

SUMMARY
Although it is established that fatty acid (FA) synthesis supports anabolic growth in cancer, the role of exogenous FA uptake remains elusive. Here we show that, during acquisition of resistance to HER2 inhibition, metabolic rewiring of breast cancer cells favors reliance on exogenous FA uptake over de novo FA synthesis. Through cDNA microarray analysis, we identify the FA transporter CD36 as a critical gene upregulated in cells with acquired resistance to the HER2 inhibitor lapatinib. Accordingly, resistant cells exhibit increased exogenous FA uptake and metabolic plasticity. Genetic or pharmacological inhibition of CD36 suppresses the growth of lapatinib-resistant but not lapatinib-sensitive cells in vitro and in vivo. Deletion of Cd36 in mammary tissues of MMTV-neu mice significantly attenuates tumorigenesis. In breast cancer patients, CD36 expression increases following anti-HER2 therapy, which correlates with a poor prognosis. Our results define CD36-mediated metabolic rewiring as an essential survival mechanism in HER2-positive breast cancer.
In Brief
The functional significance of lipid metabolism in cancer cells is not fully understood. Feng et al. show that the fatty acid transporter CD36 is essential for survival of breast cancer cells during anti-HER2 therapy, highlighting the role of lipid metabolism in acquired resistance to targeted therapy.
Graphical Abstract
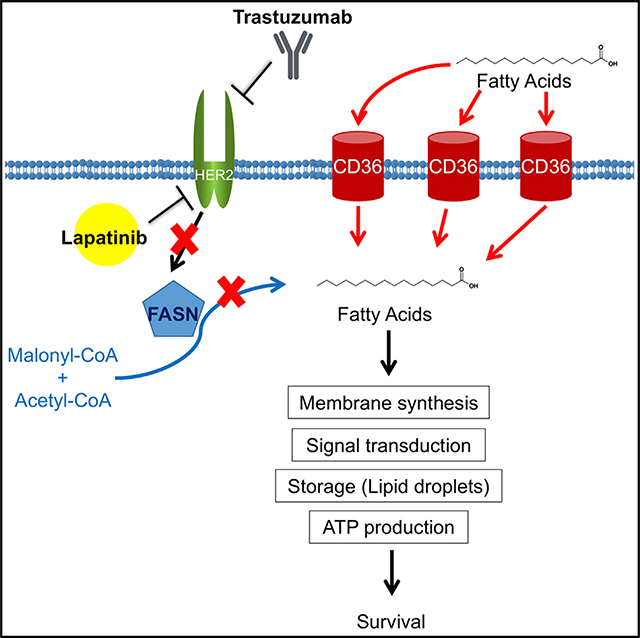
INTRODUCTION
Fatty acids (FAs) play a critical role in a variety of biological processes, including synthesis of plasma membrane phospholipids, cellular signal transduction, and energy production. As opposed to normal cells, which preferentially acquire FAs from exogenous sources, it is estimated that more than 90% of FAs in cancer cells are de novo synthesized by the enzyme FA synthase (FASN) (Ookhtens et al., 1984; Kamphorst et al., 2013). Furthermore, cancer cells often exhibit aberrant FA production, even in the presence of abundant extracellular free FAs, suggesting an inherent dependency on the biosynthetic pathway (Menendez and Lupu, 2007). Indeed, FASN overexpression is observed across cancer types and is known to promote tumor growth, increase with tumor stage, and predict a worsened prognosis in cancer patients (Menendez and Lupu, 2007). Therefore, the therapeutic potential of targeting FASN to kill cancer cells has been explored extensively (Alli et al., 2005; Menendez and Lupu, 2007). In particular, the FASN pathway is highly active in cancers overexpressing the receptor tyrosine kinase HER2 (human epidermal growth factor receptor 2, ERBB2), which promotes both FASN gene transcription and phospho-activation of FASN protein (Slamon et al., 1987; Kumar-Sinha et al., 2003; Jin et al., 2010). HER2 is overexpressed in 20% of all breast cancers, and, like FASN, its overexpression is associated with development of more aggressive tumors and poor prognoses (Kumar-Sinha et al., 2003; Yoon et al., 2007).
Current HER2-targeted therapeutic agents, such as the monoclonal antibody trastuzumab and the small molecule inhibitor lapatinib, often exhibit only transient therapeutic efficacy because of adaptations that allow tumors to evade drug sensitivity, posing a major clinical challenge (Kaufman et al., 2009; Wilken and Maihle, 2010; Nahta et al., 2006). Studies from our laboratory and others have shown that acquired lapatinib resistance is, at least in part, ascribed to activation of compensatory kinase pathways, including upregulation of ERBB family proteins and subsequent reactivation of phosphatidylinositol 3-kinase (PI3K)/AKT signaling (Garrett et al., 2011; Canfield et al., 2015). Apart from rewiring kinase activity, metabolic shifts have also been implicated in facilitating the loss of drug sensitivity in cancer cells. Metabolic reprogramming is a fundamental hallmark of cancer (Hanahan and Weinberg, 2011), and it has been reported that adjustments in metabolic preferences can arise as a result of pro-survival mechanisms that allow cancer cells to adapt and proliferate under stressful conditions, such as nutrient deprivation, hypoxia, or drug-induced cytotoxicity (Holohan et al., 2013). For instance, recent reports indicate that genes associated with glucose depletion (Komurov et al., 2012) and glutamine metabolism (Deblois et al., 2016) are upregulated concomitant with development of lapatinib resistance. In this study, we identify lipid metabolism as a critically altered pathway in lapatinib-resistant cells. Our findings position the CD36 FA transporter as a key determinant of survival in breast cancer cells that acquire resistance to HER2-targeted therapy.
RESULTS
Lapatinib-Resistant Cells Differentially Express Genes Involved in FA Metabolism
We previously produced an in vitro model of acquired tyrosine kinase inhibitor (TKI) resistance by culturing HER2-positive breast cancer cell lines in gradually increasing concentrations of the HER2/EGFR dual antagonist lapatinib over several months (Kurokawa et al., 2013). Although lapatinib-resistant cells are insensitive to lapatinib-induced apoptosis (Figure S1A), lapatinib retains the ability to exert its TKI activity in these cells to prevent downstream phosphorylation of AKT and ERK (Kurokawa et al., 2013; Figures S1B–S1D). To identify activated compensatory pathways in these cells, we performed cDNA microarray analysis of matched lapatinib-resistant and parental lapatinib-sensitive BT474 breast cancer cells (here referred to as rBT474 and BT474, respectively) before and after acute lapatinib treatment (Figure 1A). Because rBT474 cells were continuously maintained in the presence of 1 μM lapatinib, we circumvented confounding effects of chronic exposure to the drug by culturing rBT474 cells in lapatinib-free medium for 1 week prior to analysis so that intrinsic tyrosine kinase activity could be restored, as described previously (Kurokawa et al., 2013).
Figure 1. Lapatinib-Resistant BT474 Cells Differentially Express Genes Involved in Fatty Acid Metabolism.

(A) BT474 and rBT474 were harvested 0 (T0), 10 (T10), and 20 h (T20) after treatment with 1 μM lapatinib (n = 4 per treatment). Microarray analysis was performed using the Illumina Human HT-12 platform.
(B) Venn diagram representing all kinases and phosphatases that exhibit a 1.5-fold or greater change 20 h after 1 μM lapatinib treatment compared with untreated controls (FDR < 0.05).
(C) Hierarchical clustering of the top 1,000 genes with the highest variance after averaging across replicates. The color bar represents z-transformed expression values.
(D) Top 10 “Hallmark Gene Sets” upregulated in rBT474 compared with parental BT474 cells at baseline (T0). Bolded and italicized rows denote pathways directly related to lipid metabolism.
(E) Genes in the GO term “lipid metabolic process” found to be enriched in rBT474 cells and genes identified by microarray analysis of untreated cells at T0 (p < 0.01, FDR < 0.05, fold change > 1.5). CD36 is highlighted in gray.
(F) Volcano plot showing differential expression results comparing rBT474 and BT474 cells. In total, 137 of 22,013 genes were differentially expressed with FDR < 0.01. CD36 was among the most upregulated genes, with a log fold change of 2.5 (FDR = 3.5 × 10−8).
A total of 304 differentially expressed genes were identified with more than 1.5-fold change and a false discovery rate (FDR) of less than 0.05 between sensitive and resistant cells in the absence of lapatinib treatment (Tables S1 and S2). To broadly assess potential effects of lapatinib on cellular kinase signaling, we examined alterations in gene expression of kinases and phosphatases between resistant and parental cells. Although acute lapatinib exposure elicited differential expression of many kinases and phosphatases in both cell lines, few kinases were differentially expressed between lapatinib-resistant and parental cells (Figures 1B, S1F, and S1G). Although compensatory upregulation of HER family receptor tyrosine kinases is a well-established mechanism of acquired TKI resistance (Garrett et al., 2011; Canfield et al., 2015), we did not observe differential expression of ERBB kinases in the absence of lapatinib (Tables S1 and S2). Furthermore, HER2 protein levels remained similar between sensitive and resistant lines (Kurokawa et al., 2013; Canfield et al., 2015; Figure S1B). PI3K/AKT pathway hyperactivation has been implicated as a major contributor to acquired TKI resistance (Garrett et al., 2011); however, we did not observe significant differences in the expression of genes encoding PI3K or AKT proteins (Tables S1 and S2).
To assess the global gene expression changes induced by lapatinib treatment, we performed hierarchical clustering of the expression data using high-variance genes. We observed that the gene expression profile of lapatinib-sensitive cells in the absence of lapatinib (Time 0 = T0) appeared to cluster with that of lapatinib-resistant cells at all time points before and after drug treatment (Figure 1C). Furthermore, lapatinib-sensitive cells exhibited substantial changes in gene expression clustering patterns 10 (T10) or 20 h (T20) after drug treatment (Figure 1C). These results demonstrate that lapatinib elicits a dynamic response at the transcriptional level in drug-sensitive cells, whereas drug-resistant cells remain largely unaffected.
