

Abstract
Helicobacter pylori infection is a proven carcinogen for gastric cancer. Its virulence factor vacuolating cytotoxin A (VacA) promotes more severe disease and gastric colonization. VacA, by an unknown mechanism, usurps lysosomal and autophagy pathways to generate a protected reservoir for H. pylori that confers bacterial survival in vitro. Here, we show the existence of a VacA-generated intracellular niche in vivo that protects the bacteria from antibiotic treatment and leads to infection recrudescence after therapy. Furthermore, we report that VacA targets the lysosomal calcium channel TRPML1 to disrupt endolysosomal trafficking and mediate these effects. Remarkably, H. pylori that lack toxigenic VacA colonize enlarged dysfunctional lysosomes in the gastric epithelium of trpml1-null mice, where they are protected from eradication therapy. Furthermore, a small molecule agonist directed against TRPML1 reversed the toxic effects of VacA on endolysosomal trafficking, culminating in the clearance of intracellular bacteria. These results suggest that TRPML1 may represent a therapeutic target for chronic H. pylori infection.
Reporting Summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
The bacterium Helicobacter pylori infects half of the global population and is the strongest risk factor for gastric cancer development1,2. Furthermore, H. pylori eradication decreases the incidence of gastric cancer. The WHO (World Health Organization) recommends eradication of H. pylori to prevent gastric cancer in high-risk populations3. However, current therapies remain suboptimal due to increasing antibiotic resistance. In fact, the WHO placed H. pylori on its high-priority pathogen list, which aims to prioritize research and development for antibiotic-resistant organisms that pose the greatest threats to human health4.
The vacuolating cytotoxin (VacA), a key bacterial virulence factor, is a secreted toxin that inserts into host cell membranes, forming a chloride channel. VacA causes more severe disease including gastric cancer5. However, the mechanisms underlying VacA effects remain largely unknown. VacA impairs host endolysosomal trafficking, causing accumulation of dysfunctional lysosomes and autophagosomes6–8. Autophagy is implicated in protection from malignant transformation by providing safeguards to maintain homeostasis, as well as eliminating invading pathogens9. Indeed, the presence of VacA increases mitochondrial damage, reactive oxygen species and inflammation5. Furthermore, VacA increases intracellular survival of H. pylori in vitro6. However, controversy remains regarding the ability of the bacterium to invade and survive in gastric cells in vivo. Thus, understanding the mechanism by which VacA alters endolysosomal trafficking could be exploited as a potential therapeutic target for H. pylori infection.
A key player in the endolysosomal pathway is transient receptor potential membrane channel mucolipin 1 (TRPML1; also known as MCOLN1 or ML1), an endolysosomal calcium channel10,11. Interestingly, trpml1−/− cells are indistinguishable from VacA-intoxicated cells, since both display enlarged dysfunctional endolysosomes and autophagosomes12–14. Loss-of-function mutations in trpml1 cause type IV mucolipidosis (ML4), a lysosomal storage disorder. Interestingly, ML4 patients and Trpml1-deficient mice display a gastric phenotype that resembles H. pylori infection, including parietal cell defects, hypochlorhydria and hypergastrinemia15–19.
Therefore, the goal of this study was to determine whether VacA inhibits TRPML1 to disrupt endolysosomal trafficking, thereby creating a protective intracellular niche for H. pylori in vivo. Furthermore, we investigated the therapeutic efficacy of a small-molecule agonist of TRPML1 in VacA+ H. pylori-infected gastric cells in vitro and in human gastric organoids.
Results
VacA generates an intracellular reservoir in vivo leading to bacterial persistence after eradication therapy.
VacA alters endolysosomal trafficking to generate an intracellular niche for the bacteria in vitro6. However, the in vivo existence of a VacA-mediated intracellular reservoir and its biological relevance remain controversial. Therefore, we first determined whether VacA promotes intracellular H. pylori survival in vivo using a murine model. Although all H. pylori strains possess the vacA gene, genetic polymorphism exists with allelic variants in the signal (s1/s2), middle (m1/m2) and intermediate (i1/i2) regions5. These variants differ in their cytotoxicity, with s1i1 being toxigenic and strongly associated with gastric cancer, whereas s2i2 is considered non-toxigenic. Few H. pylori strains infect mice, and the widely used, mouse-adapted SS1 strain expresses non-toxigenic s2i2 VacA20,21 (Supplementary Table 1). Several SS1 isogenic mutants have been generated to study VacA function21–23. Importantly, SS1 VacA-null mutants display impaired colonization21–23 and we determined that the SS1 strain genetically engineered to produce toxigenic s1i1 VacA (SS1 s1i1) is the only SS1 isogenic VacA variant that disrupts endolysosomal trafficking in vitro21 (Supplementary Fig. 1). Therefore, to determine whether toxigenic VacA generates an intracellular niche in vivo, we compared the parental SS1 strain with isogenic SS1 s1i1 infection of wild-type mice. Both strains colonized the lumen of murine gastric glands (Fig. 1a,b). However, unlike the parental strain, SS1 s1i1 was frequently present within cells resembling parietal cells, in some cases within vacuoles (Fig. 1b). By performing co-staining for H. pylori and the H+, K+-ATPase, a parietal cell marker, we found that the majority of parental SS1 preferentially colonized antral glands with bacteria rarely detected in the corpus region (Fig. 1c). In marked contrast, numerous H. pylori were detected within parietal cells in the corpus region of SS1-s1i1-infected mice (Fig. 1d). Taken together, these results demonstrate that toxigenic VacA generates an intracellular niche for H. pylori in vivo.
Fig. 1|. VacA generates an intracellular reservoir in vivo leading to bacterial persistence after eradication therapy.
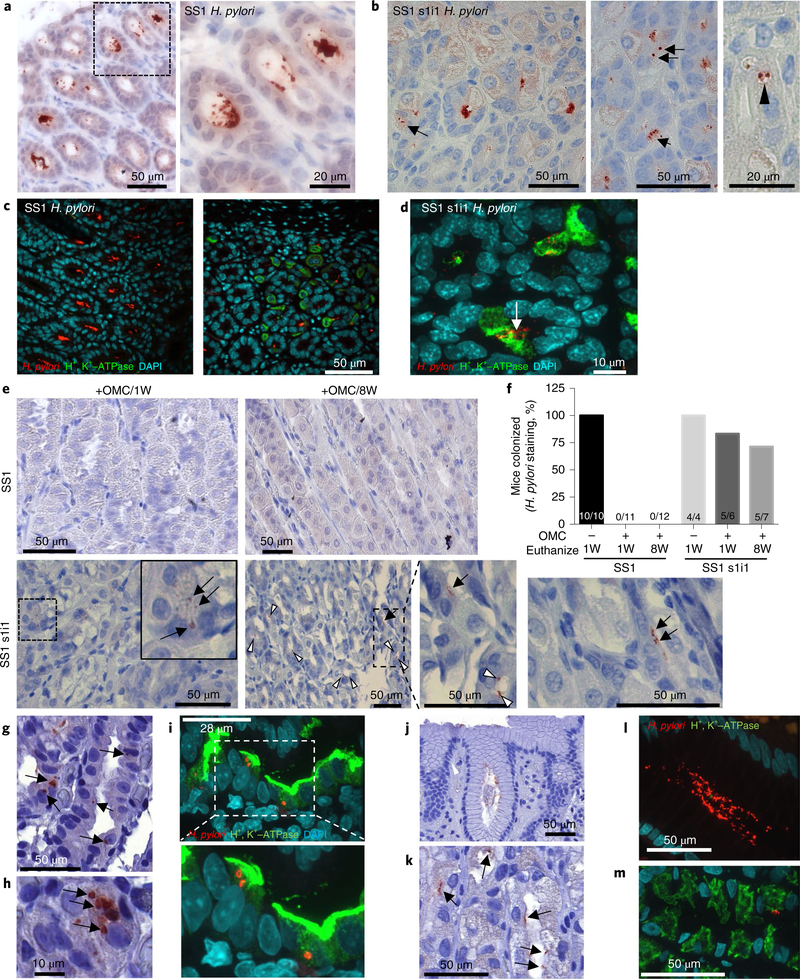
a–d, Mice were infected with SS1 parental H. pylori (SS1, n = 10) or isogenic SS1 s1i1 (SS1 s1i1, n = 22) for 10 weeks. a,b, H. pylori IHC of gastric mucosa dissected from SS1-infected (a) or SS1-s1i1-infected (b) mice. a, SS1 is restricted to the glandular lumen; a higher magnification of the outlined area is shown in the right panel. b, SS1 s1i1 is detected intracellularly (arrows) and within vacuoles (arrowhead). c,d, Double H. pylori and parietal cell marker, H+, K+-ATPase, immunofluorescence of SS1-infected (c) or SS1-s1i1-infected (d) stomachs. The experiment was repeated three independent times with similar results. e,f, Mice were infected with SS1 or SS1 s1i1 H. pylori for eight weeks, followed by seven-day eradication therapy (OMC). Mice were euthanized one week (+OMC/1W) or eight weeks (+OMC/8W) after OMC. Infected, no-OMC-treated control mice (−OMC) were euthanized along with +OMC/1W. e, Representative H. pylori IHC of gastric mucosa dissected from mice from the indicated groups. The arrows point to intracellular H. pylori; the white arrowheads point to bacteria in the glandular lumens. IHC was performed and analysed for all of the animals. f, The proportion of mice positive for H. pylori staining. The numbers within the bars denote the ratio of H. pylori positive/total mice per group. g–m, H. pylori IHC (g,h,j,k) or double H. pylori/H+, K+-ATPase immunofluorescence (i,l,m) on human corpus gastric biopsies from patients infected with VacA s1m1 (g–i, n = 6) or VacA s2m2 (j–m, n = 6) strains of H. pylori. The arrows indicate intracellular bacteria in parietal cells (g–i) or glandular bacteria (j–m).
