

Abstract
Anaplastic thyroid cancer (ATC) is an aggressive malignancy with limited treatment options. We explored novel treatment modalities by targeting epigenetic modifications using inhibitors of BET (e.g. BRD4) activity. We evaluated the efficacy in the treatment of ATC of a novel BET inhibitor, PLX51107 (PLX), currently in clinical trials for other solid tumors and hematologic malignancies, alone or combined with an MEK inhibitor, PD0325901(PD). To elucidate effects of these inhibitors on growth of ATC, we treated ATC cells derived from patient tumors (THJ-11T and THJ-16T cells) and mouse xenograft tumors with inhibitors. We found PLX and PD inhibitors singly inhibited proliferation of both human ATC cells lines, but together exhibited stronger inhibition of proliferation. In mouse xenografts, the combination treatment almost totally blocked growth in xenograft tumors derived from both ATC cells. PD effectively attenuated MEK-ERK signaling, which was further enhanced by PLX in the combined treatment in cultured cells and tumors. Importantly, the combination of PLX and PD acted synergistically to suppress MYC transcription to increase p27 in decreasing tumor cell proliferation. PLX and PD cooperated to upregulate pro-apoptotic proteins to promote apoptosis. These two inhibitors converged to reduce the binding of BRD4 to the MYC promoter to suppress the MYC expression. These findings indicate that combined treatment of BET and MEK-ERK inhibitors was more effective to treat ATC than single targeted treatment. Synergistic suppression of MYC transcription via collaborative actions on chromatin modifications suggested that targeting epigenetic modifications could provide novel treatment opportunities for ATC.
Keywords: tumorigenesis, anaplastic thyroid cancer, bromodomain and extraterminal protein inhibitor, PLX51107, PD0325901
Introduction
Anaplastic thyroid cancer (ATC) is one of the most aggressive endocrine malignancies. Patients rarely survive more than 1 year after diagnosis and have very limited treatment options. In addition to genetic alterations, aberrant expression of the MYC gene was identified as a critical oncogenic event in the development and progression of ATC (Enomoto, et al. 2017; Haugen, et al. 1993). MYC protein is frequently highly elevated in ATC tumor tissues (Enomoto et al. 2017), and the increased MYC expression has been associated with an unfavorable prognosis (Romano, et al. 1993; Terrier, et al. 1988). In a mouse model of ATC, increased expression of the Myc gene was shown to be associated with thyroid cancer progression as indicated by the loss of differentiation (Zhu, et al. 2014). In the mouse model of lung cancer, when the functions of the Myc gene were blocked by a dominant negative MYC mutant, the lung tumors could be completely eradicated (Soucek, et al. 2013). These observations suggested that MYC could be an effective target for therapeutic intervention in ATC.
So far, no effective approach is available to directly modulate the functions of the MYC protein itself. The bromodomain and extraterminal domain (BET) family of proteins interacts with acetylated histones to assemble transcription activators on specific sites of chromatin (Filippakopoulos, et al. 2012). Selective small-molecule inhibitors such as JQ1 that block the interaction of BET proteins with acetylated histones have been found to be a potent inhibitor of the MYC transcriptional program via attenuation of superenhancers (Delmore, et al. 2011). In thyroid cancer, JQ1 was shown to reduce Myc expression, arrest cell cycle progression, and inhibit tumor growth in a xenograft mouse model (Gao, et al. 2016; Mio, et al. 2016). In a preclinical mouse model of ATC, we found that JQ1 effectively suppressed MYC expression, prolonged survival, inhibited tumor growth, and attenuated transcriptional programs critical for tumor cell proliferation (Zhu, et al. 2017).
Intriguingly, despite JQ1’s effectiveness in the suppression of MYC transcription, resulting in the inhibition of tumor growth, it had no apparent inhibition on tumor cell invasion and metastasis. These observations raised the possibility that other oncogenic events could drive thyroid tumor invasion and metastasis in the progression of ATC. The MAPK-MEK signaling pathway is often over-activated in human ATC and is associated with the progression of ATC. Using both JQ1 and an MEK inhibitor, trametinib, we have shown that the combination of the two inhibitors more potently inhibited the recruitment of BET proteins to the promoter of the MYC gene to suppress the MYC transcription, tumor cell proliferation, tumor growth, and tumor cell invasion (Zhu, et al. 2018).
Recently, a new BET inhibitor PLX51107 (PLX) has been found to have more favorable pharmacokinetic properties than JQ1 and other BET inhibitors (Ozer, et al. 2018). It is currently being evaluated in clinical trials for solid tumors and hematological malignancies. PD is also an MEK inhibitor in clinical trials for several types of cancers (Barrett, et al. 2008). In the present studies, we evaluated whether combination of PLX and PD is effective for the treatment of ATC. Our results showed that the combination of the two inhibitors cooperated to inhibit the recruitment of BET proteins to the promoter of the MYC gene, resulting in suppression of the MYC transcription accompanied by reduction of tumor cell proliferation and tumor growth. Our results clearly demonstrated that the combination of BET and MEK inhibitors is an effective approach to modulate the epigenetic modifications for the treatment of ATC.
Materials and Methods
Cell culture
Two human anaplastic thyroid cancer cell lines (THJ-11T and THJ-16T) were gifts from Dr. John A. Copland III at the Mayo Foundation for Medical Education and Research (Marlow, et al. 2010). The cells were cultured in RPMI-1640 media supplemented with 10% fetal bovine serum (FBS), 1% non-essential amino acid, 1% sodium pyruvate, and 1% antibiotic-antimycotic solution (Thermo Fisher Scientific, Waltham, MA) in 5% CO2 at 37℃ in a humidified incubator.
Cell proliferation assays
PLX and PD were from Plexxikon Inc (Berkeley, CA). To evaluate the effect of the inhibitors on cell proliferation, cells were cultured in the medium with PLX, PD, or both PLX and PD at a concentration of 500 nM or DMSO in 6-well plates each with triplicates. Cell number was counted each day for 5 days using a cell counter (Countess II, Thermo Fisher Scientifid, Waltham, MA)
For the determination of EC50 of PLX, PD or the PLX and PD combined, we used MTT (3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide, Sigma, M2128) colorimetric assay. THJ-11T and THJ-16T cells were plated at the density of 2,000 cells per well in 96-well plates, followed by treating the cells with increasing concentrations of PLX and/or PD. After 48-hour treatment, the cells were incubated with MTT reagent (5 mg/mL) for 4 hours and MTT formazan generated by mitochondrial-dehydrogenases of living cells was solubilized in DMSO. Absorbance was measured using a VERSA Max ELISA reader (Molecular devices, Union City, CA) at 540 nm. The results were normalized to each DMSO-only control and dose-response curves were determined using GraphPad Prism 7.0 (San Diego, CA).
