

Abstract
The Krüppel-like zinc finger transcription factor Gli-similar 3 (GLIS3) plays a critical role in the regulation of pancreatic beta cells, with global Glis3 knockout mice suffering from severe hyperglycemia and dying by post-natal day 11. In addition, GLIS3 has been shown to directly regulate the early endocrine marker Ngn3, as well as Ins2 gene expression in mature beta cells. We hypothesize that GLIS3 regulates several other genes critical to beta cell function, in addition to Ins2, by directly binding to regulatory regions. We therefore generated a pancreas-specific Glis3 deletion mouse model (Glis3Δpanc) using a Pdx1-driven Cre mouse line. Roughly 20% of these mice develop hyperglycemia by 8-weeks and lose most of their insulin expression. However, this did not appear to be due to loss of the beta cells themselves, as no change in cell death was observed. Indeed, presumptive beta cells appeared to persist as PDX1+/INS−/MAFA−/GLUT2− cells. Islet RNA-seq analysis combined with GLIS3 ChIP-seq analysis revealed apparent direct regulation of a variety of diabetes related genes, including Slc2a2 and Mafa. GLIS3 binding near these genes coincided with binding for other islet-enriched transcription factors, indicating these are distinct regulatory hubs. Our data indicates that GLIS3 not only regulates insulin expression, but several additional genes critical for beta cell function.
Keywords: GLIS3, beta cells, RNA-seq, ChIP-seq, transcription factor, gene regulation
Introduction
GLIS3 belongs to the GLIS subfamily of Krüppel-like proteins that are most closely-related to members of the GLI and ZIC families (Jetten, 2018). It plays a critical role in the regulation of various physiological processes, including pancreas development, osteogenesis, kidney function and thyroid hormone regulation and is implicated in several major human pathologies (e.g., diabetes, hypothyroidism, cancer, cystic kidney disease) (Dimitri et al., 2011, Wen and Yang, 2017, Scoville et al., 2017, Jetten, 2018). GLIS3 contains five Krüppel-like zinc finger motifs that recognize specific DNA sequences in regulatory regions of target genes allowing it to function as a transcriptional activator and/or repressor (Kang et al., 2010, Lichti-Kaiser et al., 2012). Genetic aberrations in human GLIS3 are associated with a rare syndrome characterized by Neonatal Diabetes and congenital Hypothyroidism (NDH). Depending on the nature of the mutation, additional features have included hepatic fibrosis, congenital glaucoma, polycystic kidney disease, facial dysmorphism, and osteopenia (Senee et al., 2006, Dimitri et al., 2011, Habeb et al., 2012). Additional evidence for a role of GLIS3 in diabetes comes from genome-wide association studies (GWAS) that link GLIS3 variants to aberrant glucose regulation and reduced beta cell function, and identified GLIS3 as a risk locus for both type-1 and type-2 diabetes (Barrett et al., 2009, Dupuis et al., 2010, Cho et al., 2011, Liu et al., 2011, Rees et al., 2011).
GLIS3 protein expression in the mouse pancreas is first observed at embryonic day 13.5 in bipotent pancreatic progenitor cells (i.e. cells of capable of differentiating into ductal or endocrine cells (Zhou et al., 2007)), and its expression is maintained in both lineages into adulthood (Kang et al., 2016). Within the mature endocrine population, GLIS3 is restricted to beta cells and pancreatic polypeptide cells (Kang et al., 2016). We and others have previously demonstrated that global Glis3-null mice die by post-natal day 11 (PND11) due to neonatal diabetes (Kang et al., 2009, Watanabe et al., 2009, Yang et al., 2009). Islet size and the expression of several beta cell markers, including Ins2, the glucose transporter Slc2a2, MafA, and Nkx6.1 were dramatically decreased in Glis3-null mice. Indeed, GLIS3 is critical for Ins2 expression in mature beta cells through its interaction with other known regulators of Ins2, such as PDX1, NEUROD1, and MAFA (Kang et al., 2009, ZeRuth et al., 2013, Yang et al., 2009). However, the precise role of GLIS3 in the maintenance of functional beta cells, aside from regulating insulin expression, remains poorly defined.
To gain greater insights into the regulatory functions of GLIS3 in the pancreas, we generated a pancreas-specific Glis3 knockout mouse model. Glis3fl/fl mice were crossed with mice expressing Cre-recombinase under the control of the Pdx1 gene promoter to generate a pancreas-specific knockout model (Glis3Δpanc). We were thus able to separate the pancreatic phenotype from other organs affected by global Glis3 loss, such as the kidney and thyroid. The major objective of this study was to obtain a better understanding of GLIS3-mediated regulation of gene transcription in pancreatic islets, we therefore performed gene expression analysis on islets from these mice, as well as GLIS3 ChIP-seq analysis. In addition, we compared these results to publicly available data on the genomic binding of several other islet-enriched transcription factors, in order to determine whether GLIS3 regulates pancreatic beta cell gene transcription by binding to distinct regulatory hubs with other islet transcription factors (Hu and Tee, 2017). Our study shows that GLIS3 plays a critical role in directly regulating a number of genes associated with beta cell identity, maturation, and function, and suggests that GLIS3 performs these regulatory functions in association with other islet-enriched transcription factors.
Materials and Methods
Generation of pancreas-specific Glis3 knockout mice.
A conditional knockout allele for the Glis3 gene was generated in C57BL/6J mice using homologous recombination in mouse embryonic stem cells and subsequent blastocyst injection of the appropriate targeted ES cells (C57BL/6-Glis3<tm2Amj>). The vector contained loxP sites flanking exon 5 of Glis3, which included part of the DNA binding domain (Figure1A). Previous work has shown that deletion of any part of the DNA binding domain produces a non-functional protein (Beak et al., 2008). Mice were bred with a strain expressing Cre recombinase under the control of the Pdx1 promoter, which produces pancreas-specific deletion by e10.5 (Jackson Laboratories Stock No. 014647) (Hingorani et al., 2003). Animal studies followed guidelines outlined by the NIH Guide for the Care and Use of Laboratory Animals and protocols were approved by the Institutional Animal Care and Use Committee at the NIEHS.
Figure 1. Glis3Δpanc mice exhibit a loss of insulin-producing beta cells.



(A) Schematic of wild type and recombinant Glis3 allele. A conditional knockout allele for the Glis3 gene was generated in C57BL/6 mice containing loxP sites (triangles) surrounding exon 5 of Glis3. Exons 4–6 encode the DNA-binding domain (DBD). Mice were bred with a strain expressing Cre recombinase under the control of the Pdx1 promoter. (B) Random blood glucose levels of mice at 8-weeks of age. Glis3Δpanc mice were grouped as either Euglycemic (<250 mg/dL) or Hyperglycemic (>250 mg/dL) and compared to control littermates (WT) (N=11 WT, N=5 Glis3Δpanc euglycemic, N=4 Glis3Δpanc hyperglycemic). Immunofluorescence staining revealed a reduction in the number of INS+PDX1+ beta cells (C) and pancreatic polypeptide+ cells (D), with no apparent effect on (E) glucagon or (F) somatostatin expression. Nuclei were visualized by DAPI staining. Representative images are shown. * indicates p<0.05.
Blood Glucose measurements.
Tail blood was collected from wild type (Glis3fl/fl) and Glis3Δpanc mice at indicated time points. Glucose levels were measured with Nova Max glucose monitoring system (Nova Biomedical; Waltham, MA).
Immunohistochemical analysis.
Mouse pancreases were fixed for 4 hours in 4% paraformaldehyde in PBS and embedded in OCT (Tissue Tek; Hatfield, PA) prior to cryogenic sectioning. Primary antibodies are listed in Supplementary Table 1. DBA-Fluorescein was purchased from Vector Labs (Burlingame, CA). Alexa Fluor-conjugated secondary antibodies were purchased from Molecular Probes (Carlsbad, CA). Nuclei were stained with DAPI Prolong Diamond from Invitrogen (Carlsbad, CA). Fluorescent images were captured using a Zeiss LSM 880 confocal microscope and ZEN software.
Proliferation and TUNEL Staining.
