

Abstract
Bacterial natural products and their analogues constitute more than half of the new small molecule drugs developed over the last several decades. Despite this success, interest in natural products from major pharmaceutical companies has decreased even as genomics has uncovered the large number of biosynthetic gene clusters (BGCs) that encode for novel natural products. To date though, there is still a lack of universal strategies and enabling technologies to discover natural products at scale and speed. This review highlights several of the opportunities provided by genome sequencing and bioinformatics, challenges associated with translating genomes into natural products, and examples of successful strain prioritization and BGC activation strategies that have been used in the genomic era for natural product discovery from cultivatable bacteria.
Keywords: Genome mining, natural products, drug discovery, biosynthetic gene clusters, polyketides, nonribosomal peptides
The Role of Natural Products in Drug Discovery
Natural products (NPs) have a proven track record of success in the history of drug discovery and development, and they continue to play a significant role.[1, 2] Among all approved small molecule drugs worldwide from 1981 to 2014, more than 50% of them are of NP origin or contain a NP pharmacophore.[3] This contribution is even more remarkable considering that many major pharmaceutical companies have switched their drug discovery programs from NPs to synthetic combinatorial libraries in the last 20-30 years. This shift has been primarily due to a false perception of diminishing numbers of novel scaffolds and a high rate of rediscovery of NPs yielded by traditional strategies.[4, 5] However, the advent of low-cost and rapid genome sequencing provides an informed method for targeted NP discovery, whether for structure, novelty, or activity, thereby promising to eliminate these common pitfalls of the pre-genomic era. Specifically, genome sequencing of bacteria, prolific sources of pharmaceutically-relevant NPs, has revealed that known NPs represent just the tip of the iceberg relative to the vast diversity encoded within bacterial genomes,[6] highlighting how many more NPs have never been pursued due to low or non-existent production titers. Encouraged by this advancement and new challenges it presents, innovative strategies are continuously being developed, and novel NPs have been discovered by mining bacterial genomic information. This review will highlight the opportunities brought on by genome sequencing and advanced bioinformatics tools, the challenges that we still face, and some of the current strategies being utilized in the genome-directed discovery of new NPs from cultivable bacteria.
Opportunities of the Genomic Era
Even by the most modest models, the worldwide annual sequencing capacity is predicted to reach exa-base pairs (1018 base pairs) by the early 2020s.[7] Though much of this capacity is dedicated to sequencing human genomes, at approximately 107 bases, bacterial genomes are a small fraction of the size and can be sequenced at far greater rates. Indeed, as of May 2019, public sequencing data (from the NCBI databasei), exists for more than 211,000 bacteria, providing rich genomic diversity (Figure 1A).[8] Several thousand more genomes are represented in metagenomic (see Glossary) datasets.[8] Of the 211,705 sequenced bacterial genomes, most represent human pathogens from the Firmicutes and Gammaproteobacteria phyla. In contrast, less than 10% of publicly available sequenced bacterial genomes belong to prolific natural product-producing Actinobacteria phylum. While pathogen genomes are undoubtedly important for studying human health, the much smaller percentage of Actinobacteria demonstrates our opportunity for targeted genome sequencing of privileged bacteria for the purpose of discovering novel NPs. As our sequencing capacity increases, the cost decreases, recently falling under $1000 for a human genome and significantly less than that for a bacterial genome—enabling any lab to take advantage of the genomic era to revolutionize NP discovery.[9] Bioinformatics studies have demonstrated that within a single bacterial genome, there can be upwards of 30 biosynthetic gene clusters (BGCs), with most encoding for unknown NPs.[10] Indeed, a survey by the Joint Genomics Institute (JGI) has found that less than 0.25% of identified BGCs have been experimentally correlated to known NPs.[11] For example, in Streptomyces avermitilis, one of the most well-studied bacterial strains, 23 out of the total predicted 40 BGCs are silent under explored culturing conditions (Figure 1B).[12] Combined with estimates of the total number of bacterial species ranging from billions to one trillion,[13, 14] the field is presented with a prime opportunity for NP discovery. However, unlike S. avermitilis and numerous other Actinobacteria, many of the currently sequenced bacteria have been sequenced based on pathogenicity rather than for their biosynthetic potential, further highlighting the presented opportunity for targeted BGC discovery. We next highlight the challenges facing the community in taking full advantage of these opportunities.
Figure 1: Untapped potential of bacterial genomes.

(A) The diversity of sequenced genomes in the NCBI database sorted by phyla. (B) The genome of the model organism Streptomyces avermitilis is depicted with the locations of 40 putative BGCs indicated. Gray arrows (23) designate orphan BGCs, while blue arrows (17) link a BGC with the structure of its NP.
Challenges in Genome-Directed NP Discovery
While the opportunities now exist for discovering more NPs than ever before, the key goals of genome-guided NP discovery are: (1) expansion of NP diversity, (2) prioritization and prediction of BGCs, and (3) rapid production of NPs from silent BGCs. For each goal, however, many challenges exist that hinder or prevent efficient exploitation of these opportunities. It is unknown how many permutations exist for bacterial NPs or even how many genomes must be sequenced to achieve a plateau in BGC discovery. Random selection of BGCs will result in an increased number of NPs, but to take full advantage of NP diversity, BGCs must be prioritized by product novelty and/or activity. Once chosen, the odds of the BGC of interest even being expressed under standard conditions by the native host are poor, though this challenge may be combated somewhat by discovering alternative producers for well-conserved (and presumably valuable) BGCs. Regardless, new technologies and defined strategies are required to position ourselves for success in NP discovery in the genomic era.
Various bioinformatics tools are available for identification or classification of BGCs from genomic information,[15] but because these tools were developed based on current biosynthetic knowledge, novel families of BGCs may be missed or mischaracterized. The prediction of the NP structures from genetic information is another critical step for genome mining studies, as this step evaluates the structural novelty of NPs and facilitates their dereplication. Generally, accurate predictions can be made for the class of NP (e.g., polyketides, peptides, terpenes, etc.) and, in some cases, the core structures of the NPs (e.g., products of type I polyketide synthases [PKSs] and nonribosomal peptide synthetases [NRPSs]). However, as opposed to core biosynthetic enzymes, the function of most tailoring enzymes or non-collinear enzymes cannot be predicted precisely, so predictions for the final NP structures are still far from adequate. An additional challenge for NPs, especially those with rare or poorly-characterized self-resistance genes, which are arguably the most valuable, is the unpredictability of their bioactivities, making activity-based prioritization difficult if not impossible. For silent or low-expressing BGCs, potentially making up 90% of all BGCs, translating the results of genome mining and BGC prioritization into a NP is not always trivial.[16] Innovative strategies and generalizable synthetic biology tools need to be developed to access these BGCs and identify the encoded NPs.
