

Abstract
Processing of amyloid precursor protein (APP) by the β-secretase BACE1 is the initial step of the amyloidogenic pathway to generate amyloid-β (Aβ). Although newly synthesized BACE1 and APP are transported along the secretory pathway, it is not known whether BACE1 and APP share the same post-Golgi trafficking pathways or are partitioned into different transport routes. Here we demonstrate that BACE1 exits the Golgi in HeLa cells and primary neurons by a pathway distinct from the trafficking pathway for APP. By using the Retention Using Selective Hooks system, we show that BACE1 is transported from the trans-Golgi network to the plasma membrane in an AP-1- and Arf1/4-dependent manner. Subsequently, BACE1 is endocytosed to early and recycling endosomes. Perturbation of BACE1 post-Golgi trafficking results in an increase in BACE1 cleavage of APP and increased production of both Aβ40 and Aβ42. These findings reveal that Golgi exit of BACE1 and APP in primary neurons is tightly regulated, resulting in their segregation along different transport routes, which limits APP processing.
INTRODUCTION
BACE1 is the primary β-secretase for initiating the β-site cleavage of amyloid precursor protein (APP) to generate the membrane-bound C99 fragment (Hussain et al., 1999; Sinha et al., 1999; Yan et al., 1999; Bennett et al., 2000). The subsequent cleavage of C99 by γ-secretase leads to the release of the pathogenic amyloidogenic beta-amyloid peptide (Aβ) associated with Alzheimer’s disease (Qi-Takahara et al., 2005; Takami et al., 2009; Tomita, 2014). BACE1 cleavage of APP is the rate-limiting step along the amyloidogenic processing pathway (Thinakaran and Koo, 2008; Zhang et al., 2011) and, as both BACE1 and APP are membrane proteins, APP processing requires the convergence of these proteins to the same compartment (Kinoshita et al., 2003). Hence, the intracellular trafficking and localization of BACE1 are coordinated with the processing of APP and biogenesis of Aβ peptides.
As a type-1 transmembrane aspartyl protease, BACE1 activity is optimal at acidic pH (Shimizu et al., 2008), which is consistent with highest BACE1 activity being detected in acidic compartments along the secretory and endocytic pathways including the endosomes and trans-Golgi (Bennett et al., 2000; Huse et al., 2000, 2002; Toh et al., 2017, 2018). Modification to the trafficking and subcellular localization of BACE1 within these compartments is likely to impact upon APP processing. Indeed, previous work from our laboratory has shown that redirecting the BACE1 plasma membrane recycling pathway via the trans-Golgi network (TGN), using a chimeric BACE1/TGN38 construct (TGN38 is known to traffic from the endosomes to the TGN), leads to increased Aβ production (Chia et al., 2013). Moreover, an extended residency time of BACE1 at the early endosomes also leads to an increase in Aβ production (Toh et al., 2018). These studies highlight the importance of the TGN and early endosomes in APP processing and indicate that both the itinerary and kinetics of transport are relevant for regulating amyloidogenic processing. The intracellular itinerary of internalized BACE1 to the early endosomes followed by the recycling of BACE1 back to the plasma membrane (PM) via the recycling endosomes has been established previously (Chia et al., 2013; Toh et al., 2018). However, the post-Golgi trafficking of newly synthesized BACE1 is poorly defined, yet is likely to play a critical role in initiating the production of Aβ peptides in the TGN. The TGN is a major sorting compartment of the cell and is responsible for transport of newly synthesized membrane proteins to a variety of different destinations (De Matteis and Luini, 2008); hence the transport pathway of BACE1 from the Golgi is defined by the sorting events in the TGN.
We have previously shown that newly synthesized APP traffics directly from the TGN to the early endosomes (Toh et al., 2017), a transport step regulated by the adaptor AP-4 and the small G protein Arl5b (Burgos et al., 2010; Toh et al., 2017). The depletion of either Arl5b or AP-4 leads to the accumulation of APP, but not BACE1, in the TGN. Arl5b/AP-4 depletion also resulted in an increase in APP processing by endogenous BACE1, highlighting the importance of the TGN as a site for APP processing. As the post-Golgi export of BACE1 is independent of the Arl5b/AP-4 sorting pathway, the question arises of the identity of the transport pathway(s) used by BACE1. The use of different pathways by BACE1 and APP (Toh et al., 2017) could provide a mechanism to segregate BACE1 and APP within the TGN and to limit Aβ biogenesis.
Here we seek to define the post-Golgi export pathway of BACE1 and identify the sorting machinery involved. We have demonstrated that newly synthesized BACE1 traffics from the TGN directly to the cell surface. The adaptor AP-1 is not usually considered relevant to transport from the TGN to the cell surface. However, we considered this possibility, as we identified a previously uncharacterized acidic cluster–sorting signal in the cytoplasmic tail of BACE1. Moreover, the μ1 subunit of adaptor protein complex AP-1 was recently discovered to recognize an acidic cluster motif in the cytoplasmic tail of the model membrane cargo, CD8-furin (Navarro Negredo et al., 2017). As AP-1 is localized at the TGN, we therefore investigated whether AP-1 may regulate the post-Golgi trafficking of BACE1. We have demonstrated that AP-1 and the Arf small G proteins Arf1 and Arf4 are required for efficient Golgi export of BACE1 in both HeLa cells and primary neurons. Our findings also demonstrate that the segregation of BACE1 and APP into distinct pathways at the TGN is relevant to the regulation of APP processing and Aβ production.
RESULTS
Trafficking itinerary of newly synthesized BACE1 from the TGN
It is currently not known whether newly synthesized BACE1 shares the same post-Golgi trafficking route as APP or is segregated into a different transport pathway. The regulation of post-Golgi transport of BACE1 is likely to play a critical role in initiating the processing of APP and Aβ biogenesis. To analyze the anterograde transport of newly synthesized BACE1 from the Golgi complex, we utilized the Retention Using Selective Hooks (RUSH) system (Boncompain et al., 2012; Boncompain and Perez, 2012). The bicistronic BACE1 RUSH plasmid contains BACE1 fused to the streptavidin-binding peptide (SBP) and a GFP tag and an ER-hook protein fused to streptavidin (Figure 1A). Transiently expressed BACE1-SBP-GFP is retained in the endoplasmic reticulum (ER) compartment via interaction of its SBP with the streptavidin-fused ER-hook protein. Upon the addition of biotin, BACE1-SBP-GFP is released from the hook, thus allowing ER exit of newly synthesized BACE1-SBP-GFP (Figure 1A). The intracellular itinerary of newly synthesized BACE1-SBP-GFP in intracellular compartments was tracked in HeLa cells by fixing the cells at various time points after the addition of biotin and stained with different organelle-specific markers (Figure 1, A–C).
FIGURE 1:

Anterograde trafficking of BACE1. (A) Schematic outlining the experimental plan to investigate the post-Golgi trafficking of BACE1 using the RUSH system. BACE1-SBP-GFP is retained in the ER compartment via interaction of its SBP with streptavidin fused ER hook protein. BACE1-SBP-GFP is released from the ER upon biotin addition to allow trafficking of BACE1-SBP-GFP to its target compartment. (B) HeLa cells were transiently transfected with the BACE1-SBP-GFP and ER-hook bicistronic construct for 24 h. Transfected HeLa cells were treated with biotin (untreated represents the 0 min time point) and incubated at 37°C. Monolayers of HeLa cells were fixed at the indicated time points and then permeabilized and blocked, followed by staining with either mouse anti-EEA1 (red) and rabbit anti-GCC88 (far-red converted to red), mouse anti-Rab11 (red), or mouse anti-CD63 (red) antibodies and DAPI (blue). Inset represents magnified images. Bar represents 10 μm. (C) The percentage of BACE1-GFP at the TGN, early endosomes, recycling endosomes, and late endosomes was calculated as the percentage of total BACE1-GFP pixels that overlapped with GCC88, EEA1, Rab11, and CD63, respectively, using the OBCOL plug-in on ImageJ. The steady-state distribution of BACE1-SBP-GFP was determined from transfected HeLa cells incubated for 24 h in the continuous presence of biotin. Data are represented as the mean ± SD of three independent experiments (n = 15).
In the absence of biotin, BACE1-SBP-GFP displayed ER-like staining pattern (Figure 1B), which overlapped substantially with the ER-marker, KDEL (Supplemental Figure S1A), limited overlap (<5%) with other organelle markers (Figure 1, B and C), and no overlap with a plasma membrane marker (Supplemental Figure S1B), indicating that newly synthesized BACE1-SBP-GFP was successfully retained in the ER. After 10 min incubation with biotin, BACE1-SBP-GFP exhibited a juxtanuclear staining pattern with very little overlap with the ER marker KDEL (Supplemental Figure S1A) or endosomal markers (Figure 1C; Supplemental Figure S1C) and with 11.5 ± 3.5% of BACE1-SBP-GFP colocalized with the TGN marker GCC88 (Figure 1C; Supplemental Figure S1D), indicating arrival of the cargo at the Golgi from the ER. At 20 min, 43.8 ± 5.4% of BACE1-SBP-GFP overlapped with GCC88 and showed minimum overlap (<5%) with the endosomal/lysosomal markers EEA1, Rab11, and CD63 (Figure 1, B and C), indicating that a substantial percentage of BACE1-SBP-GFP had arrived at the TGN but not trafficked to other intracellular compartments.
At the 30-min time point, the level of BACE1-SBP-GFP that colocalized with the TGN marker had decreased to 21.7 ± 4.1%, whereas there was an increase in BACE1-SBP-GFP in the early endosome (16.7 ± 2.4%), marked by EEA1, and in the recycling endosome (11.7 ± 1.9%; Figure 1C; Supplemental Figure S1E). These data show that after 30 min of biotin addition, BACE1-SBP-GFP had begun to exit the TGN, and some cargo had arrived at the early and recycling endosomes. Given that BACE1 does not utilize the AP-4/Arl5b mediated TGN–to–early endosome trafficking pathway (Toh et al., 2017), it is unlikely that newly synthesized BACE1-SBP-GFP traffics from the TGN directly to the early endosome. Rather, the BACE1-SBP-GFP detected in the early endosomes at 30 min likely represents the arrival of internalized BACE1 from the PM. On the other hand, the BACE-SBP-GFP in the recycling endosomes at 30 min may represent either direct trafficking of BACE1 from the TGN to the recycling endosomes or, alternatively, arrival of recycled BACE1 from the early endosomes (Chia et al., 2013; Toh et al., 2018).
After 45- and 60-min time points, the colocalization of BACE1-SBP-GFP with the early endosome (EEA1) marker remained steady at ∼20%, while the level of BACE1-SBP-GFP in the recycling endosome increased to 25.9 ± 2.1% by 60 min (Figure 1, B and C; Supplemental Figure S1E). On the other hand, the level of BACE1-SBP-GFP in the TGN decreased to 15.1 ± 2.5% at 45 min and 11.7 ± 2.2% at 60 min (Figure 1, B and C; Supplemental Figure S1E). Thus, the majority of BACE1-SBP-GFP had exited the TGN by 45–60 min and was not recycled back to this compartment. Furthermore, the intracellular levels of BACE1-SBP-GFP in the early endosomes and recycling endosomes at the 60-min time point are very similar to the steady-state distribution of BACE1, as quantified from transfected cells incubated in the continuous presence of biotin (Figure 1C), indicating that the wave of BACE1 cargo from the ER had established an equilibrium throughout the cell by 60 min. There were low levels of BACE1-SBP-GFP (10.2 ± 2.3%) in CD63-positive late endosome/lysosome at both the 45- and 60-min time points (Figure 1, B and C; Supplemental Figure S1E), suggesting turnover of BACE1 in the lysosomes (Ye and Cai, 2014; Ye et al., 2017; Feng et al., 2017).
To directly analyze the arrival of newly synthesized BACE1-SBP-GFP at the PM, total internal reflection fluorescence microscopy (TIRF) was performed. BACE1-SBP-GFP at the PM was first detected at 22 min after biotin addition followed by a rapid increase in BACE1-SBP-GFP arriving at the PM (Figure 2A and Supplemental Movie S1). These data are consistent with kinetics of BACE1-SBP-GFP exiting the TGN between the 20- and 30-min time points (Figure 1, B and C; Supplemental Figure S1E). We then performed live-cell imaging with fast image acquisition to determine whether BACE1-SBP-GFP is delivered directly to the PM from the TGN or indirectly via recycling endosomes. Temporal projection of the BACE1-SBP-GFP signal shows a direct transport from a juxtanuclear compartment, likely to represent the Golgi compartment, directly to the distal edge of the cell after 20–30 min of biotin addition (Figure 2B and Supplemental Movie S2). Taken together, TIRF and fast-acquisition imaging data indicate that newly synthesized BACE1 traffics directly from the TGN to the PM.