Therefore, we focused our investigation on the inherent differences in gene expression profiles between these cell lines in the absence of lapatinib at T0. To this end, we conducted a gene set enrichment analysis (GSEA) and Gene Ontology (GO) enrichment analysis to identify differentially regulated pathways intrinsic to these cell lines (Subramanian et al., 2005). GSEA using the Molecular Signatures Database (MSigDB) “Hallmark Gene Sets” collection, which was specifically curated to minimize gene set overlap, identified two lipid metabolism-related pathways to be significantly enriched in lapatinib-resistant cells (Figure 1D). Furthermore, a panel of genes involved in two lipid metabolism-related GO terms were among the most dramatically upregulated in rBT474 cells (Figures 1E and S1G). Of particular interest, CD36 (platelet glycoprotein 4), a known membrane FA transporter capable of mediating exogenous FA uptake, was one of the most differentially expressed genes in lapatinib-resistant cells (Figures 1E and 1F). qRT-PCR was used to verify the elevated expression levels of lipid metabolism genes observed in the microarray analysis (Figure S1H).
Lapatinib-Resistant Cells Exhibit Increased Exogenous FA Uptake
Lipid metabolism has become increasingly recognized as a central node in cancer biology. Apart from aberrant FASN-mediated FA synthesis, dysregulation of FA uptake and β-oxidation pathways are gaining attention for their roles in promoting cancer cell growth and metastasis (Nieman et al., 2011; Balaban et al., 2017; Pascual et al., 2017; Ladanyi et al., 2018). Furthermore, lipid droplets are dynamic organelles that play a critical role in storage of excess FAs and the survival of cancer cells under stress conditions, such as hypoxia and nutrient starvation (Cabodevilla et al., 2013; Bensaad et al., 2014). Therefore, we investigated whether increased expression of CD36 and other lipid metabolism genes reflected a phenotypic difference between the two cell lines. We first performed lipid droplet staining in these cells. Oil red O staining revealed markedly increased lipid droplet formation in rBT474 cells (Figures 2A and 2B) and was confirmed through staining with a fluorescent boron-dipyrromethene (BODIPY) dye (BODIPY 493/503) that stains triglycerides present in lipid droplets (Figures 2C and 2D). To assess the functional consequences of increased lipid stores observed in rBT474 cells, we measured protein levels of FASN, CD36, and CPT1A, key components of the de novo FA biosynthesis, FA uptake, and β-oxidation pathways, respectively. Marked upregulation of CD36 protein levels in lapatinib-resistant cells was observed, whereas no difference was observed in levels of FASN or CPT1A (Figures 2E). Because CD36 acts at the cell surface, we confirmed enhanced plasma membrane localization of CD36 in rBT474 cells by flow cytometry (Figure 2F). To determine whether increased functional localization correlated with increased FA transport activity, we next measured exogenous FA uptake using a fluorescent analog of palmitic acid (BODIPY FL C16). Flow cytometry analysis revealed that rBT474 cells exhibited significantly enhanced uptake of the FA analog relative to BT474 parental cells at multiple time points (Figure 2G). In addition, rBT474 cells also significantly accumulated lipid droplets, compared with BT474 cells, upon supplementation of another long-chain FA, oleic acid (C18), into culture medium (Figure S2A). Importantly, the accumulation of lipid droplets was markedly attenuated by the function-blocking anti-CD36 monoclonal antibody JC63.1, demonstrating that the uptake of exogenous FAs depends on CD36. Function of CD36 as a lipid transporter was also confirmed by CD36 small interfering RNA (siRNA) knockdown, which markedly attenuated lipid droplet accumulation (Figure S2B). Pharmacologic inhibition of FASN or CPT1 alone had no effect on lipid droplet accumulation (Figure S2C). CD36 upregulation and increased lipid uptake/storage were observed in other HER2-positive breast cancer cell lines that acquired lapatinib resistance, including SKBR3 and HCC202 cells, suggesting that activation of the CD36 pathway is a general consequence of development of lapatinib resistance in HER2-positive breast cancer (Figures S2D–S2F).
Figure 2. Lapatinib-Resistant BT474 Cells Exhibit Increased Exogenous Fatty Acid Uptake but No Difference in FA Synthesis.

(A–I) rBT474 cells were cultured in lapatinib-free medium 1 week prior to analysis.
(A and B) Representative image of BT474 and rBT474 cells stained with oil red O (scale bars indicates 20 μm) from 3 experiments (A), with quantification of 200–300 cells shown in (B).
(C and D) Representative confocal microscopy image of BT474 and rBT474 stained with BODIPY 493/503 (the scale bar indicates 20 μm) from 3 experiments (C), with quantification of 200–300 cells shown in (D).
(E) Western blot of BT474 and rBT474 cell lysates for key mediators of FA synthesis, uptake, and β-oxidation.
(F) Surface CD36 protein expression in BT474 and rBT474 cells was assessed by flow cytometry. Results shown are representative of 3 experiments, with quantification shown for mean CD36-positive population ± SEM.
(G) BT474 and rBT474 cells were cultured in the presence of 2 μM BODIPY FL C16 for the indicated periods of time. Median fluorescence intensity (MFI) was measured by flow cytometry. Depicted is the MFI of three replicate samples per time point ± SD from one representative experiment of 3.
(H) BT474 and rBT474 cells were cultured in the presence of (−)-C75 or lapatinib for 2 h before an additional 4 h of exposure to 0.667 μCi/mL 14C-acetate. Incorporation was determined as described previously (Olsen et al., 2010). Results depict the mean levels of 14C incorporation ± SD from 4 experiments.
(I) BT474 and rBT47 cells were treated with 10 μg (−)-C75/mL or DMSO for 48 h. Cell proliferation was measured by MTS assay as described previously described (Canfield et al., 2015).
Data are shown as the means ± SEM from 5 experiments. Significance was assessed by non-paired Student’s t test, with significance set at *p < 0.05, ***p < 0.0005.
Next we investigated whether lapatinib-resistant cells gradually acquired resistance or were derived from an inherently resistant subpopulation expressing CD36 at high levels. Toward this end, we treated drug-naive BT474 cells with 500 nM, 1000 nM, or 2000 nM lapatinib for 8 days to intentionally select drug-tolerant persister cells. Western blot and qRT-PCR analyses of the surviving populations revealed no CD36 upregulation at any of the dosages used (Figures S2G and S2H). On the other hand, CD36 expression levels in BT474 cells gradually increased over months of continuing dose escalation of lapatinib, as measured by western blot and flow cytometry (Figures S2I and S2J). These results strongly suggest that drug-naive cells acquired high CD36 expression during chronic treatment with lapatinib rather than resistant cells arising from a small subpopulation of cells, persisters, that pre-existed with CD36 overexpression.
In contrast to CD36, there was no difference in FASN protein levels between BT474 and rBT474 cells (Figure 2E). To fully rule out the possibility that enhanced lipid accumulation in resistant cells is secondary to increased enzymatic activity of FASN, we directly measured FASN activity. In brief, lapatinib-sensitive and -resistant cells were incubated with a radiolabeled FA precursor, 14C-acetate, and its lipogenic incorporation into FAs by FASN was assayed as described previously (Olsen et al., 2010). Notably, there was no difference in the rates of de novo lipogenesis observed in either BT474 or rBT474 cells (Figure 2H). In line with previous reports of HER2 facilitating phospho-activation of FASN, HER2 inhibition by lapatinib significantly attenuated FA synthesis in both BT474 and rBT474 cells and fully mimicked the effect of an established FASN inhibitor, (−)-C75 (Figure 2H). Likewise, lapatinib treatment induced a marked reduction in mRNA levels of FASN in both lapatinib-sensitive and -resistant cells (Figure S2K). Importantly, lapatinib promoted further induction of CD36 gene expression in lapatinib-resistant cells but not in lapatinib-sensitive cells (Figure S2L). These results suggest that CD36 mediates the uptake and accumulation of exogenously acquired FAs during prolonged inhibition of the HER2-FASN axis.
FASN inhibition induces apoptosis in variety of different cancer types (Ventura et al., 2015), suggesting an inherent dependency of cancer cells on endogenous lipogenesis. Because rBT474 cells exhibit an increased ability to acquire exogenous FAs, we examined whether these cells remained sensitive to FASN inhibition. Accordingly, parental BT474 cells exhibited reduced cell viability following pharmacologic FASN inhibition, whereas rBT474 cells remained viable (Figure 2I). Considering the excessive availability of exogenous FAs supplied by the fetal bovine serum in standard tissue culture medium, rBT474 cells may remain able to withstand inhibition of FA synthesis because of an enhanced ability to acquire exogenous lipids. To directly test this hypothesis, rBT474 cells were challenged with (−)-C75 in medium depleted of lipoproteins, which carry over 90% of fatty acyl groups in serum. These FAs are found predominantly in the esterified form, and free FAs are taken up by cells via FA transporters following lipoprotein lipolysis (Goldberg et al., 2009; Discussion). Notably, lipoprotein depletion alone had no effect on rBT474 cell proliferation, but it sensitized lapatinib-resistant cells to FASN inhibition, confirming that these cells rely on exogenous FAs to survive (Figures S2M and S2N).
Lapatinib-Resistant Cells Exhibit a Preference for Glycolysis during Nutrient Abundance but Are Metabolically Plastic during Nutrient Stress
Acetyl-coenzyme A is the main precursor for FA synthesis by cancer cells and is largely derived from glucose and, to a lesser extent, from glutamine (Robey 2018). Therefore, de novo lipogenesis is attenuated in the absence of these substrates. Notably, glucose withdrawal induced significantly more apoptosis in BT474 cells than in rBT474 cells (Figure 3A). As with pharmacologic FASN inhibition, these results suggest that rBT474 cells exhibit reduced sensitivity to glucose withdrawal, in part because of an enhanced capacity for exogenous FA uptake to meet cellular metabolic demands during periods of blunted de novo lipogenesis.