Ultimately, while research groups are working to overcome these and other challenges associated with genome-guided NP discovery, the development of any universal tools or strategies will require advances in several fields, including chemistry, synthetic biology, microbiology, and computational biology. Few academic labs have the resources to span most of these fields, much less all of them at once, and thus, larger, interdisciplinary collaborations across institutes are needed.
Strategies and Selected Examples for Genome-Based Discovery of Novel NPs
Several strategies and enabling technologies have been developed to facilitate NP discovery from bacteria to address many of the initial challenges highlighted above, though many still remain. Often, the best strategy to choose depends primarily on the entry point into the discovery pipeline: whether the strain is sequenced or not and how specific the targeted structure or function is. Below, we highlight some of the most successful and generalizable strategies and examples thereof.
BGC prioritization from sequenced and unsequenced sources
While the community has acquired a substantial collection of sequenced bacterial genomes, the full biosynthetic potential mostly lies in the remaining unsequenced strains, and accessing this additional diversity is critical to the ongoing success of NP discovery programs. Several successful strategies have been developed to take advantage of one or both sources, typically focused on structural novelty (Figure 2, Key Figure). As sequencing data accumulate, the emphasis of BGC prioritization will necessarily shift from selecting an interesting gene or BGC to the prioritization of which novel gene or BGC among thousands or millions to select. Mirroring this trend, bioinformatics tools have improved significantly to the point that it is now relatively simple to group related genes by sequence (sequence similarity network [SSNs])ii,[17] genomic proximity (genome neighborhood network [GNNs])ii,[17, 18] and BGC family (BiG-SCAPE)iii[19] or to predict NP BGCs (antiSMASHiv or PRISMv).[20-22] These tools are typically utilized in combination with databases of characterized BGCs, e.g. Minimum Information about a Biosynthetic Gene Cluster (MIBiG)vi.[23] Used together, large numbers of diverse sequences can be rapidly sorted and novelty can be identified. This novelty could result in an entirely new class of NPs or diversity among a family of known clinically-relevant NPs or could be used to identify genetically-amenable and/or high-producing alternative producers of known NPs.
Figure 2, Key Figure: Strategies for natural product discovery.

The schematic depicts the general strategies that are available to identify and prioritize BGCs from sequenced (top half) and unsequenced (bottom half) sources. Partial structural predictions are possible for some enzymes, purely from sequencing data, such as for the depicted hypothetical NRPS in which each module, represented by a different color, can incorporate a corresponding amino acid into the final structure. A series of computational tools exist for identifying and comparing individual genes or BGCs, e.g. an SSN, where each color represents a different family of genes or proteins. From unsequenced strain collections, new candidate BGCs can be prioritized via rt-PCR screening based on information obtained from sequenced genomes, such as those from promising NP families (e.g., the enediynes, whose core structure is highlighted in red), or through resistance gene-guided assays (orange genes) for target prediction. Once an unsequenced strain has been selected, the accessibility of genome sequencing provides the ability to feed back into the sequenced databases and identify a specific BGC.
Key to discovering novelty is our ability to make predictions for the NP from genetic sequence. Our ever-increasing knowledge of NP biosynthesis allows the correlation of a structural motif to its corresponding biosynthetic enzyme(s) or domain(s). Using this as inspiration, methods have been developed to quickly scan the genomes of sequenced and unsequenced strains to identify BGCs encoding the type of enzymes of interest with the potential to produce NPs featuring the desired structural motif(s).[24-28] This is especially true for polyketides,[29, 30] ribosomal and non-ribosomal peptides,[22, 31] and sometimes terpenoids.[32] These predictions gain even greater importance within a family of related BGCs by showcasing natural combinatorial biosynthesis, as for the leinamycin family described later.[24]
Importantly, genome-guided NP discovery is not limited to bacteria with sequenced genomes. Instead, existing genomic data can be utilized to inform screening campaigns for unsequenced bacteria, which, upon sequencing any bacteria of interest, result in an even stronger genetic database (Figure 2). Targeting unsequenced bacteria is a very important strategy as public genomes are not currently biased towards high-level NP producers. Crucially though, there is enough public genomic data (NCBI databasei available that degenerate primers can be designed for genome mining of unsequenced bacteria with a specific chemical moiety, as with the phosphonate family of NPs highlighted below.[28] These screens often utilize both conventional and real-time PCR (rt-PCR) to identify BGCs encoding members of a selected NP family, much in the same way a BLASTvii search of a sequenced database would work.[24, 33-35] A further strategy for prioritizing bacteria, sequenced or unsequenced, is guided by resistance genes, rather than by the biosynthetic genes themselves. This strategy targets NP functions and is detailed further in a separate section.
Additionally, BGC prioritization does not have to be dependent on currently sequenced bacteria, but rather, it depends in large part on the choice of bacterial strains to be sequenced in the future. As stated, less than 10% of bacterial genomes currently in the NCBI database belong to the NP-rich Actinobacteria (Figure 1A). Thus, by balancing NP-privileged and rare taxa in future sequencing efforts, more BGC diversity can be sampled than is currently available. This diversity can be a result of phylogeny (e.g., the rare Streptosporangium versus the common but prolific Streptomyces) or ecology (e.g., bacteria from a marine sponge versus bacteria from rainforest soil). While it is impossible to predict exactly which strains will be the best NP producers solely from phylogeny or ecology, by sampling diversity using both factors, future prioritization will be better informed by a stronger genomic and BGC database.
Finally, when sequencing bacterial genomes for the purpose of NP discovery, it is critical to consider the quality of the resultant sequences. As BGCs can span over 100 kb in length and are dependent on accurate sequencing of a large number of genes for product structure and function predictions, genomes with many errors or large numbers of contigs could be considered detrimental to the discovery process. However, while a complete genome sequence is ideal, more research is needed on this topic to determine what is the minimally acceptable genome quality for NP discovery efforts.