FIGURE 2:
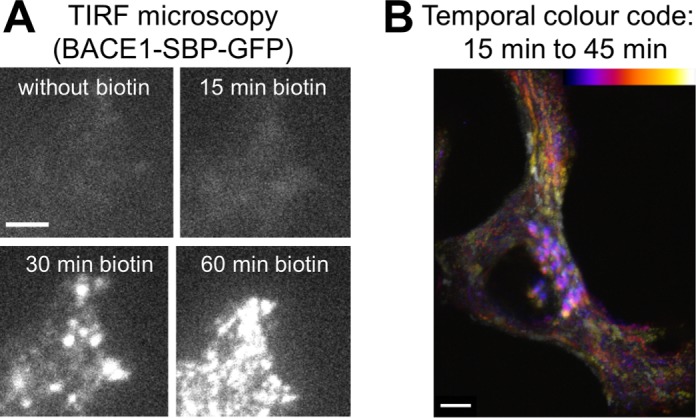
Post-Golgi trafficking of newly synthesized BACE1. HeLa cells were transiently transfected with the BACE1-SBP-GFP and ER-hook bicistronic construct for 24 h. (A) Real-time images were acquired using a TIRF microscope at the indicated time points. Biotin was added at time 0 min. Scale bar represents 5 μm. (B) Fast image acquisition of BACE1-SBP-GFP. Images were acquired every 2 s. Temporal projection was performed from 15 to 45 min using the tool “temporal-color-code” on the Fiji software. Bar represents 5 μm.
Movie S1.
TIRF microscopy of anterograde transport of BACE1 HeLa cells transfected with Str-Ii_BACE1-SBP-EGFP. Real time pictures were acquired using a TIRF microscope at the indicated time. Biotin was added at time 0. BACE1 arrival at the plasma membrane can be observed from approximately 22 min. Scale bar: 10μm.
Movie S2.
Fast image acquisition of anterograde transport of BACE1 HeLa cells transfected with Str-Ii_BACE1-SBP-EGFP. Real time pictures were acquired every 2 sec. Biotin was added at time 0. Scale bar: 5μm.
Steady-state distribution of BACE1 in HeLa cells and primary neurons
To identify the sorting machinery required for the post-Golgi trafficking of BACE1 to the PM, it was important to analyze homogeneous populations of cells expressing only modest levels of BACE1. As endogenous BACE1 in HeLa cells is very low and difficult to detect by immunofluorescence, a HeLa cell line stably expressing BACE1-GFP (HeLa-BACE1-GFP) was generated. A cell line (D5) expressing modest levels of BACE1-GFP was selected by cell sorting and cloning. HeLa D5 maintained BACE1-GFP expression over multiple passages and was used for the subsequent experiments.
We initially established the steady-state distribution of BACE1-GFP in HeLa-BACE1-GFP cells. BACE1-GFP was largely juxtanuclear, with punctate-like structures throughout the cytoplasm (Figure 3A). Dual staining showed an extensive overlap of BACE1-GFP with Rab11-positive recycling endosomes and only a low level of overlap with GCC88 (Figure 3A). Minimum overlap was detected between the Rab11 and GCC88 markers in HeLa cells (Supplemental Figure S2A), demonstrating that the majority of juxtanuclear BACE1-GFP was indeed localized to recycling endosomes. Quantitation of the steady-state distribution of BACE1-GFP revealed that 8.5 ± 1.8% localized with GCC88-marked TGN, 30.6 ± 3.6% localized with the Rab11-marked recycling endosomes, 27.4 ± 2.7% localized with the EEA1-marked early endosomes, and 9.7 ± 2.5% localized in the CD63-positive late endosomes/lysosomes (Figure 3, A and B). This distribution of BACE1-GFP is very similar to the steady-state distribution of transiently expressed untagged BACE1 in HeLa cells (Chia et al., 2013) and to the distribution of BACE1-SBP-GFP in the RUSH system in the continuous presence of biotin (Table 1).
FIGURE 3:

Steady-state distribution of BACE1 in HeLa cells and primary neurons. (A) HeLa cells stably expressing BACE1-GFP were fixed, permeabilized, and blocked, followed by staining with either rabbit anti-GCC88 (far-red converted to red/green) and mouse anti-Rab11 (red), mouse anti-EEA1 (red), or mouse anti-CD63 (red) antibodies and DAPI (blue). Insets and higher magnifications of the images of the cells marked * are also shown. Bars represents 10 μm. (B) The percentage of BACE1-GFP at the TGN, recycling endosomes, early endosomes, and late endosomes/lysosomes was calculated as the percentage of total BACE1-GFP pixels that overlapped with GCC88, Rab11, EEA1, and CD63, respectively, using the OBCOL plug-in on ImageJ. Data are represented as the mean ± SD of three independent experiments (n = 16). (C) Primary mouse cortical neurons at DIV7 were stained overnight with anti-rabbit BACE1 for endogenous BACE1 (green) and either human anti-p230/golgin-245 (red), rat anti-LAMP1 (red), mouse anti-Rab11 (red), or anti-Rab4 (red) antibodies at 4°C. Higher magnifications of the merged images are also shown. Bar represents 10 μm. (D) The percentage of endogenous BACE1 at the TGN, recycling endosomes, early endosomes, and late endosomes/lysosomes in neurons was calculated as the percentage of volume sum of BACE1 that overlapped with p230/golgin-245, Rab11, Rab4, and LAMP1, respectively, using Imaris. Data are represented as the mean ± SD of three independent experiments (n = 13–14).
TABLE 1:
Steady-state distribution of BACE1 in HeLa cells and primary mouse cortical neurons.
| HeLa cells (transient expression) | HeLa cells (stable expression) | Primary mouse cortical neurons | ||
|---|---|---|---|---|
| Organelles | Untagged-BACE1 ( Chia et al., 2013) | BACE1-SBP-GFP | BACE1-GFP | Endogenous BACE1 |
| trans-Golgi network | 4.4% | 8.5% | 5.6% | 5.6% |
| Recycling endosomes | 31.9% | 30.6% | 29.8% | 40.7% |
| Early endosomes | 31.8% | 27.4% | 21.0% | 22.4% |
| Late endosomes/lysosomes | 12.0% | 9.7% | 9.1% | 15.5% |
To determine whether the intracellular distribution of BACE1 observed in transfected HeLa cells was consistent with the distribution of BACE1 in primary neurons, we next determined the distribution of endogenous BACE1 in mouse primary cortical neurons. The TGN and recycling endosomes in primary neurons are located in close physical proximity and concentrated in the juxtanuclear position of the soma (Supplemental Figure S2B). To determine whether there is any overlap between these organelle markers, we treated neurons with nocodazole to disperse the Golgi and stained the treated cells with the TGN markers GCC88 and golgin245/p230 and the recycling endosome marker Rab11. The TGN and recycling endosome markers recognized distinct structures with only minimum overlap (Supplemental Figure S2C), demonstrating they were selective for each organelle. The majority of endogenous BACE1 colocalized with Rab11 and Rab4, and lower levels of endogenous BACE1 were detected in the late endosomes/lysosomes and Golgi (Figure 3, C and D). Although the percentages of BACE1 in each compartment show some differences between neurons and HeLa cells (Table 1), these data demonstrate that HeLa cell lines and the BACE1 RUSH system faithfully reflect the distribution of endogenous BACE1 in primary neurons.
AP-1 is required for the post-Golgi sorting of BACE1 in HeLa cells
Next, we sought to identify adaptor protein(s) that could regulate the post-Golgi transport of BACE1. In the cytoplasmic tail of BACE1, we came across a previously undetected acidic cluster sequence, DDFADDISLL, a motif presenting an extension of the previously defined DDISLL and DISLL motifs that play a role in endocytosis and endocytic sorting (He et al., 2002, 2005; Toh et al., 2018). This acidic cluster motif is similar to the acidic cluster sequences discovered in the cytosolic tail of cargoes such as furin (Voorhees et al., 1995), carboxypeptidase D (Eng et al., 1999), and the cation-independent mannose 6-phosphate receptor (Chen et al., 1997; Table 2). Given that AP-1 has been shown to be associated with the sorting of proteins with acidic cluster sequences (Navarro Negredo et al., 2017), we examined whether the depletion of AP-1 impacts the post-Golgi export of BACE1. Moreover, as Golgi-localized γ-ear–containing Arf binding proteins (GGAs) are also localized at the TGN and have been shown to play a cooperative/overlapping role with AP-1 (Doray et al., 2002; Hirst et al., 2012), the depletion of GGA1 was also performed. Immunoblotting showed that the levels of AP-1γ and GGA1 were depleted by >90% and >80%, respectively, in HeLa-BACE1-GFP treated with either AP-1γ or GGA1 small interfering RNA (siRNA) (Figure 4A). The depletion of AP-1γ resulted in an increase in the accumulation of BACE1-GFP in the juxtanuclear region that colocalizes extensively with the Golgi marker golgin-97, compared with control siRNA treatment (Figure 4, C and D). As we have used different TGN markers throughout this study, due to compatibility of the primary antibodies in dual- and triple-labeling experiments, we also analyzed the colocation of BACE1-GFP in control and AP-1γ siRNA-treated cells using antibodies to GCC88. Similar findings were observed with either TGN marker, golgin-97 or GCC88 (Supplemental Figure S3A), and moreover, we observed a high level of colocalization between golgin-97 and GCC88, as expected at the resolution used (Supplemental Figure S3B).
TABLE 2:
Acidic cluster motifs in the cytoplasmic tails of membrane cargoes.
| Cargoes | Acidic cluster motif | Reference |
|---|---|---|
| Furin | SDSEEDE | Voorhees et al., 1995 |
| Carboxypeptidase D | DETDTEEE | Eng et al., 1999 |
| Cation-independent mannose 6-phosphate receptor | DDQDSED and DDSDEDLL | Chen et al., 1997 |
| Dyslexia-associated protein KIAA0319-like protein | SESELDSDD | Navarro Negredo et al., 2017 |
| BACE1 | DDFADDISLL | — |
FIGURE 4:

Depletion of adaptor protein AP-1 results in BACE1 accumulation in the TGN. (A) HeLa cells stably expressing BACE1-GFP were transfected with either control siRNA, AP-1γ siRNA, GGA1 siRNA, AP-4ε siRNA, or Arl5b siRNA for 72 h. (B) HeLa cells stably expressing Arl5b(Q70L)-GFP were transfected with either control siRNA or Arl5b siRNA for 72 h. Cells in A and B were lysed in RIPA buffer, and 20 μg of cell extracts was subjected to SDS–PAGE on 4–12% gradient polyacrylamide gel. Proteins were transferred to a polyvinylidene fluoride (PVDF) membrane and probed with (A) mouse anti–AP-1γ, rabbit anti-GGA1, mouse anti–AP-4ε, (B) mouse anti-GFP, and (A and B) mouse anti–a-tubulin antibodies using a chemiluminescence detection system. (C) Monolayers of HeLa cells were fixed, permeabilized, and blocked, followed by staining with mouse anti–golgin-97 antibodies (red) and DAPI (blue). Higher magnification of the merged images of the cells marked * are also shown. Bar represents 10 μm. (D) The percentage of BACE1-GFP at the TGN was calculated as a percentage of total BACE1-GFP pixels that overlapped with golgin-97. Data are represented as the mean ± SD of three independent experiments (n = 15) and analyzed by one-way ANOVA using Tukey’s test. *** p < 0.001.
Quantitative analyses revealed a 3.0-fold increase in colocalization of BACE1-GFP with the TGN marker golgin-97 in AP-1γ–depleted cells compared with control siRNA-treated cells (Figure 4D). Conversely, the depletion of AP-1γ resulted in a decrease in the total cell pool of BACE1-GFP that overlapped with Rab11 (Figure 5), a reduction of magnitude similar to that of the increase found in the TGN in AP-1 knockdown cells.
FIGURE 5:
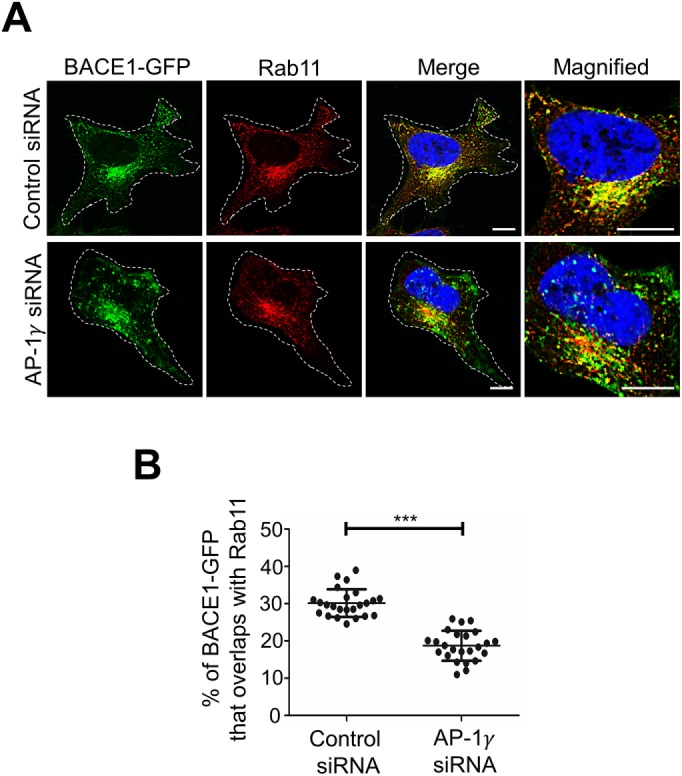
Depletion of AP-1 results in a reduction of BACE1 in the recycling endosomes. (A) HeLa cells stably expressing BACE1-GFP were transfected with either control siRNA or AP-1γ siRNA for 72 h. Cells were fixed and stained with mouse anti-Rab11 antibodies (red) and DAPI (blue). Higher magnifications of the merged images are also shown. Bar represents 10 μm. (B) Data are represented as the mean ± SD of three independent experiments (n = 24) and analyzed by an unpaired, two-tailed Student’s t test. *** p < 0.001.
