obtained that did not require further purification (138 mg, 64%). 1H NMR (500 MHz, methanol-d4) δ 8.01–7.96 (m, 2H), 7.93–7.89 (m, 2H), 7.57 (d, J = 3.7 Hz, 1H), 7.29 (d, J = 3.8 Hz, 1H), 4.36 (s, 2H), 3.16 (s, 3H). 13C NMR (126 MHz, methanol-d4) δ 144.98, 140.90, 140.20, 137.74, 131.83, 129.37, 127.31, 126.97, 44.36, 38.86. HRMS (ESI) for C12H11O2S2 ([M – NH2]+): calculated 251.0200; observed 251.0196; error = 1.6 ppm.
(6-(Benzylsulfonyl)pyridin-3-yl)methanamine Dihydrochloride (5a)
The titled compound was synthesized according to general procedures GP5 and GP2, from (i) sulfide 47 (44 mg, 0.133 mmol), mCPBA (50%; 115 mg, 0.333 mmol), and DCM (0.6 mL) at rt for 16 h and (ii) 2 M HCl in Et2O (2.0 mL), DCM (1.0 mL), rt, 20 h. Chromatography (1 N NH3 in MeOH/DCM 0 → 20%) afforded a white solid (27 mg, 77%). 1H NMR (500 MHz, methanol-d4) δ 8.76 (m, 1H), 7.92 (m, 1H), 7.75 (d, J = 8.0 Hz, 1H), 7.30–7.16 (m, 5H), 4.68 (s, 2H), 3.92 (s, 2H). 13C NMR (126 MHz, methanol-d4) δ 155.78, 150.77, 144.38, 138.22, 132.22, 129.66, 129.53, 129.19, 129.14, 124.10, 59.32, 43.72. HRMS (ESI) for C13H15N2O2S ([M + H]+): calculated 263.0849; observed 263.0848; error = 0.38 ppm.
(5-(Benzylsulfonyl)thiophen-2-yl)methanamine (5b)
5-(Benzylsulfonyl)thiophene-2-carbonitrile was synthesized according to general procedures GP4 and GP5, from (i) 5-bromothiophene-2-carbonitrile 17a (150 μL, 1.35 mmol), benzylmercaptan (174 μL, 1.49 mmol), NaH (60% in mineral oil; 59.5 mg, 1.49 mmol), and DMF (4.5 mL), 140 °C, 16 h and (ii) mCPBA (77%; 757 mg, 3.38 mmol) and DCM (4.5 mL), rt, 4 h. Chromatography (EtOAc/cyclohexane 0 → 20%) afforded a white solid (318 mg, 89%). 1H NMR (500 MHz, DMSO-d6) δ 8.07 (d, J = 4.0 Hz, 1H), 7.72 (d, J = 4.0 Hz, 1H), 7.40–7.32 (m, 3H), 7.25–7.18 (m, 2H), 4.93 (s, 2H). LCMS (ESI) m/z 286 [M + Na]+.
Compound 5b was synthesized according to general procedure GP3, from BH3 (1.0 M in THF; 1.10 mL, 1.10 mmol), 5-(benzylsulfonyl)thiophene-2-carbonitrile (151 mg, 0.570 mmol), and THF (2.8 mL) at rt for 1 h. Chromatography (MeOH/DCM 0 → 20%) afforded a white crystalline solid (23 mg, 15%). 1H NMR (500 MHz, chloroform-d) δ 7.38–7.26 (m, 3H), 7.21–7.15 (m, 3H), 6.84 (dt, J = 3.8, 1.0 Hz, 1H), 4.39 (s, 2H), 4.07 (d, J = 0.9 Hz, 2H), 1.59 (s, 2H). 13C NMR (126 MHz, chloroform-d) δ 158.50, 136.25, 135.25, 130.88, 129.00, 128.74, 128.52, 123.68, 64.15, 41.68. HRMS (ESI) for C12H14NO2S2 ([M + H]+): calculated 268.0461; observed 268.0462; error = 0.37 ppm.
(2-(Naphthalen-2-ylsulfonyl)thiazol-5-yl)methanamine hydrochloride (5c)
tert-Butyl ((2-(naphthalen-2-ylthio)thiazol-5-yl)methyl)carbamate was synthesized according to general procedures GP4, GP3, and GP6, from (i) 2-chlorothiazole-5-carbonitrile 17b (580 mg, 4 mmol), naphthalene-2-thiol (640 mg, 4 mmol), K2CO3 (800 mg, 5.8 mmol), and DMF (10 mL) at 60 °C for 24 h. Chromatography (DCM/cyclohexane 0 → 100%) 400 mg, 37%; (ii) 2-(naphthalen-2-ylthio)thiazole-5-carbonitrile (400 mg, 1.5 mmol), BH3 (1.0 M in THF; 5 mL, 5 mmol), THF (20 mL) at rt for 18 h. Chromatography (EtOAc/DCM 0 → 100%) 120 mg, 26%; (iii) (2-(naphthalen-2-ylthio)thiazol-5-yl)methanamine (120 mg, 0.4 mmol), Boc2O (109 mg, 0.5 mmol), Et3N (70 μL, 0.5 mmol), THF (5 mL), rt, 18 h. Chromatography (EtOAc/DCM 0 → 50%) to afford tert-butyl ((2-(naphthalen-2-ylthio)thiazol-5-yl)methyl)carbamate (110 mg, 74%). 1H NMR (500 MHz, DMSO-d6) δ 8.79 (d, J = 1.9 Hz, 1H), 8.29 (d, J = 8.2 Hz, 1H), 8.20 (d, J = 8.7 Hz, 1H), 8.09 (d, J = 8.1 Hz, 1H), 7.95 (dd, J = 8.6, 2.0 Hz, 1H), 7.91 (s, 1H), 7.82–7.75 (m, 1H), 7.72 (ddd, J = 8.0, 6.9, 1.3 Hz, 1H), 7.65 (t, J = 6.1 Hz, 1H), 4.36 (d, J = 6.0 Hz, 2H), 1.38 (s, 9H). LCMS (ESI) m/z 373 [M + H]+.
Compound 5c was synthesized according to general procedures GP5 and GP2, from (i) tert-butyl ((2-(naphthalen-2-ylthio)thiazol-5-yl)methyl)carbamate (110 mg, 0.296 mmol), mCPBA (77%; 340 mg, 1.52 mmol), and DCM (5.0 mL) at rt for 3 h and (ii) 4 M HCl in dioxane (2.0 mL) and dioxane (5 mL), at rt, for 16 h. The white precipitate was filtered, washed with excess dioxane, and dried under vacuum to afford the desired compound as a white solid (33 mg, 37%). 1H NMR (500 MHz, DMSO-d6) δ 8.82 (d, J = 2.0 Hz, 1H), 8.49 (s, 3H), 8.31 (d, J = 8.2 Hz, 1H), 8.23 (d, J = 8.7 Hz, 1H), 8.15 (s, 1H), 8.10 (d, J = 8.1 Hz, 1H), 7.97 (dd, J = 8.6, 2.1 Hz, 1H), 7.84–7.77 (m, 1H), 7.77–7.71 (m, 1H), 4.39 (s, 2H). HRMS (ESI) for C14H13N2O2S2 [M + H]+: calculated 305.0413; observed 305.0471; error = 19 ppm.
(5-(Naphthalen-2-ylsulfonyl)thiazol-2-yl)methanamine Hydrochloride (5d)
The titled compound was synthesized according to general procedures GP5 and GP2, from (i) tert-butyl ((5-(naphthalen-2-ylthio)thiazol-2-yl)methyl)carbamate 49 (200 mg, 0.537 mmol), mCPBA (77%; 300 mg, 1.34 mmol), and DCM (4 mL) at rt for 18 h; chromatography (EtOAc/cyclohexane 0 → 60%) and (ii) 4 M HCl in dioxane (1.6 mL) at rt for 4 h. The mixture was diluted with Et2O and the precipitate collected using filtration, washed with excess Et2O, and dried under vacuum to afford the compound as a white solid (106 mg, 52% over two steps). 1H NMR (DMSO-d6, 500 MHz) δ 8.79 (d, 1H, J = 1.8 Hz), 8.72 (br s, 3H), 8.65 (s, 1H), 8.27 (d, 1H, J = 8.4 Hz), 8.21 (d, 1H, J = 8.8 Hz), 8.09 (d, 1H, J = 8.2 Hz), 8.00 (dd, 1H, J = 8.7, 2.0 Hz), 7.78 (ddd, 1H, J = 8.2, 6.9, 1.3 Hz), 7.73 (ddd, 1H, J = 8.1, 6.9, 1.3 Hz), 4.46 (s, 2H); 13C NMR (DMSO-d6, 125 MHz) δ 170.3, 147.4, 140.2, 137.6, 134.9, 131.8, 130.4, 130.0, 129.7, 128.8, 128.2, 128.0, 121.8, 39.7; HRMS (ESI) for C14H13N2S2O2 [M + H]+: calculated 305.0402; observed 305.0413; error = 3.6 ppm.
5-(Aminomethyl)-N-benzylfuran-2-carboxamide (5e)
Benzylamine (130 μL, 1.19 mmol) was added to a solution of acyl chloride 45 (A = O; 100 mg, 0.539 mmol) and DCM (2.7 mL), and the mixture was stirred at rt for 1 h. Two molar HCl (10 mL) was added, and the aqueous phase was extracted with DCM (3 × 10 mL). The combined organic phase was dried over MgSO4 and filtered, and the solvent was removed under reduced pressure. The crude was dissolved in THF (2.7 mL), and 10% Pd/C (57.3 mg) was added. The mixture was stirred under a H2 atmosphere (balloon) at rt for 20 h and was subsequently filtered through celite. The solvent was removed under reduced pressure, and the crude was purified by chromatography (1 N NH3 in MeOH/DCM 0 → 20%) to afford compound 5e as a light brown oil (48 mg, 39%). 1H NMR (500 MHz, methanol-d4) δ 7.56 (d, J = 3.8 Hz, 1H), 7.38–7.18 (m, 5H), 6.98 (d, J = 3.8 Hz, 1H), 4.51 (s, 2H), 3.96 (s, 2H). 13C NMR (126 MHz, methanol-d4) δ 164.43, 152.94, 140.21, 138.59, 129.85, 129.56, 128.56, 128.22, 126.28, 44.41, 41.61. HRMS (ESI) for C13H14N2O2Na ([M + Na]+): calculated 253.0953; observed 253.0960; error = 2.8 ppm.