Discovery of the leinamycin family of NPs: Scaffold-directed genome mining from sequenced and unsequenced strain collections
Leinamycin (LNM) contains a unique 1,3-dioxo-1,2-dithiolane moiety spirofused to an 18-membered macrolactam ring (Figure 3A) and is a promising anticancer drug lead due to its potent activity and unprecedented mode of action.[36] However, despite being discovered from Streptomyces atroolivaceus S-140 in 1989, no additional natural producers or analogues were reported for nearly thirty years.[37] In 2002, the LNM BGC was identified and shown to encode a hybrid NRPS-PKS consisting of 2 NRPS modules and 6 PKS modules (Figure 3A).[38, 39] In 2017, to identify other LNM-like NPs, a structure-targeted genome mining strategy was applied to the publicly-available bacterial genomes (NCBI databasei) and 5000 in-house unsequenced Actinobacteria strains (Figure 3B-C).[24] As the LNM C-3 sulfur was known to be incorporated by a unique DUF-SH didomain (shown in Figure 3A), it was proposed that this genomic region could be used to identify additional lnm-type BGCs. With the DUF-SH sequence as a query, 19 lnm-type BGCs could be detected in genomes from the NCBI database. The new sequence information provided the conserved regions of the DUF-SH didomain necessary for the design of degenerate primers, and the subsequent high-throughput rt-PCR screen of the in-house strain collection resulted in the discovery of an additional 30 lnm-type BGCs, for a total of 49 BGCs.[24] Thus, the utility of untargeted public sequencing data was demonstrated for the targeted discovery of rare BGCs in an unsequenced strain library while simultaneously highlighting the incompleteness of the public database.
Figure 3: Discovery of the leinamycin family of NPs.

(A) The lnm core biosynthetic genes are depicted with the color representing the encoded enzyme type: NRPS (blue), PKS (red), and the DUF-SH didomain (green). The structure of LNM is also shown with its colors corresponding to the biosynthetic origin of the specific molecular region. The variability of the core structure is organized by module based on the predicted products of the 49 lnm-type BGCs discovered. (B) Approximately 200,000 bacterial genomes were searched in silico for sequences containing a DUF-SH didomain, resulting in the identification of 19 lnm-type BGCs and the design of degenerate primers for the PCR-guided discovery of lnm-type BGCs from unsequenced strains. (C) Rt-PCR of the DUF-SH didomain in 5,000 unsequenced Actinobacteria resulted in the further identification of 30 more lnm-type BGCs. (D) Two novel LNM-type NPs, guangnanmycin A and weishanmycin A1, were isolated as representatives from 2 of the 18 LNM-type clades. The sulfurs highlighted in green are predicted to be installed by DUF-SH didomains.
In addition to the discovery of new lnm-type BGCs, sequencing of the in-house strains confirmed the existence of as many as 18 distinct lnm-type clades, resulting in permutations of the predicted structures corresponding to products of six of the eight NRPS or PKS biosynthetic modules (Figure 3A).[24] Bioinformatics tools can predict an approximate core structure based on the sequences of these modules;[21, 22] however, it is still nearly impossible to predict the final structure of NPs from their encoding genetic information. Notably, two new LNM-like NP families, the guangnanmycins (GNMs) and the weishanmycins (WSMs), were isolated from strains Streptomyces sp. CB01883 and Streptomyces sp. CB02120-2, respectively (Figure 3D).[24] Both the GNMs and WSMs differ substantially from LNM in structure, showcasing the structural diversity produced by the varying lnm-type BGCs. This example highlights the power of genome mining as a general strategy for surveying sequenced and unsequenced strains for targeted, structure-based discovery of NPs.
Discovery of phosphonate family of NPs: High-throughput genome mining and sequencing from an unsequenced strain library
The phosphonate family of NPs have an impressive track record to function as pharmaceutically relevant NPs due to their innate similarity to phosphate-containing nutrients.[40] Much like the example of LNM reviewed above, the biosynthetic machinery responsible for this pharmacophore can be identified by the presence of a core gene, pepM, which in this case encodes for phosphoenolpyruvate phosphomutase. However, in stark contrast to the rare LNM family, many representatives of the phosphonate family were known prior to an intensive genome mining project.[41] As such, degenerate primers targeting pepM were readily established and used to screen multiple unsequenced strain collections totaling ~10,000 Actinobacteria (Figure 4A).[28] Draft genome sequencing of 403 strains identified by this screen confirmed the presence of pepM in more than two-thirds of the candidates (Figure 4B). After dereplication, a diverse collection of 192 strains encoding 78 distinct phosphonate BGCs remained (>85% novel). Bioinformatics and statistical analysis was used to determine the collection represented ~62% of a predicted 125 possible phosphonate BGCs, requiring screening of ~40,000 additional strains before saturating Actinobacteria phosphonate BGCs (Figure 4D).[28, 42]
Figure 4: Genome mining of phosphonate NPs.

(A) A PCR screen targeting the pepM gene was used to identify phosphonate BGCs from 10,000 Actinobacteria. (B) Hit strains from the PCR screen were sequenced and phosphonate BGCs identified therein. (C) Extracts from strains containing phosphonate BGCs were assayed with an engineered E. coli strain hypersensitive to phosphonates. (D) Bioinformatics and statistics were used to map the diversity of phosphonate BGCs and to extrapolate how much phosphonate diversity remains in Actinobacteria. (E) Examples of novel phosphonate compounds isolated from genome mining are shown.
In addition to the valuable genomic information, this genome mining effort also resulted in several new phosphonates. Of the 45 putative phosphonate producers generating indicative 31P NMR signals (23%), three strains that displayed positive results in response to a phosphonate-specific bioassay using an engineered E. coli with inducible hypersensitivity to phosphonate antibiotics[43] (Figure 4C) were focused on. From these strains, a rare sulfur-containing phosphonate, argolaphos B, containing a rare amino acid (N5 -hydroxyarginine), was isolated and showed potential broad-spectrum antibiotic activity (Figure 4E). Hence, here, similar to the LNM genome mining, the boundaries of a NP family’s diversity could be pushed, but instead of focusing on a single scaffold, a more relaxed strategy provided a view of the overall diversity of NPs containing a specific moiety (phosphonate) produced by a single class of bacteria (Actinobacteria).