On the other hand, the level of BACE1-GFP at the TGN was not altered in GGA1-depleted cells (Figure 4, C and D). We have previously proposed that the Golgi export of BACE1 is independent of the AP-4/Arl5b sorting machinery utilized by APP (Toh et al., 2017). As expected, the depletion of either AP-4 or Arl5b in HeLa-BACE1-GFP also did not alter the distribution of BACE1-GFP at the Golgi (Figure 4, A–C). Quantitative analyses showed no significant changes in GGA1-, AP-4ε-, and Arl5b-depleted cells (Figure 4D). AP-4 was depleted by >90% in HeLa-BACE1-GFP treated with AP-4ε siRNA (Figure 4A). As antibodies are not available to detect endogenous Arl5b, we tested the efficiency of Arl5b depletion in parallel in the same experiment by assessing the decrease in the level of GFP in HeLa cells stably expressing Arl5b(Q70L)-GFP (Figure 4B), a GFP fusion with the constitutive active form of Arl5b. Previous studies showed that silencing of GFP-Arl5b is also a readout for the silencing of endogenous Arl5b (Houghton et al., 2012). Collectively, these results revealed that AP-1 is specifically required for the export of BACE1 from the TGN and that BACE1 and APP have distinct post-Golgi sorting machineries.
Endogenous BACE1 accumulates at the Golgi in AP-1– but not AP-4–depleted primary neurons
To determine whether AP-1 is also required for the sorting of BACE1 from the TGN in a more relevant cell model, we assessed the impact of silencing AP-1 in primary mouse cortical neurons. Primary mouse cortical neurons were transduced with AP-1γ short hairpin RNA (shRNA) recombinant lentivirus to silence AP-1. Transduced neurons were detected by the expression of GFP. GFP-positive transduced neurons displayed reduced staining of AP-1γ as detected by confocal microscopy (Supplemental Figure S4). In addition, endogenous BACE1 staining displayed an increase in colocalization with the TGN marker p230/golgin-245, compared with nontransduced neurons (Figure 6A). Quantitative analyses of fluorescence intensities of 3D projections revealed an increase in colocalization of BACE1 with the TGN marker p230/golgin-245, from 10.4 ± 3.1% in nontransduced neurons to 25.2 ± 5.6% in AP-1–depleted neurons (Figure 6B). On the other hand, following transduction of primary neurons with AP-4ε shRNA recombinant lentivirus (Supplemental Figure S4B), there was no significant change in the colocalization of BACE1 with the TGN marker in AP-4–depleted neurons (Figure 6B). We also assessed whether the depletion of AP-1 in neurons impacted the export of APP from the TGN; depletion of AP-1γ in neurons did not alter the distribution of APP in the Golgi (Figure 6, C and D). AP-1 depletion specifically retains BACE1 but not APP in the TGN.
FIGURE 6:

Depletion of AP-1γ in mouse primary cortical neurons results in accumulation of BACE1 in the TGN. (A, C) Transduction of E16 primary mouse cortical neurons at DIV3 with either AP-1γ shRNA or AP-4ε shRNA recombinant lentivirus for 96 h. Neurons were fixed at DIV7 and stained with either (A) rabbit anti-BACE1 (far-red converted to green) or (C) rabbit anti-APP (Y188; red), and the TGN marker human anti-p230/golgin-245 (red/ far-red converted to green) antibodies overnight at 4°C. Nuclei was stained with DAPI (blue). Bar represents 10 μm. (B, D) Volocity software was used to calculate Manders’ coefficient M1 values of either BACE1 or APP colocalization with the TGN marker p230/golgin-245. Data are represented as the mean ± SD of three independent experiments (n = 14–15 individual cells) and analyzed by (B) one-way ANOVA using Tukey’s test and (D) unpaired, two-tailed Student’s t test. ***p < 0.001 and n.s. = not significant.
AP-1 depletion reduced the exit rate of BACE1 from the Golgi
To directly examine the impact of AP-1 depletion on the kinetics of anterograde trafficking of newly synthesized BACE1 in HeLa cells, we used the RUSH system described above. HeLa cells were transfected with AP-1γ siRNA and then transfected with BACE1-SBP-GFP. Silencing of AP-4ε was also included for comparison. Live imaging showed the arrival of BACE1-SBP-GFP within the juxtanuclear Golgi region within 15 min after biotin addition. Whereas the majority of BACE1-SBP-GFP had exited the juxtanuclear Golgi region in control and AP-4−depleted cells by 45 min, considerable BACE1-SBP-GFP fluorescence was still retained in the Golgi in AP-1–depleted cells at 45 min and also at 60 min (Figure 7, A and B).
Figure 7:

Delayed exit of RUSH-BACE1-GFP from the TGN by AP-1 depletion. (A, B) HeLa cells were transfected with either control siRNA, AP-1γ siRNA, or AP-4ε siRNA for 72 h. Cells were cotransfected with Str-Ii_BACE1-SBP-GFP after 48 h for a further 24 h before live imaging at 37°C. Biotin was added at 0 min and images at the indicated time points after biotin addition are shown in A. Bar represents 10 μm. (B) Representative curves of mean Golgi-region intensity change over time are shown. Images were taken every 1 min for a duration of 60 min. Mean intensity over the Golgi region of individual cells was measured over time after biotin addition using FIJI/ImageJ. The level of maximal Golgi-region intensity (C) and the corresponding time point (D) were recorded and quantified from the curves in B. (E) The rate of Golgi-region intensity drop was quantified from B. Linear regression of the curve starting from the maximal intensity point over the next 20 min was generated, and the slope was recorded as a measure of BACE1 Golgi exit rate. Data in C–E are presented as mean ± SD of three independent experiments (n = 18–23) and analyzed by one-way ANOVA using Tukey’s test. ** p < 0.01 and n.s. = not significant.
To further quantify and compare the residency time of BACE1-SBP-GFP in the Golgi in each condition, the average fluorescence intensity of the Golgi area was measured over time for individual cells. The maximal Golgi intensity of BACE1-SBP-GFP and the corresponding time point for the intensity to reach maximum were determined (Figure 7, C and D). There was no significant deviation between the groups, indicating that the level of BACE1-SBP-GFP expression was similar and the ER-to-Golgi trafficking of BACE1-SBP-GFP was not affected by the siRNA treatment (Figure 7, C and D). The Golgi exit rate of BACE1-SBP-GFP was calculated based on the slope of the curve in Figure 7B (see Figure 7E). AP-1–depleted cells showed a 35% reduction in slope compared with control and AP-4 siRNA-treated cells (Figure 7E). From these findings, we conclude that the Golgi exit of BACE1-SBP-GFP was delayed in AP-1–depleted cells compared with control cells and cells treated with AP-4ε siRNA
Depletion of Arf small G proteins required for the membrane recruitment of AP-1 results in the accumulation of BACE1 but not APP in the TGN
Given that the recruitment of AP-1 to TGN membranes has been reported to be regulated by both Arf1 (Stamnes and Rothman, 1993; Le Borgne and Hoflack, 1997; Lee et al., 2008) and Arf4 (Lowery et al., 2013; Nakai et al., 2013) small G proteins, we investigated whether these two Arfs are required for the post-Golgi export of BACE1. In cells treated with Arf1 or Arf4 siRNA, the levels of Arf1 and Arf4 were depleted by >90% and >81%, respectively, as assessed by immunoblotting (Figure 8A). In addition, the depletion of Arf1 resulted in an increase in Arf4 levels by ∼20%, consistent with a previous report (Reiling et al., 2013; Figure 8A). The depletion of either Arf1 or Arf4 accumulated BACE1-GFP in the juxtanuclear region that colocalizes extensively with the Golgi marker golgin-97, compared with control conditions (Figure 8B). Quantitation revealed that there was an increase in the colocalization of BACE1-GFP and golgin-97 from 8.1 ± 2.1% in control cells to 21.1 ± 2.4% and 25.7 ± 2.9% in Arf1- and Arf4-depleted cells, respectively (Figure 8D). Notably, the depletion of Arf4 resulted in a greater accumulation of BACE1-GFP at the TGN than the depletion of Arf1 (Figure 8D). To exclude an off-target effect of Arf1 siRNA and Arf4 siRNA, wild-type mCherry-tagged Arf1 and Arf4 constructs were expressed in cells treated with Arf1 siRNA and Arf4 siRNA, respectively (Figure 8, B, E, and F). Rescued cells were detected by mCherry expression. BACE1-GFP did not accumulate in the Golgi of cells expressing either the Arf1 and Arf4 rescue construct, confirming that the accumulation of BACE1-GFP in the TGN was specifically due to the depletion of Arf1 and Arf4 (Figure 8, B, E, and F).
FIGURE 8:

Depletion of small G protein Arf1 and Arf4 results in BACE1 accumulation in the TGN. HeLa cells stably expressing BACE1-GFP were transfected with either control siRNA, AP-1γ siRNA, Arf1 siRNA, Arf4 siRNA, or Arf1+4 siRNAs for 72 h. (A) Cells were lysed in RIPA buffer, and 20 μg of cell extracts was subjected to SDS–PAGE on 4–12% gradient polyacrylamide gel. Proteins were transferred to a PVDF membrane and probed with mouse anti–AP-1γ, mouse anti–α-tubulin, and either mouse anti-Arf1 or rabbit anti-Arf4 antibodies on a duplicated blot using a chemiluminescence detection system. (B) Cells transfected with Arf1 siRNA and Arf4 siRNA were also cotransfected with Arf1(WT)-mCherry and Arf4(WT)-mCherry construct, respectively, 48 h later for a further 24 h, as indicated. (B, C) Monolayers of HeLa cells were fixed, permeabilized, and blocked, followed by staining with mouse anti–golgin-97 antibodies (red/far-red converted to red) and DAPI (blue). Higher magnifications of the merged images of the cells marked * are also shown. Inset represents magnified images for Arf1 and Arf4 rescue experiments. Bar represents 10 μm. (D–F) The percentage of BACE1-GFP at the TGN was calculated as a percentage of total BACE1-GFP pixels that overlapped with golgin-97. Data are represented as the mean ± SD of three independent experiments (n = 15–25) and analyzed by one-way ANOVA using Tukey’s test in D and unpaired, two-tailed Student’s t test in E and F. *** p < 0.001 and ** p < 0.01.
Given that Arf1 and Arf4 are closely related (Donaldson and Jackson, 2011), Arf1 and Arf4 may have overlapping functions. To investigate this possibility, both Arf1 and Arf4 were depleted simultaneously. The double knockdown of Arf1+Arf4 depleted Arf1 by 82%, whereas Arf4 was depleted by only 55%, as assessed by immunoblotting (Figure 8A). A previous study showed that Arf1 depletion caused Arf4 levels to increase (Reiling et al., 2013), which may account for the modest level of Arf4 depletion. In Arf1+Arf4–depleted cells, BACE1-GFP accumulated in the TGN, as expected, but to a lesser extent than after single depletion of Arf4 (Figure 8, B and C). Quantitation revealed an increase in BACE1-GFP colocalization with golgin-97 from 8% in control cells to 17.1 ± 4.8% in Arf1+Arf4–depleted cells (Figure 8D). These data show that the double depletion of Arf1 and Arf4 did not result in a further increase in the level of BACE1-GFP in the TGN compared with single knockdowns, indicating that Arf1 and Arf4 are unlikely to have overlapping functions in regulating the export of BACE1 from the TGN.
To investigate whether AP-1γ, Arf1, and Arf4 are also required for the Golgi export of APP, AP-1γ, Arf1, and Arf4 were depleted in HeLa cells stably expressing APP695wt (HeLa-APP695wt); this cell line has been described previously (Toh et al., 2017). None of these knockdowns altered the localization of APP at the TGN from that in cells treated with control siRNA, as shown by quantitative analyses (Supplemental Figure S5). These data indicate that the AP-1/Arf1/Arf4 sorting machinery is not required for APP export from the TGN.