2-week old pancreases were stained with antibodies against PDX1 and either Ki67 or the DeadEnd™ Fluorometric TUNEL System (Promega #G3250) following manufacturer’s protocol. At least 6 sections >200 microns apart per mouse from 3 mice per genotype were examined using confocal microscopy (Zeiss LSM 880 with Airyscan), and co-localization quantified manually using ImageJ (NIH, Bethesda, MD).
Islet isolation and qRT-PCR analysis.
Pancreatic islets were isolated via collagenase injection through the common bile duct into the pancreas, followed by digestion and hand-picking. RNA was isolated using Trizol reagent (Life Technologies) followed by DNAse I digestion and additional purification via Ambion RNAqueous micro kit (Ambion). cDNA was synthesized using a High Capacity DNA Reverse Transcription Kit (Applied Biosystems; Foster City, CA). qRT-PCR was carried out in triplicate in a StepOnePlus Real Time PCR System (Applied Biosystems; Foster City, CA). All results were normalized to Gapdh expression and are shown relative to control using the 2ΔΔct method. Primer sequences are listed in Supplementary Table 1.
RNA sequencing.
Islets were isolated from 4-week old Glis3Δpanc and control littermates (Glis3fl/fl). ~10 ng of RNA was converted to cDNA using the high capacity cDNA reverse transcription kit (Applied Biosystems), and islet RNA from mice with a 78–91% removal of Glis3 (as measured by qRT-PCR of the deleted exon) were submitted for RNA-seq analysis at the NIEHS Epigenomics Core (with control littermates). Briefly, RNA was converted to cDNA using the NextFlex Rapid RNA seq kit (Illumina), and sequenced as paired-end 76mers on a NextSeq500 (Ilumina) for each sample (N=4 Glis3fl/fl, 3 Glis3Δpanc). Read pairs were filtered to retain only those with a mean base quality score of at least 20 for each end. Adapter was clipped from the end of each read via Cutadapt v1.2.1 (Martin, 2011)(parameters: -a AGATCGGAAGAG -O 5 -q 0), then read pairs were filtered to retain only those at least 30nt in length after adapter removal. Trimmed and filtered read pairs were aligned against the mm10 reference assembly via STAR v2.5 (Dobin et al., 2013) (parameters: --alignSJoverhangMin 8 --limitBAMsortRAM 55000000000 --outSAMstrandField intronMotif --outFilterIntronMotifs RemoveNoncanonical). Counts per gene were determined by Subread featureCounts v1.5.0-p1 (Liao et al., 2014) (parameters: -s0 -Sfr -p) for a set of gene models defined by RefSeq transcripts as downloaded from the UCSC Table Browser (http://genome.ucsc.edu/cgi-bin/hgTables) as of November 7, 2017. Differentially expressed genes were identified via DESeq2 v1.10.1 (Love et al., 2014) at an FDR threshold of 0.05, and a complete list is provided in Supplementary Table 2. For RNA-seq genome browser views, replicates were merged and normalized to 50 million mappable reads.
ChIP sequencing.
Islets from 8-week old GLIS3-EGFP mice (Kang et al., 2016) were isolated, flash frozen at −80°C. ChIP-seq analysis was performed by Active Motif (Carlsbad, CA). ChIP was performed using a GFP antibody and ~1,800 islets. Input and ChIP samples were run as single-end 75mers on a NextSeq500 (Illumina); 42–43 million reads each were obtained for each. Raw data is available from Gene Expression omnibus (GSE122120). For comparison to other published ChIP-seq data, raw sequencing data was obtained from GEO for: NKX2.2 (GSE79785), MAFA and NEUROD1 (GSE30298), PDX1 and FOXA2 (SRA008281), NKX6.1 (GSE40975), ISL1 and LDB1 (GSE84759) and from ArrayExpress for: PDX1 (E-MTAB-1143). For consistency, all ChIP-seq datasets were processed by the same workflow, as follows. Reads were filtered to retain only those with a mean base quality score of at least 20. Adapter was clipped from the end of each read via Cutadapt v1.2.1 (Martin, 2011)(parameters: -a AGATCGGAAGAG -O 5 -q 0), then reads were filtered to retain only those at least 30nt in length after adapter removal. Trimmed and filtered reads were aligned against the mm10 reference assembly via Bowtie v1.2 (Langmead et al., 2009), retaining only uniquely-mapped reads (parameter: -m 1). For samples with multiple sequencing runs, replicates were combined with MergeSamFiles.jar from the Picard tool suite v1.110. Duplicate reads were removed with MarkDuplicates.jar from the Picard tool suite v1.110. For visualization of mapped read depth on the UCSC Genome Browser, uniquely-mapped non-duplicate reads were extended to 200nt, converted to bedGraph format using BEDTools v2.24.0 genomeCoverageBed (Quinlan and Hall, 2010), then normalized to 10 million reads per sample. Peak calling was performed with HOMER v4.9.1 (Heinz et al., 2010), with parameters: -style factor -fdr 0.00001. The matched input sample was used as background if available. For public samples with no available matched input, a surrogate input of 20 million reads was generated by aggregating randomly-selected (uniquely-mapped non-duplicate) reads from the mouse islet input samples from GSE40975, GSE84759, E-MTAB-1143, and our current dataset. Called peaks from each dataset were re-defined as 200mers centered on the midpoint of the original peak call. A complete list of nearest genes bound by GLIS3 is included in Supplementary Table 3.
Pathway Analysis.
Pathway analysis was performed via DAVID tools (v6.8) for KEGG pathway analysis (Huang da et al., 2009b, Huang da et al., 2009a). Full list of pathways and genes are included in Supplementary Tables 4–6. Pathview analysis was performed using the online tool, as described (Luo et al., 2017).
Luciferase reporter assay.
HEK293T cells were grown in DMEM containing 10% Fetal Bovine Serum (FBS) and penicillin/streptomycin. Cells were transfected using Lipofectamine 2000 (Invitrogen) with 100 ng of luciferase reporter plasmid (pGL4.27) under the control of the MafA Region 3 regulatory sequence (Raum et al., 2006)), 50 ng of CMV-driven beta-galactosidase plasmid, and 100 ng of either CMV-driven mouse Glis3 or a control plasmid. Twenty-four hours after transfection, cells were lysed with Passive Lysis Buffer (Promega), and luciferase activity was measured with a Luciferase Assay System (Promega) and beta-galactosidase measured with a Luminescent Beta-Galactosidase Detection kit II (Takara) according to the manufacturer’s protocols. Transfections were performed in triplicate, with each experiment performed at least 3 times.
Statistical Analysis.
Where indicated, statistics were calculated using a two-tailed student t-test. Significance indicates a p-value <0.05, unless otherwise specified.
Results
Glis3Δpanc mice develop hyperglycemia due to loss of insulin expression.
In order to investigate the role of GLIS3 in the pancreas, we generated a pancreas-specific Glis3 deletion mouse model. Glis3fl/fl mice containing LoxP sites flanking exon 5 (Figure 1A), which encodes a portion of the DNA binding domain, were generated and crossed with a mouse strain expressing Cre-recombinase under regulation of the Pdx1 promoter, expressed in the early stages of pancreatic development in the common pancreatic progenitor cells. Glis3Δpanc mice did not develop neonatal diabetes like the global Glis3 mutant mice, as their blood glucose levels were not significantly different at 2-weeks (data not shown). However, by 8-weeks of age, ~20% of Glis3Δpanc mice developed severe hyperglycemia (>500 mg/dL blood glucose), while the remaining Glis3Δpanc mice exhibited glucose levels comparable to those of control littermates (Figure 1B). The development of hyperglycemia appears to relate to the efficiency of Glis3 exon 5 removal, as mice with >75% deletion displayed mild hyperglycemia at 4-weeks (Supplementary Figure 1), consistent with Pdx1-Cre mosaic expression in pancreatic progenitor cells (Hingorani et al., 2003). Another investigator utilizing a similar Glis3 knockout strategy with a Pdx1-Cre model reported hyperglycemia in only a subset of Glis3 knockout mice (Yang et al., 2017). In addition, partial Glis3 deletion in Glis3Δpanc euglycemic mice appears to phenocopy Glis3 heterozygous mice, which are euglycemic, but are more susceptible to impaired glucose tolerance in high fat diet-induced obesity (Yang et al., 2013).