(5-(Indolin-1-ylsulfonyl)-4-methylthiophen-2-yl)methanamine (5f)
The titled compound was synthesized according to general procedure GP1, from (i) sulfonyl chloride 11b (crude), Et3N (44.4 μL, 0.319 mmol), indoline (33 μL, 0.294 mmol), and DCM (1.2 mL), rt, 3 h; (ii) 7 N NH3 in MeOH (5 mL), rt for 20 h. Chromatography (1 N NH3 in MeOH/DCM 0 → 10%) afforded a compound as a light brown foam (54 mg, 73%). 1H NMR (500 MHz, chloroform-d) δ 7.59 (d, J = 8.1 Hz, 1H), 7.21–7.10 (m, 2H), 7.00 (td, J = 7.4, 0.9 Hz, 1H), 6.68 (s, 1H), 4.03 (t, J = 8.4 Hz, 2H), 3.96 (s, 2H), 2.97 (t, J = 8.3 Hz, 2H), 2.33 (s, 3H), 1.68–1.35 (br, 2H). 13C NMR (126 MHz, chloroform-d) δ 153.13, 143.82, 141.96, 132.09, 129.42, 127.97, 127.69, 125.15, 124.08, 115.79, 50.22, 41.39, 28.16, 15.56. HRMS (ESI) for C14H17N2O2S2 ([M + H]+): calculated 309.0726; observed 309.0726; error = 0 ppm.
N-Methyl-1-(5-(phenylsulfonyl)thiophen-2-yl)methanamine Hydrochloride (6a)
The titled compound was synthesized according to general procedure GP2, from tert-butyl methyl((5-(phenylsulfonyl)thiophen-2-yl)methyl)carbamate (57 mg, 0.155 mmol) and 4 M HCl in dioxane (2.5 mL) at rt for 16 h. Chromatography (EtOH/cyclohexane 50 → 100%) afforded a white solid (43 mg, quant.). 1H NMR (500 MHz, methanol-d4) δ 8.01–7.93 (m, 2H), 7.67–7.55 (m, 4H), 7.05 (d, J = 3.8 Hz, 1H), 3.93 (s, 2H), 2.38 (s, 3H). 13C NMR (126 MHz, methanol-d4) δ 154.35, 143.60, 142.54, 134.90, 134.63, 130.61, 128.22, 127.79, 50.42, 35.41. HRMS (ESI) for C12H14NO2S2 ([M + H]+): calculated 268.0461; observed 268.0457; error = 1.5 ppm.
1-(5-(Phenylsulfonyl)thiophen-2-yl)ethanamine (6b)
A mixture of sulfone 41 (177 mg, 0.665 mmol), hydroxylamine hydrochloride (76 mg, 1.10 mmol), and DIPEA (174 μL, 1.00 mmol) in EtOH (10 mL) was stirred at reflux for 18 h. After cooling to rt, the solvent was removed under reduced pressure. The crude was purified by chromatography (EtOAc/DCM 0 → 100%) to afford 1-(5-(phenylsulfonyl)thiophen-2-yl)ethanone oxime (80 mg, 43%). 1H NMR (500 MHz, DMSO-d6) δ 11.72 (s, 1H), 8.01–7.95 (m, 2H), 7.79 (d, J = 4.0 Hz, 1H), 7.75–7.70 (m, 1H), 7.68–7.62 (m, 2H), 7.41 (d, J = 4.0 Hz, 1H), 2.14 (s, 3H).
A mixture of 1-(5-(phenylsulfonyl)thiophen-2-yl)ethanone oxime (80 mg, 0.142 mmol), Zn powder (∼10 mg), and TFA (4 mL) was stirred at rt for 1 h. The mixture was diluted with DCM and filtered through celite, and the solvent was removed under reduced pressure. EtOAc was added, and the solution was washed with sat. NaHCO3, dried over MgSO4, and filtered, and the solvent was removed under reduced pressure to afford compound 6b (17 mg, 43%). 1H NMR (500 MHz, DMSO-d6) δ 7.96–7.92 (m, 2H), 7.70–7.67 (m, 2H), 7.65–7.61 (m, 2H), 7.01 (dd, J = 3.9, 1.0 Hz, 1H), 4.21 (qd, J = 6.5, 1.0 Hz, 1H), 2.40 (s, 2H), 1.30 (d, J = 6.6 Hz, 3H). LCMS (ESI) m/z 251 [M – NH2]+.
5-(Phenylsulfonyl)thiophene-2-carbonitrile (6c)
The titled compound can be obtained in two steps from the method illustrated in Scheme 3 in the manuscript. An alternative one-step method is shown here: A mixture of 5-bromothiophene-2-carbonitrile 17a (100 mg, 0.532 mmol), sodium benzenesulfinate (138 mg, 0.691 mmol), and DMF (1.0 mL) was stirred at 135 °C for 16 h. After cooling to rt, EtOAc (10 mL) was added. The organic phase was washed with H2O (2 × 10 mL) and brine (10 mL), dried over MgSO4, and filtered. The solvent was removed under reduced pressure, and the crude was purified by chromatography (EtOAc/cyclohexane 0 → 20%) to afford nitrile 6c as a white crystalline solid (67 mg, 51%). 1H NMR (500 MHz, chloroform-d) δ 8.03–7.98 (m, 2H), 7.70–7.53 (m, 5H). 13C NMR (126 MHz, chloroform-d) δ 150.24, 140.54, 137.42, 134.47, 132.39, 129.89, 127.86, 117.10, 112.37. HRMS (ESI) for C11H7NO2S2 ([M + H]+): calculated 249.9996; observed 250.0001; error = 2.0 ppm.
5-(Phenylsulfonyl)thiophene-2-carboximidamide Hydrochloride (6d)
LiHMDS (225 mg, 1.35 mmol) was added to a mixture of nitrile 6c (224 mg, 0.900 mmol) in Et2O (10 mL), and the mixture was stirred at rt for 1 h. Two molar HCl (5 mL) was added, and the mixture was stirred for a further 1 h. The aqueous layer was washed with Et2O, basified with NaOH pellets, and extracted with Et2O. The combined organic phase was dried over MgSO4 and filtered, and the solvent was removed under reduced pressure to afford compound 6d as a yellow solid (40 mg, 16%). 1H NMR (500 MHz, methanol-d4) δ 8.07 (d, J = 6.9 Hz, 2H), 7.89 (br s, 2H), 7.54–7.80 (m, 3H). 13C NMR (126 MHz, methanol-d4) δ 160.8, 151.3, 142.2, 137.2, 135.8, 135.1, 134.9, 131.2, 129.0. HRMS (ESI) for C11H10N2O2S2 [M + H]+: calculated 267.0256; observed 267.0301; error = 17 ppm.
5-(Phenylsulfonyl)thiophene-2-carboxamide (6e)
A mixture of nitrile 6c (100 mg, 0.401 mmol), InCl3 (4.4 mg, 5%), and acetaldoxime (76 mg, 1.21 mmol) in toluene (0.4 mL) was stirred at reflux for 5 h. The solvent was removed under reduced pressure, and the crude was purified by chromatography (EtOAc/cyclohexane 20 → 80%) to afford compound 6e as a yellow gel (52 mg, 50%). 1H NMR (500 MHz, CD3OD) δ 7.94–8.10 (m, 2H), 7.66–7.75 (m, 3H), 7.59–7.66 (m, 2H). 13C NMR (126 MHz, CD3OD) δ 164.8, 148.5, 148.3, 142.9, 135.2, 134.8, 130.9, 130.2, 128.7. HRMS (ESI) for C11H9NO3S2 [M + H]+: calculated 268.0097; observed 268.0187; error = 34 ppm.
5-(Naphthalen-2-ylsulfonyl)thiophene-2-carbaldehyde Oxime (6f)
DIBAL-H (1.0 M in toluene; 1.0 mL, 1.0 mmol) was added to a solution of 5-(naphthalen-2-ylsulfonyl)thiophene-2-carbonitrile 43 (104 mg, 0.347 mmol) in DCM (3 mL) at rt. The mixture was stirred at 45 °C for 45 min and cooled in an ice bath. Two molar H2SO4 was added followed by stirring for a further 1 h at rt. The aqueous phase was extracted with DCM. The organic phase was washed with H2O and sat. NaHCO3, dried over Na2SO4, and filtered, and the solvent was removed under reduced pressure. The crude was purified by column chromatography (EtOAc/cyclohexane 0 → 30%) to afford 5-(naphthalen-2-ylsulfonyl)thiophene-2-carbaldehyde as a yellow solid (96 mg, 92%). 1H NMR (500 MHz, chloroform-d) δ 9.92 (s, 1H), 8.62 (s, 1H), 8.07–7.96 (m, 2H), 7.96–7.87 (m, 2H), 7.78 (d, J = 3.8 Hz, 1H), 7.72–7.61 (m, 3H). HRMS (ESI) for C15H10O3S2 [M + H]+: calculated 303.0144; observed 303.0159; error = 5.0 ppm.
A mixture of 5-(naphthalen-2-ylsulfonyl)thiophene-2-carbaldehyde (96 mg, 0.318 mmol), hydroxylamine hydrochloride (28 mg, 0.406 mmol), and K2CO3 (132 mg, 0.957 mmol) in EtOH (6.0 mL) was stirred 70 °C for 2 h. After cooling to rt, the mixture was filtered and the solvent was removed under reduced pressure. The crude was purified by chromatography (EtOAc/cyclohexane) to afford oxime 6f as a white solid (38 mg, 38%). 1H NMR (500 MHz, chloroform-d) δ 8.61 (s, 1H), 7.95–8.08 (m, 2H), 7.87–7.95 (m, 2H), 7.58–7.74 (m, 4H), 7.41 (d, J = 4.1 Hz, 1H), 5.8 (br s, OH). 13C NMR (126 MHz, chloroform-d) δ 161.8, 147.8, 145.1, 137.9, 135.3, 132.8, 132.2, 129.9, 129.5, 129.3, 128.5, 128.0, 127.9, 122.3. HRMS (ESI) for C15H11NO3S2 [M – OH]+: calculated 300.0153; observed 300.0192; error = 13 ppm.