Western blot analysis
Western blot analyses were carried out as described previously (Zhu et al. 2014). Tumor tissues or cultured cells were washed with phosphate-buffered saline and homogenized in a solution with 50 mM Tris buffer, 150 mM NaCl, 1 mm ethylenediaminetetraacetic acid, 1% NP40 and proteinase (Roche Diagnostics, GmbH, Mannheim, Germany)/phosphatase inhibitors (Halt Phosphatase inhibitor cocktail, Thermo Scientific). After centrifuge at 14000 rpm for 5 minutes, lysates were used for Western blot analysis. The antibodies against MYC were used (ab32072) from Abcam, phosphorylated RB (S780) (#9307), RB (#9309), Bim (#2933), p-Erk½ (#9101), total-ERK (#9102), p27 (#3686), PARP (#9542) and GAPDH (#2118) from Cell Signaling. After being washed, the membranes were incubated with horseradish peroxidase-conjugated anti-mouse immunoglobulin G (Gillings, et al.) (NA9310 GE Healthcare) or anti-rabbit IgG (NA9340 GE Healthcare) as the secondary antibody and subsequently detected by means of an ECL system (Western Lightning® Plus-ECL, PerkinElmer, Waltham, MA). Band intensities were quantified by the ImageJ software (ImageJ 1.48v; Wayne Rasband, NIH).
RNA extraction and real time RT-PCR analysis
RNA extraction and real time RT-PCR analysis were carried out as previously described (Zhu et al. 2017). For the MYC gene: forward, 5’-CCTACCCTCTCAACGACAGC-3’; reverse, 5’-CTCTGACCTTTTGCCAGGAG-3’. For the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) gene: forward, 5’-CGAGATCCCTCCAAAATCAA-3’; reverse, 5’-GGTGCTAAGCAGTTGGTGGT-3’.
In vivo xenograft tumor assays
For xenograft studies, 6- to 8-week-old female athymic nude mice were used. All animal experiments were performed under protocols approved by the National Cancer Institute Animal Care and Use Committee. Five million of the THJ-11T or THJ-16 cells in 200 μl 50% Matrigel basement membrane matrix (BD Biosciences, Cat. 354234) were inoculated subcutaneously into the right flank of mice 2 to 3 weeks before treatment. The treatment with vehicle, PLX, PD, or both PLX and PD started when the median tumor size reached approximately 100-150 mm3. PLX was administered via oral gavage at a dose of 24 mg/kg/mouse per day. PD was administered via oral gavage at a dose of 1 mg/kg/mouse per day. The tumor size was measured with a caliper every 1-4 days. The mice were treated either for a short-term duration or long-term duration. For the short-treatment, mice inoculated with THJ-11T or THJ-16T cells were treated with PLX, PD or the combination of the two drugs for 24 days or 14 days, respectively. For long-term treatment, mice inoculated with THJ-11T cells were received treatment uninterrupted for 48 days in one group, or in another group, mice were treated for 20 days followed by termination of the treatment, but were continued to be monitored for the tumor progression. All mice were euthanized at the end of the treatment outlined above and the tumors were dissected for further analysis.
Flow cytometric analysis of apoptosis
THJ-11T and THJ-16T cells were treated with vehicle, PLX (500 nM), PD (500 nM), and both PLX and PD for 48 hours. After treatment, cells were collected and subjected to Annexin V and propidium iodide (PI) staining using an FITC-Annexin V Apoptosis Detection Kit I (Cat#556547, BD Bioscience, San Jose, CA), according to the manufacturer’s instructions. Apoptotic cells were analyzed by flow cytometry (LSRFortessa II, BD Biosciences, San Jose, CA) followed by FlowJo (FlowJo, AShjland OR).
Chromatin immunoprecipitation (ChIP) assay
ChIP assay was performed with slight modification as described previously (Fozzatti, et al. 2011). Briefly, cultured cells treated with DMSO, PLX, PD or combined PLX and PD were fixed in 1% of formaldehyde for 10 minutes and quenched by addition of glycine with a 0.125 M final concentration for 5 minutes. Immunoprecipitation was carried out using anti-BRD4 (A301-985A50, Bethyl Laboratories Inc) or IgG as a negative control. The primers for the MYC gene were forward, 5′-GAGCAGCAGAGAAAGGGAGA-3′; and reverse, 5′-CAGCCGAGCACTCTAGCTCT-3′.
Statistical analysis
All data are expressed as mean ± standard deviation. All tests were two-sided and p<0.05 was considered significant. GraphPad Prism version 7.0 for Mac OS X was used to perform analyses of variances.
Results
Combined treatment of PLX and PD potently inhibits cell proliferation in human ATC cells
We evaluated the effect of PLX, PD, and the combined treatment of the two inhibitors on the proliferation of two human ATC cell lines: THJ-11T and THJ-16T. These two cell lines were derived from ATC patients that have different genetic lesions (Marlow et al. 2010). We found that either PLX (500 nM) or PD (500 nM) inhibited cell proliferation in both THJ-11T and THJ-16T cells (Fig. 1A and 1B). THJ-11T cell numbers were decreased by 88.9% and 89.9% after 5 days of treatment with PLX or PD, respectively (Fig. 1A). THJ-16T cell numbers were decreased by 88.4% and 94.7% after 5 days of treatment with PLX or PD, respectively (Fig. 1B). However, combined treatment of PLX and PD inhibited cell proliferation by 95.8% and 97.3% in both THJ-11T and THJ-16T cells (Fig. 1A and 1B, respectively). The findings indicated that combined treatment was most effective in the inhibition of tumor cell proliferation. The effectiveness of the combined treatment in the inhibition of cell proliferation was further confirmed by the determination of IC50 using MTT assays (Figure 1C and 1D). PLX, PD or their combination decreased cell viability in a dose-dependent manner with IC50 values of 1.61, 10.79 and 0.73 uΜ in the THJ-11T cells (Fig. 1C), IC50 values of 1.38, 3.72 and 0.48 uM in the THJ-16T cells (Fig. 1D), respectively. In this cell-based MTT analysis, the order of efficacy in the inhibition of cell proliferation was PD<PLX<PD+PLX. It was clear that the combined treatment was most effective in the inhibiting of ATC cell proliferation.
Figure 1.