Depletion of either AP-1, Arf1, or Arf4 reduces the cell surface expression of BACE1
As depletion of AP-1, Arf1 and Arf4 resulted in the accumulation of BACE1 in the TGN, we quantified their effect on the cell surface levels of BACE1 by flow cytometry. AP-1γ–depleted HeLa-BACE1-GFP cells showed a reduction of cell surface BACE1 compared with control conditions (Figure 9A). There was a decrease in BACE1 mean fluorescence intensity of 19.8% in AP-1γ–depleted cells compared with control siRNA-treated cells (Figure 9D), whereas cell surface expression of BACE1 was unaltered in either AP-4ε− or Arl5b-depleted cells (Figure 9, B and E). Therefore, the cell surface levels of BACE1 are dependent on AP-1 mediated trafficking of BACE1 from the Golgi.
FIGURE 9:

AP-1, Arf1, and Arf4 regulate trafficking of BACE1 to the cell surface. HeLa cells stably expressing BACE1-GFP were transfected with either control siRNA or (A) AP-1γ siRNA, (B) AP-4ε siRNA or Arl5b siRNA, or (C) Arf1 siRNA, Arf4 siRNA, or Arf1+4 siRNAs for 72 h. Cells were harvested and stained with anti-BACE1 antibodies on ice for 30 min and then fixed in 4% PFA, quenched in 50 mM NH4Cl, and blocked in 5% FCS/PBS without permeabilization. Cell surface BACE1 was stained with Alexa568-conjugated IgG. Ten thousand events were analyzed per sample by flow cytometry. Data analyses and histograms were constructed using Flowjo. (D–F) Bar graphs showing arbitrary mean fluorescence intensity of cell surface BACE1 in controls and under (D) AP-1γ-, (E) AP-4ε- and Arl5b-, and (F) Arf1- and Arf4-depleted conditions. Data are represented as the mean ± SEM of three independent experiments and analyzed by one-way ANOVA using Tukey’s test. *** p < 0.001 and n.s. = not significant.
The impact of Arf1 and Arf4 depletion on the cell surface levels of BACE1 was also examined. The depletion of either Arf1 or Arf4 in HeLa-BACE1-GFP resulted in a decrease in cell surface expression of BACE1, as shown by a leftward shift in the fluorescence intensity peaks of each knockdown condition from control conditions (Figure 9C). Quantitative analysis showed a very similar decrease in BACE1 mean fluorescence intensity by 36.3% and 39.3% in cells treated with Arf1 siRNA or Arf4 siRNA, respectively, compared with control conditions (Figure 9F). The decrease in cell surface expression of BACE1 demonstrates that Arf1 and Arf4 are also required for the trafficking of BACE1 to the cell surface. These results show that cell surface expression of BACE1 is dependent on the Golgi AP-1 export pathway.
The trafficking of the transferrin receptor to the cell surface is independent of the AP-1/Arf1/Arf4 pathway
The question arises of whether the AP-1 pathway to the cell surface is used by other cargoes. AP-1 has been shown to be required for the trafficking of transferrin receptor (TfR) to the basolateral PM in polarized Madin–Darby canine kidney (MDCK) epithelial cells (Gravotta et al., 2012) and the trafficking of TfR to the somatodendritic PM in rat hippocampal neurons (Farias et al., 2012). Therefore, we assessed whether the trafficking of TfR to the cell surface in nonpolarized HeLa cells is also AP-1–dependent. The cell surface expression of TfR in AP-1–depleted HeLa-BACE1-GFP was assessed by flow cytometry. Surprisingly, the depletion of AP-1γ showed no significant change in the level of cell surface TfR from that in control cells (Supplemental Figure S6, A and C).
We also assessed the cell surface expression of TfR in either Arf1- or Arf4-depleted cells. The depletion of Arf1 showed no change and that of Arf4 a minor increase in the level of cell surface TfR, compared with that in control cells (Supplemental Figure S6 B, and D). These findings demonstrate that AP-1, Arf1, and Arf4 are not required for the trafficking of all membrane cargo to the PM; rather, these machineries are likely to be required for a subset of cargo, which includes BACE1, from the TGN to the PM.
Impact of delayed trafficking of newly synthesized BACE1 from the TGN on amyloidogenic processing of APP
To assess whether the accumulation of BACE1 at the TGN following AP-1, Arf1, and Arf4 depletion alters amyloidogenic processing of APP, we analyzed the processing of APP by endogenous BACE1 and γ-secretase in HeLa-APP695wt cells. Conditioned medium from siRNA-treated HeLa-APP695wt cells was collected and analyzed for secreted Aβ using a sandwich ELISA specific for human Aβ40. The Aβ40 levels were normalized to total cellular protein for each sample. There was a 1.7-fold increase in secreted Aβ from AP-1γ–depleted cells as compared with control cells (Figure 10A). In addition, there were 2.7-fold, 4.5-fold, and 3.5-fold increases in secreted Aβ from Arf1-, Arf4-, and Arf1+4-depleted conditions, respectively, as compared with control conditions (Figure 10B). The increase in Aβ production indicates enhanced processing of APP by endogenous BACE1 at the TGN following the depletion of AP-1, Arf1, and Arf4 sorting machinery. A higher level of secreted Aβ was detected from Arf4-depleted cells than from Arf1-depleted cells (Figure 10B), consistent with the finding that BACE1 showed a greater accumulation at the TGN in Arf4-depleted cells (Figure 8, B and D).
FIGURE 10:

APP processing and Aβ production is increased in AP-1-, Arf1-, Arf4-, and Arf1+4-depleted HeLa cells. (A, B) HeLa cells stably expressing APP695wt were transfected with either control siRNA; AP1γ siRNA (A); or Arf1 siRNA, Arf4 siRNA, or Arf1+4 siRNAs (B); 56 h after transfection the medium was changed and conditioned medium collected 16 h later and analyzed for secreted Aβ using a sandwich ELISA specific for Aβ40. The Aβ40 levels for each sample were normalized against the total cellular protein. Data are represented as the mean ± SEM of three independent experiments and analyzed by unpaired, two-tailed Student’s t test in A and one-way ANOVA using Tukey’s test in B. ***p < 0.001 and **p < 0.01. (C, D) HeLa cells stably expressing APP695wt were transfected with either control siRNA; AP-1γ siRNA (C); or Arf1 siRNA, Arf4 siRNA, or Arf1+4 siRNAs (D) for 72 h and then treated with either DMSO (carrier control) or 250 nM γ-secretase inhibitor DAPT in the last 16 h of siRNA transfection. Cells were lysed, and 20 μg of cell extracts per sample was subjected to SDS–PAGE. Proteins were transferred either onto PVDF and probed with mouse antibodies to AP-4ε or a-tubulin, or onto nitrocellulose membrane and probed with mouse W02 antibodies to β-CTF/C99. (E) Bar graph representing fold change of β-CTF/C99 levels in AP-1γ siRNA, Arf1 siRNA, Arf4 siRNA, and Arf1+4 siRNAs compared with control siRNA. Data are represented as the mean ± SD of four independent experiments and analyzed by one-way ANOVA using Tukey’s test. ***p < 0.001, **p < 0.01, and *p < 0.05.
To directly assess the level of β-CTF/C99 derived from the initial cleavage of APP by BACE1, cell extracts were probed with the W0-2 antibody, which recognizes residues 4-10 of the Aβ domain in APP and which will detect full-length APP and β-CTF/C99. Full-length APP was detected in HeLa-APP695wt cell extracts (Supplemental Figure S7), whereas in the absence of the γ-secretase inhibitor DAPT, β-CTF/C99 was not detected in control siRNA-treated cells and was only detected at low levels in cells treated with AP-1γ siRNA even after long exposures (Figure 10C; Supplemental Figure S7A). However, in the presence of DAPT, β-CTF/C99 was readily detected in both control siRNA- and AP-1γ siRNA-treated samples. Moreover, there was a 2.8-fold increase in the level of intracellular β-CTF/C99 under AP-1γ–depleted conditions from that under control conditions (Figure 10, C and E). Under Arf1 siRNA-, Arf4 siRNA-, or Arf1+4 siRNAs-depleted conditions, β-CTF/C99 was again detected only at low levels in the absence of DAPT (Figure 10D). In the presence of DAPT, there was also a 2.7-fold, 3.9-fold, and 4.1-fold increase in the level of intracellular β-CTF/C99 detected in cells treated with either Arf1 siRNA, Arf4 siRNA, or Arf1+4 siRNAs, respectively, from that in control siRNA-treated cells (Figure 10, D and E). The increase in β-CTF/C99 production directly demonstrates that the depletion of AP1γ, Arf1, Arf4, or Arf1+4 enhances BACE1 cleavage of APP at the TGN. Notably, a greater increase in the level of β-CTF/C99 was detected under Arf4-depleted conditions than under Arf1-depleted conditions (Figure 10, D and E), consistent with the level of Aβ production under these knockdown conditions (Figure 10B). The similar increase in β-CTF/C99 detected under Arf4 and Arf1+Arf4 knockdown conditions is likely due to more efficient depletion of Arf4 (>80%) in this experiment (Figure 10D) than in the previous experiment (Figure 8A). These results again indicate that Arf4 is likely to be the major small G protein regulating the post-Golgi trafficking of BACE1. Collectively, these findings highlight the relevance of AP-1, Arf1, and Arf4 trafficking machinery on the regulation of the amyloidogenic processing of APP at the TGN.
Depletion of AP-1 in neurons resulted in enhanced BACE1 processing of endogenous APP
Next, we assessed whether APP processing by endogenous BACE1 in the TGNs of primary neurons is also regulated by the AP-1 pathway. As residues 4–10 of the Aβ domain in APP, detected by W0-2 antibody, differ between human and mouse, the W0-2 antibody is not suitable for the detection of β-CTF/C99 in primary mouse cortical neurons. Instead, we employed the Y188 antibody, which was raised against the highly conserved YENPTY sorting motif of APP (residues 682–687 of APP695). The Y188 antibody detects both β-CTF/C99 (11 kDa) and α-CTF/C83 (9 kDa) in primary mouse cortical neurons (Tan and Gleeson, 2019c).
We utilized two different shRNA targets for AP-1 for the generation of recombinant lentivirus to deplete AP-1 in primary mouse cortical neurons. The depletion of AP-1 in the population of transduced neurons was then assessed by immunoblotting. AP-1γ was depleted by 43–46% in neurons transduced with AP-1γ-shRNA1 and AP-1γ-shRNA2 lentivirus as compared with nontransduced neurons (Figure 11, A and B). Based on GFP-positive neurons, the transduction efficiency of the transduced neuronal population was 35–50%, indicating that the depletion of AP-1 in GFP-positive neurons was ∼90%. In the absence of DAPT, β-CTF/C99 and α-CTF/C83 were not detected in any of the neuronal extracts (Figure 11A), consistent with previous findings (Tan and Gleeson, 2019c) and indicating that the products of α- and β-secretase cleavage are rapidly processed by γ-secretase. However, in the presence of DAPT, there were 1.88- and 1.94-fold increases in the level of intracellular β-CTF/C99 in neurons transduced with AP-1γ-shRNA1 and AP-1γ- shRNA2, respectively, compared with nontransduced neurons (Figure 11, A and C). We treated AP-1γ-shRNA1- and AP-1γ-shRNA2–transduced neurons with a BACE1 inhibitor, C3, to validate that the increase in the 11 kDa band (β-CTF/C99) was specifically due to enhanced cleavage of APP by BACE1. In the presence of C3 and DAPT, the 11-kDa band (indicated by *) was absent (Figure 11A), confirming their identity as β-CTF/C99. Moreover, there were no significant changes in the level of α-CTF/C83 (** band) in AP-1γ-shRNA1– and AP-1γ-shRNA2–transduced neurons from that in nontransduced neurons (Figure 11, A and D). Full-length APP was also detected in these immunoblots (Supplemental Figure S7B shows the entire blot). Collectively, AP-1 depletion results in an increased level of β-CTF/C99, but not α-CTF/C83, demonstrating that AP-1 depletion enhances amyloidogenic processing of APP by BACE1.