Immunofluorescent staining of pancreas sections from euglycemic and hyperglycemic Glis3Δpanc mice and control littermates at 8-weeks of age (Figure 1C–F) revealed a loss of insulin expression in PDX1+ presumptive beta cells, with a more severe loss of insulin observed in hyperglycemic animals (Figure 1C). We presume these cells are former beta cells due to their PDX1 expression and central location within the islet. Pancreatic polypeptide levels also appeared decreased (Figure 1D), consistent with our recent discovery of GLIS3 expression in pancreatic polypeptide cells (Kang et al., 2016). Glucagon and somatostatin expression appeared unaffected (Figure 1E–F), likely reflecting the lack of GLIS3 expression in the mature alpha and delta cells (Kang et al., 2016).
Glis3Δpanc islet beta cells do not undergo apoptosis or dedifferentiation.
In order to further study these PDX1+ presumptive beta cells, we stained for MAFA, a marker of mature beta cells in both mice and humans, and GLUT2, a beta cell specific glucose transporter. Expression of these markers was lost in most but not all PDX1+ cells by 8-weeks of age (Figure 2A–B). Talchai et al. suggested that, under stress conditions, beta cells may dedifferentiate into an earlier point in endocrine lineage, as marked by NGN3 expression (Talchai et al., 2012). However, we did not detect re-expression of NGN3 at 8-weeks in the PDX1+ cells (Supplementary Figure 2). Additionally, it is possible that the loss of insulin expression was due to decreased proliferation or increased apoptosis at an earlier timepoint. We therefore examined proliferation and apoptosis at 2-weeks of age, prior to the secondary effects of hyperglycemia seen at 8-weeks. Ki67+PDX1+ cells were slightly increased, indicating a slight increase in proliferation, whereas no increase in TUNEL staining was observed (Figure 2C–F). Correspondingly, we saw a slight, non-significant increase in the total number of Pdx1+ cells in Glis3Δpanc mice (data not shown). This indicates that while islet PDX1+ cells in Glis3Δpanc mice no longer express insulin and are likely non-functional, these cells remain present within the islet in a dysfunctional state.
Figure 2. Beta cells lose key markers, but do not apoptose in Glis3Δpanc mice.



Pancreas sections from 8-week-old Glis3Δpanc mice were stained for PDX1 and MAFA (A) or GLUT2 (B). Proliferation was measured via Ki67 and PDX1 double staining of 2-week old Glis3Δpanc mice (C), and quantified (D). Apoptosis was measured via TUNEL and PDX1 double staining of 2-week old Glis3Δpanc mice (E), and quantified (F). Results from N=3 mice for each genotype, error bars = SEM, * indicates p<0.05. Arrows indicate MAFA+PDX1+ (A), GLUT2+PDX1+ (B), Ki67+PDX1+ (C), or TUNEL+PDX1+ (D) cells. Representative images are shown.
GLIS3 regulates many genes critical for beta cell identity and function.
In order to establish a global network of GLIS3-regulated genes, we performed RNA-seq analysis on isolated islets from Glis3Δpanc mice at 4-weeks of age, prior to the onset of severe hyperglycemia (>500mg/dL) so as to limit its secondary effects. In total, 1,162 genes were found to be upregulated >1.5 fold, and 612 genes were found to be downregulated >1.5 fold upon loss of GLIS3, consistent with GLIS3’s role as both an activator and repressor of gene expression (Supplementary Table 2). KEGG pathway analysis indicated that the downregulated genes were involved in a variety of pathways, including Maturity Onset Diabetes of the Young (MODY) and insulin secretion, as well as a variety of metabolic pathways (Figure 3A). Examination of the insulin secretion pathway via Pathview analysis indicated that the expression of several insulin pathway-related genes was regulated by GLIS3, including downregulation of the GLP1 receptor (Glp1r) and IP3 receptor (Itpr1), along with MafA and Slc2a2 (Figure 3B). The UCSC genome browser tracks in Figure 3C–D illustrate the significant downregulation of both MafA and Slc2a2 mRNA expression, consistent with the immunofluorescent staining (Figure 2A–B). Downregulation of Ins2 was also observed, consistent with the observations of other models of Glis3 deletion (Supplementary Figure 3)(Watanabe et al., 2009, Kang et al., 2009, Yang et al., 2009, Yang et al., 2011, Yang et al., 2013, Nogueira et al., 2013, Yang et al., 2017, Amin et al., 2018).
Figure 3. Gene expression analysis of Glis3Δpanc mice.



RNA-seq analysis of Glis3Δpanc islets at 4-weeks was performed. (A) KEGG pathway analysis of genes downregulated >1.5 fold and with an FDR of <0.05. Numbers behind bars indicate number of genes in the respective pathway. (B) Modified Pathview analysis of the Insulin Secretion pathway genes, with green representing downregulated genes and red representing upregulated genes. RNA-seq analysis of (C) MafA and (D) Slc2a2, the gene that encodes the GLUT2 protein. Data displayed via the UCSC genome browser. N=3 Glis3Δpanc, N=4 WT.
GLIS3 binds to beta cell genes in mouse islets.
In order to establish which of these differentially expressed genes were directly regulated by GLIS3, we performed ChIP-seq analysis of GLIS3 in adult mouse islets. As no reliable GLIS3 antibody currently exists, we utilized a mouse model where the C-terminus of GLIS3 is fused to an enhanced GFP tag (GLIS3-GFP), thus allowing for precipitation with a GFP antibody. One of the top consensus binding sequences detected in our ChIP-seq analysis matched that of Zic/Gli/Glis family members, similar to the consensus of the GLIS3 binding site previously identified in the thyroid gland (Figure 4A and (Kang et al., 2017)), indicating the ChIP-seq was successful in detecting specific GLIS3 binding. A complete list of bound genes (i.e. nearest gene to GLIS3 binding peak) is provided in Supplementary Table 3. GLIS3 binding was most highly enriched at introns within the gene body and promoter regions, consistent with what was observed in GLIS3 ChIP-seq experiments in the thyroid (Figure 4B)(Kang et al., 2017). Similar to previous reports by our group (ZeRuth et al., 2013), GLIS3 binding was observed at the Ins2 promoter (Figure 4C), as well as an enhancer for Slc2a2 (Figure 4D). Interestingly, all of the genes examined with a known role in beta cell function display GLIS3 binding within 2kb of their transcriptional start site (Table 1). However, only some of these genes were downregulated in Glis3Δpanc mice (Table 1), indicating that GLIS3 may not be essential for regulation of a fraction of these genes, or that their regulation by GLIS3 may be dependent upon upstream signaling events absent under the tested conditions. We also combined our ChIP-seq and gene expression data to examine more closely those genes directly regulated by GLIS3 (i.e. GLIS3-bound genes that are differentially expressed in Glis3Δpanc mice). KEGG pathway analysis of these genes identified many of the same pathways as found by analyzing gene expression data alone (Figure 3A, Supplementary Figure 4). This highlights the important role GLIS3 binding plays in regulating proper beta cell function, as indicated by the hyperglycemic phenotype of the Glis3Δpanc mice.
Figure 4. Global GLIS3 binding analysis in mouse islets reveals Slc2a2 direct regulation.


(A) De novo motif analysis was performed on ChIP-seq data using HOMER. (B) Binding analysis of Glis3 ChIP-seq data indicated a preference for introns and promoters. Upstream = −10kb to −1kb relative to TSS, TSS proximal = −1kb to TSS, Gene body = TSS to TES, Intergenic = remaining sequences. (C,D) ChIP-seq binding on the Ins2 promoter, and the Slc2a2 promoter and enhancer. Peaks indicates the peaks called using HOMER.
Table 1.