Targeting resistance genes for NP discovery
As a general strategy that can be applied to either sequenced or unsequenced genomes, resistance-conferring genes can be targeted for discovery of NPs with predictable targets or modes of action. This strategy, while gaining popularity in recent years, is still limited by the need for knowledge of rare or unusual resistance mechanisms, but it has yielded NPs with an impressive array of bioactivities.[44-48] Targets can include duplicated housekeeping genes (e.g. fatty acid synthase or proteasome components),[44-46] genes encoding target eukaryotic proteins (e.g. a known herbicide target),[47] or other rare resistance genes (e.g. a gene associated with topoisomerase inhibitors).[48] By looking for resistance markers, expensive and time-consuming functional assays can be partly replaced by genetic screening, without any bias towards or prior knowledge of a final NP structure. To highlight this strategy, the resistance-based discovery of the thiotetronic acid antibiotics is discussed below.[44]
Discovery of thiotetronic acid antibiotics: Self-Resistance (target)-directed genome mining and BGC prioritization
Using a resistance-directed genome mining strategy, the genomes of 86 Salinispora strains were bioinformatically surveyed for BGCs that contained putative resistance genes encoding the protein target of the NP (Figure 5A).[44] This analysis resulted in the identification of all orthologous groups (OGs) within the Salinispora pan-genome and core genome (Figure 5A, step i). The core genes were classified by sequence similarity into clusters of orthologous groups (COGs), and any non-conserved OG from the pan-genome that fit into a COG from the core genome was considered duplicated (Figure 5A, step ii). Among the duplicated OGs, nearly 40% were associated with secondary metabolite BGCs (Figure 5A, step iii).[44]
Figure 5: Discovery of thiotetronic acid antibiotics.

(A) The tlm BGC was prioritized by searching through the Salinispora pan-genome for duplicated housekeeping genes for BGCs as depicted. First, the core OGs present in all Salinispora were identified and grouped by similarity (i). Any duplicated OGs from the core genome were then examined (ii), and those found within predicted BGCs were analyzed (iii). Further categorization and prioritization yielded a single homologue, located within the tlm BGC (iv and v). (B) The structures of several known and novel thiotetronic acid NPs isolated using this method are shown.
To this point, the findings could be generalized to any target identified among the duplicated OGs, but in this study, OGs related to lipid transport and metabolism were focused on, with the goal of finding inhibitors of bacterial fatty acid biosynthesis (Figure 5A, step iv). Salin8269, one of 12 proteins from Salinispora with homology to the fatty acid elongation enzymes FabB/F, also shows high sequence similarity to the self-resistant proteins PtmP3 and PtnP3 from the producers of fatty acid synthase inhibitors platensimycin and platencin, respectively, and is encoded within the hybrid NRPS-PKS thiolactomycin (tlm) BGC from S. pacifica CNS-863 (Figure 5A, step v).[45] The tlm BGC was cloned through a modified transformation-associated recombination (TAR)-based platform and heterologously expressed in Streptomyces coelicolor M1152. Subsequently, a group of NPs featuring a rare thiotetronic acid moiety, including the previously reported fatty acid synthase inhibitor thiolactomycin (TLM) as well as three analogues (one novel), were isolated (Figure 5B).[49, 50] A second BGC, the thiotetroamide (ttm) BGC from S. afghaniensis, was similarly cloned due to its resemblance to the tlm BGC and the presence of two putative self-resistance genes, ttmE and ttmJ, with both also showing high similarity to ptmP3 and ptnP3.[45] Heterologous expression of the ttm BGC afforded four more TLM analogues, including thiotetroamide (TTM) C, among others (Figure 5B).
This study showcases a genome mining strategy that targets BGCs with duplicated housekeeping genes that may encode protein targets of the NPs. With this strategy, it is now possible to infer the target of an uncharacterized NP by analyzing the BGC-associated self-resistance genes without a priori knowledge of the NP structure.
Activation of silent BGCs
Once one or more BGCs have been prioritized, low titers or silent BGCs can still hinder or prevent further testing without additional tools in the NP discovery pipeline. The abundance of silent orphan BGCs in bacterial genomes has inspired the development of various methods for activation,[16] which can be generally categorized into BGC-targeted and untargeted approaches. The section below reviews these approaches in brief.
Untargeted approaches aim to alter the metabolome of strains through indiscriminate techniques such as media optimization,[51, 52] addition of elicitors,[53] ribosome engineering,[51, 54] metabolic engineering,[55-57] and manipulation of global regulatory[58] and protein modification genes[59] (Box 1). Although successful for NP discovery, traditional untargeted approaches are not applicable to activate specific BGCs of interest and suffer from many pitfalls. For example, the overexpression of phosphopantetheinyl transferases (Pptases), which are responsible for post-translational covalent modification of carrier proteins in fatty acid, polyketide, and non-ribosomal peptide biosynthesis, resulted in 23 of 33 Actinobacteria strains producing new NPs.[59] However, without further investigation, the new NPs could not be identified, as any of several BGCs encoding a carrier protein could have been activated. For a targeted approach, activation of a single BGC will provide the useful genotype-phenotype link that often lacks with these untargeted approaches. Similarly, untargeted approaches such as those described in Box 1, affect global regulation or metabolic flux, which in turn can improve yields from a wide range of BGCs without requiring detailed knowledge about a specific BGC (or even a genome in some cases).[53] Despite their untargeted nature, many of these strategies can be applicable when targeting a BGC that has demonstrated low but detectable production, as the same transcriptional or translational bottlenecks that may lead to BGC silence may also result in low production.[60] In this case however, the genotype-phenotype link would have already been established, potentially via more targeted approaches.
Box 1: Strategies for BGC activation.
BGC activation is considered untargeted when the strategy is not specific to a single BGC of interest but rather affects some or all BGCs encoded within an organism. In addition to the more traditional media optimization or addition of elicitors, four untargeted approaches are detailed below (Box 1, Figure I, left panel).
A. Ribosome engineering. Some BGCs are silent at the transcriptional level while others are silent at the translational level. For those BGCs with a translational bottleneck, the bacteria can be subjected to sub-lethal levels of ribosome-targeting antibiotics, resulting in the evolution of the ribosome and improved translation.
B. Metabolic engineering. To achieve higher titers of NPs, competing and supporting pathways (often involving substrates or cofactors) can be disrupted or augmented, respectively.
C. Manipulation of global regulatory genes. Many BGCs are regulated by proteins encoded outside the BGC boundaries, and deletion or overexpression of these negative or positive regulators, respectively, may activate one or more BGCs in a bacterial host.