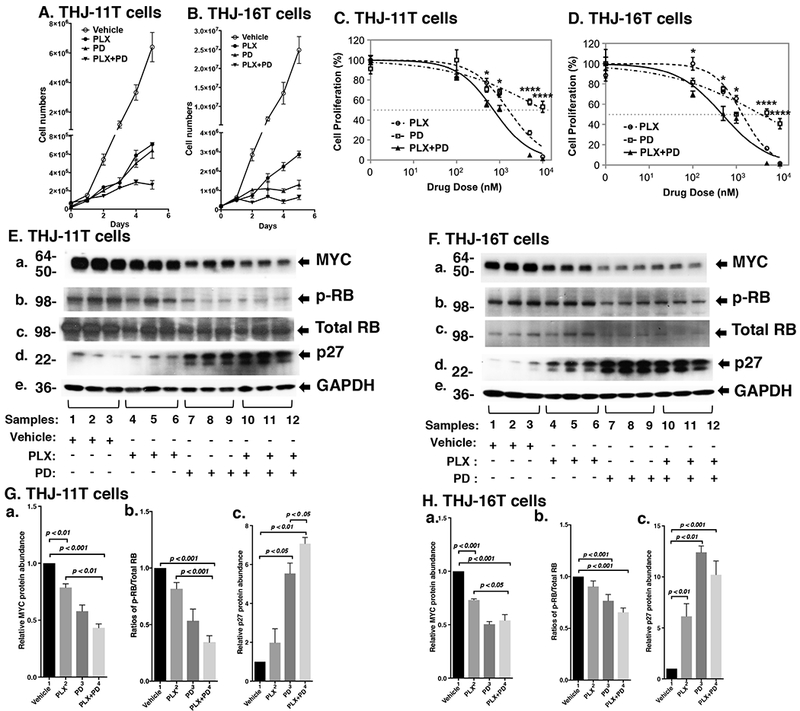
(A and B) The combined treatment of PLX and PD more potently inhibited cell proliferation in the ATC cell lines. Cell proliferation curves for the THJ-11T cells and THJ-16T cells, respectively. (C and D) Cell survival curves showing IC50 values of PLX, PD, and their combination in THJ-11T (C) and THJ-16T (D) cells following 48 hours of treatment. The experiments were performed in triplicate. Significant differences between the effects of the drug combination and its individual components were determined by One-way ANOVA test and were noted by asterisks (P < 0.05 [*] and P < 0.0001 [****]). Data are presented as mean ± SEM. (E and F) Key cell regulators are affected by PLX, PD, and the combined treatment. E and F are images of Western blot analyses for MYC (a), p-RB (b), total RB (c), p27 (d), and GAPDH (e) as loading controls. (G and H) Bar charts show the quantification of the band intensities for the comparisons among vehicle treatment, PLX treatment, PD treatment, and combined treatment with PLX and PD in both cell lines (triplicates for each treatment with three independent experiments). The p values are indicated (n=3).
We next examined what cell regulators were affected by single or combined treatment, leading to the inhibition of tumor cell proliferation. Since PLX is known to be an inhibitor of BET proteins that reduce the MYC transcription (Ozer et al. 2018), we first examined the level of MYC protein and its downstream effectors. As shown in Figure 1E-a, we found that MYC protein levels in THJ-11T cells were lowered by PLX (lanes 4-6), PD (lanes 7-9), and the combined treatment (lanes 10-12). Quantitation of the band intensities in Figure 1E-a indicated that the decreases were 21.3%, 43.4%, and 56.7%, respectively, by PLX, PD, and the combined treatment (Figure 1G-a). MYC may regulate the RB expression. While PLX (Figure 1E-b, lanes 4-6), PD (panel b, lanes 7-9, 1E-b) reduced the ratios of p-RB to total RB, the combined treatment (panel b, lanes 10-12, Figure 1E-b) reduced the ratio of p-RB to total RB more strongly. Quantitation of p-RB/total RB ratios indicate that the combined treatment was most effective (Figure 1G-b; 18.3%, 46.6%, and 65.7% for PLX, PD, and the combined treatment, respectively). The protein levels of cell cycle inhibitor p27, the downstream effector of RB, were correspondingly elevated by the three treatments (Figure 1E-d and 1G-c).
We next compared the effects of PLX and PD on the ERK signaling in THJ-11T cells. PLX did not reduce p-ERK (Figure 2A-a and Figure 2C-a). As expected, p-ERK protein levels were markedly lowered by PD (Figure 2A-a, lanes 7-9) and by the combined treatment (lanes 10-12). The ratios of p-ERK versus total ERK were decreased 85.5% and 90.7% by PD and the combined treatment, respectively (Figure 2C-a). Bcl-2-like protein 11, commonly known as BIM, is a member of the BCL-2 protein family. It promotes apoptosis (Antignani, et al. 2017; Gillings et al. 2009) and is a known downstream effector of ERK signaling (Takezawa, et al. 2011). Accordingly, we assessed BIM protein levels in THJ-11T cells treated with PLX, PD, and the combined treatment. Figure 2A-c shows that PLX, PD, and the combined treatment elevated the BIM protein levels by 1.5-fold, 2.0-fold, and 2.2-fold, respectively (Figure 2C-b). We evaluated the apoptotic marker, cleaved PARP. PLX, PD, and the combined treatment increased the ratio of cleaved PARP to PARP by 1.8-fold, 2.8-fold, and 3.2-fold, respectively (Figure 2A-d and 2C-c). These data suggested that the inhibition of THJ-11T cell proliferation by the combined treatment was mediated not only by inhibition of cell cycle progression, but also by increased apoptosis.
Figure 2.
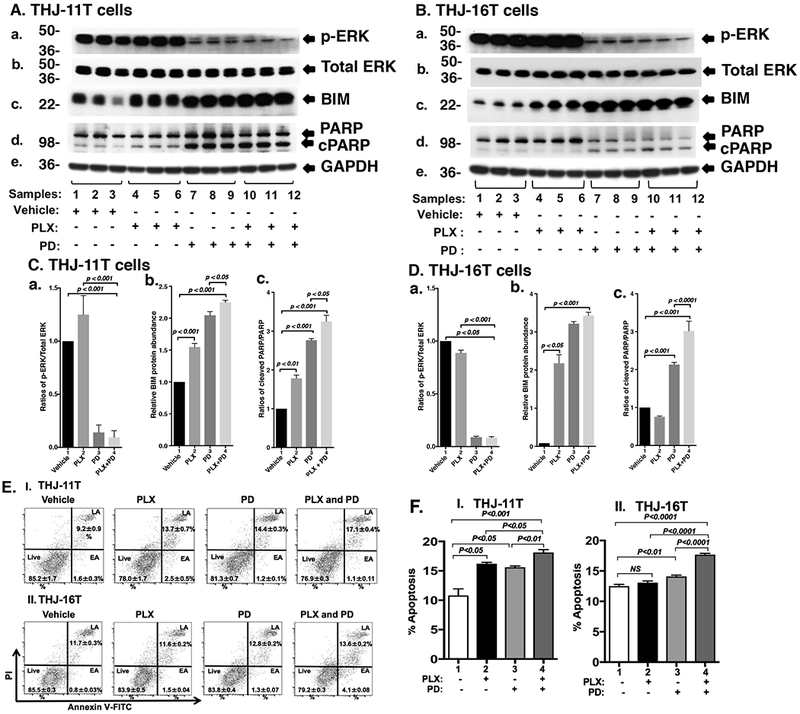
Effect of PLX, PD, and the combined treatment on MAPK signaling and apoptosis of ATC cells by Western blot analysis (A, B, C, D) and cytometric analysis (E and F). (A and B) Key cell regulators of apoptosis are affected by PLX, PD, and the combined treatment. A and B are images of Western blot analyses for p-ERK (a), total ERK (b), BIM (c), PARP (d), and GAPDH (e) as loading controls. (C and D) Band intensities were quantified for the comparisons among vehicle treatment, PLX treatment, PD treatment, and combined treatment with PLX and PD in both cell lines (triplicates for each treatment with three independent experiments). The p values are indicated (n=3). (E) THJ-11T (I) and -16T (II) cells were treated with vehicle, PLX, PD, and both PLX and PD for 48 hours. After treatment, cells were collected and subjected to Annexin V and propidium iodide (PI) staining followed by flow cytometric analysis for apoptotic cells. (F) The populations of late and early apoptotic cells were quantified and graphed as shown in (I) and (II) for THJ-11T and −16T cells, respectively. The experiment was done in triplicates. Values are shown as means ± SD. (n=3).