Islet-specific genes are bound by GLIS3
| Hormones | |||
|---|---|---|---|
| Gene | Fold Change | FDR | Glis3 ChIP-seq Signal |
| Ins1 | −2.116517315 | 0.00506878 | yes |
| Ins2 | −2.839350013 | 3.35E-13 | yes |
| Ppy | −8.154862239 | 1.87E-19 | yes |
| Gcg | −1.234894439 | NS | no |
| Sst | 1.083293292 | NS | no |
| Ghrl | 1.095231496 | NS | no |
|
| |||
| Beta cell Function | |||
|
| |||
| Slc2a2 | −5.112554821 | 9.58E-33 | yes |
| Kcnj11 | −1.299797953 | 0.01104388 | yes |
| Abcc8 | 1.444296553 | 0.00016462 | yes |
| Glp1r | −1.848329213 | 0.02182045 | yes |
| Chga | −2.13350168 | 3.53E-22 | yes |
| Ucn3 | −1.588987707 | NS | yes |
| Gjd2 | −1.120393715 | NS | yes |
|
| |||
| Transcription Factors | |||
|
| |||
| MafA | −2.693922579 | 0.00026979 | yes |
| Nkx6.1 | −2.239512577 | 1.03E-09 | yes |
| Rfx6 | 1.349799675 | 0.00446909 | yes |
| Isl1 | 1.640589346 | 4.17E-10 | yes |
| Ldb1 | −1.279981837 | 0.00500245 | yes |
| MafB | −1.496356376 | 0.00324023 | yes |
| FoxA2 | −1.281876971 | 0.045365 | yes |
| Pdx1 | −1.061699976 | NS | yes |
| Tshz1 | 1.084110443 | NS | yes |
| Mnx1 | 1.114782737 | NS | yes |
| InsM1 | 1.102778676 | NS | yes |
| Ngn3 | −1.731654424 | NS | yes |
| Nkx2.2 | −1.211137492 | NS | yes |
GLIS3 binding overlaps with that of other islet-enriched transcription factors.
Previous reports have suggested that islet enriched transcription factors often bind in close proximity to one another (Ediger et al., 2017). Moreover, immunoprecipitation assays indicated that GLIS3 is associated with PDX1, NEUROD1, and MAFA protein complexes (Yang et al., 2009). Indeed, motif analysis of GLIS3 ChIP-seq identified a number of additional motifs, including motifs for NEUROD1, FOX and RFX family members (FOXM1 and RFX5), and LHX2, which contains a homeobox DNA binding domain similar to PDX1, NKX6.1, ISL1, and NKX2.2 (Figure 4A). In order to determine whether GLIS3 co-localizes with other islet-enriched transcription factors on the same regulatory regions, we compared GLIS3 binding to that of PDX1, ISL1, NKX6.1, NKX2.2, NEUROD1, MAFA, and FOXA2 (Figure 5A). We also included the ISL1 coactivator LDB1, a critical co-regulator of ISL1 function (Hunter et al., 2013, Ediger et al., 2017). It is important to note that the quality of the ChIP-seq data compared varied greatly, as is typical among various transcription factors. Nevertheless, our analysis revealed that GLIS3 is associated with many of the same promoter/enhancer regions as the transcription factors mentioned above (Figure 5B). In addition to a global analysis, we examined the binding of these factors near the Ins2, Slc2a2, and MafA genes. Figures 5C–E demonstrate the co-localization of GLIS3 binding with that of several other critical islet transcription factors to regulatory regions near the Ins2, Slc2a2, and MafA genes, indicating potential coordination between these transcription factors. Glis3’s transcriptional regulation of MafA was further supported by luciferase assays of the MafA Region 3 upstream regulatory element. Glis3 stimulated MafA region 3 transcriptional activity, similar to what has been found for other factors (Supplementary Figure 5)(Raum et al., 2006). ChIP-seq binding data showed overlapping binding for almost all of the transcription factors examined, indicating there is likely a high level of coordination between GLIS3 and other islet enriched transcription factors in regulating genes that are critical for normal beta cell function (Figure 5F).
Figure 5. GLIS3 binding shows partial overlap with other islet enriched transcription factors.
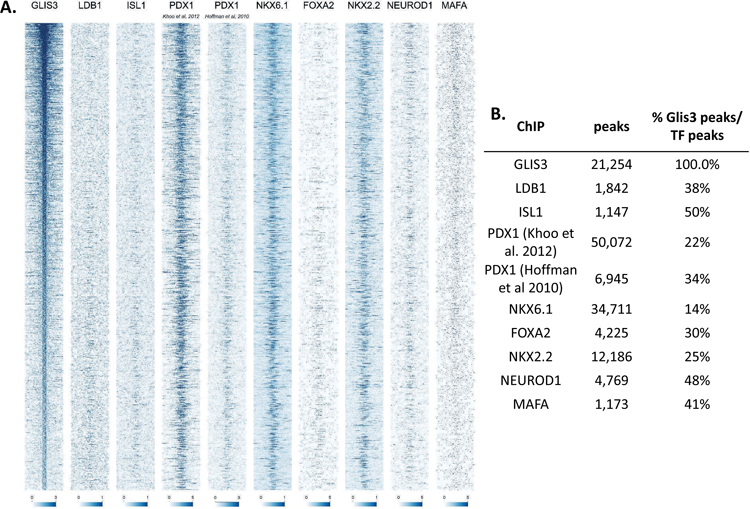
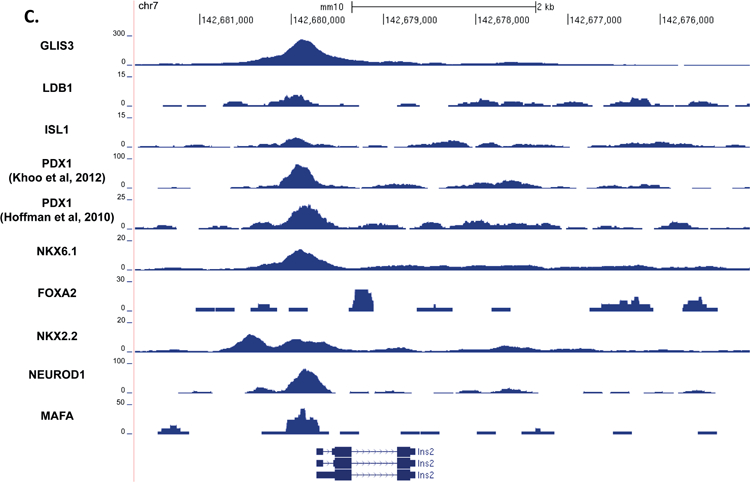
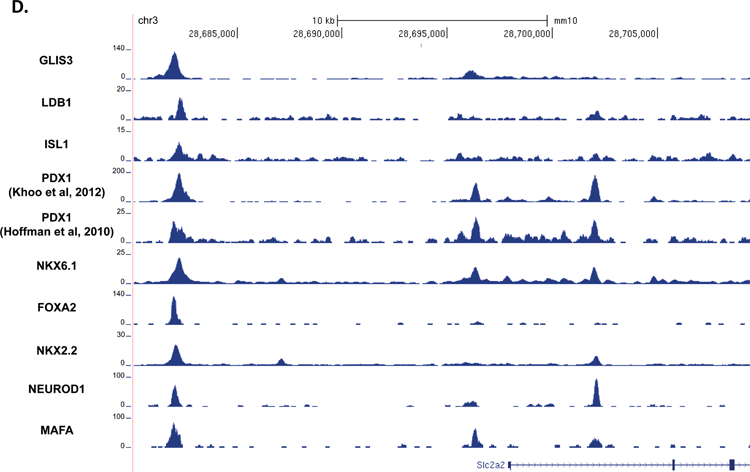
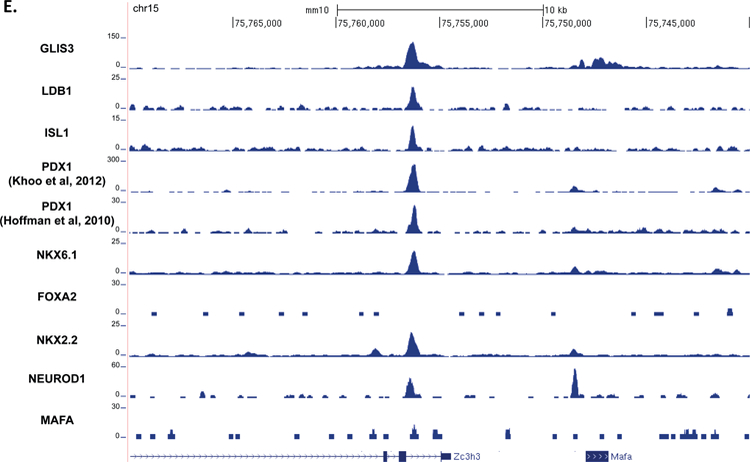
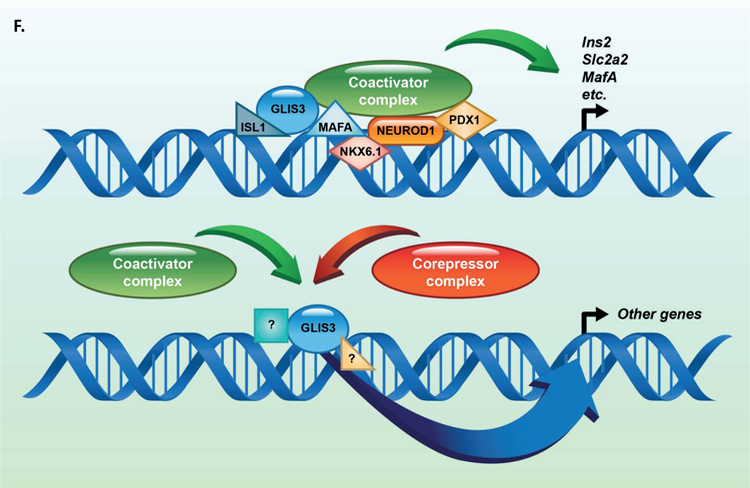
(A) Heatmap of the 2kb region centered on each of the 21,253 GLIS3 binding peaks with ChIP-seq signal normalized to 10 million reads for GLIS3, LDB1, ISL1, PDX1 (from two independent labs), NKX6.1, FOXA2, NKX2.2, NEUROD1, and MAFA. Color intensity indicates read depth, as shown by the scale below each TF panel. (B) Number of peaks called for each ChIP, as well as percentage of TF ChIP peaks colocalizing with GLIS3 peaks with signal above threshold cutoff. ChIP peak colocalization near the Ins2 (C), Slc2a2 (D), and MafA (E) genes. Data displayed via the UCSC genome browser. (F) Model of GLIS3 binding. Glis3 interacts with islet enriched transcription factors on regulatory hubs to control genes related to diabetes, and interacts with other unknown factors to recruit either coactivator or corepressor complexes to control genes related to other processes.
Discussion
Global Glis3 mutant mice have a short lifespan, develop polycystic kidney disease, hypothyroidism, and exhibit a defect in pancreatic beta cell development that leads to neonatal diabetes (Watanabe et al., 2009, Kang et al., 2009, Yang et al., 2009, Kang et al., 2017). These defects are similar to what is observed in humans with mutations in GLIS3 (Jetten, 2018). In order to study the function of GLIS3 in the pancreas while limiting secondary effects from other tissues, we developed a conditional mouse model in which GLIS3 function was impaired specifically in the pancreas. Since ubiquitous Glis3 knockout mice develop hypothyroidism that might influence the pancreatic phenotype (Chen et al., 2018), our model of Glis3 deletion allows us to examine the role of GLIS3 in beta cell function, while minimizing secondary effects caused by hyperglycemia and hypothyroidism.