Figure 3. rBT474 Cells Exhibit a Preference for Glycolysis during Nutrient Abundance but Are Metabolically Plastic during Nutrient Stress.

(A) BT474 and rBT47 cells cultured in either complete medium (control) or glucose-deficient complete medium (no glucose) for 96 h were harvested and assayed for apoptosis as in Figure 2G from 3 experiments. Significance was assessed by non-paired Student’s t test, with significance set at *p < 0.05.
(B) rBT474 cells (open symbols) uniformly exhibited lower rates of both net glucose utilization and net lactate accumulation than wild-type BT474 cells (closed symbols), both in the presence (circles) and absence (squares) of the alternative energy substrates glutamine and pyruvate. In rBT474 cells, the stoichiometric ratio between lactate accumulation and glucose disappearance approximates that expected for the quantitative conversion of 6-carbon glucose to 3-carbon lactate in the absence of either lactate reutilization or non-glycolytic sources of lactate generation (broad dashes; Δ[lactate]/Δ[glucose] = 2). In contrast, both net glucose utilization and net lactate production are higher in BT474 cells, but the corresponding ratio is closer to unity in these cells (i.e., Δ[lactate]/Δ[glucose] ~1), suggesting that approximately half of their metabolized glucose is diverted to fates other than lactate. Data are depicted as means ± SEM from 6 consecutive experiments performed in duplicate (n = 5–6 for each data point).
(C) Total hexokinase activity did not differ between BT474 and rBT474 cells under the conditions examined in (B), suggesting that differences in net glucose utilization and lactate accumulation are not attributable to differences in total cellular glucose phosphorylating capacity.
(D) A quantitative pairwise comparison of the net glucose utilization rates depicted in (B) with the total glucose-phosphorylating capacities of BT474 and rBT474 cells revealed capacities for glucose phosphorylation–the first committed step of glucose metabolism (Robey et al., 2000)–that exceeded observed rates of glucose utilization by nearly 20-fold in BT474 cells and by over 45-fold in rBT474 cells, suggesting that differences in glucose utilization reflect metabolic control rather than capacity in these cells (Robey 2018).
(E) Both rBT474 cells and BT474 cells exhibited a substantial capacity for oxidative metabolism in complete medium (circles). rBT474 cells (open symbols), but not BT474 cells (closed symbols), also increased their basal oxygen consumption in glucose- and pyruvate-deficient medium containing glutamine as its principal energy substrate, consistent with an enhanced capacity for the oxidative metabolism of non-glycolytic substrates (e.g., exogenous glutamine or endogenous substrates such as lipids or amino acids) when exogenous glucose is unavailable. Mitochondrial stress testing during Seahorse metabolic flux analysis revealed that approximately half of the basal oxygen consumption in rBT474 cells was associated with oxidative phosphorylation (49% ± 1% versus 57% ± 1% in BT474 cells, n = 5–7). The remainder could be accounted for by either mitochondrial proton leak (27% ± 5% versus 28% ± 1% in BT474 cells) or non-mitochondrial oxygen consumption (24% ± 4% versus 15% ± 1% in BT474 cells). Data are depicted as the means ± SEM from 5–6 consecutive experiments performed in triplicate (n = 3–6 for each data point).
To further characterize the metabolic phenotypes of lapatinib-resistant and -sensitive BT474 cells, we assessed basal cellular glucose utilization and associated metabolic flux. Cancer cells classically exhibit increased glycolytic flux and lactate production with a reduced dependence upon pyruvate entry into the tricarboxylic acid (TCA) cycle (Warburg, 1956; Robey 2018). Indeed, we observed robust lactate production in both BT474 and rBT474 cells cultured in complete medium containing glucose, glutamine, and pyruvate as well as in glutamine- and pyruvate-deficient medium (containing only glucose) (Figures 3B, S3A, and S3B). Interestingly, unlike lapatinib-sensitive parental cells, rBT474 cells generated lactate at a rate twice that of glucose utilization, suggesting near-quantitative conversion of 6-carbon glucose to 3-carbon lactate (Figures 3B, S3A, and S3B). This indicates that lactate is the predominant metabolic fate of glucose in rBT474 cells, whereas BT474 cells exhibit a less glycolytic phenotype involving the ability to divert glucose to alternate fates (e.g., oxidative metabolism) under basal conditions. These differences were preserved even in the presence of alternative substrates (Figure 3B). rBT474 cells also uniformly consumed less total glucose than BT474 cells under basal conditions, but reduced glucose utilization was not due to alterations in hexokinase activity (Figure 3C) and was not limited by the corresponding total cellular glucose-phosphorylating capacity of these cells (Figure 3D). The fact that only a very small percentage of the total available cellular glucose-phosphorylating capacity in each cell type needs to be invoked to account for observed rates of glucose utilization in intact cells suggests primary differences in metabolic control rather than metabolic capacity (Robey 2018). The absence of a demonstrable glycolytic reserve capacity in either cell type following oligomycin inhibition of mitochondrial oxidative metabolism (data not shown) also suggests that glycolytic metabolism is maximal in both cell types under basal conditions. As a consequence, neither cell type appears to be capable of increasing non-oxidative glucose metabolism. These findings may have particular significance for rBT474 cells given their reduced basal capacity for glucose utilization and their apparent inability to direct glucose to alternate metabolic fates (e.g., oxidative metabolism) under non-limited nutrient conditions (Figure 3B).
Under glucose- and pyruvate-free conditions, glutamine can substitute for glucose as an anaplerotic substrate to maintain TCA cycle activity and support cataplerotic acetyl-coenzyme A (CoA) generation for FA synthesis (Yang et al., 2014; Robey 2018). Neither cell type is glucose dependent in the presence of alternative substrates (Figure 3E). As expected, incubation in glutamine-containing medium lacking both glucose and pyruvate reduced extracellular acidification by both BT474 and rBT474 cells (Figure 3E), consistent with reduced contributions by glycolytic metabolism. Basal rates of acidification in the absence of glycolytic substrates were approximately two-thirds of those observed in their presence in both cell types (Figure 3E), suggesting major contributions by non-glycolytic metabolism to basal extracellular acidification in both the presence and absence of glucose. The increased acidification rates observed during metabolic flux analysis in the presence of glucose returned to near-basal levels following glycolytic inhibition by 2-deoxyglucose in both cell types (data not shown), further validating the notion that basal acidification is largely attributable to non-glycolytic metabolism and suggesting that these glucose-associated increases are largely attributable to glycolytic metabolism. Interestingly, rBT474 cells exhibited increased oxygen consumption in the absence of glycolytic substrates, suggesting the capacity to increase oxidative metabolism of non-glycolytic substrates—ostensibly exogenous glutamine and/or endogenous substrates such as lipids or proteins—when glycolytic substrates are either unavailable or insufficient to support metabolic needs (Figure 3E). This expanded metabolic repertoire in resistant cells affords flexibility to utilize alternative bioenergetic substrates under conditions of nutrient stress and helps explain both improved rBT474 cell tolerance to glucose deprivation (Figure 3A) and the inability of etomoxir inhibition of FA β-oxidation to reduce rBT474 cell proliferation in complete glucose-containing medium (Figure S3C). Taken together, our results suggest that lapatinib-resistant cells are able to survive under nutrient stress conditions because of enhanced metabolic plasticity in meeting intracellular catabolic and anabolic demands.
Genetic or Pharmacological Inhibition of CD36 Sensitizes Lapatinib-Resistant Cells
CD36 is a multifunctional protein that plays an important role in facilitating FA import into cells and is also known to be involved in a variety of cell signaling processes (Robey 2018). CD36 was among the most markedly upregulated genes in rBT474 cells in our microarray analysis, and increased CD36 expression was also confirmed by western blot. Because there were no differences in the levels of proteins involved in either FA synthesis or β-oxidation, we decided to further investigate the role of CD36 in development of lapatinib resistance. Surprisingly, siRNA-mediated knockdown of CD36 alone induced drastic levels of apoptosis in rBT474 cells compared with parental BT474 cells (Figures 4A and 4B). Furthermore, pharmacologic inhibition of CD36 with the small-molecule inhibitor sulfosuccinimidyl oleate (SSO) re-sensitized rBT474 cells to lapatinib (Figure 4C). To determine the in vivo implications of our findings, we next established orthotopic xenografts of BT474 and rBT474 cells in immunodeficient NOD scid gamma (NSG) mice. Upon development of tumors approximately 300 mm3 in size, mice were treated with lapatinib or vehicle and with JC63.1 or a control immunoglobulin A (IgA) (Figure 4D). Treatment with lapatinib alone significantly suppressed the growth of BT474-derived tumors (p < 0.05) but not of lapatinib-resistant tumors (p > 0.05) (Figure 4E). Injection of control IgA did not have any effect. Notably, JC63.1 sensitized rBT474 tumors to lapatinib (p = 0.0177), confirming a critical role of CD36 in survival of lapatinib-resistant cancer cells.
Figure 4. CD36 Is Essential for Survival of Lapatinib-Resistant Cells.

(A and B) Transient siRNA transfections were performed with Lipofectamine RNAiMAX (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions.
(A) Knockdown of CD36 was confirmed by western blot 72 h after transfection.
(B) Apoptosis was measured by Annexin V staining via flow cytometry 72 h after transfection (mean ± SEM from 3 experiments).
(C) BT474 and rBT474 cells treated with the CD36 inhibitor sulfosuccinimidyl oleate (SSO) and lapatinib. Cell proliferation was measured by live-cell imaging using an Incucyte Zoom imager at 48 and 96 h of drug treatment. The results depict the average change in confluency from 3 replicate wells ± SD from 3 experiments.
(D) Treatment scheme for xenograft experiment presented in (E).