To validate the efficacy of combined treatment of PLX and PD on ATC cells, we also evaluated the effects of the two inhibitors on another ATC cell line, THJ-16T cells. As shown in Figure 1F, Western blotting revealed that these inhibitors suppressed cell cycle progression by reducing MYC (panel a), decreased p-RB (panel b), and increased p27 protein levels (panel d). These inhibitors also attenuated the ERK signaling (panel a) to up-regulate BIM (panel c) and cleaved PARP (panel d) to promote apoptosis of THJ-16T cells (Figure 2B). The quantitative changes in the cell regulators affected by PLX, PD, and the combined treatment shown in Figure 1H and 2D indicate that the combined treatment had the highest efficacy in the inhibition of proliferation of THJ-16T cells.
Since the pro-apoptotic regulator BIM and apoptotic marker cleaved PARP were increased in both the THJ-11T and THJ-16T cells, we examined whether the combination treatment was indeed most effective in promoting tumor cell apoptosis to decrease cell growth. Flow cytometric analysis using cells stained with Annexin V and propidium iodide showed that PLX, PD, and the combined treatment increased tumor cell apoptosis in both THJ-11T (Figure 2E-I) and THJ-16T (Figure 2E-II). Quantitative analysis showed that compared with vehicle-treated cells (bar 1, Figure 2F-I), 1.5-, 1.4-, and 1.7-fold increases of apoptotic cells in early apoptosis (EA) and late apoptosis (LA) were detected in THJ-11T treated with PLX, PD, and the combined treatment, respectively (Figure 2F-I, bars 2, 3, and 4). In THJ-16T cells, compared with vehicle-treated cells (bar 1, Figure 2F-II), 1.0-, 1.1-, and 1.4-fold increases of apoptotic cells (both EA and LA) were detected in cells treated with PLX, PD, and the combined treatment, respectively (Figure 2F-II, bars 2, 3, and 4). These data further support that the combined treatment cooperated to promote tumor cell apoptosis.
PLX and PD inhibit anaplastic thyroid tumor growth in xenograft mouse models
Since combined treatment of PLX and PD effectively inhibited ATC cell proliferation, we further tested the efficacy of the inhibitors in vivo in mouse xenograft models. When the tumor growth reached 100 mm3, we started the treatment (Figure 3-I-A). Single treatment of PLX or PD blocked the tumor growth (Fig. 3-I-A-a, and b). Importantly, combined treatment more strongly suppressed the tumor growth (Figure 3-I-A-a, and -b). The extent of inhibition of the tumor growth induced by THJ-11T cells by the three treatments is shown in Figure 3-I-A-c. The quantitative analysis indicated that 58.3%, 60.0%, and 84.6% of tumor growth was suppressed by PLX, PD, and the combined treatment, respectively.
Figure 3.
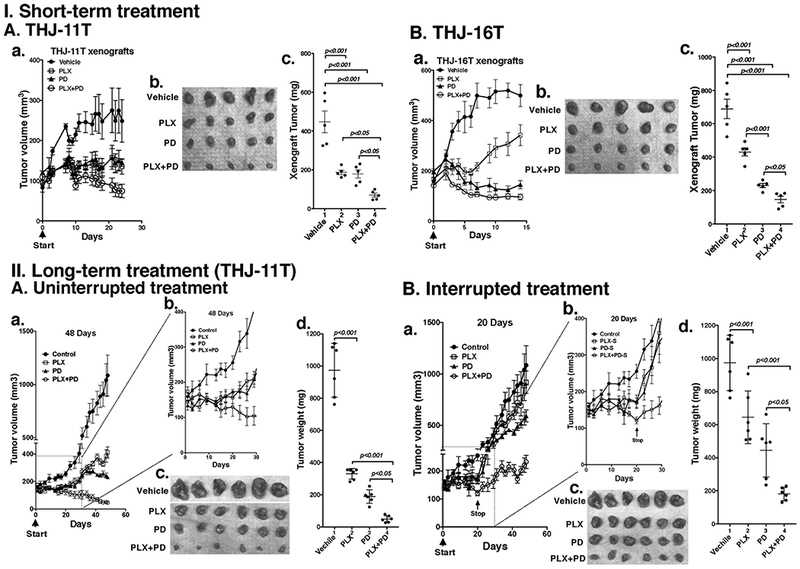
The combined treatment of PLX and PD promoted tumor regression in anaplastic thyroid tumors in athymic mice. Equal numbers of THJ-11T cells (5×106) or THJ-16T cells (5×106) were injected into the flanks of mice before treatment. (I) Short-term treatment. (A) When tumors reached the size of 100 mm3, mice were treated with vehicle, PLX, PD or the combination ot two drugs for 24 days. Tumor growth curves (a), tumor size (b), and tumor weight (c) derived from THJ-11T are shown (n=5 for each treatment). (B) When tumors reached the size of 150 mm3, mice were treated with vehicle, PLX, PD or the combination of both drugs for 14 days. Tumor growth curves (a), tumor size (b), and tumor weight (c) derived from THJ-16T are shown (n=5 for each treatment). (II). Long-term treatment. (A) When tumors reached the size of 150 mm3, mice were treated with vehicle, PLX, PD or the combination of two drugs for 48 days. Tumor growth curves treated for 48 days (a), tumor growth curves expanded from the inset shown for first 30 days (b), tumor size (c), and tumor weight (d) derived from THJ-11T xenograft tumors are shown (n=6 for each treatment). (B) When tumors reached the size of 150 mm3, mice were treated with vehicle, PLX, PD or the combination of two drugs were treated for 20 days. Then the treatement stopped followed by monitoring for additional 24 days. Tumor growth curves for duration of 48 days (a), tumor growth curves expanded for first 30 days (b), tumor size (c), and tumor weight (d) at the 48 days for THJ-11T xenograft tumors are shown (n=6 for each treatment). Note: Control mice treated with vehicle are same in the Figure 3-II-A-c and 3-II-B-c.