Next, to evaluate whether this intracellular reservoir protects the bacteria from antibiotic treatment, mice were inoculated with parental SS1 or SS1 s1i1 for eight weeks, followed by seven-day eradication therapy consisting of omeprazole, clarithromycin and metronidazole (OMC)24. Following eradication therapy, H. pylori were no longer detected in the glandular lumens of murine stomachs (Fig. 1e,f). Consistent with this finding, extracellular bacteria were not detected in gastric tissue as assessed by colony-forming units (CFUs; Supplementary Fig. 2). However, following eradication therapy, intracellular H. pylori were still detected within parietal cells in the majority of SS1s1i1-infected mice. In contrast, intracellular bacteria were not found in any of the parental-SS1-infected mice (Fig. 1e,f).
Next, to determine whether intracellular toxigenic VacA bacteria could promote persistence after eradication therapy, a group of mice were euthanized eight weeks after OMC and H. pylori colonization assessed by immunohistochemistry (IHC). H. pylori were not detected in any of the SS1-infected mice (Fig. 1e,f). In marked contrast, SS1 s1i1 H. pylori was present within parietal cells and in gastric glands in the antrum, corpus or transition zone in 71% of mice (Fig. 1e,f and Supplementary Fig. 2). The presence of intracellular bacteria remaining eight weeks after eradication therapy indicates viability and productive infection. Consistent with this, extracellular bacteria were also detected in 15% of SS1s1i1-infected mice by CFU (Supplementary Fig. 2). In mice that we detected infection by CFU, higher colonization levels were observed by IHC; therefore, the lack of CFU detection in the other SS1s1i1-infected animals is probably due to reduced sensitivity of the CFU assay. Altogether, our findings established that toxigenic VacA generates an intracellular reservoir that protects H. pylori from antibiotics allowing persistence and recolonization following eradication therapy.
Several early ultrastructural studies of human gastric tissues reported the presence of H. pylori within some gastric epithelial cells, parietal cells and immunocytes25,26. However, there are no reports associating intracellular H. pylori with toxigenic VacA expression in human samples. Therefore, we assessed the presence of intracellular H. pylori in corpus gastric biopsies obtained from H. pylori-infected patients in which the VacA genotype was known. In all gastric samples infected with bacteria expressing toxigenic s1m1 VacA, we detected intracellular H. pylori within parietal cells (Fig. 1g–i and Supplementary Fig. 3). In contrast, H. pylori was mainly restricted to the glandular lumen in individuals infected with non-toxigenic s2m2 (Fig. 1j–m and Supplementary Fig. 4). These findings support the role of toxigenic VacA in the generation of an intracellular niche for H. pylori.
VacA impairs TRPML1 activity.
Since TRPML1 inhibition or deficiency mimics the effects of VacA, we hypothesized that TRPML1 is the downstream target of VacA that results in disrupted endolysosomal trafficking and formation of the intracellular niche. TRPML1 mediates the transient, localized lysosomal calcium release that controls trafficking events between lysosomes and autophagosomes, late endosomes and the plasma membrane. Inhibition of TRPML1 results in increased levels of lysosomal calcium14. Therefore, we assessed the effects of VacA on lysosomal calcium levels as an indirect measure of TRPML1 activity. To this end, we measured the levels of the cell-impermeant calcium indicator Oregon Green 488 BAPTA-5N (Oregon Green), normalized to the calcium-and pH-insensitive endocytic probe Alexa-Fluor-586-conjugated 10 kDa dextran (Dextran Red)27,28. As the source of VacA, we used culture supernatants obtained from the H. pylori 60190 strain, which secretes high levels of toxigenic s1m1 VacA (VacA+)29. Culture supernatants from an isogenic H. pylori VacA-null mutant (VacA−) were used as a control (Supplementary Table 1 and Supplementary Fig. 1). As shown in Fig. 2a, treatment of human gastric epithelial AGS cells with VacA+ culture supernatants caused a significant increase in the Oregon Green/Dextran Red ratio in comparison with VacA−-treated control cells. The specific role of VacA on lysosomal calcium increase was confirmed by using purified VacA instead of crude supernatants30 (Supplementary Fig 5). We next employed a complementary method to assess lysosomal calcium by measuring calcium release in cells expressing the lysosomal-targeted calcium indicator GECO-ML1 following osmolysis by glycyl-L-phenylalanine 2-naphthylamide (GPN)28. Consistent with the previous result, VacA+ pretreatment of GECO-ML1-expressing cells significantly enhanced GPN-induced calcium release in comparison with VacA−-treated cells (Fig. 2b,c). The similar GECO-ML1 signal obtained for VacA+-and VacA−-treated cells in response to the ionophore ionomycin indicates that GECO-ML1 expression was comparable in both groups (Fig. 2d). Therefore, the elevated calcium content in VacA+-treated cells strongly suggests impairment in TRPML1 activity. Altogether, these results indicate that VacA disrupts TRPML1 activity, causing perturbations in lysosomal calcium homeostasis.
Fig. 2|. VacA impairs TRPML1 activity.

a, Intraluminal lysosomal Ca2+ levels in VacA−-or VacA+-treated AGS cells. AGS cells were co-loaded with Oregon Green 488 BAPTA-5N (OG BAPTA-5N), a membrane-impermeant Ca2+ indicator with negligible sensitivity to pH, and Alexa-Fluor-586-conjugated dextran (Dextran Red, as a calcium-insensitive probe) by endocytosis. The fluorescence ratio between green and red signals was compared. A representative scatter plot (mean ± s.e.m.) of the Oregon Green/Dextran Red intensity ratio of 12 fields containing 97 VacA−-and 70 VacA+-treated cells is displayed on the right. b,c, Lysosomal calcium levels assessed by GPN (200 μM)-induced GECO-ML1 response (measured as change in fluorescence (∆F) over basal fluorescence (F0); ∆F/F0) in AGS cells expressing GECO-ML1) after 4 h VacA− or VacA+ incubation. d, Estimation of GECO-ML1 expression by ionomycin (2 μM)-induced GECO-ML1 response in cells treated as in b. e,f, ML-SA1 (20 μM)-induced GECO-ML1 response (measured as change of fluorescence (∆F) over basal fluorescence (F0); ∆F/F0) in AGS cells expressing GECO-ML1) after 4 h VacA− or VacA+ incubation. c,d,f, Representative scatter plots (mean ± s.e.m.) of the number of cells measured in each assay (indicated within parentheses). All experiments were repeated three times with similar results. Two-tailed unpaired Student’s t-test was utilized for data analysis.