(E) BT474 and rBT474 xenografts were established in 6-week-old immunodeficient NSG mice. When tumors reached 300 mm3, mice were dosed with lapatinib (100 mg/kg) or vehicle twice a day by oral gavage and 10 μg JC63.1 or IgA control once every 3 days. Tumors were measured every 3 days, and mice were sacrificed after 33 days of treatment. Data are shown as average tumor volume ± SEM.
The significance of the bar graphs in (B) and (C) was assessed by unpaired Student’s t test, with the threshold of significance set at *p < 0.05. The significance of the growth curves in (E) was assessed using the “statmod” R package, with significance set at *p < 0.05 after Benjamini-Hochberg correction for multiple comparisons.
Genetic Deletion of Cd36 Attenuates MMTV-neu-Driven Mammary Tumorigenesis in Mice
MMTV-neu mice spontaneously develop mammary tumors driven by the rat homolog of HER2, neu. Daily lapatinib treatment of this mouse model significantly suppressed the tumor growth rate (Figure S4A) and prolonged the survival of tumor-bearing mice (Figure S4B) compared with vehicle treatment. To investigate the in vivo role of CD36 in breast tumor development and resistance to anti-HER2 therapy, we introduced mammary gland-specific CD36 knockout (KO) to the HER2/neu mammary tumor model by crossing three mouse lines: MMTV-neu, MMTV-Cre, and Cd36flox/flox. Cd36+/+, Cd36flox/+, or Cd36flox/flox female mice with two alleles of MMTV-neu and one allele of MMTV-Cre were produced (mCD36WT, mCD36Het, and mCD36KO, respectively) (Figure 5A). Mammary gland-specific KO of the Cd36 gene was confirmed by PCR genotyping of tail snip DNAs (Figure 5B) and western blot of isolated mammary glands (Figure 5C). Mammary-specific Cd36 KO did not have a significant effect on tumor latency (Figure 5D), suggesting that CD36 may not play a critical role in initiation of HER2-driven breast tumors. Interestingly, Cd36 gene deletion significantly attenuated tumor growth and increased the median survival time (Figure 5E). Consequently, although lapatinib treatment significantly prolonged the survival of mCD36WT mice and attenuated the tumor growth of mCD36WT and mCD36Het mice, it could no longer affect the survival or the tumor growth of mCD36KO mice (Figures 5F–5H and S4C–S4E). CD36 also functions as a thrombospondin receptor and is known to be able to inhibit tumor angiogenesis (Enciu et al., 2018). However, using CD31 immunohistochemistry as a marker of angiogenesis revealed no noticeable differences in vascularization between mCD36WT and mCD36KO tumors (Figure S4F).
Figure 5. CD36 Expression Is Induced by HER2 Inhibition In Vivo and Predicts Survival.

(A) Mammary-specific Cd36 KO mice were generated by crossing three mouse lines: MMTV-neu, MMTV-Cre, and Cd36flox/flox. Cd36+/+, Cd36flox/+, or Cd36flox/flox female mice with two alleles of MMTV-neu and one allele of MMTV-Cre were produced (mCD36WT, mCD36Het, and mCD36KO, respectively).
(B) Mammary-specific Cd36 deletion was confirmed by PCR.
(C) Deletion of Cd36 was confirmed at the protein level by western blot.
(D) Kaplan-Meier curves depicting tumor latency of MMTV-neu mice, defined as time from birth until appearance of the first palpable tumor.
(E) Kaplan-Meier curves of vehicle-treated mCD36WT, mCD36Het, and mCD36KO mice.
(F–H) Kaplan-Meier curves of mCD36WT (F), mCD36Het (G), and mCD36KO (H) CD36-deficient mice treated with lapatinib or vehicle until a total tumor volume of 2,000 mm3 was reached. Significance was measured by log-rank test conducted using Prism v7.04.
(I) qRT-PCR analysis of tumors from mice analyzed in (E) for the panel of FA metabolism genes surveyed in Figure S1H. Mean ± SEM from 8–10 mice per group. Significance was assessed by non-paired Student’s t test, with significance set at *p < 0.05.
(J) Induction of CD36 was measured by immunohistochemistry. Scale bars indicate 500 μm (control) and 677 μm (+lapatinib).
To gain an understanding of the gene expression profiles of these tumors, we performed qRT-PCR for a panel of FA metabolism genes. We found that most of the genes that were upregulated in the rBT474 cell line model of lapatinib resistance (Figure 1E) did not show significant upregulation in the genetic mouse model after prolonged lapatinib treatment (Figure 5I). Nonetheless, consistent with our observation in the cell line model, Cd36 expression was significantly induced by lapatinib (Figure 5I). Induction of CD36 was also recapitulated at the protein level, as assessed by both immunohistochemistry (Figure 5J) and western blot (Figure S4G; residual CD36 can still be seen in CD36 KO tumors, likely because of the presence of CD36-expressing stromal tissue). Importantly, the two other major FA uptake channels, FATP1 and FATP4, encoded by Slc27a1 and Slc27a4, respectively, revealed no differences in gene expression following lapatinib treatment (Figure 5I).
Interestingly, we observed that CD36 deletion from mammary tumors had no effect on the incidence of metastasis in vehicle-treated mCD36WT mice (2 of 10) compared with mCD36KO mice (3 of 10) (Figure S4H). However, lapatinib treatment of mCD36WT mice resulted in increased incidence of metastasis compared with vehicle (lapatinib, 7 of 9; vehicle, 2 of 10) (Figure S4H), although this observation may be confounded by the significantly longer survival time of lapatinib-treated mice, which would have allowed more time for metastases to develop. Nevertheless, such an increased incidence of metastasis was not observed in mCD36Het (lapatinib, 2 of 14; vehicle 8 of 16) and mCD36KO mice (lapatinib, 3 of 11; vehicle, 3 of 11) (Figure S4H), supporting recent work that highlighted the role of CD36 in initiating metastasis (Pascual et al., 2017). Taken together, our results demonstrate a unique role of CD36 in growth of HER2-positive breast tumors and a specific requirement for CD36 up-regulation in tumor survival during HER2 inhibition in vivo.
CD36 Expression Is Induced after Anti-HER2 Therapy and Correlates with a Poor Prognosis
We hypothesized that CD36 expression would enhance the growth of HER2-positive breast tumors and promote tumor cell survival during HER2-targeted therapy. Therefore, we sought to determine whether CD36 overexpression might correlate with poor clinical outcomes in human breast cancer. First, we analyzed a large tissue microarray (TMA) comprising both HER2-positive and -negative breast cancers for CD36 protein expression. CD36 levels were significantly higher in grade III (n = 181) compared with grade II (n = 398) breast cancer samples (log odds ratio = 1.89, p = 0.002) (Figure 6A). A significant association between high CD36 levels and grade III breast cancer was also validated, even after accounting for other clinical covariates, including estrogen receptor (ER) status, progesterone receptor (PR) status, HER2 status, tumor stage, and age (log odds ratio = 1.81, p = 0.005). Importantly, CD36 membranous staining significantly stratified subjects based on overall survival (OS) of patients with HER2-positive breast cancer (Figure 6B) but not HER2-negative breast cancer (Figure 6C).
Figure 6. CD36 Is Induced by HER2-Targeted Therapy and Predicts a Worsened Clinical Outcome.

(A) Tissue microarray analysis demonstrated that CD36 expression correlates with tumor grade. Scale bars indicate 200 μm.
(B and C) Overall survival of breast cancer patients with high (TMA = 2) and low (TMA = 0) CD36 expression from the tissue microarray analyzed in (A) HER2-positive (B) and HER2-negative (C) patients.
(D) CD36 expression was measured by RNA-seq in 44 pairs of tumor biopsies pre- and post-treatment with HER2-targeted therapy.
(E) Overall survival of 245 patients with pre-treatment biopsies from the NeoALLTO trial stratified by median CD36 expression.
(F and G) Survival of HER2-positive (F) and -negative (G) breast cancer patients from a publicly available TCGA dataset, stratified by median CD36 expression.
To further evaluate the expression of CD36 in HER2-positive breast cancers, we utilized the RNA sequencing (RNA-seq) dataset from the Neoadjuvant Lapatinib and/or Trastuzumab Treatment Optimisation (NeoALTTO) trial (Breast International Group [BIG] 1–06 /solti/EGF106903), a randomized clinical trial looking at the use of combined lapatinib and trastuzumab in a neoadjuvant setting (Baselga et al., 2012). When comparing the expression of CD36 in 44 pairs of pre- and post-treatment tumor biopsies, CD36 was significantly increased in post-treatment tumors following HER2-targeted therapy (log2 fold change, 1.07; adjusted p = 0.00053; Figure 6D). FATP1 (SLC27A1) was also upregulated in post-treatment samples (log2 fold change, 1.18; adjusted p = 8.8 × 10−18), whereas FATP4 (SLC27A4) was unchanged between pre- and post-treatment samples (log2 fold change, 0.20; adjusted p = 0.292) (Figures S5A and S2B). FASN remained unchanged before and after neoadjuvant HER2-targeted therapy (FASN log2 fold change, −0.65; adjusted p = 0.053; Figure S5C). Importantly, patients with CD36 expression levels greater than the median across the cohort had a significantly lower rate of OS compared with patients with lower CD36 expression levels (HR = 2.93, p = 0.02, Figure 6E; median [95% CI] follow-up time: CD36 high 3.84 [3.72–4.11] years versus CD36 low 3.89 [3.75–4.09] years). Neither FATP1 (SLC27A1) or FATP4 (SLC27A4) demonstrated significant associations with OS (HR = 0.93, p = 0.9; HR = 1.8, p = 0.2; Figures S5D and S5E). The association between CD36 expression and OS was further supported in an independent dataset from The Cancer Genome Atlas (TCGA) project. Consistent with our analysis of the NeoALTTO trial, elevated CD36 expression was significantly associated with poorer OS in HER2-positive breast cancer patients (Figure 6F). Importantly, this association between CD36 and clinical outcomes was not detected in HER2-negative breast cancer patients (Figure 6G). Independent sub-type analysis using the Kaplan-Meier plotter webtool (Lánczky et al., 2016) confirmed these conclusions in the TCGA cohort, with a difference in survival only detectable in patients with HER2-positive breast cancer but not in patients with HER2-negative/ER-positive breast cancer or triple-negative breast cancer (Figures S5F–S5H). Taken together, these results support a role of CD36 in survival of HER2-positive breast cancer in human patients.
