

Abstract
DNA-Encoded Chemical Library (DECL) synthesis must occur in aqueous media under conditions that preserve the integrity of the DNA encoding tag. While the identification of “DNA-compatible” reaction conditions is critical for the development of DECL designs that explore previously inaccessible chemical space, reports measuring such compatibility have been largely restricted to methods that do not faithfully capture the impact of reaction conditions on DNA fidelity in solution phase. Here we report a comprehensive methodology that uses soluble DNA substrates that exactly recapitulate DNA’s exposure to the chemically reactive species of DECL synthesis. This approach includes the assessment of chemical fidelity (reaction yield and purity), encoding fidelity (ligation efficiency), and readability (DNA compatibility), revealing the fate of the DNA tag during DECL chemistry from a single platform.
Keywords: quantitative PCR (qPCR), DNA-Encoded Chemical Libraries (DECLs), DNA damage, on-DNA chemistry
Graphical Abstract
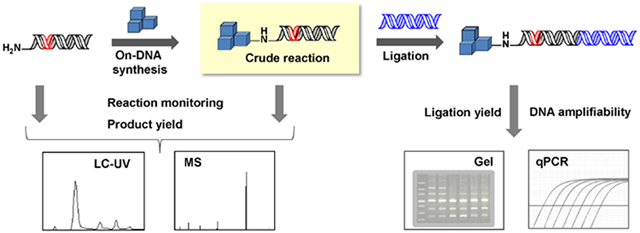
DNA-Encoded Chemical Library (DECL) technology is rapidly gaining traction as a lead finding strategy that is complementary to traditional HTS.1,2,3,4 These combinatorial chemical libraries can be designed to access new chemical space compared to conventional screening collections and are synthesized via multistep, encoded split-and-pool processes.5,6,,7,8,9 Each cycle of library chemistry begins with encoding the monomer identity and reaction conditions by the enzymatic ligation of a DNA sequence to a DNA tag, followed by “on-DNA” chemical synthesis to couple the monomer to a functional site on the DNA molecule. Since the DNA tag is eventually sequenced to deduce the identity of the attached library member, on-DNA chemical reactions must not degrade the DNA tag.
Several approaches have been developed to measure the effect of the chemical reactions used in DECL synthesis on the integrity of DNA. Researches have described the use of Sanger Sequencing to evaluate DNA integrity following on-DNA C-H activation,10 Suzuki-Miyaura coupling,11 and nitrone-olefin cycloadditions.12 qPCR has also been used for quantitating DNA post synthesis, for example, in studies of Cu(I)-catalyzed click chemistry,13 or various solid-phase reactions using magnetic “sensor” beads functionalized with a model DNA tag.14 However, the variability in the DNA substrates and assay conditions used in these studies prohibits meaningful, quantitative comparison for the purposes of on-DNA DECL reaction development. The growing scope of chemistries for DECL synthesis15,16,17 have intensified the need for quantitative methods for validating their compatibility with DNA. Here, we report a straightforward, quantitative assay format that directly reproduces on-DNA DECL synthesis.
The design and synthesis of high-quality DECLs introduces several key constraints. First, chemistry fidelity must be high to ensure high synthetic yield of the designed library member as synthesis truncates can confound library screens.18 Second, one chemistry step cannot corrupt the DNA’s eligibility as a substrate for subsequent cohesive end ligation reactions that install chemistry-encoding oligonucleotides. Third, the chemistry cannot compromise the detectability of the encoding DNA via PCR, because this is the final, crucial step of a DECL screen for generating the templates for sequencing and hit structure elucidation. We sought an approach to evaluate chemistry on the basis of these three parameters from a single assay.