TRPML1 deficiency generates a VacA–like intracellular niche for H. pylori in vivo.
On the basis of the phenotypic similarities caused by VacA and trpml1-deficiency, and the effect of VacA on lysosomal calcium release, we reasoned that VacA may target TRPML1 activity to generate an intracellular niche for H. pylori. The gastric epithelium of Trpml1−/− mice displays vacuolation in parietal cells, with mis-localization and reduced proton pump expression, as well as disrupted autophagosome maturation (Supplementary Fig. 6). We hypothesized that in the Trpml1−/− mice where TRPML1 is absent in all cells, SS1 H. pylori could establish an intracellular niche in the gastric epithelium without requiring toxigenic VacA. Therefore, we orogastrically challenged wild-type and Trpml1−/− co-housed littermate mice with parental SS1 H. pylori. The colonization density, as assessed by CFU, was similar between the two groups of mice (Fig. 3a). However, in marked contrast to infected wild-type mice where parental SS1 colonized the lumen of gastric glands (Fig. 3b,c and Supplementary Video 1), in Trpml1−/− mice, parental SS1 H. pylori were predominately present within vacuolar compartments of parietal cells (Fig. 3b–d and Supplementary Video 2). These results indicate that TRPML1 deficiency generates a VacA-like intracellular compartment for H. pylori.
Fig. 3|. TRPML1 deficiency generates a VacA-like intracellular niche for H. pylori in vivo.
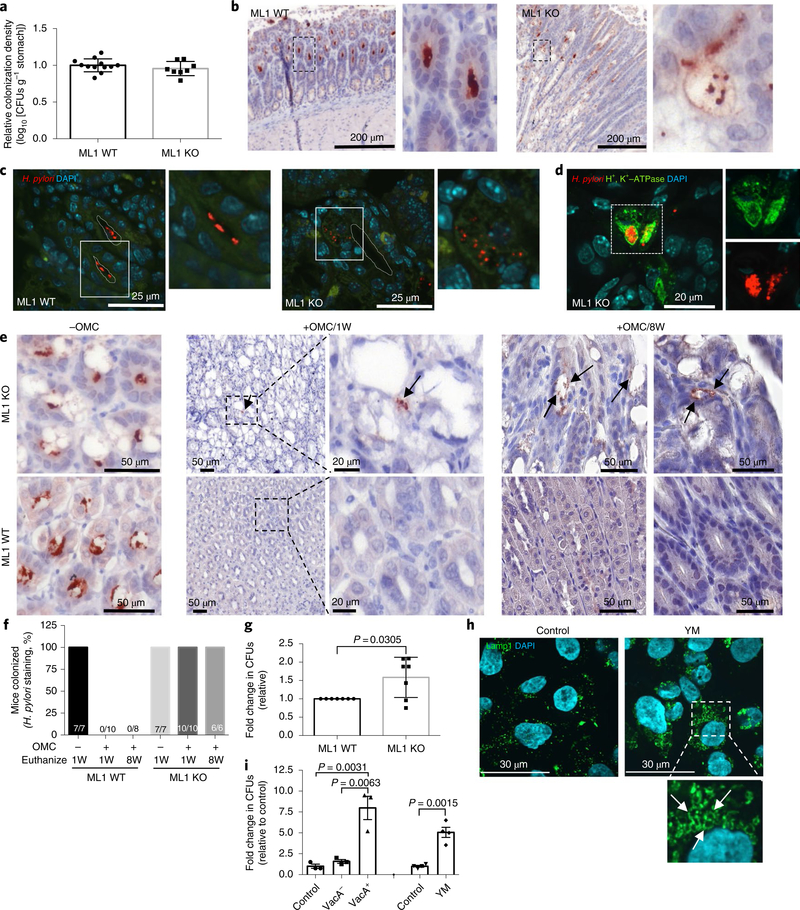
a–d, Wild-type (ML1 WT, n = 12) or Trpml1-knockout (ML1 KO, n = 8) littermate mice were infected with SS1 H. pylori for 10 weeks. a, Relative colonization density considering 1.0 as the average colonization density of WT mice per litter. The bars represent the mean value ± s.e.m. b, H. pylori IHC of gastric mucosa dissected from ML1 WT or ML1 KO H. pylori-infected mice. Higher magnification of dashed areas on the right. c, H. pylori immunofluorescence in sections from b. Higher magnifications of the outlined areas are shown on the right of each panel. The dotted line delineates the lumen of the glands. d, Double H. pylori and H+, K+-ATPase staining in the section from b. A higher magnification of the separate channels is included on the right. Infections were repeated three times with similar results with a total of 22 ML1 WT and 24 ML1 KO mice. e,f, ML1 WT or ML1 KO littermate mice were infected with SS1 H. pylori for eight weeks, followed by eradication therapy (OMC) and euthanized one week (+OMC/1W) or eight weeks (+OMC/8W) after OMC. Infected, no-OMC-treated control mice (−OMC) were euthanized along with +OMC/1W. e, Representative H. pylori IHC of gastric mucosa. The arrows point to the presence of intracellular H. pylori in parietal cell vacuoles of the ML1 KO mice. f, The proportion of mice positive for H. pylori staining. The numbers within the bars denote the ratio of H. pylori positive/total mice per group. g, Quantification of intracellular SS1 H. pylori after infection of murine gastric organoids derived from ML1 WT or ML1 KO mice. The data represent fold change CFU relative to ML1 WT control (mean ± s.e.m. of seven independent experiments). h, Lamp1 staining of AGS cells after overnight treatment with the PIKFyve inhibitor YM. The higher magnification of the outlined area shows YM-induced vacuolation (arrows). The experiment was repeated four times with similar results. i, AGS cells were infected with 60190 VacA− isogenic mutant H. pylori in the presence of YM, VacA− or VacA+ culture supernatants. Intracellular H. pylori were then quantified. The graph shows fold change in CFU (mean ± s.e.m. of three (VacA) or four (YM) independent experiments), relative to control infections. A two-tailed unpaired Student’s t-test was utilized for data analysis.
Next we determined whether the intracellular niche in Trpml1−/− mice protected H. pylori from eradication therapy. Extracellular H. pylori were not detected, as assessed by either IHC or CFU, in SS1-infected wild-type or Trpml1−/− littermate mice following eradication therapy (Fig. 3e,f and Supplementary Fig. 7). However, intracellular H. pylori were present within parietal cell vacuoles in all Trpml1−/− mice. Conversely, intracellular bacteria were not detected in wild-type mice (Fig. 3e,f and Supplementary Fig. 7).
Furthermore, eight weeks following eradication treatment, the stomach of all infected Trpml1−/− mice displayed H. pylori within parietal cell vacuoles, glandular lumens and the antral–body transition zone (Fig. 3e,f and Supplementary Fig. 7). Consistent with this finding, extracellular bacteria were also detected in 50% of the infected Trpml1−/− mice as assessed by CFU (Supplementary Fig. 7). In contrast, bacteria were not detected following eradication therapy in any of the SS1-infected wild-type mice (Fig. 3e,f and Supplementary Fig. 7). Taken together, these results indicate that TRPML1 deficiency, similar to toxigenic VacA, generates an intracellular reservoir that protects H. pylori from antibiotics allowing recolonization after eradication therapy.