DISCUSSION
Accumulating evidence strongly suggests that lipid metabolism plays significant roles in cancer cell survival, invasion, and metastasis (Buchakjian and Kornbluth, 2010; Fernald and Kurokawa, 2013; Robey 2018). Notably, cancer cells almost universally exhibit a “lipogenic phenotype” characterized by both FASN overexpression and exacerbated levels of FA biogenesis, even in the presence of abundant circulating exogenous FAs (Menendez and Lupu, 2007). Interestingly, this pattern of FASN overexpression and aberrant activation of the FA synthesis pathway appears to be unique to cancer cells because most normal cells preferentially obtain FA from extracellular sources under physiological conditions (Menendez and Lupu, 2007; Goldberg et al., 2009). Furthermore, pharmacologic inhibition of FASN has been shown to be selectively lethal to cancer cells but not to normal cells, highlighting the importance of FAs in cancer biology and simultaneously revealing a promising therapeutic window (Ventura et al., 2015). As a result, FASN has been subjected to intense investigation as a therapeutic target in cancer for decades, whereas the role of extracellular FA uptake has gone largely unexplored in cancer biology. However, this overlooked mechanism has recently started attracting attention as a re-emerging paradigm (Kuemmerle et al., 2011; Kinlaw et al., 2016; Robey 2018). For instance, malignant ovarian and breast cancer cells have been shown recently to instruct neighboring adipocytes in the tumor microenvironment (TME) to induce lipolysis of intracellular lipid stores and subsequently secrete FAs into the TME for uptake and use by cancer cells to support growth and metastasis (Nieman et al., 2011; Balaban et al., 2017). Of particular interest, one recent report identified CD36 as an important mediator of this mechanism (Ladanyi et al., 2018).
An earlier study has shown that, in response to HER2 inhibition, the endogenous FA lipogenic pathway is inhibited, and cancer cells undergo apoptosis because of the two-way cross-talk between FASN and HER2 (Vazquez-Martin et al., 2007). However, we found that cancer cells that exhibit a shift toward CD36-mediated FA uptake can escape lapatinib sensitivity and survive (Figures 7A and 7B). In addition to increased FA uptake, lapatinib-resistant cells appear to be positioned to develop drug resistance by activating a program that features metabolic plasticity and flexible use of bioenergetic substrates that allow adaptation and survival under challenging conditions, such as nutrient deprivation (Figure 3A) and drug-induced cytotoxicity (Figure 2I). Although the mechanism underlying lapatinib-induced CD36 upregulation remains to be fully elucidated, our results demonstrate that CD36 induction plays an essential role in development of resistance to HER2 inhibitors and suggests that targeting CD36 lipid transport activity holds therapeutic promise. Indeed, siRNA knockdown of CD36 caused robust apoptosis in lapatinib-resistant cells but not in sensitive cells (Figure 4B). Likewise, pharmacological inhibition of CD36 significantly suppressed the growth of resistant cells in the presence of lapatinib (Figure 4C). Moreover, a CD36 function-blocking antibody was able to resensitize lapatinib-resistant xenograft tumors to HER2-targeted therapy (Figure 4E).
Figure 7. CD36-Mediated FA Uptake Is Required for HER2-Positive Breast Tumor Cells to Acquire Resistance to HER2 Inhibitors.

(A) Cells acquire FAs endogenously through FASN and/or exogenously via CD36. HER2 activates FASN by phosphorylation and also by transcriptional induction.
(B) Inhibition of HER2 by lapatinib or trastuzumab results in suppression of FASN activity, leaving CD36 as the major source of FA acquisition.
Most importantly, although tumor-specific deletion of the Cd36 gene alone did not affect tumor latency (Figure 5D), it did approximately double the median survival time of MMTV-neu mice after tumor onset (Figure 5E). Notably, removal of one copy of CD36 was sufficient to attenuate tumor growth and prolong survival of these transgenic mice (Figure 5E), suggesting a critical role of the CD36 pathway in the growth of MMTV-neu-driven tumors. We initially expected that CD36 KO would further sensitize the tumors to lapatinib. However, this was not the case; lapatinib treatment resulted in only marginal therapeutic benefits in mCD36KO mice (Figures 5H and S4E). This may be ascribed to the difference between human breast cancer, in which the ERBB2 (HER2) gene is typically amplified, and mouse tumors driven by the activated rat homolog of HER2 (neu) under the MMTV promoter. Alternatively, when mice lacked CD36 before tumor formation, the mice might have developed some other mechanisms of resistance.
FAs are known to influence gene expression in several ways (reviewed in Jump, 2004). These include regulation of the action of specific transcription factors, such as PPARs, LXR, and HNF4, for which specific FA species are ligands, as well as nuclear factor κB (NF-κB) and SREBPs. FA may also modulate signal transduction pathways, either by metabolic effects or by remodeling the composition of plasma membrane lipid rafts, which affects the transmembrane signaling efficiency of growth factors (Swinnen et al., 2003). Additionally, mitochondrial FA β-oxidation generates a large quantity of acetyl-CoA, and this may activate gene expression epigenetically by driving histone acetylation (McDonnell et al., 2016). Our results demonstrated marked lipid accumulation, enhanced lipid metabolism, and metabolic plasticity in resistant cells, but the precise downstream effectors remain to be discovered. mCD36KO tumors did not exhibit elevated levels of FASN, FATP1 (Slc27a1), or FATP4 (Slc27a4) compared with mCD36WT tumors (Figure 5I), indicating that these compensatory pathways for free FA (FFA) acquisition pathways were not induced. The observed effect of lipoprotein depletion may provide an important clue in this regard. The fatty acyl groups carried in triglyceride-rich lipoproteins are predominantly esterified and, thus, require lipolysis for release as FFA for subsequent uptake via CD36. This could be accomplished by extracellular lipolysis mediated by lipoprotein lipase (LPL), an enzyme that is widely expressed in breast cancer. Alternatively, endocytosis of intact lipoproteins mediated by the very-low-density lipoprotein receptor (VLDLR), followed by intracellular lipolysis, could circumvent the requirement for CD36 (Kuemmerle et al., 2011). The potential compensatory roles of these pathways will require further study. Overall, our results reveal a critical role for CD36 in the development of resistance to HER2-targeted therapy as well as its potential as both a prognostic marker and a therapeutic target in breast cancer. Elucidation of the downstream actions of exogenously acquired FA and a full appreciation of the various means by which tumor cells can acquire them will help us to fully understand the mechanism of acquired resistance to anti-HER2 therapy in breast cancer.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Manabu Kurokawa (mkurokaw@kent.edu). All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
MMTV-neu (FVB-Tg(MMTV-Erbb2)NK1Mul/J; RRID:IMSR_JAX:005038), MMTV-Cre (Tg(MMTV-cre)4Mam/J) and NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ; RRID:IMSR_JAX:005557) were obtained from The Jackson Laboratory. CD36flox/flox mice were described previously (Nagendran et al., 2013). All animal studies were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) at Dartmouth College and Kent State University. Female MMTV-neu mice were treated with 100 mg/kg lapatinib (LC Laboratories; Cat# 388082–77-7) or DMSO by oral gavage BID once a palpable tumor ~100mm3 was discovered (~1 year old). Tumor growth was monitored by caliper measurement every 3 days. Mice were sacrificed when reaching maximum tumor volume permitted by IACUC protocols. For xenograft studies, 1 × 107 BT474 or rBT474 cells resuspended in Matrigel Matrix (Corning; Cat# 354234) were implanted in the mammary fat pad of 6-week old female NSG mice. Upon reaching 300mm3, mice were randomly assigned to one of four treatment groups. Mice were treated with 100 mg/kg lapatinib or DMSO by oral gavage BID in combination with 10 μg anti-CD36 function blocking antibody (Clone JC63.1; Cayman Chemical; Cat# 188150; RRID:AB_10077812) or anti-mouse IgA-isotype control (Abcam; Cat# ab37322).
Cell Lines
BT474 (RRID:CVCL_0179), SKBR3 (RRID:CVCL_0033), and HCC202 (RRID:CVCL_2062) human breast cancer cells were authenticated by Duke University Cell Culture Facility. Lapatinib resistant rBT474 cells were derived by culturing cells in the presence of gradually increasing concentrations of lapatinib for months, as described previously (Kurokawa et al., 2013). Cells were cultured in RPMI1640 (Corning) supplemented with 10% fetal bovine serum (Hyclone Laboratories) and 1% penicillin-streptomycin (Hyclone Laboratories). rBT474 cells were maintained in the presence of 1 μM lapatinib (LC Laboratories) but were cultured in the absence of lapatinib for 1 week before experiments in order to account for potential confounding effects of drug treatment. Cell lines tested negative for mycoplasma with MycoAlert Mycoplasma Detection Kit (Lonza).
METHOD DETAILS
Microarray analysis
Illumina Human HT-12 was used to interrogate gene expression from rBT474 and BT474 cell lines. Raw expression data from Illumina Imager was background corrected using detection p values, quantile normalized, and log-transformed. Genes with no detected spots across all samples as determined by presence/absence calling were removed. Illumina IDs that mapped to the same gene symbol were collapsed by selecting the probe with the highest standard deviation across all samples. Hierarchical clustering using Euclidean distance and complete linkage was performed on the normalized expression values after selecting the top 1000 genes with highest variance, scaling each gene via z-transformation, and averaging across replicate samples. The clustering analysis was implemented using the pheatmap R package. Differential expression analysis was performed using the limma R package with an intensity-trend for the prior variance.