(3-(Naphthalen-2-ylsulfonyl)thiophen-2-yl)methanamine (7a)
The titled compound was synthesized according to general procedure GP3, from BH3 (1.0 M in THF; 6.0 mL, 6.0 mmol), 3-(naphthalen-2-ylsulfonyl)thiophene-2-carbonitrile 52 (602 mg, 2.01 mmol), and THF (6.0 mL), for 2 h, at rt. Chromatography (EtOH/cyclohexane 0 → 90%) afforded a white crystalline solid (289 mg, 48%). 1H NMR (500 MHz, chloroform-d) δ 8.55 (d, J = 1.5 Hz, 1H), 8.00 (d, J = 7.9 Hz, 1H), 7.95 (d, J = 8.7 Hz, 1H), 7.90 (d, J = 8.0 Hz, 1H), 7.84 (dd, J = 8.7, 1.9 Hz, 1H), 7.69–7.60 (m, 2H), 7.42 (d, J = 5.4 Hz, 1H), 7.20 (d, J = 5.4 Hz, 1H), 4.31 (s, 2H), 1.92 (s, 2H). 13C NMR (126 MHz, chloroform-d) δ 154.77, 138.97, 135.19, 135.00, 132.29, 129.85, 129.57, 129.32, 128.68, 128.45, 128.09, 127.85, 124.02, 122.36, 40.04. HRMS (ESI) for C15H14NO2S2 ([M + H]+): calculated 304.0460; observed 304.0462; error = 0.66 ppm.
(4-(Naphthalen-2-ylsulfonyl)thiophen-3-yl)methanamine (7b)
The titled compound was synthesized according to general procedure GP3, from BH3 (1.0 M in THF; 8.2 mL, 8.2 mmol), 4-(naphthalen-2-ylsulfonyl)thiophene-3-carbonitrile 54 (0.821 g, 2.75 mmol), and THF (8.2 mL); 2 h, rt. Chromatography (EtOH/cyclohexane 0 → 100%) afforded a white crystalline solid (386 mg, 47%). 1H NMR (500 MHz, chloroform-d) δ 8.55 (br, 1H), 8.27 (d, J = 3.4 Hz, 1H), 7.97 (d, J = 7.9 Hz, 1H), 7.92 (d, J = 8.7 Hz, 1H), 7.87 (d, J = 8.0 Hz, 1H), 7.79 (dd, J = 8.7, 1.8 Hz, 1H), 7.71–7.53 (m, 2H), 7.26 (d, J = 14.5 Hz, 1H), 3.85 (s, 2H), 1.82–1.54 (m, 2H). 13C NMR (126 MHz, chloroform-d) δ 142.19, 138.80, 137.83, 135.08, 134.87, 132.08, 129.73, 129.46, 129.34, 128.86, 127.98, 127.79, 124.96, 122.48, 77.42, 77.17, 76.92, 40.35. HRMS (ESI) for C15H14NO2S2 ([M + H]+): calculated 304.0460; observed 304.0460; error = 0 ppm.
(4-(Naphthalen-2-ylsulfonyl)thiophen-2-yl)methanamine (7c)
The titled compound was synthesized according to general procedure GP3, from BH3 (1.0 M in THF; 5.0 mL, 5.0 mmol), 4-(naphthalen-2-ylsulfonyl)thiophene-2-carbonitrile 56 (501 mg, 1.68 mmol), and THF (5.0 mL), 2 h, rt. Chromatography (EtOH/cyclohexane 5 → 100%) afforded a light orange solid (256 mg, 51%). 1H NMR (500 MHz, chloroform-d) δ 8.57 (s, 1H), 8.02 (s, 1H), 8.00–7.91 (m, 2H), 7.90–7.84 (m, 2H), 7.67–7.54 (m, 2H), 7.16 (s, 1H), 3.97 (s, 2H), 1.71 (s, 2H). 13C NMR (126 MHz, chloroform-d) δ 151.36, 141.20, 138.42, 135.07, 132.24, 130.33, 129.71, 129.44, 129.20, 128.89, 127.99, 127.70, 122.56, 121.52, 41.27. HRMS (ESI) for C15H14NO2S2 ([M + H]+): calculated 304.0460; observed 304.0444; error = 5.2 ppm.
(5-(3-(Methylsulfonyl)phenylsulfonyl)thiophen-2-yl)methanamine (8a)
5-((3-(Methylsulfonyl)phenyl)sulfonyl)thiophene-2-carbonitrile was synthesized according to general procedures GP4 and GP5, from (i) 5-chlorothiophene-2-carbonitrile 17a (120 μL, 1.14 mmol), 3-(methylthio)benzenethiol (178 mg, 1.14 mmol), K2CO3 (157 mg, 1.14 mmol), and DMF (5.7 mL), 120 °C, 16 h and (ii) mCPBA (77%; 1.15 g, 5.13 mmol) and DCM (5.7 mL), rt, 16 h. Chromatography (EtOAc/DCM 5 → 30%) afforded a white solid (207 mg, 56%). 1H NMR (500 MHz, chloroform-d) δ 8.57 (t, J = 1.7 Hz, 1H), 8.28 (ddd, J = 7.9, 1.8, 1.1 Hz, 1H), 8.23 (ddd, J = 7.9, 1.6, 1.1 Hz, 1H), 7.83 (t, J = 7.9 Hz, 1H), 7.72 (d, J = 4.1 Hz, 1H), 7.60 (d, J = 4.1 Hz, 1H), 3.13 (s, 3H).
Compound 8a was synthesized according to general procedure GP3, from BH3 (1.0 M in THF; 0.88 mL, 0.88 mmol), 5-((3-(methylsulfonyl)phenyl)sulfonyl)thiophene-2-carbonitrile (96 mg, 0.293 mmol), and THF (1.5 mL); rt, 1 h. Chromatography (MeOH/DCM 0 → 20%) afforded a white solid (60 mg, 63%). 1H NMR (500 MHz, chloroform-d) δ 8.50 (t, J = 1.6 Hz, 1H), 8.24 (ddd, J = 7.9, 1.7, 1.1 Hz, 1H), 8.13 (ddd, J = 7.8, 1.7, 1.1 Hz, 1H), 7.74 (t, J = 7.9 Hz, 1H), 7.62 (d, J = 3.9 Hz, 1H), 6.91 (dt, J = 3.9, 1.0 Hz, 1H), 4.07 (d, J = 0.9 Hz, 2H), 3.09 (s, 3H), 1.65 (s, 2H). 13C NMR (126 MHz, chloroform-d) δ 160.00, 144.45, 142.31, 138.59, 134.88, 132.21, 131.73, 130.82, 126.38, 124.12, 44.45, 41.64. HRMS (ESI) for C12H11O4S3 ([M – NH2]+): calculated 314.9814; observed 314.9820; error = 1.9 ppm.
(5-(2-(Methylsulfonyl)phenylsulfonyl)thiophen-2-yl)methanamine (8b)
5-((2-(Methylsulfonyl)phenyl)sulfonyl)thiophene-2-carbonitrile was synthesized according to general procedures GP4 and GP5, from (i) 5-chlorothiophene-2-carbonitrile 17a (87.8 μL, 0.832 mmol), 2-(methylthio)benzenethiol (130 mg, 0.832 mmol), K2CO3 (172 mg, 1.25 mmol) and DMF (2.8 mL), 120 °C, 16 h and (ii) mCPBA (77%; 839 mg, 3.74 mmol) and DCM (2.8 mL), rt, 6 h. Chromatography (EtOAc/DCM 5 → 30%) afforded a white solid (46 mg, 17%). 1H NMR (500 MHz, chloroform-d) δ 8.47 (m, 1H), 8.37 (m, 1H), 7.96–7.87 (m, 3H), 7.54 (d, J = 4.1 Hz, 1H), 3.49 (s, 3H).
Compound 8b was synthesized according to general procedure GP3, from BH3 (1.0 M in THF; 0.41 mL, 0.41 mmol), 5-((2-(methylsulfonyl)phenyl)sulfonyl)thiophene-2-carbonitrile (45 mg, 0.137 mmol) and THF (0.8 mL), rt, 1 h. Chromatography (MeOH/DCM 0 → 20%) afforded compound 8b as a white solid (17 mg, 39%). 1H NMR (500 MHz, chloroform-d) δ 8.43 (dd, J = 7.5, 1.7 Hz, 1H), 8.33 (dd, J = 7.6, 1.6 Hz, 1H), 7.88 (d, J = 3.9 Hz, 1H), 7.85–7.76 (m, 2H), 6.88 (dt, J = 3.9, 0.9 Hz, 1H), 4.06 (d, J = 0.8 Hz, 2H), 3.51 (s, 3H), 1.59 (s, 2H). 13C NMR (126 MHz, chloroform-d) δ 159.33, 142.15, 139.62, 138.92, 136.80, 134.50, 134.07, 132.79, 132.44, 123.74, 45.73, 41.73. HRMS (ESI) for C12H14NO4S3 ([M + H]+): calculated 332.0080; observed 332.0108; error = 8.4 ppm.
(5-((4-(Pyrrolidin-1-ylsulfonyl)phenyl)sulfonyl)thiophen-2-yl)methanamine (8c)
A mixture of 2,2,2-trifluoro-N-((5-((4-(pyrrolidin-1-ylsulfonyl)phenyl)thio)thiophen-2-yl)methyl)acetamide 24c (91 mg, 0.202 mmol), mCPBA (77%; 100 mg, 0.444 mmol), and DCM (1.4 mL) was stirred at rt for 4 h. EtOAc (30 mL) was added. The organic phase was washed with sat. NaHCO3 (3 × 30 mL), dried over MgSO4, and filtered. The solvent was removed under reduced pressure. The crude was dissolved in 7 N NH3 in MeOH (3.0 mL), and the mixture was stirred at rt for 16 h. The solvent was removed under reduced pressure, and the crude was purified by chromatography (EtOH/cyclohexane 5 → 40%, then 100%) to afford compound 8c as a white solid (38 mg, 49%). 1H NMR (500 MHz, methanol-d4) δ 8.20–8.15 (m, 2H), 8.04–8.00 (m, 2H), 7.70 (d, J = 3.9 Hz, 1H), 7.07 (m, 1H), 4.01 (s, 2H), 3.27–3.22 (m, 4H), 1.79–1.73 (m, 4H). 13C NMR (126 MHz, methanol-d4) δ 159.72, 147.61, 143.08, 140.37, 136.09, 129.69, 129.18, 126.59, 49.15, 41.66, 26.28. HRMS (ESI) for C15H19N2O4S3 ([M + H]+): calculated 387.0502; observed 387.0489; error = 3.4 ppm.