Since the contribution of TRPML1 deficiency in other tissues or cell types could influence our observations in Trpml1−/− mice, we complemented these in vivo studies and confirmed the specificity for the requirement of TRPML1 deficiency by employing Trpml1−/− and wild-type gastric organoids. Trpml1−/− gastric organoids displayed vacuolation and autophagosome accumulation that was comparable with the gastric phenotype of Trpml1−/− mice, and phenocopied wild-type gastric organoids incubated with toxigenic VacA (Supplementary Fig. 8). Wild-type and Trpml1−/− gastric organoids were grown as two-dimensional (2D) monolayers and subsequently infected with parental SS1. Following the addition of gentamycin, a cell-impermeant antibiotic that kills extracellular bacteria, organoids were lysed to retrieve intracellular bacteria. Consistent with the in vivo data, a significant increase in intracellular H. pylori survival was detected in gastric organoids derived from Trpml1−/− mice as compared to wild-type controls (Fig. 3g).
To provide further support for the role of TRPML1 in promoting bacterial survival, we performed complementary studies with pharmacological inhibition of TRPML1 using the PIKfyve inhibitor YM201636 (YM). TRPML1 is activated by phosphatidylino-sitol-3,5-bisphosphate (PtdIns(3,5)P2), which is synthesized by PIKfyve13. In fact, deficiency in PtdIns(3,5)P2 causes the accumulation of large intracellular vacuoles and defective autophagy31. We found that YM treatment induced vacuolation in AGS cells and significantly enhanced the intracellular survival of the VacA-null isogenic mutant strain 60190 (Fig. 3h,i).
TRPML1 activation restores VacA-disrupted endolysosomal and autophagy pathways.
Next, we tested whether increasing TRPML1 activity could rescue the effects of toxigenic VacA on endolysosomal trafficking (Supplementary Fig. 9). To this end, we overexpressed TRPML1 in AGS cells and assessed VacA-mediated vacuolation by lysosomal-associated membrane protein 1 (Lamp1) immunolabelling. We confirmed that ectopic expression of TRPML1 did not affect its lysosomal localization (Supplementary Fig. 10). Remarkably, cells with enhanced TRPML1 expression failed to form large vacuoles in response to VacA, an effect not observed in the untransfected neighbouring cells (Fig. 4a). To directly induce the activity of TRPML1, we next assessed the effect of the TRPML1 small-molecule agonist ML-SA132 on VacA+-treated cells. ML-SA1 administration caused a marked reduction in the size of VacA-mediated vacuoles (Fig. 4b,e). Live imaging studies revealed that ML-SA1 induced the tubulation and fragmentation of these large vacuoles into lysosome-sized structures (Supplementary Fig. 11). We previously reported that VacA disrupts vesicular trafficking of degradative hydrolases such as cathepsin D to the lysosome, thereby impairing the maturation and function of lysosomes6–8 and autophagosomes. Therefore, we next investigated the effect of ML-SA1 on lysosomal function in VacA+-treated cells. We found that following ML-SA1 administration, VacA+-treated AGS cells recovered both the delivery of cathepsin D to lysosomes and the levels of mature cathepsin D within lysosomes (Fig. 4b,c). Furthermore, with restored delivery of cathepsin D, these compartments regained their ability to degrade the chromogenic substrate DQ-bovine serum albumin (BSA; Fig. 4d). Most hydrolases, including cathepsin D, are targeted to lysosomes by acquiring a mannose-6-phosphate (M6P) group in the trans-Golgi network, and are transported by M6P receptors (M6PR)33,34. Sortilin also contributes to the transport of cathepsin D35,36. We found mis-localization of both sorting receptors in VacA+-treated cells, which were found in large aggregates around enlarged vacuoles (Fig. 4f,g). ML-SA1 administration in VacA+-treated cells restored proper localization of both M6PR and sortilin (Fig. 4f,g). This finding was consistent with the restoration of lysosomal degradative function on ML-SA1 incubation in VacA+-treated cells (Fig. 4d).
Fig. 4|. TRPML1 activation restores VacA-disrupted endolysosomal and autophagy pathways.
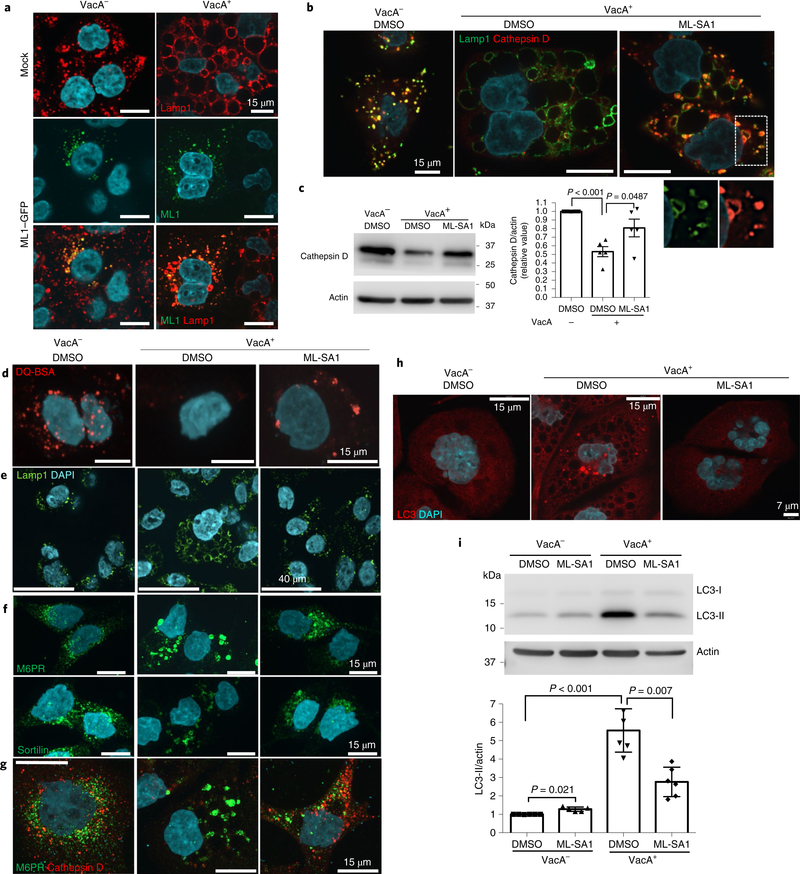
a, Lamp1 staining of control (mock-transfected, top panels) or TRPML1–GFP-transfected AGS cells, after overnight VacA− or VacA+ culture supernatant treatments. b, Lamp1 and cathepsin D staining of AGS cells after overnight VacA− or VacA+ incubation, followed by 4 h ML-SA1 (20 μM) or vehicle (DMSO) treatment. A higher magnification of the outlined area, for the separate channels, is shown at the bottom. c, Cathepsin D western blotting of cells from b using actin as a loading control. The graph shows quantification of cathepsin D normalized to actin (mean ± s.e.m. of five independent experiments). d, Chromogenic protease substrate DQ red-BSA (DQ-BSA) signal of AGS cells treated as in b. e–g, Lamp1 (e), M6PR and sortilin (f) and M6PR and cathepsin D (g) staining of AGS cells incubated for 4 h (e) or overnight (f,g) with VacA− or VacA+ followed by 3 h (e) or 4 h (f,g) ML-SA1 (20 μM) or DMSO treatment. h, LC3 staining of AGS cells treated as in b. i, LC3 western blotting of cells from h using actin as a loading control. The graph shows quantification of LC3-II normalized to actin (mean ± s.e.m. of six independent experiments). A two-tailed unpaired Student’s t-test was utilized for data analysis. All staining experiments were independently repeated a minimum of three times.