Unlike global Glis3 mutants, which die by PND11 likely due to severe hyperglycemia, Glis3Δpanc mice begin to develop hyperglycemia around 4-weeks of age dependent upon the efficiency of Glis3 deletion (Supplementary Figure 1), with almost all Glis3Δpanc mice surviving until at least 8-weeks of age. As GLIS3 is essential for Ins2 expression, this indicates that only a subset of functional beta cells (those lacking Cre expression and thus still expressing GLIS3) appear necessary for maintaining euglycemia. Unfortunately, we were unable to test for the persistence of GLIS3 expression due to the lack of a working antibody. However, our interpretation is consistent with a study by Yang et al. showing that in adult mice a ~66% deletion of Glis3 exon 4, using an tamoxifen-inducible Mouse Insulin Promoter-driven Cre (MIP-CreERT), resulted in euglycemic mice, whereas a tamoxifen inducible β-cell specific Pdx1-CreERT deletion model with a >90% deletion of Glis3, produced hyperglycemic mice (Yang et al., 2017). This conclusion is further supported by a Diptheria toxin-based beta cell ablation model, where a ~50% loss of beta cells does not cause hyperglycemia, whereas a ~99% ablation does (Thorel et al., 2010). Furthermore, the persistence of some INSULIN+ (Figure 1C) or MAFA+ cells (Figure 2A) is consistent with the mosaicism of the Pdx1-Cre that has been observed in other deletion models, such as the Pdx1-Cre deletion of MafA ({Hang, 2014 #55}, where a subset of SLC30A8+ and GLUT2+ cells persist in the islets, likely due to the continued expression of MafA in those cells.
Our analysis of Glis3Δpanc mice revealed no increase in apoptosis at 2-weeks (i.e. prior to hyperglycemia), while proliferation was very slightly increased, possibly in response to an increased demand for insulin+ cells (Figure 2D, F). This stands in contrast to a previous study of GLIS3 function in the thyroid, which demonstrated that GLIS3 does play a role in stimulating proliferation in that tissue (Kang et al., 2017). Moreover, pancreatic ducts in ubiquitous Glis3 knockout mice, as well as Glis3Δpanc mice (Supplementary Figure 6), did display a cystic phenotype that involves increased proliferation, suggesting that the effect of GLIS3 on proliferation is cell type-dependent. Previous reports suggested that GLIS3 suppresses apoptosis in a pancreatic beta cell line by regulating Srsf6 (referred to as Srp55 in (Nogueira et al., 2013)) and in human stem cells differentiated into beta like cells through the TGFβ signaling pathway (Amin et al., 2018). However, in our in vivo model, we did not observe an increase in apoptosis, Srsf6 expression was not changed, nor were apoptotic or TGFβ pathways upregulated in our gene expression analysis (Figure 2F, Supplementary Table 2, and Figure 3A). It is possible that apoptosis is increased at some later time in our model; however, it would be difficult to distinguish this from the secondary effects of hyperglycemia. Instead, we observed changes in genes involved in a variety of metabolic pathways and a downregulation of genes involved in beta cell function (Figure 3A). These results highlight the important, but underappreciated differences that exist between cell line models and in vivo mouse systems, which we believe may more accurately reflect the in vivo role of GLIS3 in humans.
In addition to a lack of apoptosis, we did not detect expression of the progenitor marker NGN3 in Glis3Δpanc pancreas sections (Supplementary Figure 2), which has been reported to be re-expressed in dedifferentiated β cells (Talchai et al., 2012). This may be expected as GLIS3 is known to function as a positive regulator of Ngn3 transcription (Kang et al., 2009, Yang et al., 2011, Kim et al., 2012). We also did not observe increased expression of several other dedifferentiation-associated genes, such as L-myc, Oct4, or Nanog, in Glis3Δpanc islets (Supplementary Table 2)(Talchai et al., 2012). These observations suggest that impaired GLIS3 function does not cause dedifferentiation of beta cells; however, we cannot rule out that GLIS3 itself may be required for dedifferentiation. Nevertheless, we did detect a ~12-fold upregulation of Aldh1a3, a gene that has been reported to be associated with not only dedifferentiation, but also with beta cell dysfunction (Supplementary Table 2)(Kim-Muller et al., 2016). The expression of Igfbp4 and Cxcl12, “disallowed” genes in pancreatic islets (Pullen et al., 2010, Thorrez et al., 2011), were also upregulated in Glis3Δpanc islets. However, the vast majority of disallowed genes were not induced, including Ldha, Oat, Cat, Arhgdib, Mct1, Maf, Zfp36l1, Itih5, Pdgfra, Lmo4, Zyx, Smad3, Acot7, Cox5a, Fam59a, Gas6, Mgst1, Nfib, Plec1, Rpl36, Tgm2, Tst, and Zdhh. These findings are consistent with the concept that Glis3Δpanc beta cells do not exhibit a dedifferentiated phenotype, but remain as non-insulin producing, dysfunctional β-like cells.