(5-((3-(Pyrrolidin-1-ylsulfonyl)phenyl)sulfonyl)thiophen-2-yl)methanamine (8d)
A mixture of 2,2,2-trifluoro-N-((5-((3-(pyrrolidin-1-ylsulfonyl)phenyl)thio)thiophen-2-yl)methyl)acetamide 24d (107 mg, 0.238 mmol), mCPBA (77%; 117 mg, 0.523 mmol), and DCM (1.6 mL) was stirred at rt for 4 h. EtOAc (30 mL) was added. The organic phase was washed with sat. NaHCO3 (3 × 30 mL), dried over MgSO4, and filtered. The solvent was removed under reduced pressure. The crude was dissolved in 7 N NH3 in MeOH (3.0 mL), and the mixture was stirred at rt for 16 h. The solvent was removed under reduced pressure, and the crude was purified by chromatography (EtOH/cyclohexane 5 → 40%, then 100%) to afford compound 8d as a white solid (49 mg, 51%). 1H NMR (500 MHz, chloroform-d) δ 8.37 (br, 1H), 8.16 (m, J = 7.0 Hz, 1H), 8.00 (m, J = 7.2 Hz, 1H), 7.69 (t, J = 7.8 Hz, 1H), 7.60 (m, 1H), 6.90 (br, 1H), 4.07 (br, 2H), 3.25 (br, 4H), 1.78 (br, 4H), 1.64 (br, 2H). 13C NMR (126 MHz, chloroform-d) δ 159.69, 144.01, 139.17, 138.94, 134.64, 131.61, 131.00, 130.42, 126.07, 124.06, 48.17, 41.64, 25.40. HRMS (ESI) for C15H19N2O4S3 ([M + H]+): calculated 387.0502; observed 387.0493; error = 2.3 ppm.
(S)-(1-((3-((5-(Aminomethyl)thiophen-2-yl)sulfonyl)phenyl)sulfonyl)pyrrolidin-2-yl)methanol (8e)
The titled compound was synthesized according to general procedures GP5 and GP3, from (i) (S)-5-((3-((2-(hydroxymethyl)pyrrolidin-1-yl)sulfonyl)phenyl)thio)thiophene-2-carbonitrile 23e (180 mg, 0.474 mmol), mCPBA (77%; 234 mg, 1.04 mmol) and DCM (3.2 mL), rt, 3 h and (ii) BH3 (1.0 M in THF; 1.42 mL, 1.42 mmol) and THF (1.42 mL), rt, 1 h. Chromatography (EtOH/cyclohexane 20 → 100%) afforded a white foam (141 mg, 72%). 1H NMR (500 MHz, chloroform-d) δ 8.37 (t, J = 1.8 Hz, 1H), 8.18 (dt, J = 7.9, 1.5 Hz, 1H), 8.02 (dt, J = 7.8, 1.4 Hz, 1H), 7.70 (t, J = 7.9 Hz, 1H), 7.60 (d, J = 3.8 Hz, 1H), 6.91 (m, 1H), 4.07 (d, J = 1.0 Hz, 2H), 3.69–3.58 (m, 3H), 3.45 (ddd, J = 10.4, 6.9, 5.2 Hz, 1H), 3.18 (dt, J = 10.4, 7.1 Hz, 1H), 2.13 (br, 3H), 1.88–1.75 (m, 2H), 1.70–1.58 (m, 1H), 1.55–1.43 (m, 1H). 13C NMR (126 MHz, chloroform-d) δ 159.51, 144.04, 139.01, 138.72, 134.73, 131.69, 131.20, 130.61, 126.10, 124.24, 65.27, 62.06, 50.01, 41.54, 28.72, 24.26. HRMS (ESI) for C16H20N2O5S3Na ([M + Na]+): calculated 439.0427; observed 439.0413; error = 3.2 ppm.
(5-((4-(Phenylsulfonyl)phenyl)sulfonyl)thiophen-2-yl)methanamine (8f)
The titled compound was synthesized according to general procedure GP3, from 5-((4-(phenylsulfonyl)phenyl)sulfonyl)thiophene-2-carbonitrile 30 (40 mg, 0.103 mmol), BH3 (1.0 M in THF; 0.5 mL, 0.5 mmol), and THF (4.0 mL), rt, 3 h. Chromatography (EtOH/cyclohexane 0 → 100%) afforded a glassy solid (8 mg, 20%). 1H NMR (500 MHz, DMSO-d6) δ 8.21–8.13 (m, 4H), 7.99 (dt, J = 7.1, 1.3 Hz, 2H), 7.75 (d, J = 3.9 Hz, 1H), 7.73–7.70 (m, 1H), 7.64 (t, J = 7.8 Hz, 2H), 7.02 (dt, J = 3.5, 1.2 Hz, 1H), 3.89 (d, J = 1.3 Hz, 2H), 2.45–2.13 (br, 2H). HRMS calcd for C17H15NO4S3 [M + H]+ 394.0236; found 394.0233; error = 0.76 ppm.
2-((4-((5-(Aminomethyl)thiophen-2-yl)sulfonyl)phenyl)sulfonyl)ethanol Hydrochloride (8g)
The titled compound was synthesized according to general procedures GP2, from tert-butyl ((5-((4-((2-hydroxyethyl)sulfonyl)phenyl)sulfonyl)thiophen-2-yl)methyl)carbamate 27 (n = 1) (52 mg, 0.113 mmol) and 4 M HCl in dioxane (3.0 mL); rt, 3 h. A white solid was obtained, which did not require further purification (39 mg, 87%). 1H NMR (500 MHz, D2O) δ 8.37–8.29 (m, 2H), 8.27–8.19 (m, 2H), 7.91 (m, 1H), 7.38 (m, 1H), 4.48 (s, 2H), 4.03–3.95 (m, 2H), 3.72–3.65 (m, 2H). 13C NMR (126 MHz, D2O) δ 146.18, 144.20, 140.94, 136.69, 131.43, 130.19, 129.18, 58.15, 55.77, 38.19. HRMS (ESI) for C13H13O5S3 ([M – NH2]+): calculated 344.9920; observed 344.9917; error = 0.87 ppm.
3-((4-((5-(Aminomethyl)thiophen-2-yl)sulfonyl)phenyl)sulfonyl)propan-1-ol Hydrochloride (8h)
The titled compound was synthesized according to general procedure GP2, from 4 M HCl in dioxane (1.5 mL) and tert-butyl ((5-((4-((3-hydroxypropyl)sulfonyl)phenyl)sulfonyl)thiophen-2-yl)methyl)carbamate 27 (n = 2) 23.5 mg, 0.0494 mmol; rt, 3 h. A white solid was obtained and did not require further purification (23 mg, quant.). 1H NMR (500 MHz, methanol-d4) δ 8.26 (d, J = 8.5 Hz, 2H), 8.15 (d, J = 8.5 Hz, 2H), 7.83 (d, J = 3.9 Hz, 1H), 7.34 (d, J = 3.9 Hz, 1H), 4.39 (s, 2H), 3.58 (t, J = 6.1 Hz, 2H), 3.37–3.32 (m, 2H), 1.89–1.80 (m, 2H). 13C NMR (126 MHz, methanol-d4) δ 148.02, 146.19, 145.44, 144.33, 136.14, 131.84, 130.66, 129.60, 60.56, 53.60, 38.46, 26.85. HRMS (ESI) for C14H18NO5S3 ([M + H]+): calculated 376.0342; observed 376.0334; error = 2.1 ppm.
(5-((4-((2-(Pyrrolidin-1-yl)ethyl)sulfonyl)phenyl)sulfonyl)thiophen-2-yl)methanamine Dihydrochloride (8i)
Pyrrolidine (12.6 μL, 0.151 mmol) was added to a solution of tert-butyl ((5-((4-(vinylsulfonyl)phenyl)sulfonyl)thiophen-2-yl)methyl)carbamate 28 (61 mg, 0.138 mmol) in DCM (0.8 mL), and the mixture was stirred at rt for 16 h. The solvent was removed under reduced pressure. Four molar HCl in dioxane (4 mL) was added to the crude and the mixture was stirred at rt for 16 h. The precipitated solid was collected on a pad of celite and washed with EtOAc. MeOH was added to dissolve the solid, and the suspension was filtered. The solvent was removed under reduced pressure to afford compound 8i as a white solid (58 mg, 85%), which did not require further purification. 1H NMR (500 MHz, D2O) δ 8.37 (d, J = 7.9 Hz, 2H), 8.27 (d, J = 7.9 Hz, 2H), 7.92 (m, 1H), 7.41 (m, 1H), 4.51 (s, 2H), 3.99 (t, J = 7.2 Hz, 2H), 3.92–2.98 (m, 6H), 2.13 (br, 4H). 13C NMR (126 MHz, D2O) δ 146.87, 145.89, 142.50, 140.91, 136.88, 131.77, 130.49, 129.53, 55.66, 51.65, 48.16, 38.08, 23.47. HRMS (ESI) for C17H23N2O4S3 ([M + H]+): calculated 415.0815; observed 415.0828; error = 3.1 ppm.
N-(2-((4-((5-(Aminomethyl)thiophen-2-yl)sulfonyl)phenyl)sulfonyl)ethyl)-acetamide Hydrochloride (8j)
A mixture of tert-butyl ((5-((4-(vinylsulfonyl)phenyl)sulfonyl)thiophen-2-yl)methyl)carbamate 28 (448 mg, 1.01 mmol) and 7 N NH3 in MeOH (10 mL) was stirred at rt for 2 h. The solvent was removed under reduced pressure to afford tert-butyl ((5-((4-((2-aminoethyl)sulfonyl)phenyl)thio)thiophen-2-yl)methyl)carbamate as a white solid (472 mg, quant.), which did not required further purification. 1H NMR (500 MHz, methanol-d4) δ 7.70 (d, J = 3.8 Hz, 1H), 7.03 (d, J = 3.9 Hz, 1H), 4.39 (s, 2H), 8.16–8.12 (m, 2H), 3.39 (t, J = 6.7 Hz, 2H), 2.96 (t, J = 6.7 Hz, 2H), 1.44 (s, 9H), 8.27–8.21 (m, 2H). LCMS (ESI) m/z 405 [M – tBu + 2H]+.