As a consequence of producing dysfunctional lysosomes, toxigenic VacA subsequently impairs autophagosome maturation, as shown by the accumulation of both LC3-positive autophagosomes (Fig. 4h) and LC3-II protein (Fig. 4i)7,8. We found that ML-SA1 administration in VacA+-treated cells restored the autophagy pathway as assessed by a reduction in both LC3 puncta and LC3-II protein levels (Fig. 4h,i). Consistent with this finding, TRPML1 overexpression also led to autophagosome maturation in VacA+-treated cells (Supplementary Fig. 12). Importantly, purified VacA caused similar effects as the culture supernatant on the endolysosomal and autophagy pathways, which were also restored by ML-SA1 administration (Supplementary Figs. 13 and 14). Finally, we measured TRPML1-mediated calcium release in cells pretreated with VacA. ML-SA1 induced an increased GECO-ML1 signal in cells pre-treated with VacA+ culture supernatant, in comparison with control VacA−-treated cells (Fig. 2e,f).
Taken together, these results indicate that TRPML1 activation by ML-SA1 restores endolysosomal trafficking defects caused by VacA, leading to the reformation of functional degradative lysosomes in AGS cells.
Validation of the VacA–TRPML1 axis in human gastric organoids.
To extend our findings to a physiological model, we generated 2D human gastric organoids and assessed the effect of toxigenic VacA and ML-SA1 on lysosomal trafficking. To this end, human gastric organoids were infected with H. pylori 60190 (VacA+ H. pylori) or treated with its culture supernatant, followed by ML-SA1 administration. Infection with isogenic 60190 VacA-null mutant H. pylori or treatment with culture supernatants was used as a control. We found that toxigenic VacA induced the formation of large vacuoles, mis-sorting of cathepsin D and accumulation of autophagosomes in the gastric monolayers (Fig. 5a–c and Supplementary Fig 15). Furthermore, the addition of ML-SA1 to organoids infected with VacA+ H. pylori or exposed to its culture supernatant reversed both the formation of large vacuoles and accumulation of autophagosomes (Fig. 5a–c and Supplementary Fig. 15). Thus, the ability of ML-SA1 to rescue the effect of VacA on endolysomal trafficking is preserved in primary human gastric epithelial cells.
Fig. 5|. Validation of the VacA–TRPML1 axis in human gastric organoids.
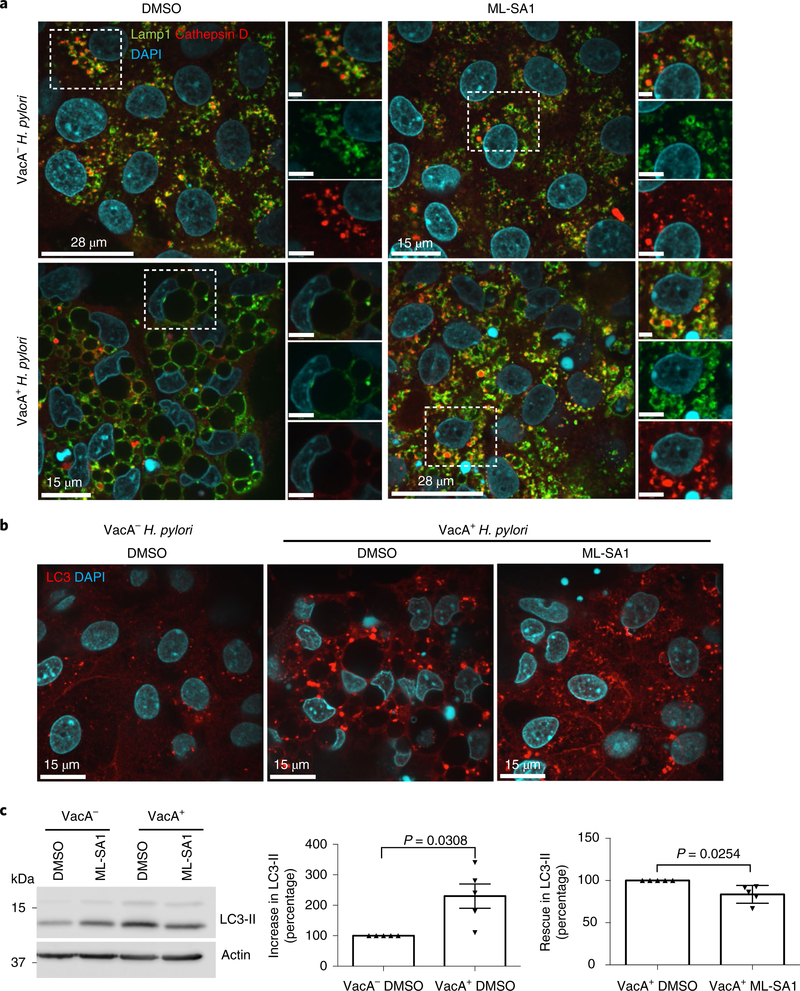
a, Lamp1 and cathepsin D staining in gastric organoid monolayers infected overnight with VacA− or VacA+ H. pylori, followed by 4 h ML-SA1 (20 μM) or DMSO (vehicle) administration. A higher magnification of the outlined area is included on the right. b, LC3 staining in organoid monolayers treated as in a. The experiments were independently repeated three times with organoids isolated from different patients. c, LC3 western blotting of 3D gastric organoids treated with VacA− or VacA+ culture supernatant followed by 4 h ML-SA1 (20 μM) or DMSO (vehicle) administration. The graphs show quantification of the increase in LC3-II in organoids treated with VacA+ and the rescue of LC3-II in VacA+-treated organoids incubated with ML-SA1. Actin was used as a loading control. The data were derived from five different patients and represent the mean ± s.e.m. A two-tailed unpaired Student’s t-test was utilized for data analysis.
TRPML1 activation eliminates both the intracellular niche and survival advantage of VacA+ H. pylori.
We next assessed the effect of TRPML1 activation during VacA+ H. pylori infection of AGS cells (Fig. 6a and Supplementary Fig. 16). Similar to the findings in cells treated with VacA+ culture supernatants (Fig. 4), we found that ML-SA1 rescued vacuolation (Fig. 6a), mis-sorting of cathepsin D (Fig. 6a) and disruption of autophagosome maturation during infection with VacA+ H. pylori (Fig. 6b,c). These findings suggest that ML-SA1 administration may eliminate the VacA-generated intracellular niche for H. pylori. In fact, gentamycin protection assays revealed that ML-SA1 activation of TRPML1 led to efficient bacterial killing such that intracellular survival of the VacA+ strain was comparable to VacA− bacteria (Fig. 6d). Importantly, these results were confirmed by using another toxigenic VacA-producing H. pylori strain, J166, and its isogenic VacA-null mutant (Supplementary Table 1 and Supplementary Fig. 17). Time-course and ML-SA1 dose–response experiments revealed that 2 h, 20 μM ML-SA1 treatment was sufficient to eliminate the toxigenic VacA-promoted bacterial survival advantage (Supplementary Fig. 18). Consistent with this finding, after 4 h ML-SA1 treatment, intracellular VacA+ H. pylori appeared to be degraded (Supplementary Fig. 19). Taken together, these findings demonstrate that activation of TRPML1 eliminates the protective intracellular niche generated by VacA, with deadly consequences for intracellular H. pylori.
Fig. 6|. TRPML1 activation eliminates both the intracellular niche and survival advantage of VacA+ H. pylori.

a–d, AGS cells infected with wild-type (VacA+) or isogenic VacA mutant (VacA−) H. pylori strains were incubated with gentamycin to kill the extracellular bacteria and treated with ML-SA1 (20 μM) or vehicle (DMSO) for an additional 4 h. a, Lamp1, cathepsin D and DAPI staining. Merge images, with higher magnification of the selected dashed areas in the separate channels, are included on the right. Note the abundant bacteria present within vacuoles (arrows). b, LC3 staining of AGS cells treated as above. Stainings were repeated a minimum of three independent times. c, LC3 western blotting of cells from b, using actin as a loading control. The graph shows quantification of LC3-II normalized to actin (mean ± s.e.m. of three independent experiments). d, Intracellular H. pylori were retrieved from AGS cells and CFUs were quantified. The graph shows the relative percentage of CFUs (mean ± s.e.m. of three independent experiments), considering 100% of the bacteria retrieved from VacA−-infected, DMSO-treated AGS cells. e, Quantification of the indicated intracellular H. pylori after overnight infection of murine gastric organoids derived from wild-type (ML1 WT) or Trpml1-null (ML1 KO) mice followed by 4 h ML-SA1 or DMSO treatment. The data represent fold change CFUs relative to wild-type, DMSO-treated controls (mean ± s.e.m. of four independent experiments). A two-tailed unpaired Student’s t-test was utilized for data analysis of c,d and one-way analysis of variance with multiple comparisons (Tukey correction) was used for comparisons among groups in e.