The effectiveness of PLX, PD, and the combined treatment was further evaluated in THJ-16T cells. While the profile of the efficacy of PLX, PD, and the combined treatment on the suppression of tumor growth induced by THJ-16T cells was similar to that of THJ-11T cells (compare Figure 3-I-A-a and 3-I-B-a), there were some minor quantitative differences in the extent of the inhibition of tumor growth. PD exhibited stronger inhibitory effects on tumor growth than PLX (Figure 3-I-B-a, -b and -c). The extent of inhibition by PLX, PD, and the combined treatment was 37.4%, 66.6%, and 78.9%, respectively (Figure 3-I-B-c). The minor differences in the sensitivities might reflect that these two cell lines were derived from patients who had different genetic lesions (Marlow et al. 2010). Despite minor quantitative sensitivities of the two tumors to the inhibitors, combined treatment resulted in strongest suppression of tumor growth.
To determine whether the treatment was effective for a longer duration, we first treated the mice inoculated with THJ-11T cells without interruption for 48 days (Figure 3-II-A). As shown Figure 3-II-A-a, the inhibitory effects of PLX and PD were durable up to 48 days. The combined treatment of PLX and PD continued to show strong efficacy in the suppression of tumor growth without an indication showing the tendency of resistance (Figure 3-II-A-c and -d). Figure 3-II-A-b, which shows the expanded inset of Figure 3-II-A-a, indicates that the inhibitory effect was similar and reproducible when compared with the short-term experiment shown in Figure 3-I-A-a. Quantiative analysis showed that the extent of inhibition by PLX, PD, and the combined treatment was 66.1%, 80.6%, and 94.9%, respectively (Figure 3-II-A-c and -d).
We further evaluated the durable response by discontinuing the treatment after the initial effective response to treatment with PLX, PD, and the combined drugs. As shown in Figure 3-II-B-a, consistent with results shown in the Figure 3-I-A-a, treatment by PLX and/or PD led to decreased tumor growth, with the combined treatment being most effective. This efficacy can be seen more clearly in the expanded inset shown in Figure 3-II-B-b. However, when the treatment stopped at the end of 20 days, the tumor growth re-initiated, but the overall growth was slower than in the control which had never been treated with PLX, PD, or the combined drugs. After the mice were drug-treatment free for an additional 28 days, the volumes of tumors in previously-treated mice were still significantly smaller than in the controls that had never been treated (Figure 3-II-B-c and -d). The extent of inhibition by PLX, PD, and the combined treatment was 33.8%, 54.3%, and 81.4%, respectively (Figure 3-II-B-c and -d). Taken together, these findings indicated that the combination of PLX with an MEK inhibitor was advantageous not only to increase the efficacy, but also to enhance the durability of the MEK inhibitor.
Combined treatment collaboratively suppresses MYC and ERK signaling to inhibit xenograft tumor growth
We next sought to identify the cell regulators affected by PLX, PD, and the combined treatment, leading to the inhibition of xenograft tumor growth. Western blotting shows that PLX or PD reduced the MYC protein level (Figure 4A-a, lanes 4-6 and 7-9, respectively). However, the combined treatment synergistically lowered MYC by 59% (lanes 10-12, Figure 4A-a, see also Figure 4E-a). While PLX did not significantly increase p27 protein abundance as a single agent, both PD and the combination treatment markedly increased p27 expression (Figure 4A-b and 4E-b). PD, but not PLX, lowered the p-ERK protein levels without changing the total ERK protein levels (Figure 4C-a). The ratio of pERK to total ERK was reduced by single treatment and by combined treatment by 50.1% and 58.9% (Figure 4G-a), suggesting the critical contribution of the MEK-ERK signaling cascade to the growth of xenograft tumors induced by THJ-11T cells. PD or combination treatment increased the protein levels of BIM, a pro-apoptotic regulator (Figure 4C-c and 4G-b). Taken together, these data suggested that the induction of apoptosis by combined treatment also contributed to the inhibition of xenograft tumor growth.
Figure 4.
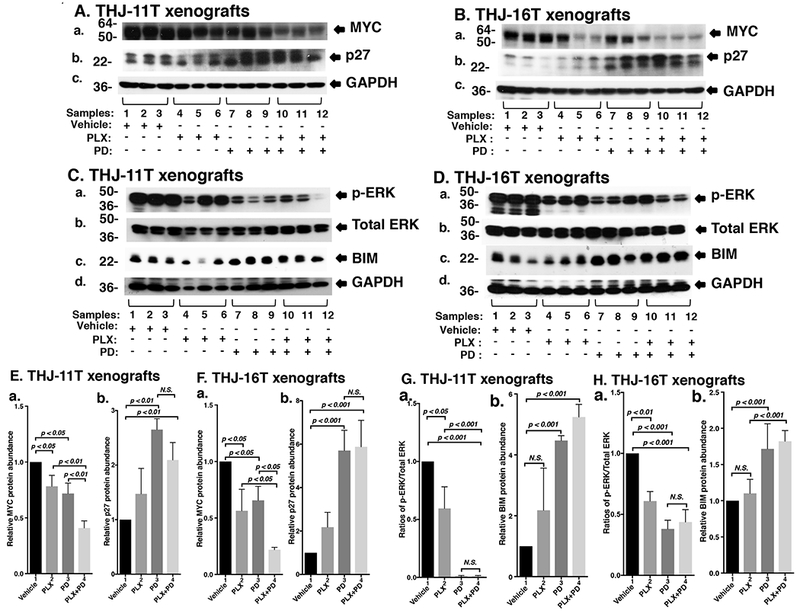
(A and B) The combined treatment of PLX and PD synergistically decreased the abundance of MYC expression and altered downstream effector p27 in xenograft tumors. Total protein extracts were prepared from xenograft tumors induced by THJ-11T cells (A) and THJ-16T cells (B). Western blot analysis was carried out for MYC (a), p27 (b), and GAPDH (c) as loading control. (C and D) The combined treatment of PLX and PD synergistically decreased the MAPK signaling and increased BIM in xenograft tumors. Total protein extracts were prepared from xenograft tumors induced by THJ-11T cells (C) and THJ-16T cells (D). Western blot analysis was carried out for p-ERK (a), total ERK (b), BIM (c),GAPDH (d) as loading control. (E and F) Band intensities in Figures 4A and 4B were quantified for the comparisons among vehicle treatment, PLX treatment, PD treatment, and combined treatment with PLX and PD in both cell lines (triplicates for each treatment with three independent experiments). The p values are indicated (n=3). (G and H) Band intensities in Figures 4C and 4D were quantified for the comparisons among vehicle treatment, PLX treatment, PD treatment, and combined treatment with PLX and PD in both cell lines (triplicates for each treatment with three independent experiments). The experiments in each treatment were analyzed in triplicates with three independent experiments. The p values are shown (n=3).