The coordinated binding of transcription factors at “super” enhancer regions, regions bound by several tissue specific transcription factors for control of tissue specific genes, has been well documented (Loven et al., 2013, Whyte et al., 2013). Within the pancreatic islet, such enhancers have linked not only the binding of islet enriched transcription factors with histone marks of enhancers, but also with mutations related to the development of diabetes and dysregulated fasting glycemia (Pasquali et al., 2014, Stitzel et al., 2010). Analysis of the consensus GLIS3 binding motif suggested co-localization with motifs of other transcription factors important for the regulation of pancreatic β cell functions, such as NEUROD1, FOX and RFX family members, and the homeobox containing Lhx2 (similar to PDX1, NKX6.1, ISL1, and NKX2.2)(Figure 4A). We therefore sought to characterize GLIS3 binding in the context of other islet-enriched transcription factors. Ediger et al. recently described the binding of ISL1 in pancreatic islets, where ISL1 largely co-localized with other islet-enriched transcription factors (Ediger et al., 2017). Indeed, we observed that 50% of ISL1 sites overlapped with GLIS3 binding (Figure 5B). In contrast, we observed that GLIS3 only co-localizes with other islet-enriched transcription factors at a fraction of their target sites. A likely explanation for this is the significantly larger number of ChIP-seq peaks called in our GLIS3 analysis, as compared to the ISL1, MAFA, NEUROD1, and FOXA2 ChIP-seq data analysis (Figure 5B). However, even when comparing GLIS3 ChIP-seq data to transcription factors with more peaks (PDX1 and NKX6.1), overlap was limited to <25%. This suggests that islet-enriched transcription factors, including GLIS3, display context-specific co-localization, possibly reflecting tissue specific function. Indeed, genes with nearby overlapping peaks for GLIS3, PDX1, and NKX6.1 were associated particularly with Maturity Onset Diabetes of Young (MODY), insulin resistance, insulin signaling, Type 2 Diabetes, and insulin secretion pathways (Supplementary Figure 7). This supports a model in which a selective set of genes directly regulated by GLIS3 are co-regulated with other islet-enriched transcription factors through their interaction on the same distinct regulatory regions in beta cell-specific genes (Figure 5F). GLIS3 and other islet-specific transcription factors may then coordinate their transcriptional regulation through co-activator protein complexes recruited to these regulatory hubs.
In summary, our study provides the first in depth characterization of the role of GLIS3 in gene regulation within the pancreatic beta cell. GLIS3 has many functions within the beta cell, from regulating aspects of several metabolic pathways and intracellular signaling pathways, to regulating other transcription factors. Indeed, some of the effects on beta cell function could be attributed to GLIS3’s direct regulation of MafA through its region 3 regulatory domain (Figure 5E). MAFA has been shown to control expression of Ins2, Slc2a2, Slc30a8, Sytl4, and Stxbp1 all of which are downregulated in Glis3Δpanc mice (Hang et al., 2014). Unlike Glis3Δpanc mice, the main defect in MafAΔpanc mice appears to be in glucose-stimulated insulin secretion, as they do not display overt hyperglycemia. Additionally, GLIS3 regulates a variety of genes not apparently regulated by MAFA, such as Ngn3 and Nkx6.1. GLIS3 is also expressed in variety of tissues which do not express MAFA, indicative of GLIS3’s role in controlling pathways not directly related to beta cell function.
Recent reports have highlighted the importance of phosphorylation in mediating NGN3’s reprogramming effects (Azzarelli et al., 2018), and MAFA’s interaction with other transcription factors (Han et al., 2016). As GLIS3 is highly phosphorylated, it is likely that GLIS3 transcriptional activity is controlled by upstream and tissue-specific signaling pathways. What these signals are, and how GLIS3 integrates them into co-regulatory interactions or activation/repression functions, remains to be discovered. Understanding how GLIS3 coordinates with other factors, and specifically which upstream signals control GLIS3 activity and guide protein-protein interactions in vivo, might provide new therapies for diabetes treatment. Future studies will hopefully build upon the findings outlined here to distinguish how GLIS3 differentially regulates disease-relevant genes in different tissues, which upstream signaling pathways control GLIS3 activity, and provide insight into how these pathways may be targeted in disease treatment.
Supplementary Material
Supplementary Figure 1. Glis3Δpanc hyperglycemia is dependent on deletion efficiency. (A) Random blood glucose levels of mice at 4-weeks of age (N=9 WT, N=8 Glis3Δpanc). (B) Random blood glucose levels of mice at 4-weeks of age compared to the efficiency of removal of Glis3 in isolated islets from the same mice. Red dots are individual Glis3Δpanc mice, and blue dots are WT mice. (C) Random blood glucose levels of mice at 4 weeks of age with less than 75% removal (<75%) or greater than 75% removal (>75%). Efficiency of removal is based on qRT-PCR for the deleted Glis3 exon. Error bars indicate SEM. * p < 0.05.
Supplementary Table 3. GLIS3 ChIP-seq in mouse islets at 8-weeks.
Supplementary Table 4. KEGG pathway analysis of Glis3Δpanc downregulated genes.
Supplementary Table 5. KEGG pathway analysis of GLIS3 direct target genes.
Supplementary Table 6. KEGG pathway analysis of genes co-bound by GLIS3, PDX1, and NKX6.1.
Supplementary Figure 2. NGN3 is not expressed in Glis3Δpanc mice. Immunostaining for NGN3 at 8-weeks of age is shown, as well as in WT mice at e15.5 as a positive control. Images shown are representative.
Supplementary Figure 3. Ins2 expression is decreased in Glis3Δpanc mice. Islet RNAseq data for Ins2 is shown in Glis3Δpanc mice and control littermates at 4-weeks of age.
Supplementary Figure 4. Pathway analysis of direct GLIS3 target genes. KEGG pathway analysis was performed on genes that were downregulated >1.5 fold, with an FDR of <0.05, and that had at least 1 nearby peak for GLIS3 binding.
Supplementary Figure 5. GLIS3 stimulates MafA Region 3 activity in 293T cells. MafA Region 3 luciferase activity was calculated relative to CMV-driven beta-galactosidase activity, and normalized to empty pGL4.27 vector. Transcriptional activity was stimulated by the addition of Glis3. Representative experiment shown, error bars indicate standard deviation for experiment run in triplicate. * p<0.01 comparing pGL4.27-MafA Reg3 with or without Glis3.
Supplementary Figure 6. Glis3Δpanc mice contain cystic ducts. Pancreas sections from 2-week and 10-week old Glis3Δpanc mice were stained with DBA-Fluorescein and DAPI. Representative images are shown.
Supplementary Figure 7. Pathway analysis of genes bound by GLIS3, PDX1, and NKX6.1. KEGG pathway analysis was performed on genes that had at least 1 nearby peak for GLIS3, PDX1, and NKX6.1 binding.
Supplementary Table 1. Primers and Antibodies.
Supplementary Table 2. Glis3Δpanc RNAseq data in 4-week islets.
Acknowledgments:
We would like to thank Molly Cook, Nicole Reeves, and the Epigenomics Core Laboratory at NIEHS for their assistance in high throughput sequencing. We would also like to thank Dr. Chad Hunter at the University of Alabama-Birmingham for assistance in review of this manuscript, and the Cell Biology group at NIEHS for thoughtful comments throughout.
Funding: This research was supported by the Intramural Research Program of the NIEHS, NIH Z01-ES-101585.