We designed a multifunctional oligonucleotide substrate capable of assaying all aspects of DNA compatibility for DECL reaction development. This substrate, NH2-HPT 1 (Figure 1A), contains a primary amine for on-DNA synthesis (e.g., attachment of scaffolds, first-cycle building blocks) through various amine-focused chemistries (e.g., acylation, Buchwald, SNAr, reductive amination). This amine-handle is attached to the widely used covalent DNA duplex headpiece (hp-DNA9). The hp-DNA is further elaborated with a 33-bp dsDNA fragment (dsT) that approximates a common length and ligation junction found in the DNA encoding tag of an industrial DECL. A DNA fragment of this size (> 25 kDa) would limit mass spectral measurement of chemical synthesis yield. Thus the 33-bp sequence (dsT) contains a ClaI restriction endonuclease sequence, allowing enzymatic cleavage of a minimal-mass fragment for chemical analysis. Following the enzymatic cleavage site is a forward PCR primer binding site and a terminal dinucleotide overhang ligation junction. This substrate (1) was synthesized via enzymatic ligation of the appropriate oligonucleotides using standard DECL procedures. (see Experimental Procedures and Supporting Information)
Figure 1.

Assay reagent and workflow schema of the solution-phase DNA compatibility assessment platform (A) The NH2-HPT substrate 1 contains a primary amine for on-DNA synthesis displayed from the covalent DNA duplex headpiece, hp-DNA. A ClaI restriction endonuclease recognition sequence (teal) separates the hp-DNA from the forward primer binding site (blue nucleobases) and ligation junction. (B) NH2HPT substrate 1 is used in on-DNA chemical synthesis to couple monomer 2. Crude reaction mixture is ligated to a double-stranded oligonucleotide containing the reverse primer binding site (dsR) to furnish templates for PCR amplification.
Substrate 1 is used in a newly developed workflow for assaying on-DNA chemistry compatibility. The workflow (Figure 1B) begins with on-DNA chemical synthesis that couples monomer 2 to substrate 1, generating a mixture of chemical products 3 for LC-MS analysis. Restriction endonucleolytic cleavage (ClaI) liberates a smaller hp-DNA portion if necessary (see Supporting Information). Separately, mixture 3 is used in a post-chemistry enzymatic ligation that introduces TaqMan™ probe and reverse PCR primer binding sites to furnish templates 4 for PCR amplification. Ligation efficiency is determined by gel electrophoresis and crude ligation product is analyzed via qPCR to determine the amount of amplifiable DNA remaining after exposure to chemical synthesis conditions compared to a purified, untreated control (FL, see Supporting Information). The ability to co-screen on-DNA chemical synthesis yield and overall DNA damage using one substrate streamlines analysis while providing a comprehensive understanding of DNA tag integrity under conditions that directly recapitulate on-DNA DECL synthesis.
We undertook studies to define the analytical sensitivity and dynamic range of the PCR method using a model chemical synthesis step. In these studies, substrate 1 was treated with increasing concentrations of methyl methanesulfonate (MMS), which alkylates DNA in the presence of heat by transfer of methyl groups via SN2 reaction from MMS to N3 in adenine and N7 in guanine.19 MMS-induced damage affecting the encodability (% enzymatic ligation efficiency) and detectability (% PCR amplifiability) of the DNA tag was determined as a function of [MMS] and relative to an untreated control. Gel electrophoresis analysis of ligation products confirmed that MMS treatment reduces yield of the ligation products (FL, 103 bp) with increasing [MMS] (Figure 2). Apart from for the product band (FL) and the reagent bands (NH2-HPT, 39 bp; dsR, 62 bp), we did not observe unanticipated oligonucleotide side products.
Figure 2.

Platform validation. The ligation efficiency of the substrates NH2-HPT to dsR, yielding the product FL was determined in the presence of varying [MMS].
Next, we determined if the crude ligation products could be quantitated in qPCR without additional purification. Four ligation reaction mixtures containing the crude FL were analyzed in qPCR to determine the MMS-induced DNA damage. DNA damage increases with increasing [MMS] (Figure 3A). Test sample PCR amplification efficiencies ranged 94–98%. Assays met acceptance criteria for all analysis parameters (see Supporting Information), including linear regression analysis demonstrating excellent agreement of standards (R2 > 0.98). High amplification efficiency with good reaction sensitivity (CT = 12.1–25.5 for 10−3 – 10−7 pmol of 100-mM MMS-treated target) demonstrated that high-quality data are attainable by directly using the crude ligation product without purification (Figure 3B). Under the tested conditions, the PCR protocol demonstrated a broad working range (15–80% amplifiable DNA) of interest in reaction development for DECL synthesis.
Figure 3.