We also evaluated how the growth of xenograft tumors induced by THJ-16T cells was inhibited by different inhibitor treatment (Figures 4B and 4D). We found that the patterns of changes in MYC (Figure 4B-a, 4F-a), p27 (Figure 4B-b, 4F-b), p-ERK (Figure 4D-a, 4H-a) and BIM (Figure 4D-c and 4H-b) in the tumors induced by THJ-16T cells were similar to those in tumors induced by THJ-11T cells, further supporting the critical role of MAPK signaling in the inhibition of tumor growth.
PLX and PD converge to inhibit binding of BRD4 to the promoter of the MYC gene
Since MYC protein levels were decreased by PLX, PD, and combined treatment, we evaluated whether they inhibited the expression of MYC at the mRNA levels. We found that the expression of MYC mRNA was inhibited by PLX, PD, and the combined treatment by 44.0%, 74.0%, and 85.5%, respectively, in THJ-11T cells (Figure 5A). We also determined the MYC mRNA levels in THJ-16T cells. As found in THJ-11T cells, the expression of MYC mRNA was suppressed similarly in the cultured THJ-16T cells in vitro by PLX, PD, and the combined treatment (55.6%, 62.9%, and 90.4%, respectively, Figure 5B), These results indicate that, indeed, the PLX and PD acted collaboratively to suppress MYC expression at the transcription level in both cultured cells. We further examined whether the treatment reduced MYC expression in the xenograft tumors induced by THJ-11T and THJ-16T cells. We observed that the combination treatment reduced MYC mRNAs in both THJ-11T and THJ-16T xenograft tumors (Figure 5C and 5D). These data confirmed that the combination treatment would be effective to reduce MYC expression to inhibit tumor growth similarly observed in the inhibition of cell proliferation in vitro.
Figure 5.
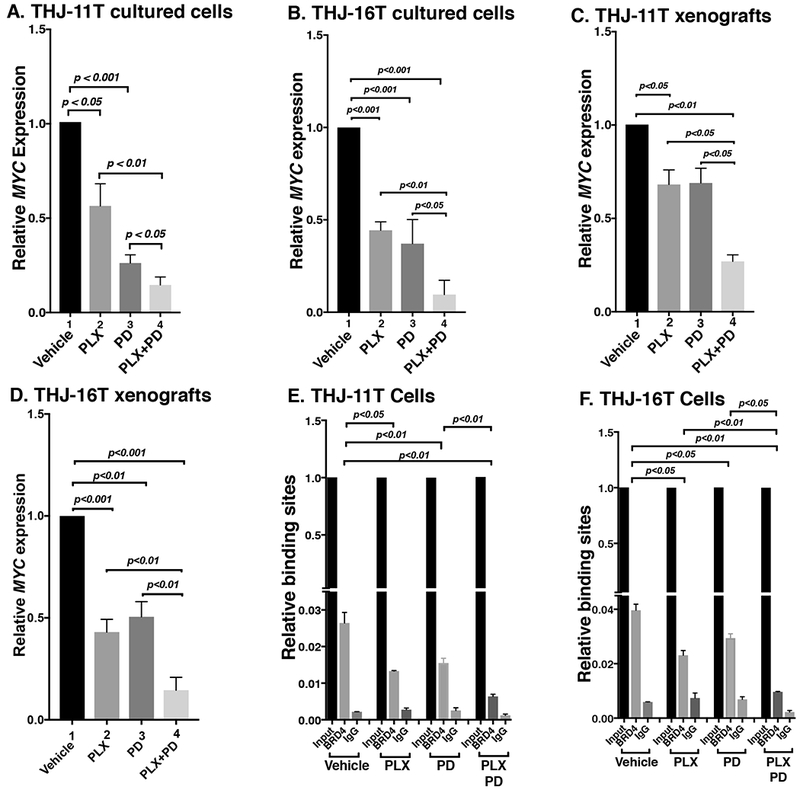
(A and B) The combined treatment of PLX and PD synergistically decreased the MYC mRNA expression in ATC cell lines. The mRNA level of the MYC gene was measured by real-time RT–PCR in THJ-11T cells (A) and THJ-16T cells (B). (C and D) The combined treatment of PLX and PD synergistically decreased the MYC mRNA expression in ATC xenograft tumors. Quantitative PCRs were performed using 200 ng of total RNA. GAPDH was used as a reference. (E and F) The combined treatment of PLX and PD synergistically reduced the recruitment of BRD4 on the promoter of the MYC gene in THJ-11T cells (E) and THJ-16T cells (F). ChIP assay was carried out using normal rabbit IgG or BRD4 antibody. The experiments were carried out in triplicates. The p values are shown (n=3).
We next investigated whether PLX and PD acted collaboratively to suppress MYC transcription, as we previously reported that JQ1 and trametinib converged to inhibit MYC transcription (Zhu et al. 2018). We therefore carried out chromatin immunoprecipitation assay (ChIP) to evaluate the effects on the binding of BRD4 to the MYC promoter by the inhibitors in both THJ-11T and THJ-16T cells. Compared with the control (vehicle-treated cells), 49.6%, 41.3%, and 75.8% inhibitions of the binding to BRD4 to the MYC promoter were detected in THJ-11T cells treated with PLX, PD, and the combined treatment, respectively (Figure 5E). Similar profiles in the inhibition of binding to BRD4 by the three treatments in THJ-16T cells were detected (41.7%, 25.8%, and 75.6%, respectively, by PLX, PD, and the combined treatment, Figure 5F). These results suggested that PLX and PD collaborated to block BRD4 binding to acetyl-lysines on the promoter of the MYC gene. These results suggested that the collaboratively inhibitory actions of PLX and PD could converge to alter epigenetic modifications to inhibit MYC transcription.
Discussion
Aberrant epigenetic modifications have been shown to be as critical as genetic mutations in the development and progression of cancer. Although mutations of the MYC gene are not common in solid tumors, aberrant expression of the MYC gene is common in cancer (1) and is known to be a driver in the development of many cancers (Dang 2012). Targeting epigenetic modifications has emerged as a potential strategy to treat cancers. JQ1 is the prototype of BET inhibitors targeting the interaction between BET proteins and acetylated histones that has proved effective in the treatment of hematological cancer (Ott, et al. 2012). ATC often has increased expression of MYC protein. In a mouse model of anaplastic thyroid cancer (Thrb PV/PVKrasG12D mice), treatment with JQ1 inhibited thyroid tumor growth. However, JQ1 had no apparent effect on the tumor invasion and metastasis in Thrb PV/PVKrasG12D mice (Zhu et al. 2017). These findings suggested that other oncogenic events may drive metastasis progression other than MYC signaling in the ATC model of Thrb PV/PVKrasG12D mice.