FIGURE 11:

Depletion of AP-1γ in mouse primary cortical neurons results in increased APP processing. (A) E16 primary mouse cortical neurons were transduced at DIV3 with either AP-1γ-shRNA1 or AP-1γ-shRNA2 lentivirus (LV) for 96 h and treated with DMSO (carrier control), 2 μM DAPT, and/or 2 μM BACE1 inhibitor C3 in the last 16–20 h of transduction. Neurons were lysed in RIPA buffer and cell extracts (10 μg) subjected to SDS–PAGE, as described in Materials and Methods. Proteins transferred onto PVDF membrane were probed with mouse antibodies to either AP-1γ or α-tubulin. Proteins transferred onto nitrocellulose membrane were probed with rabbit anti-APP (Y188) antibodies to C99*/C83**. (B–D) Bar graph representing percentage of AP-1 depletion (B) and fold change of β-CTF/C99 (C) and α-CTF/C83 (D) in AP-1γ shRNA1 or AP-1γ shRNA2 lentiviral transduction compared with nontransduced controls. Levels of α-CTF/C83 and β-CTF/C99 were normalized to α-tubulin. Data are represented as the mean ± SD of three independent experiments and analyzed by one-way ANOVA using Tukey’s test. **p < 0.01, *p < 0.05, and n.s. = not significant. (E, F) Primary mouse cortical neurons were transduced at DIV3 with either AP-1γ-shRNA1 or AP-1γ-shRNA2 lentivirus, and 80 h after transduction the medium was changed and spent medium containing secreted Aβ was collected 16 h later and analyzed with a sandwich ELISA specific for either Aβ40 (E) or Aβ42 (F). The Aβ40 or Aβ42 levels for each sample were normalized against the total cellular protein. Data are represented as the mean ± SEM of three independent experiments and analyzed by one-way ANOVA using Tukey’s test. ***p < 0.001, **p < 0.01, and *p < 0.05. (G) Model of anterograde transport pathway of newly synthesized APP and BACE1. Both BACE1 (green bars) and APP (red bars) are synthesized in the ER and transported through the Golgi. Upon arrival at the TGN, APP and BACE1 are sorted for TGN export by distinct transport machinery. BACE1 is transported from the TGN directly to the PM via an AP-1-/Arf1-/Arf4-dependent pathway. APP is transported via an AP-4-/Arl5b-dependent pathway from the TGN directly to the early endosomes and then to either the PM or the late endosomes/lysosomes.
Processing of APP by BACE1 and γ-secretase in neurons results in the production of the two major species of amyloid peptides, Aβ40 and the more highly cytotoxic Aβ42. To determine the levels of Aβ40 and Aβ42 exported into the cell media of treated neurons, conditioned medium from shRNA-treated neurons was collected and analyzed for secreted Aβ40 (Figure 11E) and Aβ42 (Figure 11F) using mouse Aβ40– and Aβ42–specific ELISAs. We confirmed that these Aβ-ELISAs were specific for the individual Aβ peptides (unpublished data). In nontransduced neurons, the secreted Aβ40:Aβ42 ratio was 5.3:1, consistent with Aβ40 representing the major species from neuronal tissues (Schieb et al., 2011). In AP-1–silenced neurons, there was an increase in levels of both Aβ species: a 3.0-fold increase for Aβ40 and a 3.7-fold increase for Aβ42. Therefore, the Golgi-associated APP processing events in primary neurons result in the generation of the cytotoxic Aβ42 species.
DISCUSSION
There is considerable evidence from genome-wide association study (GWAS) analysis and experimental animal models that dysfunctional membrane trafficking contributes to aberrant Aβ production and AD. Many studies have also indicated that Aβ can be generated in both the endosomal and secretory pathway (Tan and Gleeson, 2019b). We and others have shown that the TGN is a major site for Aβ production along the secretory pathway (Burgos et al., 2010; Toh et al., 2017). As newly synthesized APP and BACE1 are cotransported along the secretory pathway, it is important to understand how the trafficking of these two membrane cargos is regulated to control the cleavage of APP by BACE1. We previously showed that newly synthesized APP is transported from the TGN directly to the early endosomes by an AP-4–dependent pathway. Here we have examined the sorting and trafficking of newly synthesized BACE1 at the Golgi in both HeLa and primary neurons and have shown that 1) newly synthesized BACE1 traffics from the TGN to the PM; 2) the AP complex, AP-1, and Arf1 and Arf4 small G proteins are essential for TGN-to-PM trafficking of BACE1; 3) the post-Golgi trafficking of APP is independent of the AP-1-/Arf1-/Arf4-regulated pathway; 4) the depletion of either AP-1, Arf1, or Arf4 enhanced the amyloidogenic processing of APP; and 5) in primary neurons the depletion of AP-1 increased the levels of both secreted Aβ40 and Aβ42, demonstrating that the cytotoxic Aβ42 can be generated in the secretory pathway. Collectively, our findings indicate that the AP-1-/Arf1-/Arf4-dependent transport pathway is crucial for the efficient export of BACE1 from the Golgi to the PM, which in turn limits the processing of APP at the TGN.
We used the RUSH-system to track the anterograde itinerary of newly synthesized BACE1 (BACE1-SBP-GFP), as it provides an effective approach to synchronize protein trafficking along the anterograde pathway (Boncompain et al., 2012; Chen et al., 2017). BACE1-SBP-GFP was transported to the Golgi within 10–15 min following release from the ER by addition of biotin, and then trafficked from the TGN to the PM. Our data strongly indicate that BACE1 is transported directly from the TGN to the PM. This conclusion is based on quantitative analysis of confocal live imaging, TIRF analysis, and fast image acquisition, which showed that BACE1-SBP-GFP–containing vesicles were transported from a juxtanuclear compartment directly to the distal edge of the cell. Also, that BACE1 was detected arriving at the plasma membrane 20 min after addition of biotin, whereas it was first detected in recycling endosomes at a later time point (30 min), implies a direct Golgi-to-PM transport step. Arrival of BACE1 at the early and recycling endosomes at the 30-min time point most likely represents internalized BACE1 from the PM (Chia et al., 2013). The intracellular distribution of BACE1-SBP-GFP at an extended time point was very similar to the steady-state distribution of BACE1 in either stable or transiently transfected HeLa cells, with or without a GFP tag (Chia et al., 2013), indicating that the SBP and GFP tags are unlikely to interfere with the trafficking of BACE1-SBP-GFP in the RUSH system.
The current studies involved silencing of trafficking machinery in a stable cell line expressing only modest levels of BACE1-GFP, to minimize the possibility of saturation of the post-Golgi transport machinery (Chia et al., 2013). The physiological relevance of our studies in HeLa cells was confirmed by the demonstration that endogenous BACE1 in neurons was found mainly in the recycling endosomes and early endosomes under steady-state conditions, with minor levels in the Golgi and late endosomes/lysosomes, a distribution similar to that in transfected HeLa cells; and by the observation that silencing AP-1, but not AP-4, in neurons resulted in the accumulation of endogenous BACE1 in the TGN. Confirmation of the findings in HeLa cells by the behavior of endogenous BACE1 in primary neurons demonstrates that the post-Golgi transport pathways of BACE1 are common between the cell types.
A key finding of this work is that AP-1 is required for the efficient post-Golgi sorting of BACE1 in both HeLa cells and neurons. BACE1 was retained in the Golgi/TGN following AP-1 knockdown, as revealed by localization studies of fixed cells with defined markers and by the analysis of the kinetics of BACE1 transport through the Golgi in live cells using RUSH. In addition, the reduced levels of BACE1 at the cell surface in AP-1 knockdown cells, as assessed by FACS of nonpermeabilized cells, are also consistent with the retention of BACE1 in the Golgi. In contrast, GGA1, which is also found at the Golgi, is not required for the post-Golgi export of BACE1. Rather, GGA1, which can interact with a DISLL motif in the cytoplasmic tail of BACE1, is required for the endosomal sorting of BACE1 (He et al., 2002, 2005; Toh et al., 2018). Interestingly, the DISLL motif is part of an acidic cluster (DDFADDISLL) motif in the cytoplasmic tail BACE1 that has properties similar to those of other acidic cluster sequences in proteins that are known to associate with AP-1 (Table 2). The basic patch (Lys274, Lys298, Lys302, Arg303, and Arg304) of the AP-1μ subunit has been shown to be required for the binding of acidic cluster sequences (Navarro Negredo et al., 2017). Based on the findings in this paper, AP-1 is likely to interact with BACE1 via the acidic motif at the TGN and recruit this membrane cargo into a transport pathway that directs BACE1 to the PM. A previous study analyzing the AP-2–mediated endocytosis of BACE1-mutated D495 in the acidic cluster motif (DDFAD495DISLL) in BACE1 to R495 showed that the D495 residue is essential for AP-2–mediated endocytosis of BACE1 (Prabhu et al., 2012). However, an additional effect of the D495R mutation, which was not alluded to by these authors, was the accumulation of D495R BACE1 mutant in the juxtanuclear region of the cell (Prabhu et al., 2012), suggesting that the acidic D495 residue could also be important for the post-Golgi trafficking of BACE1. Direct evidence that AP-1μ interacts with the acidic cluster motif of BACE1 remains to be formally shown.
The recruitment of AP-1 to the TGN is known to be mediated by Arf1 and Arf4 small G proteins (Stamnes and Rothman, 1993; Lee et al., 2008; Nakai et al., 2013) and we have shown that both these small G proteins are also required for the sorting of BACE1 at the Golgi. The depletion of either Arf1 or Arf4 resulted in the accumulation of BACE1-GFP in the TGN. In contrast to BACE1, the depletion of either AP-1, Arf1, or Arf4 did not affect the distribution of APP. We have previously shown that depletion of Arf1 or Arf4 does not affect recruitment of AP-4 to the TGN (Toh et al., 2017). These findings further support the conclusion that the post-Golgi sorting machinery and trafficking pathways that regulate APP and BACE1 differ.
Depletion of AP-1, Arf1, or Arf4 resulted in a reduction of BACE1 at the cell surface. Nonetheless, substantial levels of BACE1 were present at the surface in these depleted cells, which could be due to either the incomplete knockdown of the transport machinery or transport by an AP-1–independent mechanism. On the other hand, depletion of either AP-1, Arf1, or Arf4 did not reduce the level of TfR at the cell surface, consistent with the findings from the Bonifacino laboratory, which showed that TfR is loaded into tubular carriers derived from the TGN in nonpolarized HeLa cells by an AP-1–independent process (Chen et al., 2017). Thus, it is clear there are multiple transport pathways to the PM, and the AP-1 pathway may be selective for a subset of membrane cargoes that includes BACE1.
AP-1 has been reported to facilitate membrane cargo trafficking along a number of transport pathways. Although not generally considered to be a cargo adaptor for transport to the PM, a previous study of a reovirus membrane protein also reported transport to the plasma membrane from the Golgi by an AP-1–dependent pathway (Parmer and Duncan, 2016). AP-1B, which is a distinct isoform of AP-1 bearing a μ1B subunit, is important for transport of cargo from the recycling endosomes to the PM in a subset of polarized epithelial cells; however, AP-1B is absent in nonepithelial cells (see Tan and Gleeson, 2019a). AP-1 is known to regulate the TGN-to-endosomal pathway required for the delivery of newly synthesized lysosomal enzymes bound to mannose 6-phosphate receptors (MPRs) from the TGN to the late endosomes (Hille-Rehfeld, 1995; Meyer et al., 2000). A key question arising from our study is how AP-1 regulates the trafficking of cargoes from the TGN to the PM as well as the TGN to late endosomes. The GGA proteins, which are also recruited to the TGN by Arf1, have been shown to function cooperatively with AP-1 to regulate the trafficking of cargoes from the TGN to the endosomes (Doray et al., 2002; Hirst et al., 2012). These studies suggest that the combination of AP-1 and GGA is likely to regulate the sorting of cargoes to the endosomes from the TGN. On the other hand, GGAs are not required for the trafficking of BACE1 to the PM. Therefore, cargo that interacts selectively with AP-1 at the TGN may be transported from the Golgi directly to the PM. In addition to these anterograde pathways, AP-1 has also been reported to regulate retrograde transport in the Golgi in yeast cells (Casler et al., 2019).
An important component of the current study was determining the consequences of retention of BACE1 in the Golgi for APP processing. Using stably transfected HeLa-APP695wt cells, we showed that Golgi export of endogenous BACE1 by the AP-1 pathway is essential for the regulation of amyloidogenic processing. Upon the depletion of either AP-1, Arf1, or Arf4, there was increased processing of APP, as demonstrated by a 2.8- to 4.1-fold increase of intracellular β-CTF/C99 and a 1.7- to 4.5-fold increase in secreted Aβ secretion compared with control conditions. Moreover, AP-1 depletion in primary neurons resulted in a 1.9-fold increase in the level of intracellular β-CTF/C99 and 3.1- and 3.7-fold increases in secreted Aβ40 and Aβ42, respectively. These findings demonstrate that the AP-1 pathway is critical for regulating processing events under physiological conditions. In addition, although Aβ40 was the predominant species in neurons, as expected from previous reports (Schieb et al., 2011), the finding that Aβ42 is generated via TGN-mediated processing of APP is an important observation, as Aβ42 is more cytotoxic than Aβ40 (Lane et al., 2018) and has not, to our knowledge, been demonstrated previously to be produced from APP processing events within the secretory pathway.