Analysis of dose-dependent damage of NH2HPT by methyl methane sulfonate (MMS). (A) Amplifiable NH2HPT was quantified by qPCR following exposure to varying [MMS]. (B) The average CT values versus 10-fold serial dilutions of the MMS-treated NH2HPT samples increase with increasing [MMS] and indicate a high degree of linearity for the samples over the dynamic range of the assay (for additional details, see Supporting Information).
The new substrates and assay workflow were used to study three on-DNA solution-phase chemistry transformations that are commonly used in DECL builds20 (see Supporting Information for details). Of these reactions (Table 1), acylation21 with HATU-coupling conditions is performed at room temperature and is considered “DNA-compatible.” The DNA-compatibility outcome of this acylation reaction (RT, mild reagents) was compared with two additional chemical transformations that have the potential to damage DNA due to the reaction temperature and duration (e.g., BOC deprotection at 90 °C),22 or exposure to a transition metal catalyst with heat (e.g., Buchwald Pd precatalyst, 80 °C).23,24,25,26 NH2-HPT was exposed to the test reaction conditions using the validated workflow. Acylation proceeded with high yield (92% product), minimal impact on encodability (94% ligation efficiency), and no detectable loss in DNA (~ 100% amplifiability). Boc deprotection conditions did not affect the DNA tag (92% amplifiable DNA remaining), even after prolonged incubation at an elevated temperature. However, enzymatic ligation efficiency suffered a ~30% loss. Even though the on-DNA chemistry under Buchwald conditions (Pd precatalyst tBuXPhos Pd G3) proceeded with good yield (82% product), the ligation efficiency was decreased by ~20%, along with a 30% drop in the amplifiable DNA remaining.
Table 1.
Application of the Compatibility Assessment Platform to Evaluate Solution-phase DECL Chemistries.a
| Reaction | Yield (%) | Ligation Efficiency (%) | Amplifiable DNA Remaining (%) |
|---|---|---|---|
| 92 | 94 | 102 | |
 |
100 | 71 | 92 |
| 82 | 81 | 73 |
See Supporting Information for details.
Chemistry conditions: Acylation – HATU/DIPEA, DMA, 250 mM borate buffer, pH 9.5, 2 h, RT; BOC deprotection - 250 mM borate buffer, pH 9.5, 18 h, 90 °C; Buchwald amination – water/DMA, tBuXPhos Pd G3, NaOH, 1h, 80 °C.
Investigation of these on-DNA chemistry highlights the comprehensive and quantitative nature of this new reaction development platform. While acylation proceeded uneventfully, Boc deprotection specifically decrease ligation efficiency while leaving amplifiability unaffected. The decreased ligation efficiency could result from damage27 to the single-stranded 2-base overhang or loss of the 5’-phosphate, thus compromising the DNA’s ability to serve as a substrate of T4 DNA ligase. Amplifiability results nonetheless suggest that this chemistry is suitable to deploy in DECL. Exposure to the Buchwald catalyst also suppressed ligation yields in addition to amplifiability. The DNA damage observed in the presence of Pd is likely due to depurination along with phosphodiester bond cleavage, resulting in DNA lesions.28,29 Like previous quantitative assays, this new approach does not directly reveal the mechanism of DNA loss, but suspect reagents can be evaluated independently to identify starting points for reaction optimization that would not otherwise be detectable using conventional oligonucleotide-LCMS analysis.
This study demonstrated the robustness of our platform in effectively screening for DNA damage under standard conditions of DECL synthesis. In addition to quantitating DNA amplifiability in PCR, the assay protocol generates information on damage affecting the enzymatic ligation of the ssDNA overhang and the reaction efficiency of the synthetic transformation. Moreover, all this information can be obtained in a single work stream, using a single substrate NH2-HPT. Since DNA compatibility screening is performed on a single substrate (1), using a validated and uniform workflow, the results will be readily reproduced and comparable across different DECL reaction development and synthesis campaigns.
Although the importance of identifying DNA-compatible chemistry has been a main focus in encoded reaction development for the past several years, a straightforward and quantitative method has not been available until now for realizing this goal. Instead, the current practice has been to report the chemical yield as a guide for optimizing DECL-chemistries, sometimes in conjunction with PCR.30 Moreover, ligation efficiency is not typically evaluated when optimizing reactions for DNA compatibility. Herein we have addressed these shortcomings in a more standardized and streamlined bioanalytical workflow that will serve as the foundation for future, deep analyses of ongoing DECL reaction development efforts.