Ac2O (18.1 μL, 0.191 mmol) was added to a solution of afford tert-butyl ((5-((4-((2-aminoethyl)sulfonyl)phenyl)thio)thiophen-2-yl)methyl)carbamate (80 mg, 0.174 mmol) and Et3N (29.1 μL, 0.209 mmol) in DCM (1.2 mL), and the mixture was stirred at rt for 1 h. DCM (20 mL) was added. The organic solution was washed with H2O (20 mL) and brine (20 mL), dried over MgSO4, and filtered, and the solvent was removed under reduced pressure. Four molar HCl in dioxane (5.0 mL) was added to the crude, and the mixture was stirred at rt for 16 h. The precipitated solid was filtered through a pad of celite and washed with EtOAc. MeOH was added to dissolve the solid, and the suspension was filtered. The solvent was removed under reduced pressure to afford compound 8j as a white solid (42 mg, 55%), which did not require further purification. 1H NMR (500 MHz, methanol-d4) δ 8.26 (d, J = 8.5 Hz, 2H), 8.15 (d, J = 8.6 Hz, 2H), 7.83 (d, J = 3.9 Hz, 1H), 7.34 (d, J = 3.8 Hz, 1H), 4.39 (s, 2H), 3.57–3.44 (m, 4H), 1.69 (s, 3H). 13C NMR (126 MHz, methanol-d4) δ 173.41, 148.05, 146.22, 145.69, 144.29, 136.17, 131.85, 130.69, 129.58, 54.98, 38.43, 34.81, 22.23. HRMS (ESI) for C15H19N2O5S3 ([M + H]+): calculated 403.0451; observed 403.0453; error = 0.50 ppm.
(5-((4-((2-Methoxyethyl)sulfonyl)phenyl)sulfonyl)thiophen-2-yl)methanamine Hydrochloride (8k)
A mixture of tert-butyl ((5-((4-(vinylsulfonyl)phenyl)sulfonyl)thiophen-2-yl)methyl)carbamate 28 (65.9 mg, 0.149 mmol), K2CO3 (24.6 mg, 0.178 mmol), and MeOH (1 mL) was stirred at rt for 2 h before it was diluted with EtOAc (20 mL). The organic layer was washed with 1:1 H2O/brine (20 mL), dried over MgSO4, and filtered, and the solvent was removed under reduced pressure. Four molar HCl in dioxane (5 mL) was added to the crude, and the mixture was stirred at rt for 3 h. The precipitated solid was filtered through a pad of celite and washed with EtOAc. MeOH was added to dissolve the solid, and the suspension was filtered. The solvent was removed under reduced pressure to afford compound 8k as a white solid (30 mg, 54%) which did not require further purification. 1H NMR (500 MHz, methanol-d4) δ 8.22 (d, J = 8.6 Hz, 2H), 8.13 (d, J = 8.6 Hz, 2H), 7.83 (d, J = 3.9 Hz, 1H), 7.33 (d, J = 3.9 Hz, 1H), 4.39 (s, 2H), 3.72 (t, J = 5.6 Hz, 2H), 3.55 (t, J = 5.6 Hz, 2H), 3.11 (s, 3H). 13C NMR (126 MHz, methanol-d4) δ 147.77, 146.76, 146.08, 144.50, 136.04, 131.78, 130.69, 129.24, 66.83, 58.69, 56.78, 38.43. HRMS (ESI) for C14H18NO5S3 ([M + H]+): calculated 376.0342; observed 376.0333; error = 2.4 ppm.
(5-((3-(tert-Butyl)-5-(methylsulfonyl)phenyl)sulfonyl)thiophen-2-yl)methanamine (9a)
To a solution of carbaldehyde 33 (590 mg, 1.66 mmol) in THF (50 mL) at 0 °C was added sodium borohydride (141 mg, 3.72 mmol). The reaction mixture was stirred at rt for 2 h, before it was quenched with ice. The pH of the solution was adjusted to 4–5 with 1 M HCl, and the aqueous phase was extracted with EtOAc. The combined organic phase was washed with brine, dried over Na2SO4, and filtered, and the solvent was removed under reduced pressure. The crude was dissolved in DCM (50 mL). mCPBA (77%; 742 mg, 3.31 mmol) was added in small portions at 0 °C. The mixture was stirred at rt for 12 h, and the solvent was subsequently removed under reduced pressure. The crude was purified by chromatography (EtOAc/cyclohexane 0 → 60%) to afford (5-((3-(tert-butyl)-5-(methylsulfonyl)phenyl)sulfonyl)thiophen-2-yl)methanol (500 mg, 78% over two steps). 1H NMR (500 MHz, chloroform-d) δ 8.32 (t, J = 1.4 Hz, 1H), 8.27 (t, J = 1.7 Hz, 1H), 8.13 (t, J = 1.6 Hz, 1H), 7.65 (d, J = 3.8 Hz, 1H), 6.99 (d, J = 3.8 Hz, 1H), 4.87 (s, 2H), 3.10 (s, 3H), 2.24 (br, 1H), 1.40 (s, 9H). 13C NMR (126 MHz, chloroform-d) δ 155.4, 155.3, 143.8, 142.0, 140.2, 134.3, 129.1, 128.7, 125.0, 123.7, 60.1, 44.4, 35.8, 31.0. HRMS (ESI) for C16H19O4S3 ([M – OH]+): calculated 371.0445; observed 371.0486.
To a solution of (5-((3-(tert-butyl)-5-(methylsulfonyl)phenyl)sulfonyl)thiophen-2-yl)methanol (500 mg, 1.29 mmol) in THF (15 mL) at 0 °C was added PPh3 (406 mg, 1.54 mmol) and DEAD (0.24 mL, 1.54 mmol). The mixture was stirred at 0 °C for 10 min, followed by the addition of DPPA (0.35 mL, 1.54 mmol). The reaction was warmed to rt over 16 h, and the solvent was removed under reduced pressure. The crude was dissolved in EtOH (50 mL), and Pd/C (10%; 150 mg) was added. The mixture was stirred at rt under H2 pressure (balloon) for 12 h and then filtered through celite. The solvent was removed under reduced pressure, and the crude was purified by chromatography (EtOH/cyclohexane 0 → 100%) to afford compound 9a as an orange gum (262 mg, 52%). 1H NMR (500 MHz, chloroform-d) δ 8.28 (t, J = 1.4 Hz, 1H), 8.23 (t, J = 1.6 Hz, 1H), 8.09 (t, J = 1.6 Hz, 1H), 7.60 (d, J = 3.8 Hz, 1H), 6.90 (d, J = 3.8 Hz, 1H), 4.06 (s, 2H), 3.08 (s, 3H), 1.70 (br, 2H), 1.36 (s, 9H). 13C NMR (126 MHz, chloroform-d) δ 159.4, 155.2, 143.9, 141.8, 138.6, 134.5, 128.9, 128.4, 123.9, 123.5, 44.3, 41.4, 35.6, 30.9. HRMS (ESI) for C16H19O4S3 ([M – NH]2+): calculated 371.0445; observed 371.0428; error = 4.6 ppm.
(5-((3-(tert-Butoxy)-5-(methylsulfonyl)phenyl)sulfonyl)thiophen-2-yl)methanamine (9b)
NaOtBu (65.8 mg, 0.684 mmol) was added to a degassed mixture of 2-ethylhexyl 3-((3-(tert-butoxy)-5-(methylthio)phenyl)thio)propanoate 60 (282 mg, 0.684 mmol) and 5-bromothiophene-2-carbonitrile 17a (76.1 μL, 0.684 mmol) in DMF (3.4 mL), and the mixture was stirred at 60 °C for 3h. The cooled mixture was diluted with EtOAc (50 mL), washed with H2O (3 × 50 mL) and brine (50 mL), dried over MgSO4, and filtered. The solvent was removed under reduced pressure, and the crude was purified by chromatography (EtOAc/pet ether 0 → 10%) to afford 5-((3-(tert-Butoxy)-5-(methylthio)phenyl)thio)thiophene-2-carbonitrile as a colorless oil (85 mg, 37%). 1H NMR (300 MHz, chloroform-d) δ 7.51 (d, J = 3.9 Hz, 1H), 7.12 (d, J = 3.9 Hz, 1H), 6.94 (t, J = 1.7 Hz, 1H), 6.79 (t, J = 1.9 Hz, 1H), 6.72 (t, J = 1.9 Hz, 1H), 2.44 (s, 3H), 1.33 (s, 9H). LCMS (ESI) m/z 280 [M – tBu + 2H]+.
5-((3-(tert-Butoxy)-5-(methylsulfonyl)phenyl)sulfonyl)thiophene-2-carbonitrile was synthesized according to general procedure GP5, from 5-((3-(tert-Butoxy)-5-(methylthio)phenyl)thio)thiophene-2-carbonitrile (41 mg, 0.122 mmol), mCPBA (70–75%; 145 mg, 0.612 mmol) and DCM (1.2 mL), rt, 16 h. Chromatography (EtOAc/cyclohexane 0 → 60%) afforded a white solid (43 g, 88%). 1H NMR (500 MHz, chloroform-d) δ 8.17 (t, J = 1.6 Hz, 1H), 7.81 (dd, J = 2.2, 1.7 Hz, 1H), 7.76 (dd, J = 2.2, 1.6 Hz, 1H), 7.71 (d, J = 4.0 Hz, 1H), 7.60 (d, J = 4.1 Hz, 1H), 3.10 (s, 3H), 1.46 (s, 9H). LCMS (ESI, -ve) m/z 398 [M – H]−.
Compound 9b was synthesized according to general procedure GP3, from BH3 (1.0 M in THF; 0.53 mL, 0.53 mmol), 5-((3-(tert-Butoxy)-5-(methylsulfonyl)phenyl)sulfonyl)thiophene-2-carbonitrile (70 mg, 0.175 mmol) and THF (0.53 mL), 2 h, rt. Chromatography (EtOH/cyclohexane 30, then 100%) afforded a white foam (60 mg, 85%). 1H NMR (500 MHz, methanol-d4) δ 8.14 (t, J = 1.6 Hz, 1H), 7.81 (m, 1H), 7.79–7.74 (m, 2H), 7.15 (m, 1H), 4.10 (s, 2H), 3.20 (s, 3H), 1.44 (s, 9H). 13C NMR (126 MHz, methanol-d4) δ 158.93, 157.22, 146.05, 144.77, 140.99, 136.12, 127.67, 126.97, 126.78, 120.47, 82.68, 43.97, 41.02, 28.83. HRMS (ESI) for C16H22NO5S3 ([M + H]+): calculated 404.0655; observed 404.0651; error = 0.99 ppm.