D. Manipulation of protein modification genes. The addition of certain genes, such as a phosphopantetheinyl transferase, may result in the post-translational activation of key proteins encoded by silent BGCs.
When targeting a specific BGC, several options are available for activation in addition to the untargeted strategies. Like the untargeted strategies, these targeted strategies can affect multiple levels of silence, and they are described below (Box 1, Figure I, right panel).
E. Heterologous expression. The targeted BGC can be expressed in a heterologous host to disrupt native regulatory networks and to enable improved genetic amenability.
F. Promoter exchange. Promoters from the targeted BGC can be replaced with non-native promoters to remove transcriptional regulation.
G. BGC refactoring. The targeted BGC can be reorganized with predictable transcriptional (promoters, terminators) and translational (ribosome binding sites) elements.
H. Manipulation of BGC-specific regulatory genes. Deletion or overexpression of BGC-specific negative or positive regulators, respectively, can be used to overcome transcriptional silence.
Box 1, Figure I: Strategies for BGC activation.
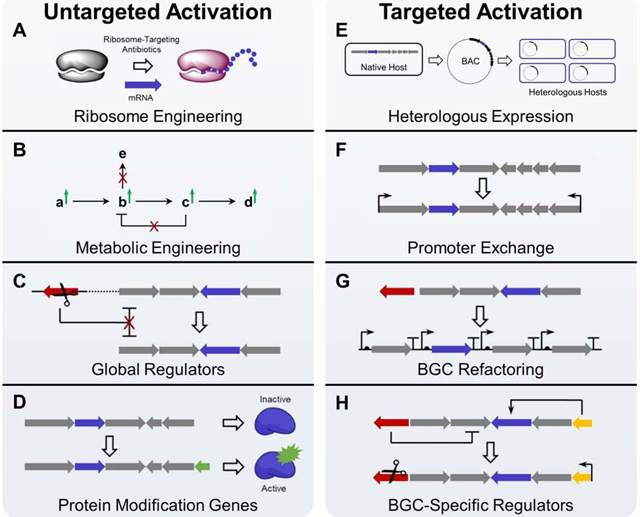
Untargeted strategies are illustrated in the left panel (parts A-D), and targeted strategies are illustrated in the right panel (parts E-H). Genes are color-coded as follows: core biosynthetic genes (blue), negative regulatory genes (red), protein modification genes (green), and positive regulatory genes (orange).
Alternatively, targeted approaches are designed to activate specific BGCs by a variety of sequence-dependent techniques including heterologous expression, promotor exchange, BGC refactoring, and BGC-specific regulator manipulation (Box 1).[58, 61-65] While typically lower throughput, targeted approaches often can take advantage of both the vast genomic information and cutting-edge genome editing technologies such as CRISPR[66, 67] and recombineering.[68, 69] Two other especially popular targeted activation strategies involve isolation of a single BGC from its native environment: BGC refactoring[61, 70, 71] and heterologous expression[44, 58, 72-74] (Box 1). As both strategies are predicated primarily on breaking transcriptional regulatory networks, they are often used in conjunction. Heterologous expression is possibly the most popular targeted activation strategy, likely because of two other key advantages: (1) the BGC of interest is introduced into a characterized environment, making identification of any new NPs simpler, and (2) the new host has often been domesticated[12] and is generally more genetically amenable.[60] However, BGC-specific regulators and low titers due to poor transcriptional or translational throughput are both major hurdles, even in a heterologous host.
BGC refactoring is synthetic biology’s most thorough (and time-intensive) answer to BGC activation. In this process, known genetic elements (promoters, ribosome binding sites, and terminators) are coupled with each gene from the targeted BGC, yielding a new BGC that contains as little or as much regulation as desired with controlled transcription and translation rates.[75, 76] While refactoring is potentially powerful, genetic elements are not universal in bacteria, so genetic toolboxes must be developed for each genetic system.[77] Additionally, refactoring of a single BGC requires both the assembly and balancing of potentially dozens of genetic parts, and this hypervariability results in extensive troubleshooting during and after assembly. Alternatively, intermediate steps such as BGC-specific regulator manipulation[62] or promoter exchanges[46, 78] can also be utilized in order to overcome regulatory or transcriptional challenges, usually with far less effort than full BGC refactoring.
Ultimately, both targeted and untargeted activation approaches have their own merits, and depending on the chosen overall strategy for discovery, either may be preferred. However, as genomic data continue to increase and the targeted activation tools become more developed and mainstream, it is expected that researchers will continue to shift towards targeted approaches to better access their prioritized BGCs.
Concluding Remarks and Future Perspectives
NP discovery, especially over the past several decades, has struggled to reach the heights of its golden age in the 1950s and 1960s, but the field appears poised for a renaissance driven by advancements in genomics, bioinformatics, and synthetic biology.[2] Many of the reasons commonly listed for the decline in NP discovery, such as rediscovery, now can be alleviated by genomic information. Dropping sequencing costs, the availability of powerful bioinformatics tools, and the realization of near limitless BGCs have provided the impetus needed for the field to re-focus on novel NP discovery. However, several pertinent questions remain and are addressed below (see Outstanding Questions).
Outstanding Questions.
How many genomes must be sequenced before BGC discovery plateaus?
How should strains be prioritized for genome sequencing to most efficiently discover novel NPs?
Is it possible to develop tools for targeting structural and functional diversity from genomic information?
What strategy is most effective for the prioritization of BGCs for targeted expression?
Is it possible to develop a universal strategy to translate targeted BGCs into natural products at both scale and speed?
Does the quality of genome sequences matter, i.e. draft versus fully assembled genomes, especially with regards to natural product discovery?
How can we encourage better collaboration between chemists, synthetic biologists, microbiologists, and computational biologists towards developing the necessary technologies for a natural product discovery pipeline?
Genome sequencing has significantly advanced the field, but the rate of discovery of NPs cannot keep pace with the sequencing of their encoding BGCs. Eventually, the rate of BGC discovery will begin to plateau, though that point has not yet been determined. Without BGC sequencing as the rate-limiting step, prioritization of BGCs for NP discovery has become one of the key strategic questions facing the field moving forward. How can novelty as a BGC/NP trait be screened and sorted for? The solution will not come from a single source, but rather, it is a question of developing multidisciplinary collaborations between chemists, synthetic biologists, microbiologists, and computational biologists, among others, to address the wide range of smaller challenges. Of special interest is the construction of a public BGC database to assess total NP diversity and to survey microbial biodiversity. Likewise, the development of platform technologies that can be rapidly embraced by the entire field and utilized at both scale and speed (such as synthetic biology tools in non-model hosts or standardized computational pipelines) is critically important for realizing the genetic information all the way through to the point of discovery. Fortunately, despite all these questions, the field of NP discovery is making great progress in the genomic era and shows strong signs of continuing to develop.