EXPERIMENTAL PROCEDURES
Unless otherwise specified, all of the on-DNA reactions were carried out in 0.5 mL Eppendorf® Safe-Lock microcentrifuge tubes (Cat # EP022363719). Oligonucleotide purity (A260/A280 ~1.8) and concentration (in ug/mL) were determined using a NanoDrop™ 2000 Spectrophotometer (Thermo Scientific).
Preparation of the Substrate (NH2-HPT).
The duplex oligonucleotide dsT (1 equiv., 150 nmol, 50 μM in annealing buffer, 300 μL) was combined with the headpiece hp-DNA (2 equiv., 300 nmol, 1 mM in nuclease free water, 3000 μl), T4 DNA-ligase 10X buffer (400 μL, x1), T4 DNA ligase (1250 U/nmol dsT) and additional nuclease-free water to adjust the final reaction volume to 4 mL (approximate reaction concentrations of dsT and hp-DNA, 37.5 μM and 75 μM respectively). Following incubation (18 h, RT) enzyme was inactivated (10 min, 65 °C). The crude reaction is worked up by adding 5 M NaCl (15% reaction volume) followed by cold ethanol (x 3 reaction volume). The contents were chilled (1h, −20 °C), then centrifuged (12,000 RPM, 2 min) and the supernatant was discarded. The resulting DNA pellet was dissolved in water (~5 mg/mL) and purified by preparative-HPLC (see Supporting Information). The HPLC fractions of the purified NH2-HPT was lyophilized overnight, re-dissolved in water, and separated into 10-nmol aliquots and stored as lyophilized solids (−20 °C).
Preparation of the Full-Length Control Oligomer (FL) –
The duplex oligonucleotide NH2-HPT (1 equiv., 20 nmol, 1 mM in nuclease free water, 20 μL) was combined with the oligonucleotide dsR (1.5 equiv., 30 nmol, 50 μM in annealing buffer, 200 μL), T4 DNA ligase 10X buffer (26.7 μL, x1), T4 DNA ligase (1250 U/nmol NH2-HPT) and additional nuclease-free water to adjust the final reaction volume to ~270 μL (approximate reaction concentrations of NH2-HPT and dsR, 75 μM and 112 μM respectively). Following incubation (18 h, RT), enzyme was inactivated (10 min, 65 °C) and purified by preparative gel-electrophoresis (4% agarose; supporting information). The purified FL oligomer was lyophilized and stored as 5-pmol aliquots (−20 °C).
Workflow for Evaluating DNA Damage using the DNA Compatibility Platform.
On-DNA chemistries were performed by exposing the NH2-HPT (10 nmol, 1 mM solution) to test chemistry conditions (~60–70% water or aqueous buffer with organic solvent, chemical reagents, 25 °C–90 °C, on benchtop or Eppendorf Thermal Cycler). After the desired reaction time, the crude reaction mixture containing the chemically-modified-NH2-HPT is precipitated (5M NaCl/EtOH, −20 °C). The resulting DNA pellet is dissolved in nuclease-free water (1 mM solution, 10 μL) and an aliquot is analyzed by LCMS for the efficiency of chemical transformation (% yield by LC-UV260nm; see supporting information)
Next, the NH2-HPT (1 equiv., 1 nmol, 250 mM in nuclease free water, 4 μL) is combined with the duplex oligonucleotide dsT (1 equiv., 1 nmol, 50 μM in annealing buffer, 20 μL), T4 DNA ligase 10X buffer (3 μL, x1), T4 DNA ligase (0.63 μL; 1250 U/nmol) and additional nuclease free water to adjust the final reaction volume to 30 μL (approximate reaction concentrations of NH2-HPT and dsR, 33.3 μM). Following incubation (2 h, RT), enzyme is inactivated (10 min, 65 °C), DNA is quantitated spectrophotometrically (NanoDrop™) and all samples are normalized to 0.25 pmol/μL in TE buffer and analyzed by E-gel™ (4%, agarose). Band intensities were quantified by image analysis to obtain fraction ligated (see Supporting Information).