Gene Ontology Enrichment Analysis
Gene Ontology (GO) enrichment analysis was conducted using the “GO biological process” gene sets of the Molecular Signatures Database (MSigDB) v6.2. The gene set interrogated was defined as genes upregulated in resistant BT474 cells as compared to sensitive parental cells at baseline (T0), with the following cutoffs: Fold Change > 1.5; p value < 0.01; False Discovery Rate (FDR) < 0.05. The top 20 GO terms are listed in order of decreasing statistical significance as defined by FDR. p value was calculated from the hypergeometric distribution and FDR represents hypergeometric p value after Benjamini-Hochberg correction for multiple hypothesis testing. Bold font and italicized rows denote GO terms related to lipid metabolism.
TMA samples
NCI Cancer Diagnosis Program (CDP) Stage II Breast Prognostic TMA (Case Sets 14–17) and NCI Cancer Diagnosis Program (CDP) Stage III Breast Prognostic TMA (Case Sets 18–19) were analyzed. Association between CD36 expression and tumor grades were analyzed by Univariate Logistic Regression and multivariable logistic regression.
Patients Samples
245 pre-treatment and 44 post-treatment RNA samples with matching clinical and survival data were taken from the NeoALTTO trial (Baselga et al., 2012; Fumagalli et al., 2017). Illumina paired-end sequencing libraries were successfully constructed and subjected to sequencing on the Illumina HiSeq 2500 system as described previously (Fumagalli et al., 2017). Read pairs were trimmed using Trimmomatic (Bolger et al., 2014). alignment was performed using STAR (Dobin et al., 2013), and gene expression abundances were estimated using RSEM (Li and Dewey, 2011). Differential expression between the 44 paired pre- and post-treatment samples was modeled using DESeq2 (Love et al., 2014) with patient ID included as a covariate to adjust for baseline characteristics shared between matched pre- and post-treatment samples.
TCGA analysis
Level 3 mRNA expression data from the Agilent 244k Custom Gene Expression G4502A-07–3 platform was downloaded from The Cancer Genome Atlas data portal. A Kaplan-Meier estimator was fitted to CD36 expression values across HER2-positive and -negative samples. Patient samples that exhibited values greater than the median CD36 expression were labeled as high CD36, and samples less than the median CD36 expression were labeled as low. p value was calculated using the log-rank test.
Quantitative RT-PCR
RNA was isolated using an SV Total RNA Isolation System kit (Promega). cDNA was synthesized using a qScript cDNA synthesis kit (Quanta). RT-qPCR was performed using either iTaq Universal SYBR® Green Supermix (Bio-Rad) or TaqMan™ Universal PCR Master Mix (Applied Biosciences) on a CFX96 Real-Time PCR Detection System (Bio-Rad). TaqMan™ probes were purchased from Applied Biosciences. Target gene mRNA levels were normalized to Cyclophilin A (PPIA) and were compared using the delta-delta Ct method. qPCR primers were purchased from IDT. Probes and primer sequences listed in Figure S3.
Lipid Staining
Cells were seeded onto coverslips (NeuVitro) in a 12-well plate at a density of 8×104 cells/well. Cells were washed in PBS and fixed with 10% formalin for 10 min. Cells were washed once with water and once with 60% isopropanol. Cells were dried for 5 min then stained with Oil Red O (Cayman Chemical) working solution for 15 min. Cells were washed with 60% isopropanol and stained with hematoxylin for 30 s. Coverslips were mounted in 70% glycerol and were imaged by brightfield microscopy. Cells were seeded onto coverslips at a density of 3 × 104 cells/well. Cells were washed with DPBS, fixed in 4% paraformaldehyde for 15 min, then stained with 1:1000 BODIPY 493/503 (Invitrogen) for 15 min. Cells were washed in PBS then stained with 1:15,000 DAPI. Coverslips were mounted with ProLong Gold Antifade Mountant (Invitrogen). Slides were imaged by confocal microscopy.
Western Blot
Cells were lysed in RIPA buffer supplemented with protease and phosphatase inhibitors (Roche). Lysates were cleared by centrifugation at 20,000 g for 20 min at 4°C and 30 μg (for cell lines) or 50 μg (for tumor tissue) protein was loaded per lane on 8% polyacrylamide gels. Proteins were transferred onto PVDF membranes (Millipore) and were blocked in 3% BSA for 30 min at room temperature. Membranes were incubated with primary antibodies overnight at 4°C with gentle agitation. Membranes were developed using Pierce™ ECL (Thermo Scientific) and X-ray film.
Flow Cytometry
Cells were seeded at a density of 3 × 105 cells per well of a 6-well plate. Cells were harvested by trypsinization and washed in PBS. Cells were blocked with Human TruStain FcX (BioLegend; Cat# 422301) for 20 min and were then stained with anti-CD36-FITC antibody and analyzed on a MACSQuant (Miltenyi Biotec) flow cytometer.
BODIPY-C16 uptake assay
Uptake assays were performed as described previously (Li et al., 2005). Cells were seeded at a density of 3 × 105 cells per well of a 6-well plate. Cells were cultured in serum free media supplemented with 2 μM BODIPY FL C16 (Invitrogen; Cat# D3821) for the indicated times. Cells were harvested by trypsinization and analyzed by flow cytometry. Median fluorescence intensity was compared between samples at various time points.
Lipogenesis Assay
Cells were seeded at a density of 3 × 105 cells per well of a 6-well plate. Cells were pre-treated with 50 μM (−)-trans-C75 (Cayman Chemical), 1 μM lapatinib, or DMSO for 2 h prior to a 6 h incubation with 0.667 μCi/mL [1-14C]-acetic acid (PerkinElmer; Cat# NEC084H001MC). Cells were harvested by trypsinization and lipids were extracted as previously described (Olsen et al., 2010). 14C incorporation was measured was measured by scintillation counting (3 replicates per sample for n = 3 experiments). Average disintegrations per min (DPM) was normalized to protein concentration.
Viability Assays
Cell viability was assessed after 48-hour treatment using CellTiter 96 Aqueous One Solution Cell Proliferation Assay (Promega; Cat# G3582). The absorbance at 490 nm (OD 490 nm) was normalized against DMSO treated controls. Annexin V (Alexa Fluor® 647 conjugate; Invitrogen; Cat# A23204) staining was performed according to manufacturer’s instructions. Live cell imaging was performed using an IncucyteZoom (Essen BioScience) and a Cytation5 (BioTek) multimodal plate reader. 1×104 cells were seeded into wells of a 96-well plate. Cells were imaged (3 wells per treatment for n = 3 experiments) prior to and 48 h after treatment with sulfosuccinimidyl oleate (SSO; Cayman Chemical; Cat# 11211). Percent confluency was calculated at each time point for each well. Proliferation is presented as the average fold change in percent confluency of replicate wells for each treatment.
Seahorse Metabolic Assays
Rates of net oxygen consumption (OCR) and net extracellular acidification (ECAR) were determined concurrently using a Seahorse XF24–3 metabolic flux analyzer and standard Seahorse XF Mito Stress Test and Seahorse XF Glycolysis Stress Test kits according to the manufacturer’s instructions (Agilent Technologies, Santa Clara, CA, USA). Cells were routinely assayed in unbuffered bicarbonate-, phenol red-, and serum-free Dulbecco’s Modification of Eagle’s Medium (DMEM) containing 2 mM glutamine in the presence or absence of glucose (5.6 mM) and/or pyruvate (1 mM). Complete test media containing DMEM = 5.6 mM glucose, 2 mM glutamine, 1 mM pyruvate, and 0 mM lactate – or dropout media lacking various combinations of the principal energy substrates (i.e., glucose, pyruvate, and glutamine) – were prepared fresh before use (Corning Mediatech, Manassas, VA, USA).
Glucose Utilization and Lactate Production Assays
Glucose utilization was assayed as the net disappearance of glucose from the culture medium, and lactate production was assayed in parallel as the net accumulation of lactate in the same samples as described previously (Robey et al., 2000). Cells were routinely tested in defined basal DMEM containing 5.6 mM glucose and lacking phenol red which interferes with the colorimetric coupled enzymatic assays used to measure both glucose and lactate concentrations. All experiments were performed in the presence of non-limiting physiological concentrations of glucose and under conditions of linear net glucose disappearance and lactate accumulation. All measured of medium glucose and lactate content were also normalized for cell protein content to control for differences in cell number.
Hexokinase Assays
Total hexokinase activity was measured astheglucose-phosphorylating capacity of fresh whole cell lysates using a standard glucose 6-phosphate dehydrogenase-coupled assay as described previously (Robey et al., 2000). All data were expressed as specific hexokinase activity in units per g of total cellular protein where 1 unit is defined as the amount of enzyme activity resulting in the coupled formation of 1 mol NADPH per min at 25°C.
Transfection
Transient siRNA transfections were performed with Lipofectamine RNAiMAX (Invitrogen; Cat# 13778150) according to the manufacturer’s instructions. Cells were harvested for western blot or Annexin V staining 72 h after transfection.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
Two-tailed Student’s t test was used to determine statistical significance between groups. Kaplan-Meier survival curves and log rank tests were conducted using GraphPad Prism v7.04. Xenograft growth curve analysis was conducted using the “statmod” R package with significance set at *p < 0.05 after Benjamini-Hochberg correction for multiple comparison. Analyses were run in R version 3.4.2. Further statistical details of experiments can be found in the figure legends.