We next validated our results by infecting gastric organoids obtained from wild-type and Trpml1-deficient mice. In wild-type gastric organoids, ML-SA1 significantly reduced intracellular survival of SS1 s1i1 H. pylori, but had no effect on parental SS1 (Fig. 6e). In addition, ML-SA1 had no effect on intracellular survival of H. pylori in gastric organoids obtained from Trpml1-deficient mice, demonstrating that the findings with ML-SA1 treatment were specific to its effects on TRPML1 (Fig. 6e). Collectively, these results confirm in a physiological model of the gastric epithelium that ML-SA1 promotes intracellular bacterial killing of H. pylori in a toxigenic VacA-dependent and TRPML1-mediated manner.
Discussion
In this study, we show, by infecting mice with a genetically modified H. pylori strain that expresses a toxigenic VacA, the presence of intracellular bacteria, mainly in parietal cells of the gastric epithelium. Importantly, bacteria within this intracellular niche were protected from antibiotic treatment, which led to infection recrudescence post-therapy. Furthermore, we found numerous intracellular bacteria within parietal cells in human gastric biopsies from patients infected with H. pylori strains containing the s1m1 toxigenic variant of VacA. The presence of intraepithelial H. pylori in human gastric biopsies has previously been reported by several groups25,37. Here we show that this is a VacA-dependent phenomenon. The fraction of intracellular versus intraluminal H. pylori has been reported to be approximately 1%25. Although this represents a small percentage of overall bacteria, our in vivo studies indicate that this proportion is biologically relevant as bacteria within this reservoir resist antibiotic therapy and recolonize the stomach following eradication therapy. Thus, understanding the molecular mechanisms by which VacA generates this niche could open new avenues for the development of more effective therapeutics. Here we described a mechanistic interplay between VacA and the endolysosmal calcium channel TRPML1 that appears to underlie the intracellular survival of H. pylori. These findings collectively raise the possibility that TRPML1 activity may serve as a potential therapeutic target in treating chronic H. pylori infection.
TRPML1 is essential in maintaining adequate ion homeostasis and membrane trafficking in the endolysosomal pathway by mediating release of calcium from late endosomes and lysosomes11,38. Here, using complementary approaches we demonstrated that VacA+-derived vacuoles accumulated intraluminal calcium as a consequence of reduced TRPML1 activity. In all model organisms, TRPML1 disruption causes accumulation of large lysosome-like organelles and autophagic vacuoles with severely compromised autophagic flux. The consequences of TRPML1 loss cause varying effects from embryonic lethality in worms to severe neuronal and retinal degeneration in flies and zebrafish11,38. In all cases, the exogenous expression of wild-type TRPML1 rescues or delays the onset of these phenotypes11. In addition to the neurologic and ophthalmologic phenotypes, Trpml1-deficient mice and ML4 patients display severe gastric pathology15–19. Loss of TRPML1 causes reduced levels and mis-localization of the gastric proton pump, causing hypochlorhydria and hypergastrinemia, both characteristic features of chronic H. pylori infection. Furthermore, Trpml1-deficient gastric epithelium displays dramatic parietal cell vacuolation15,16,18. These vacuoles represent very enlarged dysfunctional lysosomes that resemble the VacA intracellular niche. In fact, here we show that unlike parietal cells in wild-type mice, colonization of these Trpml1−/− vacuoles by H. pylori no longer requires toxigenic VacA. Interestingly, in Trpml1−/− mice, intravacuolar bacteria persist even after conventional eradication therapy, further confirming that these vacuoles constitute a protected reservoir responsible for recrudescence of infection. There could be additional downstream effects of the VacA–TRPML1 axis including alteration in parietal cell H+, K+-ATPase as has recently been described39. Furthermore, intracellular bacteria could contribute to more intimate interactions with the host cell. Indeed, current evidence suggests that strains expressing s1i1 have an increased risk for intestinal metaplasia21.
The reasons for preferential intracellular colonization of parietal cells by toxigenic VacA+ H. pylori are currently unknown. However, within the gastric epithelium, TRPML1 expression levels are highest in parietal cells18. Furthermore, parietal cells may lack expression of other members of the TRPML family that could compensate for TRPML1 inhibition as was reported for enterocytes40. This hypothesis is consistent with the dramatic vacuolization that is observed only in parietal cells in Trpml1−/− gastric epithelium.
Notably, we found that TRPML1 overexpression prevented VacA-mediated vacuolation. More importantly, TRPML1 activation by the synthetic agonist ML-SA1 reversed all VacA toxic effects. Following TRPML1 reactivation, the VacA-mediated large vacuoles showed tubulation and reformation of functional, degradative lysosomes due to restored vesicular trafficking and localization of sorting receptors, resulting in delivery of cathepsin D to the newly formed lysosomes. This led to the destruction of the intracellular protective niche, the restoration of the autophagic pathway and the elimination of the VacA-mediated intracellular survival advantage. Thus, ML-SA1 administration resulted in effective intracellular bacteria killing and restored normal homeostatic endolysosomal trafficking in both gastric cell lines and in human gastric organoids. Unfortunately, due to its lack of solubility and potency, ML-SA1 is not suitable for in vivo administration.
The mechanism by which VacA impairs TRPML1 activity is currently unknown, but the regulation of the channel itself is not well understood. Since lysosomes contain a high density of proton pumps, ion co-transporters and exchangers in a small-sized vesicle, an increase in the permeability of one ion may alter the concentration gradients of other ions, thereby affecting ion homeostasis41. Consistent with this, we recently reported that the big conductance calcium-activated potassium (BK) channel positively regulates TRPML1 calcium release by providing counter-cation influx to maintain lysosomal membrane potential27. Interestingly, VacA chloride channel function is essential for its toxigenic activity, as either point mutations that abolish pore formation, or the chloride channel blocker NPPB eliminates VacA effects on vesicular trafficking and generation of the intracellular niche6,42,43. These findings suggest that TRPML1 activity may also require a lysosomal chloride gradient. Lysosomal chloride is much greater than cytoplasmic levels, and this gradient is maintained by the sole lysosomal Cl−/H+ exchanger ClC744. VacA, forming a chloride channel on the lysosomal membrane, will dissipate this gradient. Supporting this possibility, mutation or loss of ClC7 leads to lysosomes with reduced luminal chloride and causes a lysosomal storage disorder characterized by drastically enlarged lysosomes, lysosome storage and neuronal and retinal degeneration45. In addition, ClC7-deficient mice display increased levels of LC3-II, indicative of autophagosome accumulation44,45. Additional studies will be required to determine the exact mechanism involved in VacA-mediated TRPML1 inhibition.
In summary, we have determined that H. pylori VacA usurps TRPML1 activity, thereby inhibiting lysosomal and autophagic killing to promote an intracellular niche that allows bacterial survival. By activating TRPML1, we reverse the detrimental effects of VacA and eliminate the intracellular H. pylori reservoir, providing evidence that TRPML1 could serve as a therapeutic target for infection.
Methods
Data reporting.
The investigators were not blinded during experiments due to the different treatments administered. Data blinding was performed during sample processing, with the limitation that trpml1−/− mice are distinguishable.
Bacterial culture.