For the qPCR assay, a 96-well compound source plate is prepared by step-wise dilution of each sample from 5 × 10−2 – 5 × 10−12. A second 96-well plate is prepared with 15 μL/well of a qPCR-cocktail [2X TaqMan™ Universal PCR Master Mix (ThermoFisher Scientific, Cat # 4304437), nuclease-free water and 20X Custom TaqMan Gene Expression Assay, FAM (ThermoFisher Scientific, Cat # 4331348)], followed by addition of 10 uL/well of the sample dilutions ranging from 5 × 10−4 – 5 × 10−10. A non-template control (nuclease-free water) is included on each plate to detect background amplification and contamination. Each sample is analyzed in quadruplicate (5 μL/well) in a 384-well PCR plate using an automated liquid hander.
Reaction of NH2-HPT with Methyl Methanesulfonate (MMS).
Aliquots of the lyophilized NH2-HPT (10 nmol) were dissolved in 10 μL of aqueous MMS (25, 50, 75 and 100 mM, Vortex mixture, room temperature). After incubation (1 h, 60 °C) in a Thermal Cycler, the reaction mixtures were precipitated (5M NaCl/EtOH, −20 °C). The NH2-HPT pellets were dissolved in nuclease-free water (1 mM solution, 10 μL) and an aliquot analyzed by LCMS (supporting information). A second aliquot (1 nmol) was ligated to the oligomer dsR (described above) and assayed in qPCR.
qPCR Analysis.
The TaqMan (5′-exonuclease) qPCR assay was performed using a Quantstudio 6k Flex Real-Time PCR system (Thermo Fisher Scientific, Waltham, MA). PCR amplification was performed with the following qPCR parameters: 50 °C, 2 min; 95 °C, 10 min; [95 °C, 15 s; 60 °C, 60 s] × 40 cycles. Run quality was assessed using the QuantStudio™ Real-Time PCR Software (Thermo Fisher Scientific, Waltham, MA). Linear regression analysis was performed for each sample to determine the R2 value and assess qPCR performance. Assay acceptance criteria consists of a slope of −3.1 to −3.6, which is indicative of PCR efficiency between 90 and 110 percent and an R2 value >0.98. DNA concentration was then used to determine the percent damage using the following equation: 100 – [([DNA] observed/ [DNA] expected) × 100].
Supplementary Material
Table 2.
Sequence Information of the Oligomer Constructs, Primers and TaqMan probe
| Namea | Duplex Sequences (5’−3’) | Length (bases) | Molecular weight (Da) |
|---|---|---|---|
| hp-DNA | /5Phos/GAGTCA/iSp9//iUniAmM//iSp9/TGACTCCC | 6 + (2-base overhang) | 4937.23 |
| dsT | /5Phos/ATCGATCCTGCTCGCTTCGCTACATGGACAAAG | 33 | 10,153.6 |
| dsT complement | /5Phos/TTGTCCATGTAGCGAAGCGAGCAGGATCGATGG | 33 | 10,338.7 |
| dsR | /5Phos/AGCCGACGACGACTTCCCCGCGGTCTAAACCTCAAGCCTCGGTATCAGGGATATGCTCAGTG | 62 | 19,104.3 |
| dsR complement | CACTGAGCATATCCCTGATACCGAGGCTTGAGGTTTAGACCGCGGGGAAGTCGTCGTCGGCTCT | 62 + (2-base overhang) | 19,759.8 |
| Forward Primer | CCTGCTCGCTTCGCTACATGGACAAAG | 27 | 8220.4 |
| Reverse Primer | CACTGAGCATATCCCTGATACCG | 23 | 6968.6 |
| TaqMan Probe | /56FAM/CCGACGACGACTTCCCCGCG-NFQ-MGB | 20 | 6023.9 |
DNA Constructs.
Duplex oligonucleotides dsT and dsR (Integrated DNA Technologies, Inc. Coralville, IA) were purchased as desalted lyophilate and used without further purification. Oligonucleotide ligation substrates were 5′-phosphorylated (/5Phos/). The amino-modified headpiece DNA (hp-DNA) was SEC purified at the manufacturer (LGC Biosearch Technologies, Petaluma, CA) and obtained as a lyophilized solid. Primers and TaqMan probe sequence were custom ordered (Life Technologies Corporation US).