In our previous study, we examined the MEK inhibitor trametinib in combination with JQ1 in the treatment of ATC. We found that, indeed, combined treatment not only further enhanced the inhibition of tumor cell proliferation by a single agent, but also more effectively suppressed tumor cell invasion. However, JQ1 is not used in the treatment of patients with cancer. PLX51107 (PLX) is a newly developed BET inhibitor that has favorable pharmacokinetics properties than JQ1 and other BET inhibitors (Ozer et al. 2018). PLX is being tested in clinical trials to treat hematological malignancy and solid tumors . We therefore tested whether the combination of PLX and PD could improve inhibition of ATC tumors. We found that the combination of two inhibitors exhibited more potent inhibition in cancer cell proliferation and tumor growth. These findings indicated the combination treatment, via targeting different pathways, could lead to synergistic effects for more effective treatment. MYC is known to play a pivotal role in cancer biology as exemplified in our present study and in reports from many others. Its regulation is usually under tight transcriptional control, not only by a whole host of transcription factors that bind to its proximal promoter and enhancers, but also by distal super enhancers that connect via looping to the MYC proximal promoter (Dang 2012; Loven, et al. 2013). BRD4 and other regulators bind thousands of enhancers associated with active genes as well as a small set of exceptionally large super-enhancers associated with genes such as MYC. Interaction of BRD4 with acetylated lysine residues of histones on the MYC promoter and super enhancers results in chromatin remodeling and assembly of transcription activators to initiate MYC transcription. Selective small-molecule inhibitors (e.g., JQ1), block the interaction of BET proteins with acetylated histones on chromatin, resulting in the silencing of the MYC transcription as shown in our work (Enomoto et al. 2017; Zhu et al. 2017). In the present studies, we showed that PLX, a second generation of BETi whose efficacy was evaluated in clinical trials for solid tumors, acute myeloid leukemia, myelodysplastic syndrome, non-Hodgkin’s lymphoma (ClinicalTrials.gov 2018) was effective to target BRD4 to reduce MYC transcription. Interestingly, in the present studies, we found that PD also suppressed MYC transcription, leading to a lowering of MYC protein levels. Moreover, ChIP analysis demonstrated that PD also acted to inhibit the binding of BRD4 to the MYC promoter, suggesting that actions of PLX and PD could converge on chromatin by epigenetic modifications to collaboratively suppress MYC transcription (see Figure 6). Thus, the present studies uncovered a novel epigenetic action of MEK inhibitors in suppressing MYC transcription, leading to inhibition of thyroid tumor growth. Although it is not known how the two inhibitors cooperated to target BET proteins in the inhibition of MYC transcription, these results raised the possibility of the cooperation of the BET inhibitors and MEK inhibitors through at least two different mechanisms to inhibit tumor growth: (1) cooperation in the inhibition of MYC transcription via chromatin remodeling and (2) cooperation via MAPK signaling (Figure 6).
Figure 6.
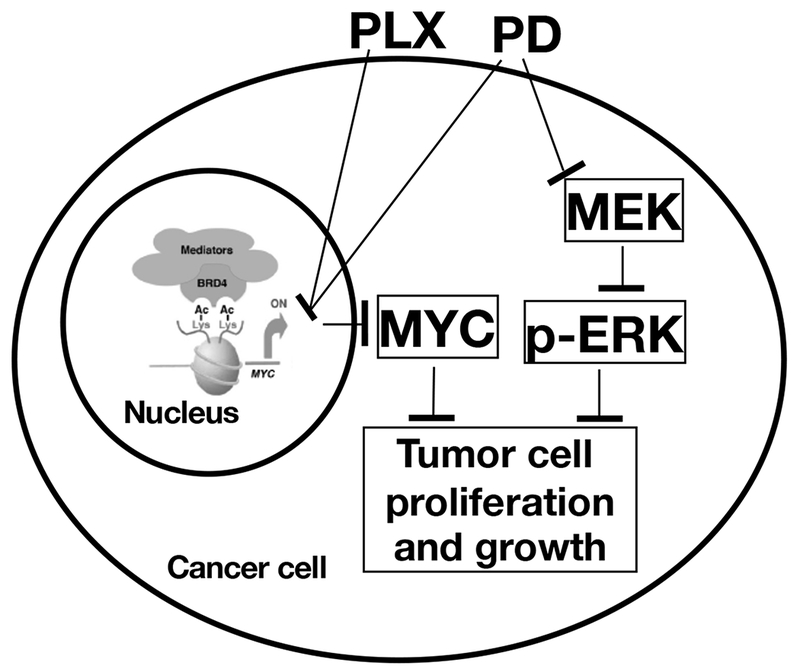
Proposed mechanisms of PLX- and PD-induced inhibition of anaplastic thyroid cancer cell proliferation and growth.
ATC has frequent mutations in BRAF and RAS that activate MEK/ERK signaling and drive distant metastasis (Landa, et al. 2016; Pozdeyev, et al. 2018). Further, using the next generation sequencing, previously unknown mutations of genes involved in the chromatin modifiers have been uncovered (e.g., SWI/SNF subunits and histone methyltransferases) and transcription-associated regulators (e.g., MED12, RBM10, and EIF1AX) (Landa et al. 2016). We have recently shown elevated levels of the MYC protein in human ATC tumors, as well as in THJ-11T and THJ-16T cells, as compared with differentiated thyroid cancer cells (Enomoto et al. 2017). The current findings showed that PLX in single treatment or combined treatment with PD resulted in markedly decreased MYC expression, leading to the suppression of tumor growth. Together, these results clearly indicated that MYC was one of the critical drivers to promote ATC progression.
The effect of combined treatment was assessed in two different ATC cell lines from patients with different genetic lesions (Marlow et al. 2010). Despite these genetic differences, combined treatment with PLX and PD seemingly had similar inhibitory effects on cell proliferation, xenograft tumor growth, treatment-induced alterations of tumor growth, and apoptotic signalings with only minor quantitative differences. Abnormal epigenetic modifications such as genes encoding components of the SWI/SNF chromatin remodeling complex have been identified to be frequently altered in ATC (Landa et al. 2016). These observations suggest that targeting chromatin modifications could be therapeutically beneficial to treat ATC since anaplastic thyroid tumors are heterogenous with quite different mutational and epigenetic profiles. Our present studies demonstrated that using PLX synergistically enhanced by an MEK-specific inhibitor was most effective in blocking proliferation and tumor growth of ATC in vitro and in vivo. Therefore, targeting epigenetic modifications via a combination of BET inhibitors and MEK inhibitors could potentially be effective for the treatment of ATC, a disease for which current treatment options are very limited.