Footnotes
Declaration of Interest: We have no conflict of interest that could be perceived as prejudicing the impartiality of the research reported
References
- AMIN S, COOK B, ZHOU T, GHAZIZADEH Z, LIS R, ZHANG T, KHALAJ M, CRESPO M, PERERA M, XIANG JZ, et al. 2018. Discovery of a drug candidate for GLIS3-associated diabetes. Nat Commun, 9, 2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AZZARELLI R, RULANDS S, NESTOROWA S, DAVIES J, CAMPINOTI S, GILLOTIN S, BONFANTI P, GOTTGENS B, HUCH M, SIMONS B, et al. 2018. Neurogenin3 phosphorylation controls reprogramming efficiency of pancreatic ductal organoids into endocrine cells. Sci Rep, 8, 15374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BARRETT JC, CLAYTON DG, CONCANNON P, AKOLKAR B, COOPER JD, ERLICH HA, JULIER C, MORAHAN G, NERUP J, NIERRAS C, et al. 2009. Genome-wide association study and meta-analysis find that over 40 loci affect risk of type 1 diabetes. Nat Genet, 41, 703–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BEAK JY, KANG HS, KIM YS & JETTEN AM 2008. Functional analysis of the zinc finger and activation domains of Glis3 and mutant Glis3(NDH1). Nucleic Acids Res, 36, 1690–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHEN C, XIE Z, SHEN Y & XIA SF 2018. The Roles of Thyroid and Thyroid Hormone in Pancreas: Physiology and Pathology. Int J Endocrinol, 2018, 2861034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHO YS, CHEN CH, HU C, LONG J, ONG RT, SIM X, TAKEUCHI F, WU Y, GO MJ, YAMAUCHI T, et al. 2011. Meta-analysis of genome-wide association studies identifies eight new loci for type 2 diabetes in east Asians. Nat Genet, 44, 67–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DIMITRI P, WARNER JT, MINTON JA, PATCH AM, ELLARD S, HATTERSLEY AT, BARR S, HAWKES D, WALES JK & GREGORY JW 2011. Novel GLIS3 mutations demonstrate an extended multisystem phenotype. Eur J Endocrinol, 164, 437–43. [DOI] [PubMed] [Google Scholar]
- DOBIN A, DAVIS CA, SCHLESINGER F, DRENKOW J, ZALESKI C, JHA S, BATUT P, CHAISSON M & GINGERAS TR 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics, 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUPUIS J, LANGENBERG C, PROKOPENKO I, SAXENA R, SORANZO N, JACKSON AU, WHEELER E, GLAZER NL, BOUATIA-NAJI N, GLOYN AL, et al. 2010. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet, 42, 105–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EDIGER BN, LIM HW, JULIANA C, GROFF DN, WILLIAMS LT, DOMINGUEZ G, LIU JH, TAYLOR BL, WALP ER, KAMESWARAN V, et al. 2017. LIM domain-binding 1 maintains the terminally differentiated state of pancreatic beta cells. J Clin Invest, 127, 215–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HABEB AM, AL-MAGAMSI MS, EID IM, ALI MI, HATTERSLEY AT, HUSSAIN K & ELLARD S 2012. Incidence, genetics, and clinical phenotype of permanent neonatal diabetes mellitus in northwest Saudi Arabia. Pediatr Diabetes, 13, 499–505. [DOI] [PubMed] [Google Scholar]
- HAN SI, TSUNEKAGE Y & KATAOKA K 2016. Phosphorylation of MafA enhances interaction with Beta2/NeuroD1. Acta Diabetol, 53, 651–60. [DOI] [PubMed] [Google Scholar]
- HANG Y, YAMAMOTO T, BENNINGER RK, BRISSOVA M, GUO M, BUSH W, PISTON DW, POWERS AC, MAGNUSON M, THURMOND DC, et al. 2014. The MafA transcription factor becomes essential to islet beta-cells soon after birth. Diabetes, 63, 1994–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEINZ S, BENNER C, SPANN N, BERTOLINO E, LIN YC, LASLO P, CHENG JX, MURRE C, SINGH H & GLASS CK 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell, 38, 576–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HINGORANI SR, PETRICOIN EF, MAITRA A, RAJAPAKSE V, KING C, JACOBETZ MA, ROSS S, CONRADS TP, VEENSTRA TD, HITT BA, et al. 2003. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell, 4, 437–50. [DOI] [PubMed] [Google Scholar]
- HU Z & TEE WW 2017. Enhancers and chromatin structures: regulatory hubs in gene expression and diseases. Biosci Rep, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUANG DA W, SHERMAN BT & LEMPICKI RA 2009a. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res, 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUANG DA W, SHERMAN BT & LEMPICKI RA 2009b. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc, 4, 44–57. [DOI] [PubMed] [Google Scholar]
- HUNTER CS, DIXIT S, COHEN T, EDIGER B, WILCOX C, FERREIRA M, WESTPHAL H, STEIN R & MAY CL 2013. Islet alpha-, beta-, and delta-cell development is controlled by the Ldb1 coregulator, acting primarily with the islet-1 transcription factor. Diabetes, 62, 875–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JETTEN AM 2018. GLIS1–3 transcription factors: critical roles in the regulation of multiple physiological processes and diseases. Cell Mol Life Sci, 75, 3473–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KANG HS, KIM YS, ZERUTH G, BEAK JY, GERRISH K, KILIC G, SOSA-PINEDA B, JENSEN J, PIERREUX CE, LEMAIGRE FP, et al. 2009. Transcription factor Glis3, a novel critical player in the regulation of pancreatic beta-cell development and insulin gene expression. Mol Cell Biol, 29, 6366–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KANG HS, KUMAR D, LIAO G, LICHTI-KAISER K, GERRISH K, LIAO XH, REFETOFF S, JOTHI R & JETTEN AM 2017. GLIS3 is indispensable for TSH/TSHR-dependent thyroid hormone biosynthesis and follicular cell proliferation. J Clin Invest, 127, 4326–4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KANG HS, TAKEDA Y, JEON K & JETTEN AM 2016. The Spatiotemporal Pattern of Glis3 Expression Indicates a Regulatory Function in Bipotent and Endocrine Progenitors during Early Pancreatic Development and in Beta, PP and Ductal Cells. PLoS One, 11, e0157138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KANG HS, ZERUTH G, LICHTI-KAISER K, VASANTH S, YIN Z, KIM YS & JETTEN AM 2010. Gli-similar (Glis) Kruppel-like zinc finger proteins: insights into their physiological functions and critical roles in neonatal diabetes and cystic renal disease. Histol Histopathol, 25, 1481–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIM-MULLER JY, FAN J, KIM YJ, LEE SA, ISHIDA E, BLANER WS & ACCILI D 2016. Aldehyde dehydrogenase 1a3 defines a subset of failing pancreatic beta cells in diabetic mice. Nat Commun, 7, 12631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KIM YS, KANG HS, TAKEDA Y, HOM L, SONG HY, JENSEN J & JETTEN AM 2012. Glis3 regulates neurogenin 3 expression in pancreatic beta-cells and interacts with its activator, Hnf6. Mol Cells, 34, 193–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LANGMEAD B, TRAPNELL C, POP M & SALZBERG SL 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol, 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIAO Y, SMYTH GK & SHI W 2014. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics, 30, 923–30. [DOI] [PubMed] [Google Scholar]
- LICHTI-KAISER K, ZERUTH G, KANG HS, VASANTH S & JETTEN AM 2012. Gli-similar proteins: their mechanisms of action, physiological functions, and roles in disease. Vitam Horm, 88, 141–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIU C, LI H, QI L, LOOS RJ, QI Q, LU L, GAN W & LIN X 2011. Variants in GLIS3 and CRY2 are associated with type 2 diabetes and impaired fasting glucose in Chinese Hans. PLoS One, 6, e21464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOVE MI, HUBER W & ANDERS S 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol, 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOVEN J, HOKE HA, LIN CY, LAU A, ORLANDO DA, VAKOC CR, BRADNER JE, LEE TI & YOUNG RA 2013. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell, 153, 320–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LUO W, PANT G, BHAVNASI YK, BLANCHARD SG JR. & BROUWER C 2017. Pathview Web: user friendly pathway visualization and data integration. Nucleic Acids Res, 45, W501–W508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MARTIN M 2011. Cutadapt removes adapater sequences from high throughput sequencing reads. EMBnet journal, 17, 10–12. [Google Scholar]