AKNOWLEDGEMENTS
The authors wish to acknowledge the extended Pfizer DEL team. This work was funded by Pfizer Inc.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website Detailed experimental procedures, analytical characterization data, gel-electrophoresis and qPCR data
REFERENCES
- 1.Favalli N; Bassi G; Scheuermann J; Neri D DNA-Encoded Chemical Libraries: Achievements and Remaining Challenges. FEBS Lett. 2018, 592 (12), 2168–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ding Y; O’Keefe H; DeLorey JL; Israel DI; Messer JA; Chiu CH; Skinner SR; Matico RE; Murray-Thompson MF; Li F; Clark MA; Cuozzo JW; Arico-Muendel C; Morgan BA Discovery of Potent and Selective Inhibitors for ADAMTS-4 through DNA-Encoded Library Technology (ELT). ACS Med. Chem. Lett 2015, 6 (8), 888–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Belyanskaya SL; Ding Y; Callahan JF; Lazaar AL; Israel DI Discovering Drugs with DNA-Encoded Library Technology: From Concept to Clinic with an Inhibitor of Soluble Epoxide Hydrolase. ChemBioChem. 2017, 18 (9), 837–842. [DOI] [PubMed] [Google Scholar]
- 4.Yuen LK; Dana S; Liu Y; Bloom SJ; Thorsell A-G; Neri D; Donato AJ; Kireev D; Schüler H; Franzini RM A Focused DNA-Encoded Chemical Library for the Discovery of Inhibitors of NAD+-Dependent Enzymes. J. Am. Chem. Soc 2019, 141 (13), 5169–5181. [DOI] [PubMed] [Google Scholar]
- 5.Kunig V; Potowski M; Gohla A; Brunschweiger A; DNA-encoded libraries – an efficient small molecule discovery technology for the biomedical. Biol. Chem 2018; 399 (7), 691–710. [DOI] [PubMed] [Google Scholar]
- 6.Franzini RM; Neri D; Scheuermann J DNA-Encoded Chemical Libraries: Advancing beyond Conventional Small-Molecule Libraries. Acc. Chem. Res 2014, 47 (4), 1247–1255. [DOI] [PubMed] [Google Scholar]
- 7.Zimmermann G; Neri D DNA-encoded chemical libraries: foundations and applications in lead discovery. Drug Discov. Today 2016, 21 (11): 1828–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Clark MA et al. Design, synthesis and selection of DNA-encoded small-molecule libraries. Nat. Chem. Biol 2009, 5 (9), 647–654. [DOI] [PubMed] [Google Scholar]
- 9.Satz AL What Do You Get from DNA-Encoded Libraries? ACS Med. Chem. Lett 2018, 9 (5), 408–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang X; Sun H; Liu J; Dai D; Zhang M; Zhou H; Zhong W; Lu X Ruthenium-Promoted C−H Activation Reactions between DNA Conjugated Acrylamide and Aromatic Acids. Org. Lett 2018, 20 (16), 4764–4768. [DOI] [PubMed] [Google Scholar]
- 11.Li J-Y, Huang H Development of DNA-Compatible Suzuki-Miyaura Reaction in Aqueous Media. Bioconjugate Chem. 2018. 29 (11), 3841–3846. [DOI] [PubMed] [Google Scholar]
- 12.Gerry CJ; Yang Z; Stasi M; Schreiber SL A DNA-Compatible [3+2] Nitrone–Olefin Cycloaddition Suitable for DEL Syntheses. Org. Lett 2019, 21 (5), 1325–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abel GR Jr; Calabrese ZA; Ayco J; Hein JE; Ye T Measuring and Suppressing the Oxidative Damage to DNA During Cu(I)-Catalyzed Azide−Alkyne Cycloaddition. Bioconjugate Chem. 2016, 27 (3), 698–704. [DOI] [PubMed] [Google Scholar]
- 14.Malone ML; Paegel BM What is a “DNA-Compatible” Reaction? ACS Comb. Sci 2016, 18 (4), 182–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang X; Hui Sun H; Liu J; Zhong W; Zhang M; Zhou H; Dai D; Lu X Palladium-Promoted DNA-Compatible Heck Reaction. Org. Lett, .2019, 21 (3), 719–723. [DOI] [PubMed] [Google Scholar]