(5-((3-(Methylsulfonyl)-5-((trimethylsilyl)ethynyl)phenyl)sulfonyl)thiophen-2-yl)methanamine Hydrochloride (9c)
A mixture of tert-butyl ((5-((3-bromo-5-(methylsulfonyl)phenyl)sulfonyl)thiophen-2-yl)methyl)carbamate 34 (170 mg, 0.333 mmol), Pd(PPh3)4 (38.5 mg, 10%), CuI (12.7 mg, 20%), trimethylsilylacetylene (70.6 μL, 0.500 mmol), Et3N (0.5 mL), and 1,4-dioxane (0.5 mL) was degassed with argon and then stirred at rt for 16 h. The solvent was removed under reduced pressure, and the crude was dissolved in DCM (1.0 mL). Four molar HCl in dioxane (1.0 mL) was added, and the mixture was stirred at rt for 16 h. EtOAc (2.0 mL) was added to precipitate the solids, which were filtered, washed with EtOAc, and dried under vacuum to afford compound 9c as white powder (80 mg, 88%). 1H NMR (500 MHz, methanol-d4) δ 8.42 (H, 1H), 8.29–8.21 (m, 2H), 7.88 (d, J = 3.7 Hz, 1H), 7.33 (d, J = 3.8 Hz, 1H), 4.39 (s, 2H), 3.21 (s, 3H), 0.28 (s, 9H). 13C NMR (126 MHz, methanol-d4) δ 146.34, 145.18, 144.58, 143.96, 136.34, 136.15, 135.52, 131.87, 127.82, 126.41, 101.83, 54.75, 43.85, 38.40. HRMS (ESI) for C17H22NO4S3Si ([M + H]+): calculated 428.0475; observed 428.0477; error = 0.47 ppm.
(5-((3-(1-Methyl-1H-pyrazol-4-yl)-5-(methylsulfonyl)phenyl)sulfonyl)thiophen-2-yl)methanamine (9d)
The titled compound was synthesized according to general procedures GP7 and GP2, from (i) tert-butyl ((5-((3-bromo-5-(methylsulfonyl)phenyl)sulfonyl)thiophen-2-yl)methyl)carbamate 34 (100 mg, 0.196 mmol), Pd(PPh3)4 (22.6 mg, 10%), (1-methyl-1H-pyrazol-4-yl)boronic acid pinacol ester (48.9 mg, 0.235 mmol), Cs2CO3 (76.7 mg, 0.235 mmol), and 1,4-dioxane (1.3 mL), 90 °C, 16 h and (ii) 4 M HCl in dioxane (1.0 mL) and DCM (1.0 mL), rt, 3 h. The crude was basified with 2 N NH3 in MeOH and purified by chromatography (MeOH/DCM 0 → 25%) to afford a light yellow solid (57 mg, 71%). 1H NMR (500 MHz, methanol-d4/chloroform-d) δ 8.29 (t, J = 1.7 Hz, 1H), 8.27–8.22 (m, 2H), 8.07 (s, 1H), 7.91 (s, 1H), 7.69 (d, J = 3.9 Hz, 1H), 7.04 (d, J = 3.7 Hz, 1H), 4.04 (s, 2H), 3.95 (s, 3H), 3.18 (s, 3H). 13C NMR (126 MHz, methanol-d4/chloroform-d) δ 157.51, 145.17, 143.55, 139.75, 137.56, 137.06, 135.61, 129.87, 128.63, 128.57, 126.43, 123.50, 120.52, 44.25, 41.01, 39.31. HRMS (ESI) for C16H18N3O4S3 ([M + H]+): calculated 412.0454; observed 412.0444; error = 2.4 ppm.
(5-((3-(Methylsulfonyl)-5-(pyridin-4-yl)phenyl)sulfonyl)thiophen-2-yl)methanamine (9e)
The titled compound was synthesized according to general procedures GP7 and GP2, from (i) tert-butyl ((5-((3-bromo-5-(methylsulfonyl)phenyl)sulfonyl)thiophen-2-yl)methyl)carbamate 34 (100 mg, 0.196 mmol), Pd(PPh3)4 (45.2 mg, 20%), pyridine-4-boronic acid (57.8 mg, 0.470 mmol), Cs2CO3 (76.7 mg, 0.235 mmol), and 1,4-dioxane (1.3 mL), 90 °C, 16 h and (ii) 4 M HCl in dioxane (0.6 mL) and 1,4-dioxane (0.6 mL), rt, 16 h. The crude was basified with 2 N NH3 in MeOH and purified by chromatography (MeOH/DCM 0 → 20%) to afford a yellow solid (35 mg, 44%). 1H NMR (500 MHz, methanol-d4/chloroform-d) δ 8.72–8.69 (m, 2H), 8.55 (t, J = 1.6 Hz, 1H), 8.50 (t, J = 1.6 Hz, 1H), 8.45 (t, J = 1.6 Hz, 1H), 7.73–7.70 (m, 3H), 7.04 (d, J = 3.9 Hz, 1H), 4.04 (s, 2H), 3.23 (s, 3H). 13C NMR (126 MHz, methanol-d4/chloroform-d) δ 158.47, 150.06, 145.33, 145.14, 143.36, 141.22, 138.23, 135.26, 130.28, 130.21, 126.22, 125.35, 122.13, 43.68, 40.64. HRMS (ESI) for C17H17N2O4S3 ([M + H]+): calculated 409.0345; observed 409.0340; error = 1.2 ppm.
(5-((4′-Methyl-5-(methylsulfonyl)-[1,1′-biphenyl]-3-yl)sulfonyl)thiophen-2-yl)methanamine (9g)
The titled compound was synthesized according to general procedures GP7 and GP2, from (i) tert-butyl ((5-((3-bromo-5-(methylsulfonyl)phenyl)sulfonyl)thiophen-2-yl)methyl)carbamate 34 (100 mg, 0.196 mmol), Pd(PPh3)4 (22.6 mg, 10%), p-tolyl boronic acid (32.0 mg, 0.235 mmol), Cs2CO3 (76.7 mg, 0.235 mmol), and 1,4-dioxane (1.3 mL), 90 °C, 16 h and (ii) 4 M HCl in dioxane (1.0 mL) and DCM (1.0 mL), rt, 3 h. The crude was basified with 2 N NH3 in MeOH and purified by chromatography (acetone/DCM 0 → 40%) to afford a light yellow foam (20 mg, 24%). 1H NMR (500 MHz, chloroform-d) δ 8.45–8.39 (m, 2H), 8.30 (t, J = 1.6 Hz, 1H), 7.67 (d, J = 3.9 Hz, 1H), 7.54 (d, J = 8.2 Hz, 2H), 7.33 (d, J = 8.0 Hz, 2H), 6.93 (d, J = 3.9 Hz, 1H), 4.09 (s, 2H), 3.14 (s, 3H), 2.44 (s, 3H), 1.61 (s, 2H). 13C NMR (126 MHz, chloroform-d) δ 144.95, 144.56, 142.77, 139.79, 138.73, 134.92, 134.43, 130.27, 130.15, 129.73, 127.25, 124.34, 124.16, 44.59, 41.71, 21.36. HRMS (ESI) for C19H20NO4S3 ([M + H]+): calculated 422.0549; observed 422.0561; error = 2.8 ppm.
(5-((3′,5′-Dimethyl-5-(methylsulfonyl)-[1,1′-biphenyl]-3-yl)sulfonyl)thiophen-2-yl)methanamine Hydrochloride (9h)
The titled compound was synthesized according to general procedures GP7 and GP2, from (i) tert-butyl ((5-((3-bromo-5-(methylsulfonyl)phenyl)sulfonyl)thiophen-2-yl)methyl)carbamate 34 (100 mg, 0.196 mmol), Pd(PPh3)4 (22.6 mg, 10%), (3,5-dimethylphenyl)boronic acid (35.3 mg, 0.235 mmol), Cs2CO3 (76.7 mg, 0.235 mmol), and 1,4-dioxane (1.3 mL), 90 °C, 16 h and (ii) 4 M HCl in dioxane (0.7 mL) and 1,4-dioxane (0.7 mL), rt, 16 h. The crude was basified with 2 N NH3 in MeOH and purified by chromatography (EtOH/cyclohexane 0 → 100% then 100%) to afford a white solid (23 mg, 27%). 1H NMR (500 MHz, methanol-d4/chloroform-d) δ 8.41–8.35 (m, 2H), 8.30 (t, J = 1.6 Hz, 1H), 7.68 (d, J = 3.9 Hz, 1H), 7.22 (br, 2H), 7.09 (br, 1H), 7.03 (d, J = 3.8 Hz, 1H), 4.04 (s, 2H), 3.17 (s, 3H), 2.38 (s, 6H). 13C NMR (126 MHz, methanol-d4/chloroform-d) δ 145.50, 144.81, 143.06, 139.62, 139.57, 137.51, 135.39, 131.47, 130.63, 130.52, 126.17, 125.50, 124.57, 44.43, 40.89, 21.44. HRMS (ESI) for C20H22NO4S3 ([M + H]+): calculated 436.0705; observed 436.0700; error = 1.1 ppm.
(5-((2′-Ethyl-5-(methylsulfonyl)-[1,1′-biphenyl]-3-yl)sulfonyl)thiophen-2-yl)methanamine Hydrochloride (9i)
The titled compound was synthesized according to general procedures GP7 and GP2, from (i) tert-butyl ((5-((3-bromo-5-(methylsulfonyl)phenyl)sulfonyl)thiophen-2-yl)methyl)carbamate 34 (100 mg, 0.196 mmol), Pd(PPh3)4 (22.6 mg, 10%), (2-ethylphenyl)boronic acid (35.3 mg, 0.235 mmol), Cs2CO3 (76.7 mg, 0.235 mmol), and 1,4-dioxane (1.3 mL), 90 °C, 16 h and (ii) 4 M HCl in dioxane (0.7 mL) and 1,4-dioxane (0.7 mL), rt, 16 h. The crude was basified with 2 N NH3 in MeOH and purified by chromatography (EtOH/cyclohexane 0 → 100% then 100%) to afford a white foam (42 mg, 49%). 1H NMR (500 MHz, chloroform-d) δ 8.49 (t, J = 1.7 Hz, 1H), 8.21 (t, J = 1.6 Hz, 1H), 8.09 (t, J = 1.6 Hz, 1H), 7.66 (d, J = 3.8 Hz, 1H), 7.42 (td, J = 7.6, 1.3 Hz, 1H), 7.37 (m, 1H), 7.30 (td, J = 7.4, 1.4 Hz, 1H), 7.18 (dd, J = 7.6, 1.2 Hz, 1H), 6.94 (d, J = 3.9 Hz, 1H), 4.10 (s, 2H), 3.14 (s, 3H), 2.53 (q, J = 7.5 Hz, 2H), 1.61 (s, 2H), 1.12 (t, J = 7.6 Hz, 3H). 13C NMR (126 MHz, chloroform-d) δ 159.97, 145.25, 144.31, 142.19, 141.50, 138.66, 137.63, 134.95, 132.72, 132.24, 129.87, 129.41, 129.33, 126.37, 124.59, 124.15, 44.53, 41.73, 26.17, 15.73. HRMS (ESI) for C20H22NO4S3 ([M + H]+): calculated 436.0705; observed 436.0727; error = 5.0 ppm.