- NOGUEIRA TC, PAULA FM, VILLATE O, COLLI ML, MOURA RF, CUNHA DA, MARSELLI L, MARCHETTI P, CNOP M, JULIER C, et al. 2013. GLIS3, a susceptibility gene for type 1 and type 2 diabetes, modulates pancreatic beta cell apoptosis via regulation of a splice variant of the BH3-only protein Bim. PLoS Genet, 9, e1003532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PASQUALI L, GAULTON KJ, RODRIGUEZ-SEGUI SA, MULARONI L, MIGUEL-ESCALADA I, AKERMAN I, TENA JJ, MORAN I, GOMEZ-MARIN C, VAN DE BUNT M, et al. 2014. Pancreatic islet enhancer clusters enriched in type 2 diabetes risk-associated variants. Nat Genet, 46, 136–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PULLEN TJ, KHAN AM, BARTON G, BUTCHER SA, SUN G & RUTTER GA 2010. Identification of genes selectively disallowed in the pancreatic islet. Islets, 2, 89–95. [DOI] [PubMed] [Google Scholar]
- QUINLAN AR & HALL IM 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics, 26, 841–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAUM JC, GERRISH K, ARTNER I, HENDERSON E, GUO M, SUSSEL L, SCHISLER JC, NEWGARD CB & STEIN R 2006. FoxA2, Nkx2.2, and PDX-1 regulate islet beta-cell-specific mafA expression through conserved sequences located between base pairs −8118 and −7750 upstream from the transcription start site. Mol Cell Biol, 26, 5735–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REES SD, HYDRIE MZ, O’HARE JP, KUMAR S, SHERA AS, BASIT A, BARNETT AH & KELLY MA 2011. Effects of 16 genetic variants on fasting glucose and type 2 diabetes in South Asians: ADCY5 and GLIS3 variants may predispose to type 2 diabetes. PLoS One, 6, e24710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCOVILLE DW, KANG HS & JETTEN AM 2017. GLIS1–3: emerging roles in reprogramming, stem and progenitor cell differentiation and maintenance. Stem Cell Investig, 4, 80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SENEE V, CHELALA C, DUCHATELET S, FENG D, BLANC H, COSSEC JC, CHARON C, NICOLINO M, BOILEAU P, CAVENER DR, et al. 2006. Mutations in GLIS3 are responsible for a rare syndrome with neonatal diabetes mellitus and congenital hypothyroidism. Nat Genet, 38, 682–7. [DOI] [PubMed] [Google Scholar]
- STITZEL ML, SETHUPATHY P, PEARSON DS, CHINES PS, SONG L, ERDOS MR, WELCH R, PARKER SC, BOYLE AP, SCOTT LJ, et al. 2010. Global epigenomic analysis of primary human pancreatic islets provides insights into type 2 diabetes susceptibility loci. Cell Metab, 12, 443–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TALCHAI C, XUAN S, LIN HV, SUSSEL L & ACCILI D 2012. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell, 150, 1223–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THOREL F, NEPOTE V, AVRIL I, KOHNO K, DESGRAZ R, CHERA S & HERRERA PL 2010. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature, 464, 1149–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THORREZ L, LAUDADIO I, VAN DEUN K, QUINTENS R, HENDRICKX N, GRANVIK M, LEMAIRE K, SCHRAENEN A, VAN LOMMEL L, LEHNERT S, et al. 2011. Tissue-specific disallowance of housekeeping genes: the other face of cell differentiation. Genome Res, 21, 95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WATANABE N, HIRAMATSU K, MIYAMOTO R, YASUDA K, SUZUKI N, OSHIMA N, KIYONARI H, SHIBA D, NISHIO S, MOCHIZUKI T, et al. 2009. A murine model of neonatal diabetes mellitus in Glis3-deficient mice. FEBS Lett, 583, 2108–13. [DOI] [PubMed] [Google Scholar]
- WEN X & YANG Y 2017. Emerging roles of GLIS3 in neonatal diabetes, type 1 and type 2 diabetes. J Mol Endocrinol, 58, R73–R85. [DOI] [PubMed] [Google Scholar]
- WHYTE WA, ORLANDO DA, HNISZ D, ABRAHAM BJ, LIN CY, KAGEY MH, RAHL PB, LEE TI & YOUNG RA 2013. Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell, 153, 307–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG Y, BUSH SP, WEN X, CAO W & CHAN L 2017. Differential Gene Dosage Effects of Diabetes-Associated Gene GLIS3 in Pancreatic beta Cell Differentiation and Function. Endocrinology, 158, 9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG Y, CHANG BH & CHAN L 2013. Sustained expression of the transcription factor GLIS3 is required for normal beta cell function in adults. EMBO Mol Med, 5, 92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG Y, CHANG BH, SAMSON SL, LI MV & CHAN L 2009. The Kruppel-like zinc finger protein Glis3 directly and indirectly activates insulin gene transcription. Nucleic Acids Res, 37, 2529–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG Y, CHANG BH, YECHOOR V, CHEN W, LI L, TSAI MJ & CHAN L 2011. The Kruppel-like zinc finger protein GLIS3 transactivates neurogenin 3 for proper fetal pancreatic islet differentiation in mice. Diabetologia, 54, 2595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZERUTH GT, TAKEDA Y & JETTEN AM 2013. The Kruppel-like protein Gli-similar 3 (Glis3) functions as a key regulator of insulin transcription. Mol Endocrinol, 27, 1692–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHOU Q, LAW AC, RAJAGOPAL J, ANDERSON WJ, GRAY PA & MELTON DA 2007. A multipotent progenitor domain guides pancreatic organogenesis. Dev Cell, 13, 103–14. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Glis3Δpanc hyperglycemia is dependent on deletion efficiency. (A) Random blood glucose levels of mice at 4-weeks of age (N=9 WT, N=8 Glis3Δpanc). (B) Random blood glucose levels of mice at 4-weeks of age compared to the efficiency of removal of Glis3 in isolated islets from the same mice. Red dots are individual Glis3Δpanc mice, and blue dots are WT mice. (C) Random blood glucose levels of mice at 4 weeks of age with less than 75% removal (<75%) or greater than 75% removal (>75%). Efficiency of removal is based on qRT-PCR for the deleted Glis3 exon. Error bars indicate SEM. * p < 0.05.
Supplementary Table 3. GLIS3 ChIP-seq in mouse islets at 8-weeks.
Supplementary Table 4. KEGG pathway analysis of Glis3Δpanc downregulated genes.
Supplementary Table 5. KEGG pathway analysis of GLIS3 direct target genes.
Supplementary Table 6. KEGG pathway analysis of genes co-bound by GLIS3, PDX1, and NKX6.1.
Supplementary Figure 2. NGN3 is not expressed in Glis3Δpanc mice. Immunostaining for NGN3 at 8-weeks of age is shown, as well as in WT mice at e15.5 as a positive control. Images shown are representative.
Supplementary Figure 3. Ins2 expression is decreased in Glis3Δpanc mice. Islet RNAseq data for Ins2 is shown in Glis3Δpanc mice and control littermates at 4-weeks of age.
Supplementary Figure 4. Pathway analysis of direct GLIS3 target genes. KEGG pathway analysis was performed on genes that were downregulated >1.5 fold, with an FDR of <0.05, and that had at least 1 nearby peak for GLIS3 binding.
Supplementary Figure 5. GLIS3 stimulates MafA Region 3 activity in 293T cells. MafA Region 3 luciferase activity was calculated relative to CMV-driven beta-galactosidase activity, and normalized to empty pGL4.27 vector. Transcriptional activity was stimulated by the addition of Glis3. Representative experiment shown, error bars indicate standard deviation for experiment run in triplicate. * p<0.01 comparing pGL4.27-MafA Reg3 with or without Glis3.
Supplementary Figure 6. Glis3Δpanc mice contain cystic ducts. Pancreas sections from 2-week and 10-week old Glis3Δpanc mice were stained with DBA-Fluorescein and DAPI. Representative images are shown.
Supplementary Figure 7. Pathway analysis of genes bound by GLIS3, PDX1, and NKX6.1. KEGG pathway analysis was performed on genes that had at least 1 nearby peak for GLIS3, PDX1, and NKX6.1 binding.
Supplementary Table 1. Primers and Antibodies.
Supplementary Table 2. Glis3Δpanc RNAseq data in 4-week islets.