DATA AND CODE AVAILABILITY
The accession number for the microarray data reported in this paper is GEO: GSE136304. RNA-seq data from NeoALLTO patient tumors was previously published (Fumagalli et al., 2017). The RNA-seq dataset and the Clinical Data from the NeoALTTO trial have not been deposited in a public repository because of contractual and legal constraints. The RNASeq data and the Clinical Data can be requested by directly contacting altto@bigagainstbc.org.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE Antibodies | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-FASN | BD Biosciences | Cat# 610962; RRID:AB_398275 |
| Rabbit monoclonal anti-CPT1A | Cell Signaling Technology | Cat#12252 |
| Goat polyclonal anti-CD36 | R&D Systems | Cat# AF1955; RRID:AB_355073 |
| Rabbit polyclonal anti-CD36 | Novus Biologicals | Cat#NB400–144; RRID:AB_10003498 |
| Rabbit polyclonal anti-β-Actin | Santa Cruz Biotechnology | Cat# SC1616-R; RRID:AB_630836 |
| Rabbit monoclonal anti-HER2 | Cell Signaling Technology | Cat# 2165; RRID:AB_560966 |
| Rabbit polyclonal anti-p-HER2 (Tyr1221/1222) | Cell Signaling Technology | Cat# 2249S; RRID:AB_2099241 |
| Rabbit monoclonal anti-pAKT (Thr308) | Cell Signaling Technology | Cat# 2965; RRID:AB_2255933 |
| Rabbit polyclonal anti-AKT | Cell Signaling Technology | Cat# 9272; RRID:AB_329827 |
| Rabbit monoclonal anti-pERK1/2 (Thr202/Tyr204) | Cell Signaling Technology | Cat# 4370; RRID:AB_2315112 |
| Rabbit monoclonal anti-ERK | Cell Signaling Technology | Cat#4695; RRID:AB_390779 |
| Mouse monoclonal anti-CD36, FITC | eBioscience | Cat# 11–0369-41; RRID:AB_10719431 |
| Mouse monoclonal anti-CD36 (Clone JC63.1; Function Blocking) | Cayman Chemical | Cat# 188150; RRID:AB_10077812 |
| Mouse monoclonal mouse IgA isotype control | Abcam | Cat# ab37322 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Oil Red O | Cayman Chemical | Cat# 14419 |
| BODIPY 493/503 (4,4-Difluoro-1,3,5,7,8-Pentamethyl-4-Bora-3a,4a- Diaza-s-Indacene) | Invitrogen | Cat# D3922 |
| BODIPY FL C16 (4,4-Difluoro-5,7-Dimethyl-4-Bora-3a,4a-Diaza-s- Indacene-3-Hexadecanoic Acid) | Invitrogen | Cat# D3821 |
| [1-14C]-Acetic Acid, sodium salt | PerkinElmer | Cat# NEC084H001MC |
| Annexin V Alexa Fluor® 647 conjugate | Invitrogen | Cat# A23204 |
| (−)-trans-C75 | Cayman Chemical | Cat# 9000784 |
| Sulfosuccinimidyl Oleate (SSO) | Cayman Chemical | Cat# 11211 |
| Etomoxir (sodium salt) | Cayman Chemical | Cat# 11969 |
| Lapatinib | LC Laboratories | Cat# 388082–77-7 |
| SV Total RNA Isolation System | Promega | Cat# Z3105 |
| qScript cDNA synthesis kit | Quanta | Cat# 95047–100 |
| iTaq Universal SYBR® Green Supermix | Bio-Rad | Cat# 1725124 |
| Lipofectamine RNAiMAX Transfection Reagent | Invitrogen | Cat# 13778150 |
| Critical Commercial Assays | ||
| HumanHT-12 v4 BeadChip | Illumina | Cat# BD-103–0204 |
| CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS) | Promega | Cat# G3582 |
| Seahorse XF Mito Stress Test Kit | Agilent | Cat# 103015–100 |
| Seahorse XF Glycolysis Stress Test Kit | Agilent | Cat# 103020–100 |
| Matrigel Matrix | Corning | Cat# 354234 |
| Deposited Data | ||
| Microarray data | This paper | GSE136304 |
| RNaseq data | Fumagalli et al., 2017 | N/A |
| Experimental Models: Cell Lines | ||
| Human: BT474 | ATCC | RRID:CVCL_0179 |
| Human: rBT474 | Kurokawa et al., 2013 | N/A |
| Human: SKBR3 | ATCC | RRID:CVCL_0033 |
| Human: rSKBR3 | Kurokawa et al., 2013 | N/A |
| Human HCC202 | ATCC | RRID:CVCL_2062 |
| Human HCC202R | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: FVB-Tg(MMTV-Erbb2)NK1Mul/J | The Jackson Laboratory | RRID:IMSR_JAX:005038 |
| Mouse: Tg(MMTV-cre)4Mam/J | The Jackson Laboratory | RRID:IMSR_JAX:003553 |
| Mouse: CD36F/F | Maria Febbraio | N/A |
| Mouse: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ | The Jackson Laboratory | RRID:IMSR_JAX:005557 |
| Oligonucleotides | ||
| CD36 Human siRNA Oligo Duplex | Origene | Cat# SR319610 |
| Trilencer-27 Universal scrambled negative control siRNA duplex | Origene | Cat# SR30004 |
| qPCR Primer Sequences | This Paper | Table S3 |
| Software and Algorithms | ||
| GraphPad Prism v7 | GraphPad | https://www.graphpad.com/ |
| R: A Language and Environment for Statistical Computing | R Foundation for Statistical Computing | https://www.r-project.org/ |
| Molecular Signatures Database | Broad Institute | http://software.broadinstitute.org/gsea/msigdb/index.jsp |
Highlights.
Lapatinib-resistant breast cancer cells upregulate CD36 expression for survival
Targeting CD36 re-sensitizes lapatinib-resistant cells to HER2-inhibition
Deletion of Cd36 significantly attenuates MMTV-neu-driven mammary tumor growth
CD36 is induced by HER2-targeted therapy and predicts poor clinical prognosis
ACKNOWLEDGMENTS
We would like to thank members of the Dartmouth Genomics Shared Resource (Craig Tomlinson, Joanna Hamilton, and Carol Ringelberg) for performing the microarray experiment. We would also like to thank Reem Chamseddine, Wilson “Jay” Davis, Leslie Lupien, and Nancy Kuemmerle for valuable discussions. We would additionally like to thank the sponsors of the NeoALTTO trial, Novartis in collaboration with the Breast International Group (BIG), the Spanish Breast Cancer Cooperative Group SOLTI, and the BrEAST Data Center, which was responsible for data management. This study was supported by National Institutes of Health (NIH) research project grant R01 CA58961 (to W.B.K.); NCI career development award R00 CA140948; NIH R03 CA208384; a Mary Kay Foundation research grant (to M.K.); NIH Centers of Biomedical Research Excellence (COBRE) grant GM103534 (to C.C.); Cancer Center core grant P30 CA23108 (to the Norris Cotton Cancer Center); Hitchcock Foundation pilot grants (to M.K. and R.B.R.); and a Rosalin Borison Memorial pre-doctoral fellowship (to M.U.). This research was also supported by The Dartmouth Clinical and Translational Science Institute under award number KL2TR001088 from the National Center for Advancing Translational Sciences (NCATS) of the NIH (to C.C.). C.S. is funded by the Fonds de la Recherche Scientifique (FNRS) and the Breast Cancer Research Foundation (BCRF).
DECLARATION OF INTERESTS
L.P. reports personal fees and a research grant from AstraZeneca to conduct a clinical trial and personal fees and a research grant from Merck, Seattle Genetics, Astra Zeneca, Novartis, Genentech/Roche, Pieris, Eisai, Almac, Syndax, Immunomedics, Athenex, Biotheranostics, Celgene, and Boehringer Ingelheim outside of the submitted work. L.P. is also involved in collaborative research with Nanostring, Biotheranostics, and Foundation Medicine (not involving any financial transactions). A provisional patent application related to this work was submitted (provisional patent application number 62/ 804,440).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.11.008.