ACKNOWLEDGEMENTS
Research on natural product discovery, biosynthesis, engineering and drug discovery in the Shen lab is currently supported by NIH grants CA106150, GM114353, GM115575, and the Natural Products Library Initiative at The Scripps Research Institute. E.K. is a recipient of an NIH postdoctoral fellowship GM134688. This is manuscript no. XXXX from The Scripps Research Institute.
GLOSSARY
- antiSMASH
a bioinformatics tool that identifies and annotates secondary metabolite BGCs from bacterial and fungal sequences.
- Biosynthetic gene cluster (BGC)
a physically clustered set of genes that together encode the proteins responsible for the biosynthesis of a NP. This genetic organization is far more common in bacteria than in most eukaryotic genomes.
- BiG-SCAPE
a bioinformatics tool used to group BGCs into sequence similarity networks for exploration and classification.
- Combinatorial biosynthesis
the exploitation of biosynthetic pathways using combinatorial strategies to produce NPs with altered structures.
- Core genome
all genes that are conserved throughout strains in a given species.
- CRISPR
a DNA editing tool that works by utilizing the specificity of the Cas9 enzyme to cleave DNA at a very specific site as dictated by a synthetic guide RNA (sgRNA).
- Degenerate primers
a mix of oligonucleotides with similar sequences that cover all possible nucleotide combinations for a given protein sequence.
- Dereplication
the step in natural product discovery used to prevent rediscovery of natural products.
- Elicitors
small molecules that trigger the production of a secondary metabolite.
- Genome mining
a search for a specific DNA sequence, often associated with a specific gene or BGC. This search may be facilitated by either bioinformatics, for sequenced sources, or PCR, for unsequenced sources.
- Genome neighborhood network (GNN)
a bioinformatics tool used for visualizing the protein families encoded by genes in proximity to the genes analyzed in an SSN.
- Hybrid NRPS-PKS
biosynthetic assembly line-like enzyme(s) in which NRPS and PKS modules are both present, leading to a product with both amino acid- and acyl-derived moieties.
- Metabolome
the collection of small molecules from a single biological sample, typically a single organism.
- Metagenomic
relating to the total DNA from an environmental sample.
- NRPSs
(nonribosomal peptide synthetases) assembly line-like enzymes that can be divided in multi-domain modules, with each module responsible for the nonribosomal incorporation and tailoring of a single amino acid into the scaffold of a small molecule product.
- Orphan BGCs
BGCs that have not been experimentally correlated with a NP.
- Orthologous group
a group of genes or proteins with the same function in different species that are related by a common ancestor.
- Pan-genome
all genes from every strain of a particular species.
- PKSs
(polyketide synthases) assembly line-like enzymes that can be divided in multi-domain modules, with each module responsible for the decarboxylative incorporation and tailoring of a single acyl-CoA substrate (or analogue) into the scaffold of a small molecule product.
- rt-PCR
(or qPCR) a technique that couples amplification of targeted DNA with real-time quantification of the amplified DNA through the use of fluorescently labeled reporters.
- Sequence similarity network (SSN)
a bioinformatics tool used for visualizing large sets of sorted protein sequences with different stringency levels.
- Silent BGC
a BGC that does not produce a NP under tested culture conditions.
- Tailoring enzyme
an enzyme that acts to decorate a NP scaffold with additional moieties, e.g. hydroxyl or methyl groups.
- Transformation-associated recombination (TAR)
a technique that takes advantage of yeast’s propensity for initiating homologous recombination. This is an especially powerful tool for cloning large BGCs from genomic DNA.
Footnotes
DISCLAIMER STATEMENT
The authors declare no competing financial interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
RESOURCES
REFERENCES
- 1.Newman DJ and Cragg GM (2016) Natural Products as Sources of New Drugs from 1981 to 2014. J Nat Prod 79 (3), 629–661. [DOI] [PubMed] [Google Scholar]
- 2.Shen B (2015) A New Golden Age of Natural Products Drug Discovery. Cell 163 (6), 1297–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Newman DJ and Cragg GM (2016) Natural Products as Sources of New Drugs from 1981 to 2014. Journal of Natural Products 79 (3), 629–661. [DOI] [PubMed] [Google Scholar]
- 4.Katz L and Baltz RH (2016) Natural product discovery: past, present, and future. J Ind Microbiol Biotechnol 43 (2-3), 155–76. [DOI] [PubMed] [Google Scholar]
- 5.Pye CR et al. (2017) Retrospective analysis of natural products provides insights for future discovery trends. Proceedings of the National Academy of Sciences 114 (22), 5601–5606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cimermancic P et al. (2014) Insights into secondary metabolism from a global analysis of prokaryotic biosynthetic gene clusters. Cell 158 (2), 412–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stephens ZD et al. (2015) Big Data: Astronomical or Genomical? PLoS Biol 13 (7), e1002195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mukherjee S et al. (2018) Genomes OnLine database (GOLD) v.7: updates and new features. Nucleic Acids Research 47 (D1), D649–D659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mardis ER (2017) DNA sequencing technologies: 2006-2016. Nat Protoc 12 (2), 213–218. [DOI] [PubMed] [Google Scholar]
- 10.Mukherjee S et al. (2017) 1,003 reference genomes of bacterial and archaeal isolates expand coverage of the tree of life. Nature Biotechnology 35, 676. [DOI] [PubMed] [Google Scholar]
- 11.Hadjithomas M et al. (2017) IMG-ABC: new features for bacterial secondary metabolism analysis and targeted biosynthetic gene cluster discovery in thousands of microbial genomes. Nucleic Acids Res 45 (D1), D560–D565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ikeda H et al. (2014) Genome mining of the Streptomyces avermitilis genome and development of genome-minimized hosts for heterologous expression of biosynthetic gene clusters. J Ind Microbiol Biotechnol 41 (2), 233–50. [DOI] [PubMed] [Google Scholar]