The trafficking of BACE1 to the PM via the AP-1 pathway may serve as a regulatory mechanism to segregate BACE1 from APP at the TGN, thus limiting the amyloidogenic processing of APP in this compartment. This conclusion is also consistent with our earlier study where enhanced APP cleavage was detected at the TGN upon the redirection of BACE1 recycling to this compartment using a BACE1/TGN38 chimera (Chia et al., 2013). Only low levels of β-CTF/C99 were detectable in AP-1-/Arf1-/Arf4-depleted cells in the absence of the γ-secretase inhibitor, indicating that γ-secretase processes β-CTF/C99 rapidly. Given that γ-secretase is also localized in the Golgi compartment (Rechards et al., 2003), the liberation of Aβ peptide from β-CTF/C99 by γ-secretase is also likely to occur in the TGN. The secretion of Aβ peptide from the cell is proposed to occur through various anterograde trafficking pathways (reviewed in Toh and Gleeson, 2016). We propose that the loss of the AP-1/Arf1/Arf4 transport machinery results in accumulation of BACE1 in the TGN, which results in a loss in the segregation of BACE1 from newly synthesized APP arriving at the TGN (see Figure 11G). The increase in convergence between BACE1 and APP in the TGN then leads to increased processing of APP by BACE1 in this compartment.
In conclusion, a functional AP-1/Arf1/Arf4 pathway is required for the export of BACE1 from the Golgi directly to the PM. On the other hand, the trafficking of APP from the TGN is independent of this pathway. Rather, the trafficking of APP from the TGN is regulated by the AP-4/Arl5b transport machinery directly to the endosomal pathway. The anterograde pathways for BACE1 and APP are illustrated in Figure 11G. The requirement for distinct transport machinery for post-Golgi transport of APP and BACE1 provides a mechanism to sort and segregate APP and BACE1 into separate pathways. Therefore, the convergence between APP and BACE1 along the anterograde pathway would be limited, which in turn would minimize the extent of amyloidogenic APP processing and Aβ production.
MATERIALS AND METHODS
Plasmids and antibodies
Streptavidin fused to the ER retention signal Ii (human major histocompatibility complex class II invariant chain) and full-length wild-type BACE1 fused with a streptavidin-binding peptide (SBP) and EGFP-tag were inserted into pIRESneo3 (Str-Ii_BACE1FL-SBP-EGFP). Constructs encoding human wild-type Arf1 and Arf4 tagged with mCherry at their C-termini, pcDNA3/hArf1(WT)-mCherry (Addgene plasmid: #79419) and pCAG/hArf4(WT)-mCherry (Addgene plasmid: #79406), respectively, were gifts from Kazuhisa Nakayama (Department of Physiological Chemistry, Graduate School of Pharmaceutical Sciences, Kyoto University, Japan). pGIPZ-shRNA lentiviral constructs against mouse AP-1γ (V2LMM_44992 and V3LMM_442644) were purchased from Dharmacon, GE Healthcare. pGIPZ-shRNA lentiviral constructs against mouse AP-4ε, pCMV-VSV-G, and psPAX2 were previously described (Tan and Gleeson, 2019c).
Rat monoclonal antibodies to mouse LAMP1 (Clone 1D4B, #553792) and mouse monoclonal antibodies to AP-1γ (Clone 88, #610385), AP-4ε (Clone 32, #612019), EEA1 (Clone 14, #610456), Rab4 (Clone 7, #610889), and Rab11 (Clone 47, #610656) were purchased from BD Biosciences. Mouse monoclonal antibodies to human golgin-97 (CDF4, #A21270) were purchased from Thermo Fisher Scientific. Mouse monoclonal antibodies to human CD63 (MX-49.129.5, #sc-5275) were purchased from Santa Cruz Biotechnology. Mouse monoclonal antibodies to Aβ4-10 (W0-2, #MABN10) were from Merckmillipore (Merck). Rabbit monoclonal antibodies to GGA1 (#ab170956) and APP (Clone Y188, #ab32136), and mouse monoclonal antibodies to Arf1 (ARFS3F1, #ab11038) were obtained from Abcam (Cambridge, UK). Rabbit monoclonal antibodies to β-secretase (BACE1; D10E5, #5606) were from Cell Signaling Technology. Rabbit polyclonal antibodies to BACE1 (N-terminus 46–62, Clone EE17, #B0681), mouse monoclonal antibodies to APP (Clone NAB228), and mouse monoclonal antibodies to α-tubulin (Clone DM1A, #T9026) were bought from Sigma-Aldrich (Merck). Rabbit polyclonal anti-GCC88 antibodies (Luke et al., 2003) and human autoantibodies to p230/golgin-245 (Kooy et al., 1992) have been described. Rabbit polyclonal antibodies to Arf4 (#11673-1-AP) were from ProteinTech. Mouse monoclonal antibodies to human transferrin receptor (TfR/OKT9; Schneider et al., 1982) were purified from supernatants from a hybridoma. Can Get Signal immunoreaction enhancer solution (Toyobo Life Science, Japan) was used to dilute mouse anti-Rab11 antibodies for immunofluorescence analysis.
Goat anti-mouse immunoglobulin G (IgG)-Alexa Fluor 488 nm, goat anti-mouse IgG-Alexa Fluor 568 nm, goat anti-mouse IgG-Alexa Fluor 647 nm, goat anti-rabbit IgG-Alexa Fluor 488 nm, goat anti-rabbit IgG-Alexa Fluor 568 nm, goat anti-rabbit IgG-Alexa Fluor 647 nm, goat anti-rat IgG-Alexa Fluor 568 nm, and goat anti-human IgG-Alexa Fluor 647 nm (for human anti–p230/golgin-245 antibodies) conjugated secondary antibodies for immunofluorescence were obtained from Thermo Fisher Scientific. Horseradish peroxidase (HRP)-conjugated rabbit anti-mouse Ig and HRP-conjugated swine anti-rabbit Ig were bought from DAKO Corporation (Santa Clara, CA).
RNAi and transient transfections
The silencing of AP-1 γ-adaptin, AP-4 ε-adaptin, GGA1, Arl5b, Arf1, and Arf4 was conducted using short-interfering RNA (siRNA) duplexes manufactured by Sigma-Proligo (Australia). Cell monolayers were transfected with siRNA using DharmaFECT1 (Thermo Fisher Scientific) according to the manufacturer’s protocol and incubated for 72 h in a humidified 10% CO2 incubator at 37°C. AP-1γ siRNA (Camus et al., 2007), GGA1 siRNA (Toh et al., 2018), Arl5b siRNA (Houghton et al., 2012), Arf1 siRNA (Southon et al., 2011), and AP-4ε siRNA and Arf4 siRNA (Toh et al., 2017) have been described previously.
Transient transfection of Arf1-mCherry and Arf4-mCherry plasmids was conducted for 24 h using FuGENE 6 (Promega) per manufacturer’s protocol.
Generation of HeLa cells stably expressing BACE1-GFP (HeLa-BACE1-GFP)
Parental HeLa cells were transfected with the pBACE1-EGFP-N3 construct using FuGENE 6 (Promega). Stably expressing cells were selected in C-DMEM supplemented with 1.5 mg/ml G418 for 2 wk. The resultant polyclonal HeLa-BACE1-GFP cells were harvested and GFP-positive cells were single-cell sorted into 96-well plates using a BD Influx cell sorter (BD Bioscience) at the Murdoch Children Research Institute (MCRI), Victoria, Australia. HeLa-BACE1-GFP stable clones were validated by immunofluorescence, immunoblotting, and flow cytometry. Cells were maintained in C-DMEM supplemented with 1 mg/ml G418 and cultured in a humidified 10% CO2 incubator at 37°C.
Cell culture
Authentic and mycoplasma-tested HeLa cells (Scherer et al., 1953) were maintained as semiconfluent monolayers in DMEM (Thermo Fisher Scientific, Australia) supplemented with 10% vol/vol fetal calf serum (FCS; Life Technologies, Thermo Fisher Scientific), 2 mM l-glutamine, 100 U/μl penicillin and 0.1% wt/vol streptomycin (complete DMEM/C-DMEM). HeLa-APP695wt cell line was generated as previously described (Toh et al., 2017) and maintained in C-DMEM supplemented with 500 ng/ml puromycin (Invitrogen, Thermo Fisher Scientific). HEK293T packaging cells were maintained as semiconfluent monolayers in C-DMEM without penicillin and streptomycin (C-DMEM–P/S). All cells were cultured in a humidified 10% CO2 incubator at 37°C.
Primary mouse cortical neuronal cultures
All experiments carried out on animals (Ethics ID 1613960) were in accordance with animal ethics guidelines (approval number 1212502.1) approved by the Animal Ethics Committee, the University of Melbourne. Primary cortical neurons were prepared from the collected embryos of pregnant mice (C57BL/6) at gestational day 15–16 and cultured as previously described (Tan and Gleeson, 2019c). Primary cortical neurons were plated at a density of 1.2 × 105 cells/well (12-well plates) and 5 × 105 cells/well (six-well plates), and cultured in neurobasal medium supplemented with 2.5% B-27, 0.25% GlutaMAX, and 100 U/μl penicillin and 0.1% streptomycin (complete NBM; Life Technologies).
Generation of lentivirus and transduction of primary mouse cortical neuronal cultures
Recombinant lentiviruses were generated and used for transduction of primary mouse cortical neurons as previously described (Tan and Gleeson, 2019c). Briefly, lentivirus was generated via calcium phosphate transfection of the HEK293T packaging cells with lentiviral plasmids at a ratio of 7 pCMV-VSV-G:3 psPAX2:10 pGIPZ-shRNA. Cells were transfected overnight in a humidified 10% CO2 incubator at 37°C. The existing medium was then replaced with fresh medium and cells were incubated for a further 48 h to allow the production of viral particles. Virus-containing medium was harvested, centrifuged to pellet cell debris, and the viral supernatant filtered using Steriflip-HV sterile centrifuge tube top filter unit (Merck). PEG-it virus precipitation solution (Integrated Sciences, Australia) was added to the filtered viral medium at 1× final and incubated overnight at 4°C. Viral particles were pelleted by centrifugation and resuspended in DMEM/NBM supplemented with 10 mM HEPES (Life Technologies, Thermo Fisher Scientific) at 1/100 of its original harvested volume. Concentrated virus was allocated into cryovials, snap frozen on dry ice, and stored at –80°C.
Primary mouse cortical neurons were transduced at day 3 in vitro (DIV 3) with 10–15 μl/well (12-well plate) of either AP-1γ-shRNA or AP-4ε-shRNA lentivirus diluted with fresh C-NBM to a final volume of 200 μl/well. Transduced neurons were incubated for 24 h (DIV 4) in a 37°C incubator with 10% CO2, followed by replacement of lentiviral medium with conditioned C-NBM (half fresh C-NBM and half C-NBM recovered from neuronal culture) and incubated for a further 72 h (DIV 7).
Immunofluorescence analyses
Monolayers of cultured mammalian cells and primary mouse cortical neurons were fixed in 4% paraformaldehyde (PFA; Wako Pure Chemical Industries, Japan) for 15 min at room temperature (RT), quenched in 50 mM NH4Cl/phosphate-buffered saline (PBS) for 10 min at RT, permeabilized with 0.1% vol/vol Triton X-100/PBS for 4 min at RT, and blocked in blocking solution (5% vol/vol FCS and 0.02% vol/vol sodium azide, in PBS) for 30 min to reduce nonspecific binding. All staining was conducted using the above method except for mouse anti-Rab11, anti-Rab4, and mouse anti-AP1γ antibodies. For Rab11 staining, cells were fixed with 10% trichloroacetic acid (TCA) on ice for 15 min, quenched in 30 mM glycine/PBS for 10 min at RT, and then permeabilized and blocked as above. For AP-1γ staining, cells were fixed in 4% PFA for 15 min, quenched in 50 mM NH4Cl/PBS for 10 min, permeabilized in 0.1% vol/vol Triton X-100/PBS for 15 min, and then blocked with 1% wt/vol BSA/0.3% vol/vol Triton X-100/PBS for 1 h. To stain for Rab-4 in neurons, cells were fixed in cold 100% methanol and incubated at -20°C for 5 min and blocked as above. Cultured cells were stained with primary antibodies diluted in blocking solution, with the exception of mouse anti-Rab11 antibodies diluted in Can Get Signal immunoreaction enhancer solution (Toyobo Life Science, Japan) for 1 h at RT. Staining of neurons with primary antibodies was conducted overnight at 4°C. Cultured cells and neurons were washed with PBS and stained with fluorophore-conjugated secondary antibodies diluted in blocking solution for 30 min at RT. Coverslips were washed with PBS and then with distilled water and mounted in Mowiol (10% wt/vol Hopval 5-88, 25% wt/vol glycerol, 0.1 M Tris in milli-Q water) on a microscope glass slide. For staining the TGN in HeLa cells, mouse anti–golgin-97 was routinely used, with the exception where primary antibodies to Rab11, EEA1, and APP were used, in which case rabbit anti-GCC88 was used instead. As mouse anti–golgin-97 and rabbit anti-GCC88 antibodies are specific for their respective human antigens, and do not cross-react with the mouse homologues, human autoantibodies to p230/golgin-245 were used for TGN staining in primary mouse cortical neurons.