H. pylori strain SS1 (cagA+, vacA+ s2i2m2) and the isogenic SS1 strains engineered to produce the different VacA alleles as well as VacA-null SS1 strain were obtained from J. Atherton (University of Nottingham School of Medicine). H. pylori strains 60190 and J166 (cagA+, vacA+ s1m1i1) and the respective isogenic vacA− mutants were provided by R.P.Jr (Vanderbilt University School of Medicine, Nashville). Supplementary Table 1 outlines the genotypes of each of these bacterial strains. Bacteria were grown on blood agar plates containing 5% sheep blood for 2–3 days, and then transferred to Brucella broth (Difco Laboratories) supplemented with 10% FBS (Wisent Inc.) for 16–24 h. All cultures were incubated at 37 °C under microaerophilic conditions (5% O2, 10% CO2, 85% N2).
Preparation of conditioned culture media supernatant or purified toxin.
Broth cultures of H. pylori were grown to an optical density of 1.0 at 600 nm. H. pylori culture supernatants were first filtered through a 0.22 μm-cutoff membrane filter and then concentrated 10 times using a 30-kDa-cutoff Amicon Ultra centrifugal filter (Ultracel, Millipore). In most experiments, conditioned culture media supernatant (CCMS) was utilized at 1× final concentration. When indicated, 5× final concentration was administered. VacA purified toxin was provided by S. R. Blanke (Department of Microbiology, University of Illinois)30, activated by incubation in acidified Ham’s F-12 culture medium, pH 2, for 30 min at 37 °C and neutralized before using at 35 nM or 200 nM as indicated.
Immunofluorescence.
Cells grown on glass coverslips were fixed for 20 min in 4% paraformaldehyde (Electron Microscopy Sciences), permeabilized with ice-cold methanol or 0.1% Triton X-100 in phosphate-buffered saline (PBS, Wisent Inc.) for 15 min, and then blocked in 1–5% BSA (Sigma-Aldrich) prepared in PBS for 1 h. Primary antibody incubations were performed overnight at 4 °C in blocking solution. The following primary antibodies were utilized in this study: LC3 (1:200, NB600–1384, Novus Biologicals), Lamp1 (1:300, H4A3, Developmental Studies Hybridoma Bank), cathepsin D (1:1,000, 219631, Calbiochem, EMD Millipore), sortilin (1:500, ab16640, Abcam), M6PR (1:500, 2G11, MAI-066, Thermo Scientific), H. pylori (1:200, B047101–2, DAKO), H+, K+-ATPase β-subunit (1:500, D032–3, MBL International) and rabbit anti-VacA polyclonal antibody (1:500, provided by S. R. Blanke). Secondary antibody incubations were performed for 1–2 h at room temperature in blocking solution. The secondary antibodies utilized in this study were: Alexa-Fluor-568-conjugated goat anti-rabbit (1:1,000, A-11036), Alexa-Fluor-488-conjugated goat anti-rabbit (1:1,000, A-11034) and Alexa-Fluor-488-conjugated goat anti-mouse (1:1,000, A-11029, all from Thermo Fisher Scientific). To visualize nuclei and, when appropriate, H. pylori, 4ʹ,6-diamidino-2-phenylindole (DAPI) staining (1 μg ml−1, Thermo Fisher Scientific) was performed for 10 min in PBS. Coverslips were mounted using Dako Fluorescence Mounting Medium (Agilent Technologies). For the internal/external H. pylori immunostaining, infected and fixed AGS cells were incubated with the anti-H. pylori in blocking buffer overnight at 4 °C, followed by Alexa-Fluor-488-conjugated secondary antibody. Cells were then permeabilized for 15 min with 0.1% Triton X-100 in PBS, and the intracellular H. pylori was detected by incubation with anti-H. pylori antibody in blocking buffer, followed by Alexa-Fluor-568-conjugated secondary antibody. Intracellular bacteria are stained red while extracellular are stained green-yellow.
Microscopy.
All fluorescent images were acquired at ×63 magnification using a Quorum spinning-disc confocal microscope, controlled by Volocity acquisition software (Perkin Elmer). For video construction, tissue sections were imaged on a Quorum spinning-disc Confocal Microscope using a 40× water objective. The video was then created by a 3D reconstruction of confocal z-sections acquired each 0.20 μm and deconvolved utilizing Volocity software.
Western blotting.
Cell lysates were prepared in RIPA buffer on ice for 30 min. Protein samples were run on SDS–PAGE gels and transferred to nitrocellulose membranes for immunoblotting. Membranes were blocked in 5% skimmed milk prepared in Tris-buffered saline containing 0.1% Tween 20 (TBS-T) for 1 h. Primary antibody incubations were performed overnight at 4 °C in blocking solution. The following primary antibodies were utilized in this study: β-actin (1:5,000, clone AC-15, A5441, Sigma-Aldrich), LC3 (1:1,000, NB600–1384, Novus Biologicals), H+, K+-ATPase β-subunit (1:1,000, D032–3, MBL International), cathepsin D (1:2,000, 219631, Calbiochem, EMD Millipore), p62 (1:1,000, 610832, BD Bioscience) and rabbit anti-VacA polyclonal antibody that recognizes all VacA isoforms (1:1,000, provided by S. R. Blanke). Secondary antibody incubations were performed for 1–2 h at room temperature in blocking solution. The secondary antibodies utilized in this study were: HRP-conjugated goat anti-rabbit (1:5,000, 111–035-144, Cedarlane) or HRP-conjugated goat-anti mouse (1:5,000, 115–035-003, Cedarlane). Western blot visualization and densitometric analyses were conducted using a Li-Cor Odyssey Fc imaging system.
VacA intoxication assay.
Human gastric epithelial AGS cells (obtained from the ATCC, tested negative for mycoplasma) were cultured in Ham’s F-12 (Wisent Inc.) supplemented with 10% FBS (Wisent Inc.) at 37 °C in a humidified atmosphere with 5% CO2. AGS cells were plated on a 6-well plate for protein extraction and western blotting analysis, or on glass coverslips in a 24-well plate for immunolabelling. AGS cells were incubated in the presence of VacA+ or VacA− CCMS (1× final concentration) or acid-activated purified toxin (35 mM) for 4 h or overnight. ML-SA1 (20 μM, Sigma Aldrich) or the same volume of dimethylsulfoxide (DMSO, vehicle, Sigma Aldrich) was added for an additional 3 h to 4 h. Vacuolation was detected by Lamp1 or VacA staining. In other experiments, cells were loaded with Alexa-Fluor-586-conjugated 10 kDa dextran (0.1 mg ml−1, Molecular Probes, Life Technologies) for 2 h and chased overnight to allow lysosomal accumulation of the dye before CCMS administration. When indicated, cells were transfected using Fugene HD (Promega Corporation) according to the manufacturer’s instructions with TRPML1–GFP construct (gift from H. Xu, University of Michigan) for 24 h before VacA+ or VacA− CCMS incubation. To assess the degradative capacity of lysosomes, VacA+-and ML-SA1-treated AGS cells were loaded with DQ red-BSA (10 μg ml−1, Molecular Probes, Life Technologies) for 4 h before fixation and imaging. Alternatively, the PIKfyve inhibitor YM201636 (80 nM) was added to the culture medium overnight before fixation.
Cell infections.
Overnight H. pylori broth cultures were grown. The bacterial number was estimated from the optical density of the culture (OD 600 nm of 1 = 2 × 108 bacteria). Bacteria were pelleted and resuspended to an OD of 1 in cell culture media. For the infections, AGS cells were seeded onto 6-well plates and grown to 80%–90% confluence and bacteria were added at a multiplicity of infection of 50:1. After 4 h incubation, unattached bacteria were removed by 4 washes with PBS. To prevent any extracellular bacterial growth, the culture medium was supplemented with 100 μg ml−1 gentamicin (Wisent Inc.) for 1 h, monolayers were washed 4 times with PBS, and then the gentamicin was reduced to 10 μg ml−1 for the remaining infection time (3 h to 19 h). Intracellular bacteria were retrieved from invaded cells by a 12-min incubation, at 37 °C, with 0.1% saponin (from Quillaja bark, Sigma Aldrich) in cell culture medium. Serial dilutions of bacterial suspensions were prepared in Brucella broth and drop-plated (50 μl drop) onto Columbia blood agar plates for CFU determinations. When indicated, VacA− or VacA+ CCMS (1× final concentration) or PIKfyve inhibitor YM201636 (80 nM) was added to the culture medium at the time of infection. ML-SA1 (20 μM, Sigma Aldrich) or the same volume of DMSO (vehicle) was added to the cells for the last 3 or 4 h of infection time, except for the time– and dose–response studies where the indicated ML-SA1 doses at the indicated times were assessed.