Acknowledgments
This research was supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health. We are grateful to Plexxikon Inc. Berkeley, CA for providing us with PLX51107 and PD0325901 for our studies.
Funding: Supported by the Intramural Research Program of the Center for Cancer Research, National Cancer Institute, National Institutes of Health.
Footnotes
Declaration of Interest: Authors have nothing to declare
References
- Antignani A, Segal D, Simon N, Kreitman RJ, Huang D & FitzGerald DJ 2017. Essential role for Bim in mediating the apoptotic and antitumor activities of immunotoxins. Oncogene 36 4953–4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett SD, Bridges AJ, Dudley DT, Saltiel AR, Fergus JH, Flamme CM, Delaney AM, Kaufman M, LePage S, Leopold WR, et al. 2008. The discovery of the benzhydroxamate MEK inhibitors CI-1040 and PD 0325901. Bioorg Med Chem Lett 18 6501–6504. [DOI] [PubMed] [Google Scholar]
- ClinicalTrials.gov 2018. A Study of PLX51107 in Advanced Malignancies. https://ClinicalTrials.gov/show/NCT02683395.
- Dang CV 2012. MYC on the path to cancer. Cell 149 22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, Kastritis E, Gilpatrick T, Paranal RM, Qi J, et al. 2011. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell 146 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enomoto K, Zhu X, Park S, Zhao L, Zhu YJ, Willingham MC, Qi J, Copland JA, Meltzer P & Cheng SY 2017. Targeting MYC as a Therapeutic Intervention for Anaplastic Thyroid Cancer. J Clin Endocrinol Metab 102 2268–2280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert JP, Barsyte-Lovejoy D, Felletar I, Volkmer R, Muller S, Pawson T, et al. 2012. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 149 214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fozzatti L, Lu C, Kim DW & Cheng SY 2011. Differential recruitment of nuclear coregulators directs the isoform-dependent action of mutant thyroid hormone receptors. Mol Endocrinol 25 908–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Wu X, Zhang X, Hua W, Zhang Y, Maimaiti Y, Gao Z & Zhang Y 2016. Inhibition of BRD4 suppresses tumor growth and enhances iodine uptake in thyroid cancer. Biochem Biophys Res Commun 469 679–685. [DOI] [PubMed] [Google Scholar]
- Gillings AS, Balmanno K, Wiggins CM, Johnson M & Cook SJ 2009. Apoptosis and autophagy: BIM as a mediator of tumour cell death in response to oncogene-targeted therapeutics. FEBS J 276 6050–6062. [DOI] [PubMed] [Google Scholar]
- Haugen DR, Akslen LA, Varhaug JE & Lillehaug JR 1993. Demonstration of a TGF-alpha-EGF-receptor autocrine loop and c-myc protein over-expression in papillary thyroid carcinomas. Int J Cancer 55 37–43. [DOI] [PubMed] [Google Scholar]
- Landa I, Ibrahimpasic T, Boucai L, Sinha R, Knauf JA, Shah RH, Dogan S, Ricarte-Filho JC, Krishnamoorthy GP, Xu B, et al. 2016. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J Clin Invest 126 1052–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI & Young RA 2013. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marlow LA, D’Innocenzi J, Zhang Y, Rohl SD, Cooper SJ, Sebo T, Grant C, McIver B, Kasperbauer JL, Wadsworth JT, et al. 2010. Detailed molecular fingerprinting of four new anaplastic thyroid carcinoma cell lines and their use for verification of RhoB as a molecular therapeutic target. J Clin Endocrinol Metab 95 5338–5347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mio C, Lavarone E, Conzatti K, Baldan F, Toffoletto B, Puppin C, Filetti S, Durante C, Russo D, Orlacchio A, et al. 2016. MCM5 as a target of BET inhibitors in thyroid cancer cells. Endocr Relat Cancer 23 335–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott CJ, Kopp N, Bird L, Paranal RM, Qi J, Bowman T, Rodig SJ, Kung AL, Bradner JE & Weinstock DM 2012. BET bromodomain inhibition targets both c-Myc and IL7R in high-risk acute lymphoblastic leukemia. Blood 120 2843–2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozer HG, El-Gamal D, Powell B, Hing ZA, Blachly JS, Harrington B, Mitchell S, Grieselhuber NR, Williams K, Lai TH, et al. 2018. BRD4 Profiling Identifies Critical Chronic Lymphocytic Leukemia Oncogenic Circuits and Reveals Sensitivity to PLX51107, a Novel Structurally Distinct BET Inhibitor. Cancer Discov 8 458–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozdeyev N, Gay LM, Sokol ES, Hartmaier R, Deaver KE, Davis S, French JD, Borre PV, LaBarbera DV, Tan AC, et al. 2018. Genetic Analysis of 779 Advanced Differentiated and Anaplastic Thyroid Cancers. Clin Cancer Res 24 3059–3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano MI, Grattone M, Karner MP, Moiguer S, Tetelbaum F, Romano LA, Illescas E, Padin R, Cueva F & Burdman JA 1993. Relationship between the level of c-myc mRNA and histologic aggressiveness in thyroid tumors. Horm Res 39 161–165. [DOI] [PubMed] [Google Scholar]
- Soucek L, Whitfield JR, Sodir NM, Masso-Valles D, Serrano E, Karnezis AN, Swigart LB & Evan GI 2013. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev 27 504–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takezawa K, Okamoto I, Nishio K, Janne PA & Nakagawa K 2011. Role of ERK-BIM and STAT3-survivin signaling pathways in ALK inhibitor-induced apoptosis in EML4-ALK-positive lung cancer. Clin Cancer Res 17 2140–2148. [DOI] [PubMed] [Google Scholar]
- Terrier P, Sheng ZM, Schlumberger M, Tubiana M, Caillou B, Travagli JP, Fragu P, Parmentier C & Riou G 1988. Structure and expression of c-myc and c-fos proto-oncogenes in thyroid carcinomas. Br J Cancer 57 43–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Enomoto K, Zhao L, Zhu YJ, Willingham MC, Meltzer P, Qi J & Cheng SY 2017. Bromodomain and Extraterminal Protein Inhibitor JQ1 Suppresses Thyroid Tumor Growth in a Mouse Model. Clin Cancer Res 23 430–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Holmsen E, Park S, Willingham MC, Qi J & Cheng SY 2018. Synergistic effects of BET and MEK inhibitors promote regression of anaplastic thyroid tumors. Oncotarget 9 35408–35421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Zhao L, Park JW, Willingham MC & Cheng SY 2014. Synergistic signaling of KRAS and thyroid hormone receptor beta mutants promotes undifferentiated thyroid cancer through MYC up-regulation. Neoplasia 16 757–769. [DOI] [PMC free article] [PubMed] [Google Scholar]