Images were acquired sequentially for multicolor imaging on a laser confocal scanning microscope (Leica TCS SP8 confocal imaging system) using a 63 × 1.4 NA HCX PL APO CS oil immersion objective and a Leica HyD photodetector. GFP and Alexa Fluor 488 were excited using the 488-nm line source of an argon laser. Alexa Fluor 568 and Alexa Fluor 647 were excited with 543- and 633-nm helium–neon (HeNe) lasers, respectively. 4′,6-Diamidino-2-phenylindole (DAPI) was excited with a 405-nm UV laser. Images for HeLa cells were acquired at 0.3-μm single section and images for primary neurons were acquired as Z-stacks of 5–6 sections at 0.2 μm per section.
Anterograde transport assay of BACE1 using the RUSH system
HeLa cells were cultured as monolayers in 12-well or 24-well and were transfected with calcium phosphate (Jordan et al., 1996). Transfected cells were incubated for 24 h at 37°C with 10% CO2. d-biotin (Sigma-Aldrich, Merck) at 40 μM final concentration was added to transfected cells to allow the synchronous release of BACE1-SBP-GFP from the streptavidin-Ii hook protein in the ER.
For live imaging, an inverted Nikon Eclipse Ti microscope (with optical autofocus system and a motorized piezo stage) was used with a 100× oil-immersed objective. Round (24-mm) coverslips with HeLa cell monolayers were clipped in a metal chamber, maintained in Leibovitz’s medium (Thermo-Fisher Scientific), and incubated at 37°C during the imaging. Images with z-stacks (11 steps, step size 0.4 μm) were captured every 1 or 2 min for 60 min after addition of 40 μM D-biotin to the medium with an Andor Ixon Ultra (EM-CCD) camera and using Metamorph software (Molecular Devices). Laser power and acquisition settings were chosen to avoid photobleaching, and settings were identical for control and siRNA treated samples. For fast acquisition, images were collected in real time every 2 s. Temporal projections (Figure 2B) were performed with the tool “temporal-color-code” on the Fiji software for the entire movie (15 min of biotin to 45 min of biotin 1–601).
TIRF experiments were performed using the same device. Images were collected every 10 s from time point 1 to time point 170 and then every 2 s until the end.
Flow cytometry for cell surface expression analyses
Control, AP1γ-, AP4ε-, Arl5b-, Arf1-, or Arf4-depleted HeLa-BACE1-GFP cells were lifted from a six-well plate with 5 mM EDTA/PBS at 37°C. Live cells were stained in suspension with either rabbit anti-BACE1 (EE17) antibodies or mouse anti-TfR/OKT9 antibodies for 30 min on ice. Cells were washed twice with ice-cold PBS, fixed with 4% PFA for 15 min at RT, quenched with 50 mM NH4Cl for 10 min at RT, and blocked for 30 min at RT. To detect antibody-bound complexes, cells were incubated with Alexa Fluor conjugated secondary antibodies for 30 min. Cells were washed with PBS and resuspended in 2 mM EDTA/PBS. Cells were analyzed by medium flow rate in a BD LSRFortessa flow cytometry (BD Biosciences) and 10, 000 events were collected with a forward scatter threshold of 5000. Data were analyzed using FlowJo V10.
Aβ ELISAs
Aβ40-specific human ELISA (enzyme-linked immunosorbent assay; #KHB3481), Aβ40-specific mouse ELISA (#KMB3481), and Aβ42-specific mouse ELISA (#KMB3441) kits were purchased from Invitrogen, Thermo Fisher Scientific. Aβ ELISA kits were used to measure either secreted human Aβ40 levels in 16 h conditioned medium, or secreted mouse Aβ40 and Aβ42 levels in 20 h conditioned medium following the manufacturer’s protocol. Levels of secreted Aβ were normalized to total cellular protein concentrations measured by Bradford assay.
γ-Secretase inhibitor and BACE1 inhibitor (C3) treatment
HeLa cell monolayers were treated with either 0.01% vol/vol dimethyl sulfoxide (DMSO) (carrier control) or 250 nM γ-secretase inhibitor N-(N-[3,5-difluorophenacetyl]-l-alanyl)-S-phenylglycine t-butyl ester (DAPT; Sigma-Aldrich, Merck, USA). Primary mouse cortical neurons were treated with either 0.01% vol/vol DMSO (carrier control), 2 μM DAPT, or 2 μM β-secretase/BACE1 inhibitor C3 (Calbiochem, Merck). HeLa cells and neurons were incubated for 16 and 20 h, respectively, in a 37°C incubator with 10% CO2.
Cell extracts for immunoblotting
Cellular lysates were extracted using radioimmunoprecipitation (RIPA) lysis buffer (50 mM Tris-HCl, pH 7.3, 150 mM NaCl, 0.1 mM EDTA, 1% wt/vol sodium deoxycholate, 1% vol/vol Triton X-100, 0.2% wt/vol NaF, and 100 μM Na3VO4) supplemented with 1X cOmplete Mini Protease Inhibitor Mixture (Roche Applied Science, Sigma, and Merck). Cell lysates were incubated 10 min on ice followed by centrifugation at 16,000 × g at 4°C for 10 min. Protein concentration was determined by the Bradford protein assay (Bio-Rad) using bovine serum albumin (BSA) protein standards (Pierce BSA standard, Thermo Fisher Scientific).
β-CTF/C99 and α-CTF/C83 blots
The detection of β-CTF/C99 and/or α-CTF/C83 in HeLa-APP695wt cells and primary mouse cortical neurons was previously described (Tan and Gleeson, 2019c). Briefly, cell extracts were prepared in 2× reducing sample buffer containing 10% β-mercaptoethanol and boiled at 100°C. Samples were resolved on 12% NuPAGE Bis–Tris SDS–PAGE polyacrylamide gel (Invitrogen, Thermo Fisher Scientific) at 125 V for ∼2.5 h. Transfer of proteins onto 0.2-μm nitrocellulose membrane (BIO-RAD) was conducted at 400 mA for 1 h on ice. The membrane was incubated and blocked with 10% wt/vol skim milk/PBSTween-20 for 1 h at RT to reduce nonspecific binding. The membrane was incubated overnight at 4°C with either W0-2 (detection of β-CTF/C99) or Y188 (detection of β-CTF/C99 and α-CTF/C83) antibodies diluted 1/6000 in PBS Tween-20. The membrane was then incubated with 1/500 diluted secondary antibodies conjugated with horseradish peroxidase (HRP) for 1 h and washed as above.
Quantitative analyses
Quantitation of the colocalization between either BACE1-GFP or Str-Ii_BACE1FL-SBP-EGFP with endogenous organelle markers in HeLa cells was conducted using an organelle-based colocalization (OBCOL) plug-in (Woodcroft et al., 2009) in FIJI/ImageJ (National Institutes of Health public domain software). Quantitation of the average Golgi intensity was carried out using FIJI/ImageJ. Time-lapse videos of cells with z-stacks were projected with maximum intensity projection, and the frames were aligned with the “Linear Stack Alignment with SIFT” plug-in. A region of interest (ROI) was drawn around the Golgi apparatus, and the average fluorescence intensity over time was measured and recorded for individual cells. The rate of Golgi intensity drop was calculated using linear regression from the maximum Golgi intensity point. The percentage of colocalization of cargo with various organelles was calculated by dividing the total cargo pixels that overlapped with each organelle marker by the total cargo pixels within each cell. All analysis was carried out on the indicated number of cells over three independent experiments.
Owing to the superiority in image analysis of z-stack images, Imaris microscopy image analysis software (Oxford Instruments) was used over OBCOL to determine the colocalization of endogenous BACE1 with organelle markers in primary cortical neurons at steady-state. Prior to Imaris analyses, confocal images of primary cortical neurons in z-stacks were deconvoluted with Huygens software (Scientific Volume Imaging) using the default conservative deconvolution setting. Imaris thresholding parameters used were grain size/surface detail 0.0847 μm, diameter of largest sphere 0.2 μm, and number of voxels above 10. Imaris surface–surface colocalization tool was used to obtain volume sums of BACE1, the organelle marker, and colocalization between BACE1 and the organelle marker. To obtain the percentage of colocalization, the volume sum of colocalization between BACE1 and a specific organelle marker was divided by the total volume sum of BACE1. To determine the levels of colocalization of either endogenous BACE1 or APP with endogenous p230/golgin-245 based on pixel intensity in shRNA-treated neurons, Volocity imaging software (Perkin Elmer, UK) was used to calculate Manders’ coefficient M1 values (Manders et al., 1993) of either endogenous BACE1 or APP with endogenous p230/golgin-245. All analyses were carried out on the indicated number of cells over three independent experiments.
Generation of graphs and statistical analyses
The generation of graphs and statistical analyses was conducted using Prism (GraphPad Software). Data for fold change of α-CTF/C83 and β-CTF/C99 levels were plotted as bar graphs. Data from Aβ ELISAs and flow cytometry experiments were plotted as bar graphs and analyzed by an unpaired, two-tailed Student’s t test. Data from quantitation of colocalization, average BACE1 fluorescence intensity, and the rate of decrease in BACE1 fluorescence intensity at the Golgi were plotted as dot plots and analyzed by an unpaired, two-tailed Student’s t test. Data sets involving multiple comparisons to the control were analyzed by one-way analysis of variance (ANOVA) using Tukey’s multiple comparison test. A p value of <0.05 (*) was considered significant, a p value of <0.01 (**) was considered highly significant, and a p value of <0.001 (***) was considered very highly significant.
Supplementary Material
Acknowledgments
Confocal microscopy was performed at the Biological Optical Microscopy Platform (BOMP) at the University of Melbourne. This work was supported by funding from the National Health and Medical Research Council of Australia (APP1163862). J.Z.A.T. and J.W. are supported by University of Melbourne International Postgraduate Awards. We gratefully acknowledge Fiona Houghton for reagents and expert technical advice, Wei Hong Toh and other members of the Gleeson laboratory for valuable discussions, Allison Van De Meene for maintenance of the spinning disk, and Ellie Cho for help with the quantitation.
Abbreviations used:
- Aβ
beta-amyloid peptide
- AD
Alzheimer’s disease
- APP
amyloid precursor protein
- BACE1
beta site APP-cleaving enzyme
- DIV
day in vitro
- ER
endoplasmic reticulum
- GFP
green fluorescent protein
- PM
plasma membrane
- RUSH
Retention Using Selective Hooks
- TfR
transferrin receptor
- TGN
trans-Golgi network
- TIRF
total internal reflection fluorescence microscopy
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E19-09-0487) on November 20, 2019.