(5-((3-(1-Methyl-1H-pyrazol-4-yl)-4-(methylsulfonyl)phenyl)sulfonyl)thiophen-2-yl)methanamine Dihydrochloride (9j)
The titled compound was synthesized according to general procedures GP7 and GP2, from (i) tert-butyl ((5-((3-bromo-4-(methylsulfonyl)phenyl)sulfonyl)thiophen-2-yl)methyl)carbamate 37 (150 mg, 0.294 mmol), Pd(PPh3)4 (34.0 mg, 10%), (1-methyl-1H-pyrazol-4-yl)boronic acid pinacol ester (73.0 mg, 0.353 mmol), Cs2CO3 (115 mg, 0.353 mmol), and 1,4-dioxane (2.0 mL), 90 °C, 16 h and (ii) 4 M HCl in dioxane (1.5 mL) and DCM (1.5 mL), rt, 3 h. A white solid was obtained (111 mg, 92%). 1H NMR (500 MHz, D2O) δ 8.32 (d, J = 8.5 Hz, 1H), 8.16 (dd, J = 8.6, 2.2 Hz, 1H), 8.05 (d, J = 1.9 Hz, 1H), 7.92 (s, 1H), 7.84 (d, J = 4.0 Hz, 1H), 7.74 (s, 1H), 7.33 (d, J = 4.0 Hz, 1H), 4.44 (s, 2H), 3.96 (s, 3H), 2.96 (s, 3H). 13C NMR (126 MHz, D2O) δ 144.87, 144.70, 142.07, 140.32, 139.70, 135.77, 134.00, 132.82, 131.38, 130.83, 130.08, 126.31, 116.55, 41.49, 38.31, 37.18. HRMS (ESI) for C16H18N3O4S3 ([M + H]+): calculated 412.0454; observed 412.0441; error = 3.2 ppm.
(5-((6-(Methylsulfonyl)-[1,1′-biphenyl]-3-yl)sulfonyl)thiophen-2-yl)methanamine (9k)
The titled compound was synthesized according to general procedures GP7 and GP2, from (i) tert-butyl ((5-((3-bromo-4-(methylsulfonyl)phenyl)sulfonyl)thiophen-2-yl)methyl)carbamate 37 (150 mg, 0.294 mmol), Pd(PPh3)4 (34.0 mg, 10%), phenylboronic acid (43.1 mg, 0.353 mmol), Cs2CO3 (115 mg, 0.353 mmol), and 1,4-dioxane (2.0 mL), 90 °C, 16 h. Chromatography (EtOAc/DCM 0 → 100%), white foam, 169 mg and (ii) 4 M HCl in dioxane (1.0 mL) and DCM (1.0 mL), rt, 3 h. The crude was basified with 2 N NH3 in MeOH and purified by chromatography (EtOH/cyclohexane 0 → 100%) to afford a white solid (66 mg, 55%). 1H NMR (500 MHz, chloroform-d) δ 8.37 (d, J = 8.3 Hz, 1H), 8.11 (dd, J = 8.3, 1.9 Hz, 1H), 7.97 (d, J = 2.0 Hz, 1H), 7.63 (d, J = 3.9 Hz, 1H), 7.55–7.41 (m, 5H), 6.93 (d, J = 3.8 Hz, 1H), 4.10 (s, 2H), 2.61 (s, 3H), 1.63 (s, 2H). 13C NMR (126 MHz, chloroform-d) δ 160.04, 146.65, 143.39, 142.92, 138.54, 136.79, 134.97, 131.05, 130.11, 129.70, 129.38, 128.43, 126.64, 124.12, 43.13, 41.67. HRMS (ESI) for C18H18NO4S3 ([M + H]+): calculated 408.0392; observed 408.0373; error = 4.9 ppm.
(5-((4′-Methyl-6-(methylsulfonyl)-[1,1′-biphenyl]-3-yl)sulfonyl)thiophen-2-yl)methanamine Hydrochloride (9l)
The titled compound was synthesized according to general procedures GP7 and GP2, from (i) tert-butyl ((5-((3-bromo-4-(methylsulfonyl)phenyl)sulfonyl)thiophen-2-yl)methyl)carbamate 37 (150 mg, 0.294 mmol), Pd(PPh3)4 (34.0 mg, 10%), p-tolyl boronic acid (48.0 mg, 0.353 mmol), Cs2CO3 (115 mg, 0.353 mmol), and 1,4-dioxane (2.0 mL), 90 °C, 16 h and (ii) 4 M HCl in dioxane (1.5 mL) and DCM (1.5 mL), rt, 3 h. A beige solid obtained (83 mg, 62% over). 1H NMR (500 MHz, methanol-d4) δ 8.37 (d, J = 8.4 Hz, 1H), 8.22 (dd, J = 8.4, 2.0 Hz, 1H), 7.91 (d, J = 2.0 Hz, 1H), 7.83 (d, J = 3.9 Hz, 1H), 7.36–7.30 (m, 5H), 4.40 (s, 2H), 2.70 (s, 3H), 2.43 (s, 3H). 13C NMR (75 MHz, methanol-d4) δ 147.21, 146.24, 145.44, 144.69, 144.29, 140.60, 136.19, 135.39, 132.22, 131.88, 131.03, 130.92, 129.94, 127.81, 43.41, 38.43, 21.30. HRMS (ESI) for C19H20NO4S3 ([M + H]+): calculated 422.0549; observed 422.0531; error = 4.3 ppm.
Pan Assay Interference Compound (PAINS) Assessment
To identify compounds that may demonstrate some degree of promiscuity in biochemical screening, the PAINS filters as described by Baell and Holloway23 were curated as SMARTS and scripted as a flagging protocol deployed in Vortex (version 2018.09.76561.53-s, 2018, https://www.dotmatics.com/products/vortex) and Pipeline Pilot (Dassault Systèmes BIOVIA, BIOVIA Pipeline Pilot, Release 2018, San Diego: Dassault Systèmes, 2018). The 480 patterns were used to recognize structures that may result in nonspecific binding to multiple biological targets by virtue of being comprised of one or more fragments established to be of concern. No LOX inhibitor in this study showed any potential PAINS liability when screened against this PAINS filter.
LOX Protein Preparation and Enzyme Assay
LOX enzyme was extracted from pig skin by the method of Shackleton and Hulmes.24 LOX catalytic activity was determined using a horseradish peroxidase (HRP)-coupled fluorescent assay previously described,19 with cadaverine hydrochloride as a substrate, BAPN as positive control, and a preincubation time of 20 min with nine dilutions from a top concentration of 100 μM. LOXL2 was purchased from R&D System. LOXL2 catalytic activity was determined using the Promega ROS-Glo assay kit with cadaverine hydrochloride as a substrate, BAPN as positive control, and a preincubation time of 20 min at the same concentrations as above.
High-Throughput Screening
HTS was performed by Evotec AG on 270 000 diverse compounds and 5000 fragments using the enzyme assay described above.
Amine Oxidase Assays
Methods for the determination of catalytic activity of DAO, MAO-A, and MAO-B have been previously described.19 All amine oxidase assays were performed with concentrations as above. MAO-A and MAO-B enzymes were purchased from Promega and Sigma, respectively. The catalytic activity of MAO-A and MAO-B was determined using the Promega MAO-Glo assay kit (substrate included), with clorgyline and deprenyl as positive controls, respectively. DAO was purchased from Sigma and the catalytic activity was determined using the Promega ROS-Glo assay kit, with aminoguanidine as the positive control. SSAO was purchased from Sigma. SSAO catalytic activity was determined using the Promega MAO-Glo assay kit, with Mofegiline as positive control.
Assessment of Compound 9f as a Substrate for Amine Oxidases
The catalytic activities of MAO-A, MAO-B, and SSAO with compound 9f as a substrate were determined using the respective enzymes described above, and the hydrogen peroxide produced was quantified using the HRP-coupled fluorescent method as in the LOX activity assay.
MLM Stability Assay
Mouse liver microsomes (BALB/c) were purchased from Tebu-bio, and the assay was performed by methods previously described.19 Inhibitors at 10 μM concentration incubated with the microsomes were assessed at 0, 15, and 30 min. Control samples containing no microsomes and no cofactors were also assessed at 0 and 30 min. Samples were extracted by protein precipitation and centrifugation for 20 min in a refrigerated centrifuge (4 °C) at 3700 rpm. The supernatant was analyzed by LCMS/MS for % metabolized over time.
In Vivo PK
All procedures involving animals were performed in accordance with national Home Office regulations under the Animals (Scientific Procedures) Act 1986 and within guidelines set out by the Institute’s Animal Ethics Committee and the United Kingdom Coordinating Committee for Cancer Research’s ad hoc Committee on the Welfare of Animals in Experimental Neoplasia.25 Female BALB/c or CD1 mice (Charles River Laboratories) at 6 weeks of age were used for the PK analyses. The mice were dosed orally by gavage (50 mg/kg in dimethyl sulfoxide (DMSO)/water 1:19 v/v; n = 21) or intravenously in the tail vein (10 mg/kg in DMSO/Tween20/saline 10:1:89 v/v/v; n = 24). Samples were taken at seven (po) or eight (iv) time points between 5 min and 24 h. Three mice were used per time point per route. They were placed under halothane or isoflurane anesthesia, and blood for plasma preparation was taken by terminal cardiac puncture into heparinized syringes. Plasma samples were snap-frozen in liquid nitrogen and then stored at −80 °C prior to analysis.
In Vivo Anti-metastatic Efficacy
LOX inhibitor treatment was carried out in mouse GEMM breast cancer model where MMTV-PyMT female mice were randomized as described previously.19 Mice were treated daily by oral gavage with 70 mg/kg compound 9f (n = 5) in vehicle (5% DMSO/2.5% Tween20 in water) or control with vehicle alone (n = 7). Lungs were collected at the end of the experiment, as previously described.19
Histology and Immunohistochemistry
All mouse tissue samples were fixed in 10% formalin (Sigma) and embedded in paraffin. For spontaneous lung metastasis in MMTV-PyMT animals, the number of metastasis in the lung parenchyma was counted and the size was measured.
Commercial ADME-T Services
hERG inhibition was determined using the “hERG Human Potassium Ion Channel Cell Based Antagonist Qpatch Assay” by Eurofins Ltd. Cell permeability was determined using the “Caco-2 permeability assay” by Cyprotex Ltd.