- 16.Du H-C; Simmons N; Faver JC; Yu Z; Palaniappan M; Riehle K; Matzuk MM A Mild, DNA-Compatible Nitro Reduction Using B2(OH)4. Org Lett. 2019, 21 (7), 2194–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kölmel DK; Loach RP; Knauber T; Flanagan ME Employing Photoredox Catalysis for DNA-Encoded Chemistry: Decarboxylative Alkylation of α-Amino Acids. Chem. Med. Chem 2018, 13 (20), 2159–2165. [DOI] [PubMed] [Google Scholar]
- 18.Satz AL Simulated Screens of DNA Encoded Libraries: The Potential Influence of Chemical Synthesis Fidelity on Interpretation of Structure–Activity Relationships. ACS Comb. Sci 2016, 18 (7), 415–424. [DOI] [PubMed] [Google Scholar]
- 19.Schwartz TR; Kmiec EB Using methyl methanesulfonate (MMS) to stimulate targeted gene repair activity in mammalian cells. Gene Ther. Mol. Biol 2005, 9, 193–202. [Google Scholar]
- 20.Satz AL; Cai Z; Chen Y; Goodnow R; Gruber F; Kowalczyk A; Petersen A; Naderi-Oboodi G; Orzechowski L; Strebel Q DNA Compatible Multistep Synthesis and Applications to DNA Encoded Libraries. Bioconjugate Chem. 2015, 26 (8), 1623–1632. [DOI] [PubMed] [Google Scholar]
- 21.Li Y; Gabriele E; Samain F; Favalli N; Sladojevich F; Scheuermann J; Neri D Optimized Reaction Conditions for Amide Bond Formation in DNA-Encoded Combinatorial Libraries. ACS Comb. Sci 2016, 18 (4), 438–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gates KS Chemical Reactions of DNA Damage and Degradation In Reviews of Reactive Intermediate Chemistry; Platz, Moss MS, Jones RA, Jr M, Eds.; Wiley-Interscience: Hoboken, NJ, 2007; Chapter 8, pp 333–378. [Google Scholar]
- 23.Nicholas Favalli N; Bassi G; TZanetti T; Scheuermann J; Neri D Screening of Three Transition Metal-Mediated Reactions Compatible with DNA-Encoded Chemical Libraries. Helv. Chim. Acta 2019, 102 (4), e1900033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ding Y; Clark MA Robust Suzuki–Miyaura Cross-Coupling on DNA-Linked Substrates. ACS Comb. Sci 2015, 17 (1), 1–4. [DOI] [PubMed] [Google Scholar]
- 25.Lu X; Roberts SE; Franklin’ GJ; Davie CP On-DNA Pd and Cu promoted C–N cross-coupling reactions. Med. Chem. Commun, 2017, 8, 1614–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beato EP; Priego J; Gironda-Martínez A; González F; Benavides J; Blas J; Martín-Ortega MD; Toledo MA; Ezquerra J; Torrado A Mild and Efficient Palladium-Mediated C−N Cross-Coupling Reaction between DNA-Conjugated Aryl Bromides and Aromatic Amines. ACS Comb. Sci 2019, 21 (2), 69–74. [DOI] [PubMed] [Google Scholar]
- 27.Greer S; Zamenhof S Studies on depurination of DNA by heat. J. Mol. Biol 1962, 4 (3), 123–141. [DOI] [PubMed] [Google Scholar]
- 28.Iverson BL; and Dervan PB Adenine specific DNA chemical sequencing reaction. Nucleic Acids Res 1987, 15 (19), 7823–7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shearman CW; Loeb LA Depurination decreases fidelity of DNA synthesis in vitro. Nature, 1977, 270, 537–538. [DOI] [PubMed] [Google Scholar]
- 30.Dickson’ P.; Kodadek, T. Chemical composition of DNA-encoded libraries, past present and future. Org. Biomol. Chem 2019, 17 (19), 4676–4688. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.