- 13.Louca S et al. (2019) A census-based estimate of Earth's bacterial and archaeal diversity. PLoS Biol 17 (2), e3000106–e3000106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Locey KJ and Lennon JT (2016) Scaling laws predict global microbial diversity. Proceedings of the National Academy of Sciences 113 (21), 5970–5975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weber T and Kim HU (2016) The secondary metabolite bioinformatics portal: Computational tools to facilitate synthetic biology of secondary metabolite production. Synthetic and Systems Biotechnology 1 (2), 69–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nett M et al. (2009) Genomic basis for natural product biosynthetic diversity in the actinomycetes. Nat Prod Rep 26 (11), 1362–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao S et al. (2014) Prediction and characterization of enzymatic activities guided by sequence similarity and genome neighborhood networks. Elife 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rudolf JD et al. (2016) Genome neighborhood network reveals insights into enediyne biosynthesis and facilitates prediction and prioritization for discovery. J Ind Microbiol Biotechnol 43 (2-3), 261–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Navarro-Muñoz J et al. (2018) A computational framework for systematic exploration of biosynthetic diversity from large-scale genomic data. bioRxiv, 445270. [Google Scholar]
- 20.Blin K et al. (2017) antiSMASH 4.0-improvements in chemistry prediction and gene cluster boundary identification. Nucleic acids research 45 (W1), W36–W41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Skinnider MA et al. (2015) Genomes to natural products PRediction Informatics for Secondary Metabolomes (PRISM). Nucleic Acids Res 43 (20), 9645–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rottig M et al. (2011) NRPSpredictor2--a web server for predicting NRPS adenylation domain specificity. Nucleic Acids Res 39 (Web Server issue), W362–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Epstein SC et al. (2018) A standardized workflow for submitting data to the Minimum Information about a Biosynthetic Gene cluster (MIBiG) repository: prospects for research-based educational experiences. Stand Genomic Sci 13, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pan G et al. (2017) Discovery of the leinamycin family of natural products by mining actinobacterial genomes. Proc Natl Acad Sci U S A 114 (52), E11131–e11140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hindra et al. (2014) Strain prioritization for natural product discovery by a high-throughput real-time PCR method. J Nat Prod 77 (10), 2296–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yan X et al. (2016) Strain Prioritization and Genome Mining for Enediyne Natural Products. MBio 7 (6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Owen JG et al. (2015) Multiplexed metagenome mining using short DNA sequence tags facilitates targeted discovery of epoxyketone proteasome inhibitors. Proc Natl Acad Sci U S A 112 (14), 4221–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ju KS et al. (2015) Discovery of phosphonic acid natural products by mining the genomes of 10,000 actinomycetes. Proc Natl Acad Sci U S A 112 (39), 12175–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eng CH et al. (2017) ClusterCAD: a computational platform for type I modular polyketide synthase design. Nucleic Acids Research, gkx893–gkx893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Helfrich EJN et al. (2019) Automated structure prediction of trans-acyltransferase polyketide synthase products. Nat Chem Biol 15 (8), 813–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Agrawal P et al. (2017) RiPPMiner: a bioinformatics resource for deciphering chemical structures of RiPPs based on prediction of cleavage and cross-links. Nucleic acids research 45 (W1), W80–W88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chow JY et al. (2015) Computational-guided discovery and characterization of a sesquiterpene synthase from Streptomyces clavuligerus. Proc Natl Acad Sci U S A 112 (18), 5661–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yan X et al. (2018) Discovery of Alternative Producers of the Enediyne Antitumor Antibiotic C-1027 with High Titers. Journal of Natural Products 81 (3), 594–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shen B et al. (2015) Enediynes: Exploration of microbial genomics to discover new anticancer drug leads. Bioorg Med Chem Lett 25 (1), 9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yan X et al. (2016) Strain Prioritization and Genome Mining for Enediyne Natural Products. 7 (6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Viswesh V et al. (2010) Characterization of DNA Damage Induced by a Natural Product Antitumor Antibiotic Leinamycin in Human Cancer Cells. Chemical Research in Toxicology 23 (1), 99–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hara M et al. (1989) Leinamycin, a New Antitumor Antibiotic from Streptomyces - Producing Organism, Fermentation and Isolation. Journal of Antibiotics 42 (12), 1768–1774. [DOI] [PubMed] [Google Scholar]
- 38.Cheng YQ et al. (2002) Identification and Localization of the Gene Cluster Encoding Biosynthesis of the Antitumor Macrolactam Leinamycin in Streptomyces atroolivaceus S-140. J Bacteriol 184 (24), 7013–7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tang GL et al. (2004) Leinamycin biosynthesis revealing unprecedented architectural complexity for a hybrid polyketide synthase and nonribosomal peptide synthetase. Chem. Biol. 11 (1), 33–45. [DOI] [PubMed] [Google Scholar]
- 40.Yu X et al. (2013) Diversity and abundance of phosphonate biosynthetic genes in nature. Proceedings of the National Academy of Sciences of the United States of America 110 (51), 20759–20764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ju KS et al. (2014) Genomics-enabled discovery of phosphonate natural products and their biosynthetic pathways. J Ind Microbiol Biotechnol 41 (2), 345–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Doroghazi JR et al. (2014) A roadmap for natural product discovery based on large-scale genomics and metabolomics. Nat Chem Biol 10 (11), 963–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eliot AC et al. (2008) Cloning, expression, and biochemical characterization of Streptomyces rubellomurinus genes required for biosynthesis of antimalarial compound FR900098. Chem Biol 15 (8), 765–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang X et al. (2015) Identification of Thiotetronic Acid Antibiotic Biosynthetic Pathways by Target-directed Genome Mining. ACS Chem Biol 10 (12), 2841–2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Peterson RM et al. (2014) Mechanisms of self-resistance in the platensimycin- and platencin-producing Streptomyces platensis MA7327 and MA7339 strains. Chem Biol 21 (3), 389–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yeh HH et al. (2016) Resistance Gene-Guided Genome Mining: Serial Promoter Exchanges in Aspergillus nidulans Reveal the Biosynthetic Pathway for Fellutamide B, a Proteasome Inhibitor. ACS Chem Biol 11 (8), 2275–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yan Y et al. (2018) Resistance-gene-directed discovery of a natural-product herbicide with a new mode of action. Nature 559 (7714), 415–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Panter F et al. (2018) Self-resistance guided genome mining uncovers new topoisomerase inhibitors from myxobacteria. Chem Sci 9 (21), 4898–4908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sasaki H et al. (1982) Thiolactomycin, a New Antibiotic .2. Structure Elucidation. Journal of Antibiotics 35 (4), 396–400. [DOI] [PubMed] [Google Scholar]