REFERENCES
- Bennett BD, Babu-Khan S, Loeloff R, Louis JC, Curran E, Citron M, Vassar R. (2000). Expression analysis of BACE2 in brain and peripheral tissues. J Biol Chem , 20647–20651. [DOI] [PubMed] [Google Scholar]
- Boncompain G, Divoux S, Gareil N, de Forges H, Lescure A, Latreche L, Mercanti V, Jollivet F, Raposo G, Perez F. (2012). Synchronization of secretory protein traffic in populations of cells. Nat Methods , 493–498. [DOI] [PubMed] [Google Scholar]
- Boncompain G, Perez F. (2012). Synchronizing protein transport in the secretory pathway. Curr Protoc Cell Biol Chapter 15, Unit 15 19. [DOI] [PubMed] [Google Scholar]
- Burgos PV, Mardones GA, Rojas AL, daSilva LL, Prabhu Y, Hurley JH, Bonifacino JS. (2010). Sorting of the Alzheimer’s disease amyloid precursor protein mediated by the AP-4 complex. Dev Cell , 425–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camus G, Segura-Morales C, Molle D, Lopez-Verges S, Begon-Pescia C, Cazevieille C, Schu P, Bertrand E, Berlioz-Torrent C, Basyuk E. (2007). The clathrin adaptor complex AP-1 binds HIV-1 and MLV Gag and facilitates their budding. Mol Biol Cell , 3193–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casler JC, Papnikou E, Barrero JJ, Glick BS. (2019). Maturation-driven transport and AP-1–dependent recycling of a secretory cargo in the Golgi. J Cell Biol , 1582–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HJ, Yuan J, Lobel P. (1997). Systematic mutational analysis of the cation-independent mannose 6-phosphate/insulin-like growth factor II receptor cytoplasmic domain. An acidic cluster containing a key aspartate is important for function in lysosomal enzyme sorting. J Biol Chem , 7003–7012. [DOI] [PubMed] [Google Scholar]
- Chen Y, Gershlick DC, Park SY, Bonifacino JS. (2017). Segregation in the Golgi complex precedes export of endolysosomal proteins in distinct transport carriers. J Cell Biol , 4141–4151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chia PZ, Toh WH, Sharples R, Gasnereau I, Hill AF, Gleeson PA. (2013). Intracellular itinerary of internalised beta-secretase, BACE1, and its potential impact on beta-amyloid peptide biogenesis. Traffic , 997–1013. [DOI] [PubMed] [Google Scholar]
- De Matteis MA, Luini A. (2008). Exiting the Golgi complex. Nat Rev Mol Cell Biol , 273–284. [DOI] [PubMed] [Google Scholar]
- Donaldson JG, Jackson CL. (2011). ARF family G proteins and their regulators: roles in membrane transport, development and disease. Nat Rev Mol Cell Biol , 362–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doray B, Ghosh P, Griffith J, Geuze HJ, Kornfeld S. (2002). Cooperation of GGAs and AP-1 in packaging MPRs at the trans-Golgi network. Science , 1700–1703. [DOI] [PubMed] [Google Scholar]
- Eng FJ, Varlamov O, Fricker LD. (1999). Sequences within the cytoplasmic domain of gp180/carboxypeptidase D mediate localization to the trans-Golgi network. Mol Biol Cell , 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farias GG, Cuitino L, Guo X, Ren X, Jarnik M, Mattera R, Bonifacino JS. (2012). Signal-mediated, AP-1/clathrin-dependent sorting of transmembrane receptors to the somatodendritic domain of hippocampal neurons. Neuron , 810–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng T, Tammineni P, Agrawal C, Jeong YY, Cai Q. (2017). Autophagy-mediated regulation of BACE1 protein trafficking and degradation. J Biol Chem , 1679–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravotta D, Carvajal-Gonzalez JM, Mattera R, Deborde S, Banfelder JR, Bonifacino JS, Rodriguez-Boulan E. (2012). The clathrin adaptor AP-1A mediates basolateral polarity. Dev Cell , 811–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Chang WP, Koelsch G, Tang J. (2002). Memapsin 2 (beta-secretase) cytosolic domain binds to the VHS domains of GGA1 and GGA2: implications on the endocytosis mechanism of memapsin 2. FEBS Lett , 183–187. [DOI] [PubMed] [Google Scholar]
- He X, Li F, Chang WP, Tang J. (2005). GGA proteins mediate the recycling pathway of memapsin 2 (BACE). J Biol Chem , 11696–11703. [DOI] [PubMed] [Google Scholar]
- Hille-Rehfeld A. (1995). Mannose 6-phosphate receptors in sorting and transport of lysosomal enzymes. Biochim Biophys Acta , 177–194. [DOI] [PubMed] [Google Scholar]
- Hirst J, Borner GH, Antrobus R, Peden AA, Hodson NA, Sahlender DA, Robinson MS. (2012). Distinct and overlapping roles for AP-1 and GGAs revealed by the “knocksideways” system. Curr Biol , 1711–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houghton FJ, Bellingham SA, Hill AF, Bourges D, Ang DK, Gemetzis T, Gasnereau I, Gleeson PA. (2012). Arl5b is a Golgi-localised small G protein involved in the regulation of retrograde transport. Exp Cell Res , 464–477. [DOI] [PubMed] [Google Scholar]
- Huse JT, Liu K, Pijak DS, Carlin D, Lee VM, Doms RW. (2002). Beta-secretase processing in the trans-Golgi network preferentially generates truncated amyloid species that accumulate in Alzheimer’s disease brain. J Biol Chem , 16278–16284. [DOI] [PubMed] [Google Scholar]
- Huse JT, Pijak DS, Leslie GJ, Lee VM, Doms RW. (2000). Maturation and endosomal targeting of beta-site amyloid precursor protein-cleaving enzyme. The Alzheimer’s disease beta-secretase. J Biol Chem , 33729–33737. [DOI] [PubMed] [Google Scholar]
- Hussain I, Powell D, Howlett DR, Tew DG, Meek TD, Chapman C, Gloger IS, Murphy KE, Southan CD, Ryan DM, et al (1999). Identification of a novel aspartic protease (Asp 2) as beta-secretase. Mol Cell Neurosci , 419–427. [DOI] [PubMed] [Google Scholar]
- Jordan M, Schallhorn A, Wurm FM. (1996). Transfecting mammalian cells: optimization of critical parameters affecting calcium-phosphate precipitate formation. Nucleic Acids Res , 596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita A, Fukumoto H, Shah T, Whelan CM, Irizarry MC, Hyman BT. (2003). Demonstration by FRET of BACE interaction with the amyloid precursor protein at the cell surface and in early endosomes. J Cell Sci , 3339–3346. [DOI] [PubMed] [Google Scholar]
- Kooy J, Toh BH, Pettitt JM, Erlich R, Gleeson PA. (1992). Human autoantibodies as reagents to conserved Golgi components. Characterization of a peripheral, 230-kDa compartment-specific Golgi protein. J Biol Chem , 20255–20263. [PubMed] [Google Scholar]
- Lane CA, Hardy J, Schott JM. (2018). Alzheimer’s disease. Eur J Neurol , 59–70. [DOI] [PubMed] [Google Scholar]
- Le Borgne R, Hoflack B. (1997). Mannose 6-phosphate receptors regulate the formation of clathrin-coated vesicles in the TGN. J Cell Biol , 335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee I, Doray B, Govero J, Kornfeld S. (2008). Binding of cargo sorting signals to AP-1 enhances its association with ADP ribosylation factor 1-GTP. J Cell Biol , 467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowery J, Szul T, Styers M, Holloway Z, Oorschot V, Klumperman J, Sztul E. (2013). The Sec7 guanine nucleotide exchange factor GBF1 regulates membrane recruitment of BIG1 and BIG2 guanine nucleotide exchange factors to the trans-Golgi network (TGN). J Biol Chem , 11532–11545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luke MR, Kjer-Nielsen L, Brown DL, Stow JL, Gleeson PA. (2003). GRIP domain-mediated targeting of two new coiled-coil proteins, GCC88 and GCC185, to subcompartments of the trans-Golgi network. J Biol Chem , 4216–4226. [DOI] [PubMed] [Google Scholar]
- Manders EMM, Verbeek FJ, Aten JA. (1993). Measurement of co-localization of objects in dual-colour confocal images. J Microsc , 375–382. [DOI] [PubMed] [Google Scholar]
- Meyer C, Zizioli D, Lausmann S, Eskelinen EL, Hamann J, Saftig P, von Figura K, Schu P. (2000). mu1A-Adaptin-deficient mice: lethality, loss of AP-1 binding and rerouting of mannose 6-phosphate receptors. EMBO J , 2193–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakai W, Kondo Y, Saitoh A, Naito T, Nakayama K, Shin HW. (2013). ARF1 and ARF4 regulate recycling endosomal morphology and retrograde transport from endosomes to the Golgi apparatus. Mol Biol Cell , 2570–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro Negredo P, Edgar JR, Wrobel AG, Zaccai NR, Antrobus R, Owen DJ, Robinson MS. (2017). Contribution of the clathrin adaptor AP-1 subunit micro1 to acidic cluster protein sorting. J Cell Biol , 2927–2943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmer HB, Duncan R. (2016). A novel tribasic Golgi export signal directs cargo protein interaction with activated Rab11 and AP-1–dependent Golgi–plasma membrane trafficking. Mol Biol Cell , 1320–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhu Y, Burgos PV, Schindler C, Farias GG, Magadan JG, Bonifacino JS. (2012). Adaptor protein 2-mediated endocytosis of the beta-secretase BACE1 is dispensable for amyloid precursor protein processing. Mol Biol Cell , 2339–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi-Takahara Y, Morishima-Kawashima M, Tanimura Y, Dolios G, Hirotani N, Horikoshi Y, Kametani F, Maeda M, Saido TC, Wang R, Ihara Y. (2005). Longer forms of amyloid beta protein: implications for the mechanism of intramembrane cleavage by gamma-secretase. J Neurosci , 436–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rechards M, Xia W, Oorschot VM, Selkoe DJ, Klumperman J. (2003). Presenilin-1 exists in both pre- and post-Golgi compartments and recycles via COPI-coated membranes. Traffic , 553–565. [DOI] [PubMed] [Google Scholar]
- Reiling JH, Olive AJ, Sanyal S, Carette JE, Brummelkamp TR, Ploegh HL, Starnbach MN, Sabatini DM. (2013). A CREB3-ARF4 signalling pathway mediates the response to Golgi stress and susceptibility to pathogens. Nat Cell Biol , 1473–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer WF, Syverton JT, Gey GO. (1953). Studies on the propagation in vitro of poliomyelitis viruses. IV. Viral multiplication in a stable strain of human malignant epithelial cells (strain HeLa) derived from an epidermoid carcinoma of the cervix. J Exp Med , 695–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schieb H, Kratzin H, Jahn O, Mobius W, Rabe S, Staufenbiel M, Wiltfang J, Klafki HW. (2011). Beta-amyloid peptide variants in brains and cerebrospinal fluid from amyloid precursor protein (APP) transgenic mice: comparison with human Alzheimer amyloid. J Biol Chem , 33747–33758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C, Sutherland R, Newman R, Greaves M. (1982). Structural features of the cell surface receptor for transferrin that is recognized by the monoclonal antibody OKT9. J Biol Chem , 8516–8522. [PubMed] [Google Scholar]
- Shimizu H, Tosaki A, Kaneko K, Hisano T, Sakurai T, Nukina N. (2008). Crystal structure of an active form of BACE1, an enzyme responsible for amyloid beta protein production. Mol Cell Biol , 3663–3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, Doan M, Dovey HF, Frigon N, Hong J, et al (1999). Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature , 537–540. [DOI] [PubMed] [Google Scholar]
- Southon A, Greenough M, Hung YH, Norgate M, Burke R, Camakaris J. (2011). The ADP-ribosylation factor 1 (Arf1) is involved in regulating copper uptake. Int J Biochem Cell Biol , 146–153. [DOI] [PubMed] [Google Scholar]
- Stamnes MA, Rothman JE. (1993). The binding of AP-1 clathrin adaptor particles to Golgi membranes requires ADP-ribosylation factor, a small GTP-binding protein. Cell , 999–1005. [DOI] [PubMed] [Google Scholar]
- Takami M, Nagashima Y, Sano Y, Ishihara S, Morishima-Kawashima M, Funamoto S, Ihara Y. (2009). gamma-Secretase: successive tripeptide and tetrapeptide release from the transmembrane domain of beta-carboxyl terminal fragment. J Neurosci , 13042–13052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan JZA, Gleeson PA. (2019a). Cargo sorting at the trans-Golgi network for shunting into specific transport routes: role of Arf small G proteins and adaptor complexes. Cells , E531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan JZA, Gleeson PA. (2019b). The role of membrane trafficking in the processing of amyloid precursor protein and production of amyloid peptides in Alzheimer’s disease. Biochim Biophys Acta Biomembr , 697–712. [DOI] [PubMed] [Google Scholar]
- Tan JZA, Gleeson PA. (2019c). The trans-Golgi network is a major site for alpha-secretase processing of amyloid precursor protein in primary neurons. J Biol Chem , 1618–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thinakaran G, Koo EH. (2008). Amyloid precursor protein trafficking, processing, and function. J Biol Chem , 29615–29619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toh WH, Chia PZC, Hossain MI, Gleeson PA. (2018). GGA1 regulates signal-dependent sorting of BACE1 to recycling endosomes which moderates Abeta production. Mol Biol Cell , 191–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toh WH, Gleeson PA. (2016). Dysregulation of intracellular trafficking and endosomal sorting in Alzheimer’s disease: controversies and unanswered questions. Biochem J , 1977–1993. [DOI] [PubMed] [Google Scholar]
- Toh WH, Tan JZ, Zulkefli KL, Houghton FJ, Gleeson PA. (2017). Amyloid precursor protein traffics from the Golgi directly to early endosomes in an Arl5b- and AP4-dependent pathway. Traffic , 159–175. [DOI] [PubMed] [Google Scholar]
- Tomita T. (2014). Molecular mechanism of intramembrane proteolysis by gamma-secretase. J Biochem , 195–201. [DOI] [PubMed] [Google Scholar]
- Voorhees P, Deignan E, van Donselaar E, Humphrey J, Marks MS, Peters PJ, Bonifacino JS. (1995). An acidic sequence within the cytoplasmic domain of furin functions as a determinant of trans-Golgi network localization and internalization from the cell surface. EMBO J , 4961–4975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodcroft BJ, Hammond L, Stow JL, Hamilton NA. (2009). Automated organelle-based colocalization in whole-cell imaging. Cytometry A , 941–950. [DOI] [PubMed] [Google Scholar]
- Yan R, Bienkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM, Brashier JR, Stratman NC, Mathews WR, Buhl AE, et al (1999). Membrane-anchored aspartyl protease with Alzheimer’s disease beta-secretase activity. Nature , 533–537. [DOI] [PubMed] [Google Scholar]
- Ye X, Cai Q. (2014). Snapin-mediated BACE1 retrograde transport is essential for its degradation in lysosomes and regulation of APP processing in neurons. Cell Rep , 24–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye X, Feng T, Tammineni P, Chang Q, Jeong YY, Margolis DJ, Cai H, Kusnecov A, Cai Q. (2017). Regulation of synaptic amyloid-beta generation through BACE1 retrograde transport in a mouse model of Alzheimer’s disease. J Neurosci , 2639–2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YW, Thompson R, Zhang H, Xu H. (2011). APP processing in Alzheimer’s disease. Mol Brain , 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.