Acknowledgments
This work was supported by the CRUK Manchester Institute (C5759/A27412, C5759/A12328), CRUK Manchester Institute Drug Discovery Unit (C480/A17098), the Division of Cancer Therapeutics at The Institute of Cancer Research (C309/A11566, C309/A8274, C107/A10433), and the Wellcome Trust (1003X, 103021/Z/13/Z). We thank Dr. Maggie Liu, Dr. Amin Mirza, and Meirion Richards for assistance with NMR and MS instrumentation. We would also like to thank Dr. Simon Pearce for his advice on statistics.
Glossary
Abbreviations
- AMT
aminomethylenethiophene
- BAPN
3-aminopropionitrile
- DAO
diamine oxidase
- dba
dibenzylideneacetone
- CL
clearance
- Cmax
maximal concentration
- DIPEA
N,N-diisopropylethylamine
- DPPA
diphenyl phosphoryl azide
- GEMM
genetically engineered mouse model
- HMDS
hexamethyldisilazane
- HRP
horseradish peroxidase
- LOX
lysyl oxidase
- LOXL
lysyl oxidase like
- LTQ
lysine tyrosylquinone
- MAO
monoamine oxidase
- mCPBA
meta-chloroperbenzoic acid
- MLM
mouse liver microsome
- MMTV
mouse mammary tumor virus
- PAINS
pan assay interference compounds
- PyMT
polyomavirus middle T-antigen
- ROS
reactive oxygen species
- SSAO
semicarbazide-sensitive amine oxidase
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.9b00335.
Molecular formula strings of all final inhibitors (CSV)
Experimental procedures and characterization data for synthetic intermediates, compounds 11a, 11b, 15, 16, 19, 22c, 22d, 22e, 23c, 23d, 23e, 24c, 24d, 26, 27, 28, 29, 30, 32, 33, 36, 37, 38, 39, 41, 43, 45, 47, 49, 52, 54, 56, 58, 59, and 60 (PDF)
The authors declare the following competing financial interest(s): H.T., L.L., D.N.-D., D.S, C.C, R.Ma. and C.S. have filed a patent application which includes the therapeutic use of aminomethylenethiophene inhibitors such as CCT365623. All authors L.L., D.N.-D, D.S., F.L., C.C., R.Mc., G.S., L.D., M.A., M.B., L.J., A.Z., T.C., D.M., N.B., R.K., L.F., R.L., M.C., H.T., R.Ma. and C.S. could benefit financially if any of the described inhibitors such as CCT365623 is licenced, as part of a reward scheme for employees at ICR and CRUK MI.
Supplementary Material
References
- Finney J.; Moon H.-J.; Ronnebaum T.; Lantz M.; Mure M. Human Copper-Dependent Amine Oxidases. Arch. Biochem. Biophys. 2014, 546, 19–32. 10.1016/j.abb.2013.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan H. M.; Ryvkin F.. Lysyl Oxidase and Lysyl Oxidase-Like Enzymes. In The Extracellular Matrix: An Overview, 1st ed.; Mecham R. P., Ed.; Springer, 2011; pp 303–335. [Google Scholar]
- Lucero H. A.; Kagan H. M. Lysyl Oxidase: an Oxidative Enzyme and Effector of Cell Function. Cell. Mol. Life Sci. 2006, 63, 2304–2316. 10.1007/s00018-006-6149-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan H. M.; Li W. Lysyl Oxidase: Properties, Specificity, and Biological Roles Inside and Outside of the Cell. J. Cell. Biochem. 2003, 88, 660–672. 10.1002/jcb.10413. [DOI] [PubMed] [Google Scholar]
- Wang T.-H.; Hsia S.-M.; Shieh T.-M. Lysyl Oxidase and the Tumor Microenvironment. Int. J. Mol. Sci. 2016, 18, 62–73. 10.3390/ijms18010062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox T. R.; Gartland A.; Erler J. T. Lysyl Oxidase, a Targetable Secreted Molecule Involved in Cancer Metastasis. Cancer Res. 2016, 76, 188–192. 10.1158/0008-5472.CAN-15-2306. [DOI] [PubMed] [Google Scholar]
- Trackman P. C. Lysyl Oxidase Isoforms and Potential Therapeutic Opportunities for Fibrosis and Cancer. Expert Opin. Ther. Targets 2016, 3, 935–945. 10.1517/14728222.2016.1151003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perryman L.; Erler J. Lysyl Oxidase in Cancer Research. Future Oncol. 2014, 10, 1709–1717. 10.2217/fon.14.39. [DOI] [PubMed] [Google Scholar]
- Barker H. E.; Cox T. R.; Erler J. T. The Rationale for Targeting the LOX Family in Cancer. Nat. Rev. Cancer 2012, 12, 540–552. 10.1038/nrc3319. [DOI] [PubMed] [Google Scholar]
- Nishioka T.; Eustace A.; West C. Lysyl Oxidase: From Basic Science to Future Cancer Treatment. Cell Struct. Funct. 2012, 37, 75–80. 10.1247/csf.11015. [DOI] [PubMed] [Google Scholar]
- Siddikuzzaman; Grace V. M. B.; Guruvayoorappan C. Lysyl Oxidase: a Potential Target for Cancer Therapy. Inflammopharmacology 2011, 19, 117–129. 10.1007/s10787-010-0073-1. [DOI] [PubMed] [Google Scholar]
- Johnston K. A.; Lopez K. M. Lysyl Oxidase in Cancer Inhibition and Metastasis. Cancer Lett. 2018, 417, 174–181. 10.1016/j.canlet.2018.01.006. [DOI] [PubMed] [Google Scholar]
- Erler J. T.; Bennewith K. L.; Nicolau M.; Dornhofer N.; Kong C.; Le Q. T.; Chi J. T. A.; Jeffrey S. S.; Giaccia A. J. Lysyl Oxidase is Essential for Hypoxia-Induced Metastasis. Nature 2006, 440, 1222–1226. 10.1038/nature04695. [DOI] [PubMed] [Google Scholar]
- Tang S. S.; Trackman P. C.; Kagan H. M. Reaction of Aortic Lysyl Oxidase with beta-Aminopropionitrile. J. Biol. Chem. 1983, 258, 4331–4338. [PubMed] [Google Scholar]
- Pinnell S. R.; Martin G. R. The Cross-Linking of Collagen and Elastin: Enzymatic Conversion of Lysine in Peptide Linage to α-Aminoadipic-δ-Semialdehyde (Allysine) by an Extract from Bone. Proc. Natl. Acad. Sci. U.S.A. 1968, 61, 708–714. 10.1073/pnas.61.2.708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J.; Lucas M. C.; Leonte L. E.; Garcia-Montolio M.; Singh L. B.; Findlay A. D.; Deodhar M.; Foot J.; Jarolimek W.; Timpson P.; Erler J. T.; Cox T. R. Pre-clinical Evaluation of Small Molecule LOXL2 Inhibitors in Breast Cancer. Oncotarget 2017, 8, 26066–26078. 10.18632/oncotarget.15257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowbottom M. W.; Bain G.; Calderon I.; Lasof T.; Lonergan D.; Lai A.; Huang F.; Darlington J.; Prodanovich P.; Santini A. M.; King C. D.; Goulet L.; Shannon K. E.; Ma G. L.; Nguyen K.; MacKenna D. A.; Evans J. F.; Hutchinson J. H. Identification of 4-(Aminomethyl)-6-(trifluoromethyl)-2-(phenoxy)pyridine Derivatives as Potent, Selective, and Orally Efficacious Inhibitors of the Copper-Dependent Amine Oxidase, Lysyl Oxidase-Like 2 (LOXL2). J. Med. Chem. 2017, 60, 4403–4423. 10.1021/acs.jmedchem.7b00345. [DOI] [PubMed] [Google Scholar]
- Hutchinson J. H.; Rowbottom M. W.; Lonergan D.; Darlington J.; Prodanovich P.; King C. D.; Evans J. F.; Bain G. Small Molecule Lysyl Oxidase-like 2 (LOXL2) Inhibitors: The Identification of an Inhibitor Selective for LOXL2 over LOX. ACS Med. Chem. Lett. 2017, 8, 423–427. 10.1021/acsmedchemlett.7b00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang H. R.; Leung L.; Saturno G.; Viros A.; Smith D.; Di Leva G.; Morrison E.; Niculescu-Duvaz D.; Lopes F.; Johnson L.; Dhomen N.; Springer C.; Marais R. Lysyl Oxidase Drives Tumour Progression by Trapping EGF Receptors at the Cell Surface. Nat. Commun. 2017, 8, 14909 10.1038/ncomms14909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rückle T.; Biamonte M.; Grippi-Vallotton T.; Arkinstall S.; Cambet Y.; Camps M.; Chabert C.; Church D. J.; Halazy S.; Jiang X.; Martinou I.; Nichols A.; Sauer W.; Gotteland J.-P. Design, Synthesis, and Biological Activity of Novel, Potent, and Selective (Benzoylaminomethyl)thiophene Sulfonamide Inhibitors of c-Jun-N-Terminal Kinase. J. Med. Chem. 2004, 47, 6921–6934. 10.1021/jm031112e. [DOI] [PubMed] [Google Scholar]
- Chakraborty T. K.; Tapadar S.; Kiran Kumar S. Cyclic Trimer of 5-(Aminomethyl)-2-furancarboxylic Acid as a Novel Synthetic Receptor for Carboxylate Recognition. Tetrahedron Lett. 2002, 43, 1317–1320. 10.1016/S0040-4039(01)02367-X. [DOI] [Google Scholar]
- Sasaki T.; Ando T.; Yamamoto Y.; Imai T.; Kubota D.; Noguchi K.; Hori N.; Shitara E.; Atsumi K.; Yasuda S.. Intermediate for 2-Substituted Carbapenem Derivative and Production Process. WO2004/055027, 2004.
- Baell J. B.; Holloway G. A. New Substructure Filters for Removal of Pan Assay Interference Compounds (PAINS) from Screening Libraries and for Their Exclusion in Bioassays. J. Med. Chem. 2010, 53, 2719–2740. 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Shackleton D. R.; Hulmes D. J. Purification of Lysyl Oxidase from Piglet Skin by Selective Interaction with Sephacryl S-200. Biochem. J. 1990, 266, 917–919. [PMC free article] [PubMed] [Google Scholar]
- Workman P.; Balmain A.; Hickman J. A.; McNally N. J.; Rohas A. M.; Mitchison N. A.; Pierrepoint C. G.; Raymond R.; Rowlatt C.; Stephens T. C.; et al. UKCCCR Guidelines for the Welfare of Animals in Experimental Neoplasia. Lab. Anim. 1988, 22, 195–201. 10.1258/002367788780746467. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.