- 50.Hayashi T et al. (1984) Inhibition of Fatty-Acid Synthesis by the Antibiotic Thiolactomycin. Journal of Antibiotics 37 (11), 1456–1461. [DOI] [PubMed] [Google Scholar]
- 51.Liu L et al. (2018) Ribosome engineering and fermentation optimization leads to overproduction of tiancimycin A, a new enediyne natural product from Streptomyces sp. CB03234. 45 (3), 141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shi J et al. (2016) Titer improvement and pilot-scale production of platensimycin from Streptomyces platensis SB12026. J Ind Microbiol Biotechnol 43 (7), 1027–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu F et al. (2019) A genetics-free method for high-throughput discovery of cryptic microbial metabolites. Nat Chem Biol 15 (2), 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang Y et al. (2015) Activation and enhancement of Fredericamycin A production in deepsea-derived Streptomyces somaliensis SCSIO ZH66 by using ribosome engineering and response surface methodology. Microbial Cell Factories 14 (1), 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jung WS et al. (2014) Characterization and engineering of the ethylmalonyl-CoA pathway towards the improved heterologous production of polyketides in Streptomyces venezuelae. Appl Microbiol Biotechnol 98 (8), 3701–3713. [DOI] [PubMed] [Google Scholar]
- 56.Dayem LC et al. (2002) Metabolic engineering of a methylmalonyl-CoA mutase-epimerase pathway for complex polyketide biosynthesis in Escherichia coli. Biochemistry 41 (16), 5193–201. [DOI] [PubMed] [Google Scholar]
- 57.Murli S et al. (2003) Metabolic engineering of Escherichia coli for improved 6-deoxyerythronolide B production. J Ind Microbiol Biotechnol 30 (8), 500–9. [DOI] [PubMed] [Google Scholar]
- 58.Peng Q et al. (2018) Engineered Streptomyces lividans Strains for Optimal Identification and Expression of Cryptic Biosynthetic Gene Clusters. Frontiers in Microbiology 9 (3042). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang B et al. (2017) Activation of Natural Products Biosynthetic Pathways via a Protein Modification Level Regulation. ACS Chem Biol 12 (7), 1732–1736. [DOI] [PubMed] [Google Scholar]
- 60.Zhang MM et al. (2016) Engineering microbial hosts for production of bacterial natural products. Nat Prod Rep 33 (8), 963–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ren H et al. (2018) Rapid Discovery of Glycocins through Pathway Refactoring in Escherichia coli. ACS Chem Biol 13 (10), 2966–2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Smanski MJ et al. (2009) Engineered Streptomyces platensis strains that overproduce antibiotics platensimycin and platencin. Antimicrob Agents Chemother 53 (4), 1299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Liu Y et al. (2019) A CRISPR–Cas9 Strategy for Activating the Saccharopolyspora erythraea Erythromycin Biosynthetic Gene Cluster with Knock-in Bidirectional Promoters. ACS Synth Biol 8 (5), 1134–1143. [DOI] [PubMed] [Google Scholar]
- 64.Chen Y et al. (2011) Improvement of the enediyne antitumor antibiotic C-1027 production by manipulating its biosynthetic pathway regulation in Streptomyces globisporus. J Nat Prod 74 (3), 420–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Alberti F et al. (2019) Triggering the expression of a silent gene cluster from genetically intractable bacteria results in scleric acid discovery. Chem Sci 10 (2), 453–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cobb RE et al. (2015) High-efficiency multiplex genome editing of Streptomyces species using an engineered CRISPR/Cas system. ACS Synth Biol 4 (6), 723–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhang MM et al. (2017) CRISPR-Cas9 strategy for activation of silent Streptomyces biosynthetic gene clusters. Nature Chemical Biology 13, 607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sharan SK et al. (2009) Recombineering: a homologous recombination-based method of genetic engineering. Nature protocols 4 (2), 206–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thomason LC et al. (2014) Recombineering: genetic engineering in bacteria using homologous recombination. Curr Protoc Mol Biol 106, 1.16.1–39. [DOI] [PubMed] [Google Scholar]
- 70.Shao Z et al. (2013) Refactoring the Silent Spectinabilin Gene Cluster Using a Plug-and-Play Scaffold. ACS Synthetic Biology 2 (11), 662–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bauman KD et al. Refactoring the Cryptic Streptophenazine Biosynthetic Gene Cluster Unites Phenazine, Polyketide, and Nonribosomal Peptide Biochemistry. Cell Chemical Biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fang L et al. (2017) Heterologous erythromycin production across strain and plasmid construction. Biotechnol Prog. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Alberti F et al. (2017) Heterologous expression reveals the biosynthesis of the antibiotic pleuromutilin and generates bioactive semi-synthetic derivatives. Nat Commun 8 (1), 1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tan GY et al. (2017) Heterologous Biosynthesis of Spinosad: An Omics-Guided Large Polyketide Synthase Gene Cluster Reconstitution in Streptomyces. ACS Synth Biol 6 (6), 995–1005. [DOI] [PubMed] [Google Scholar]
- 75.D'Agostino PM and Gulder TAM (2018) Direct Pathway Cloning Combined with Sequence- and Ligation-Independent Cloning for Fast Biosynthetic Gene Cluster Refactoring and Heterologous Expression. ACS Synth Biol 7 (7), 1702–1708. [DOI] [PubMed] [Google Scholar]
- 76.Ren H et al. (2017) A plug-and-play pathway refactoring workflow for natural product research in Escherichia coli and Saccharomyces cerevisiae. Biotechnol Bioeng 114 (8), 1847–1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Englund E et al. (2016) Evaluation of promoters and ribosome binding sites for biotechnological applications in the unicellular cyanobacterium Synechocystis sp. PCC 6803. Scientific Reports 6, 36640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Montiel D et al. (2015) Yeast homologous recombination-based promoter engineering for the activation of silent natural product biosynthetic gene clusters. Proc Natl Acad Sci U S A 112 (29), 8953–8. [DOI] [PMC free article] [PubMed] [Google Scholar]