REFERENCES
- Alli PM, Pinn ML, Jaffee EM, McFadden JM, and Kuhajda FP (2005). Fatty acid synthase inhibitors are chemopreventive for mammary cancer in neu-N transgenic mice. Oncogene 24, 39–46. [DOI] [PubMed] [Google Scholar]
- Balaban S, Shearer RF, Lee LS, van Geldermalsen M, Schreuder M, Shtein HC, Cairns R, Thomas KC, Fazakerley DJ, Grewal T, et al. (2017). Adipocyte lipolysis links obesity to breast cancer growth: adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab. 5, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baselga J, Bradbury I, Eidtmann H, Di Cosimo S, de Azambuja E, Aura C, Gómez H, Dinh P, Fauria K, Van Dooren V, et al. ; NeoALTTO Study Team (2012). Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): a randomised, open-label, multicentre, phase 3 trial. Lancet 379, 633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensaad K, Favaro E, Lewis CA, Peck B, Lord S, Collins JM, Pinnick KE, Wigfield S, Buffa FM, Li JL, et al. (2014). Fatty acid uptake and lipid storage induced by HIF-1α contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 9, 349–365. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchakjian MR, and Kornbluth S (2010). The engine driving the ship: metabolic steering of cell proliferation and death. Nat. Rev. Mol. Cell Biol 11, 715–727. [DOI] [PubMed] [Google Scholar]
- Cabodevilla AG, Sánchez-Caballero L, Nintou E, Boiadjieva VG, Picatoste F, Gubern A, and Claro E (2013). Cell survival during complete nutrient deprivation depends on lipid droplet-fueled β-oxidation of fatty acids. J. Biol. Chem 288, 27777–27788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canfield K, Li J, Wilkins OM, Morrison MM, Ung M, Wells W, Williams CR, Liby KT, Vullhorst D, Buonanno A, et al. (2015). Receptor tyrosine kinase ERBB4 mediates acquired resistance to ERBB2 inhibitors in breast cancer cells. Cell Cycle 14, 648–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deblois G, Smith HW, Tam IS, Gravel SP, Caron M, Savage P, Labbé DP, Bégin LR, Tremblay ML, Park M, et al. (2016). ERRα mediates metabolic adaptations driving lapatinib resistance in breast cancer. Nat. Commun 7, 12156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enciu AM, Radu E, Popescu ID, Hinescu ME, and Ceafalan LC (2018). Targeting CD36 as Biomarker for Metastasis Prognostic: How Far from Translation into Clinical Practice? BioMed Res. Int 2(018, 7801202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernald K, and Kurokawa M (2013). Evading apoptosis in cancer. Trends Cell Biol. 23, 620–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli D, Venet D, Ignatiadis M, Azim HA Jr., Maetens M, Rothé F, Salgado R, Bradbury I, Pusztai L, Harbeck N, et al. (2017). RNA sequencing to predict response to neoadjuvant anti-HER2 therapy: a secondary analysis of the NeoALTTO randomized clinical trial. JAMA Oncol. 3, 227–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett JT, Olivares MG, Rinehart C, Granja-Ingram ND, Sánchez V, Chakrabarty A, Dave B, Cook RS, Pao W, McKinely E, et al. (2011). Transcriptional and posttranslational up-regulation of HER3 (ErbB3) compensates for inhibition of the HER2 tyrosine kinase. Proc. Natl. Acad. Sci. USA 108, 5021–5026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg IJ, Eckel RH, and Abumrad NA (2009). Regulation of fatty acid uptake into tissues: lipoprotein lipase- and CD36-mediated pathways. J. Lipid Res 50 (Suppl), S86–S90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Holohan C, Van Schaeybroeck S, Longley DB, and Johnston PG (2013). Cancer drug resistance: an evolving paradigm. Nat. Rev. Cancer 13, 714–726. [DOI] [PubMed] [Google Scholar]
- Jin Q, Yuan LX, Boulbes D, Baek JM, Wang YN, Gomez-Cabello D, Hawke DH, Yeung SC, Lee MH, Hortobagyi GN, et al. (2010). Fatty acid synthase phosphorylation: a novel therapeutic target in HER2-overex-pressing breast cancer cells. Breast Cancer Res. 12, R96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jump DB (2004). Fatty acid regulation of gene transcription. Crit. Rev. Clin. Lab. Sci 41, 41–78. [DOI] [PubMed] [Google Scholar]
- Kamphorst JJ, Cross JR, Fan J, de Stanchina E, Mathew R, White EP, Thompson CB, and Rabinowitz JD (2013). Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc. Natl. Acad. Sci. USA 110, 8882–8887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman B, Trudeau M, Awada A, Blackwell K, Bachelot T, Salazar V, DeSilvio M, Westlund R, Zaks T, Spector N, and Johnston S (2009). Lapatinib monotherapy in patients with HER2-overexpressing relapsed or refractory inflammatory breast cancer: final results and survival of the expanded HER2+ cohort in EGF103009, a phase II study. Lancet Oncol. 10, 581–588. [DOI] [PubMed] [Google Scholar]
- Kinlaw WB, Baures PW, Lupien LE, Davis WL, and Kuemmerle NB (2016). Fatty Acids and Breast Cancer: Make Them on Site or Have Them Delivered. J. Cell. Physiol 231, 2128–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komurov K, Tseng JT, Muller M, Seviour EG, Moss TJ, Yang L, Nagrath D, and Ram PT (2012). The glucose-deprivation network counteracts lapatinib-induced toxicity in resistant ErbB2-positive breast cancer cells. Mol. Syst. Biol 8, 596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuemmerle NB, Rysman E, Lombardo PS, Flanagan AJ, Lipe BC, Wells WA, Pettus JR, Froehlich HM, Memoli VA, Morganelli PM, et al. (2011). Lipoprotein lipase links dietary fat to solid tumor cell proliferation. Mol. Cancer Ther 10, 427–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar-Sinha C, Ignatoski KW, Lippman ME, Ethier SP, and Chinnaiyan AM (2003). Transcriptome analysis of HER2 reveals a molecular connection to fatty acid synthesis. Cancer Res. 63, 132–139. [PubMed] [Google Scholar]
- Kurokawa M, Kim J, Geradts J, Matsuura K, Liu L, Ran X, Xia W, Ribar TJ, Henao R, Dewhirst MW, et al. (2013). A network of substrates of the E3 ubiquitin ligases MDM2 and HUWE1 control apoptosis independently of p53. Sci. Signal 6, ra32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladanyi A, Mukherjee A, Kenny HA, Johnson A, Mitra AK, Sundaresan S, Nieman KM, Pascual G, Benitah SA, Montag A, et al. (2018). Adipocyte-induced CD36 expression drives ovarian cancer progression and metastasis. Oncogene 37, 2285–2301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lánczky A, Nagy Á, Bottai G, Munkácsy G, Szabó A, Santarpia L, and Győrffy B (2016). miRpower: a web-tool to validate survival-associated miRNAs utilizing expression data from 2178 breast cancer patients. Breast Cancer Res. Treat 160, 439–46. [DOI] [PubMed] [Google Scholar]
- Li H, Black PN, and DiRusso CC (2005). A live-cell high-throughput screening assay for identification of fatty acid uptake inhibitors. Anal Biochem. 336, 11–19. [DOI] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonnell E, Crown SB, Fox DB, Kitir B, Ilkayeva OR, Olsen CA, Grimsrud PA, and Hirschey MD (2016). Lipids reprogram metabolism to become a major carbon source for histone acetylation. Cell Rep. 17, 1463–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez JA, and Lupu R (2007). Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 7, 763–777. [DOI] [PubMed] [Google Scholar]
- Nagendran J, Pulinilkunnil T, Kienesberger PC, Sung MM, Fung D, Febbraio M, and Dyck JR (2013). Cardiomyocyte-specific ablation of CD36 improves post-ischemic functional recovery. J. Mol. Cell. Cardiol 63, 180–188. [DOI] [PubMed] [Google Scholar]
- Nahta R, Yu D, Hung MC, Hortobagyi GN, and Esteva FJ (2006). Mechanisms of disease: understanding resistance to HER2-targeted therapy in human breast cancer. Nat. Clin. Pract. Oncol 3, 269–280. [DOI] [PubMed] [Google Scholar]
- Nieman KM, Kenny HA, Penicka CV, Ladanyi A, Buell-Gutbrod R, Zillhardt MR, Romero IL, Carey MS, Mills GB, Hotamisligil GS, et al. (2011). Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med 17, 1498–1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen AM, Eisenberg BL, Kuemmerle NB, Flanagan AJ, Morganelli PM, Lombardo PS, Swinnen JV, and Kinlaw WB (2010). Fatty acid synthesis is a therapeutic target in human liposarcoma. Int. J. Oncol 36, 1309–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ookhtens M, Kannan R, Lyon I, and Baker N (1984). Liver and adipose tissue contributions to newly formed fatty acids in an ascites tumor. Am. J. Physiol 247, R146–R153. [DOI] [PubMed] [Google Scholar]
- Pascual G, Avgustinova A, Mejetta S, Martín M, Castellanos A, Attolini CS, Berenguer A, Prats N, Toll A, Hueto JA, et al. (2017). Targeting metastasis-initiating cells through the fatty acid receptor CD36. Nature 541, 41–45. [DOI] [PubMed] [Google Scholar]
- Robey RB (2018). Metabolic dysregulation in environmental carcinogenesis and toxicity In Translational Toxicology and Therapeutics: Windows of Developmental Susceptibility in Reproduction and Cancer, Waters M and Hughes C, eds. (John Wiley & Sons; ), pp. 511–606. [Google Scholar]
- Robey RB, Raval BJ, Ma J, and Santos AV (2000). Thrombin is a novel regulator of hexokinase activity in mesangial cells. Kidney Int. 57, 2308–2318. [DOI] [PubMed] [Google Scholar]
- Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, and McGuire WL (1987). Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science 235, 177–182. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinnen JV, Van Veldhoven PP, Timmermans L, De Schrijver E, Brusselmans K, Vanderhoydonc F, Van de Sande T, Heemers H, Heyns W, and Verhoeven G (2003). Fatty acid synthase drives the synthesis of phospholipids partitioning into detergent-resistant membrane microdomains. Bio-chem. Biophys. Res. Commun 302, 898–903. [DOI] [PubMed] [Google Scholar]
- Vazquez-Martin A, Colomer R, Brunet J, and Menendez JA (2007). Pharmacological blockade of fatty acid synthase (FASN) reverses acquired autoresistance to trastuzumab (Herceptin by transcriptionally inhibiting ‘HER2 super-expression’ occurring in high-dose trastuzumab-conditioned SKBR3/Tzb100 breast cancer cells. Int. J. Oncol 31, 769–776. [PubMed] [Google Scholar]
- Ventura R, Mordec K, Waszczuk J, Wang Z, Lai J, Fridlib M, Buckley D, Kemble G, and Heuer TS (2015). Inhibition of de novo palmitate synthesis by fatty acid synthase induces apoptosis in tumor cells by remodeling cell membranes, inhibiting signaling pathways, and reprogramming gene expression. EBioMedicine 2, 808–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O (1956). On the origin of cancer cells. Science 123, 309–314. [DOI] [PubMed] [Google Scholar]
- Wilken JA, and Maihle NJ (2010). Primary trastuzumab resistance: new tricks for an old drug. Ann. N Y Acad. Sci 1210, 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C, Ko B, Hensley CT, Jiang L, Wasti AT, Kim J, Sudderth J, Calvaruso MA, Lumata L, Mitsche M, et al. (2014). Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol. Cell 56, 414–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon S, Lee MY, Park SW, Moon JS, Koh YK, Ahn YH, Park BW, and Kim KS (2007). Up-regulation of acetyl-CoA carboxylase α and fatty acid synthase by human epidermal growth factor receptor 2 at the translational level in breast cancer cells. J. Biol. Chem 282, 26122–26131. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the microarray data reported in this paper is GEO: GSE136304. RNA-seq data from NeoALLTO patient tumors was previously published (Fumagalli et al., 2017). The RNA-seq dataset and the Clinical Data from the NeoALTTO trial have not been deposited in a public repository because of contractual and legal constraints. The RNASeq data and the Clinical Data can be requested by directly contacting altto@bigagainstbc.org.