Animal studies.
C57/Bl6 mice were obtained from Jackson Laboratories and bred in the facility. trpml1 heterozygous mice were obtained from S. Slaugenhaupt (Center for Human Genetic Research, Massachusetts General Hospital at Harvard Medical School). rpml1−/− and wild-type littermates were obtained by mating trpml1 heterozygotes. At weaning time, tail-tip samples were collected, and genomic DNA was prepared and subsequently genotyped by PCR using the KAPA HotStart mouse genotyping kit (KAPA Biosystems). To characterize the gastric phenotype, stomachs were dissected from 9-week-old trpml1−/− and normal littermates. Half of the stomach was formalin-fixed, paraffin-embedded and sectioned to be stained with haematoxylin–eosin. The other half was used to isolate epithelial cells for protein extraction and western blotting analysis. Briefly, half stomachs were incubated with 0.04% bleach in water for 20 min at room temperature before 1 h incubation in 5 mM EDTA. After vigorous shaking, the remaining stomach was removed and the epithelial cells were collected by centrifugation.
For the infection studies, 8-week-old littermates were inoculated with SS1 or SS1 s1i1 strains of H. pylori (109 bacteria in 100 μl of Brucella broth by oral gavage, 3 times in a 5-day period). After eight weeks of infection, mice were euthanized. Alternatively, infected mice were treated intragastrically, twice daily, for 7 days with triple therapy consisting of omeprazole (400 μmol kg−1 day−1) and, after a 30-min period to allow a change in stomach acid pH to occur, metronidazole (14.2 mg kg−1 day−1) and clarithromycin (7.15 mg kg−1 day−1). Mice were euthanized one week, or eight weeks after the cessation of treatment. Dissected stomachs were carefully halved longitudinally along the greater and lesser curvatures and rinsed in sterile PBS. Half of each stomach was homogenized with a pellet pestle in Brucella broth containing 15% glycerol (750 μl) on ice, serially diluted and plated on Glaxo Selective Supplement A plates (blood agar base 2 (Sigma Aldrich) supplemented with 10% defibrinated horse blood (Cedarlane), 10 μg ml−1 vancomycin, 20 μg ml−1 bacitracin, 4 μg ml−1 amphotericin B, 2.5 IU ml−1 polymyxin B sulfate and 1.07 μg ml−1 nalidixic acid (all supplements from Sigma Aldrich)) for CFU determination. The other half of the stomach was formalin-fixed, paraffin-embedded and sectioned, to perform IHC for H. pylori and immunofluorescence staining for H. pylori and H+, K+-ATPase. Wild-type mice were randomly allocated to the different infection groups with an equal distribution of sex and age littermates in each group. For the experiments performed with wild-type and trpml1−/− co-housed littermates, an equal number of sex-matched mice was not always obtained within each litter per genotype group. Data blinding was performed during sample processing, with the limitation that trpml1−/− mice are distinguishable. No statistical consideration was used to choose the mouse sample size. All animal procedures were approved by the University of Toronto Ethical Review Board and performed according institutional guidelines. Mice were housed in ventilated cages and allowed water and food ad libitum.
Intraluminal lysosomal Ca2+ determination.
Lysosomal Ca2+ was evaluated by an Oregon Green 488 BAPTA-5N and Alexa-Fluor-586-conjugated dextran (10 kDa) fluorescence assay. AGS cells were loaded with the membrane-impermeant Oregon Green 488 BAPTA-5N (a pH-insensitive Ca2+ indicator; 10 μM) (Molecular Probes, Life Technologies) and Alexa-Fluor-586-conjugated 10 kDa dextran (Ca2+ insensitive, endocytic probe; 0.25 mg ml−1) (Molecular Probes, Life Technologies) for 2 h. After 2 washes with warm media, the chemicals were chased for 2.5 h (to allow lysosomal accumulation of dyes through endocytosis) in the presence of VacA− or VacA+ CCMS (5× final concentration) or acid-activated purified toxin (200 nM). Cell culture medium was replaced with live-cell imaging buffer (Life Technologies) and Oregon Green images were randomly captured in fields focused on the Dextan Red signal. Luminal Ca2+ concentration in lysosomes (indicated by fluorescence intensity of Oregon Green 488 BAPTA-5N) was then normalized by the endocytic ability of cells (indicated by the fluorescence intensity of dextran). The experiment was repeated four times with similar results.
GECO Ca2+ imaging.
AGS cells were transfected with GECO-ML1 for 24–48 h, trypsinized and plated onto glass coverslips. Most experiments were carried out within 0.5 to 2 h after plating, when cells still exhibited a round morphology. Measurements were performed 4 h after treatment with VacA− or VacA+ CCMS (1× final concentration). The fluorescence intensity at 470 nm (F470 nm) was monitored using the EasyRatioPro system. Lysosomal Ca2+ release was measured under a ‘low’ external Ca2+ solution (145 mM NaCl, 5 mM KCl, 3 mM MgCl2, 10 mM glucose, 1 mM EGTA and 20 mM HEPES, pH 7.4). Ca2+ concentration in the nominally free Ca2+ solution is estimated to be 1 to 10 μM. With 1 mM EGTA, the free Ca2+ concentration is estimated to be <10 nM, based on the Maxchelator software (http://maxchelator.stanford.edu/). The experiments were repeated three times independently. When indicated, ML-SA1 (Tocris), ionomycin (Cayman Chemical Company) or GPN (Santa Cruz Biotechnology) was added.
Immunohistochemistry.
Paraffin-embedded gastric sections were prepared from the indicated mice. After de-paraffination and rehydration of the sections, antigen retrieval was performed in 10 mM sodium citrate buffer pH 6.0 in a pressure cooker for 15 min at 110 °C. Endogenous peroxidase was then blocked by 10 min incubation in 10% H2O2 (Sigma Aldrich) in methanol (v/v), slides were rehydrated and H. pylori staining was performed using polyclonal rabbit anti-H. pylori antibody B0471 (1:200, Dako Corporation) and the Histostain-SP Rabbit Primary (AEC) kit (Life Technologies) following the manufacturer’s instructions. Gastric corpus biopsy sections from patients infected with different strains of H. pylori (six cases/each VacA genotype; only the s and m regions of VacA were genotyped) were obtained from R. Peek and stained for H. pylori as above.
Generation and maintenance of murine wild-type and trpml1−/− gastric organoids.
Eight-to twelve-week-old mice were euthanized by cervical dislocation. The oesophagus and duodenums were tied off and stomachs were extracted. The forestomach was cut and the entire stomach was inverted, retied and inflated with PBS. Inverted stomachs were incubated in 5 mM EDTA in PBS at 4 °C for 2 h with rocking, after which the glands were removed by mechanical shaking in 55 mM d-sorbitol, 44 mM sucrose (both from Sigma Aldrich) in PBS. The glands were collected by centrifugation at 500g for 10 min and embedded in Matrigel (BD Biosciences). After Matrigel polymerization, embedded glands were overlaid with organoid media consisting of Advanced Dulbecco’s modified Eagle medium (DMEM)/F12 (Life Technologies) growth medium containing 50% conditioned Wnt3a medium, 25% conditioned Rspo1 medium and 10% conditioned noggin medium, supplemented with 2 mM l-glutamine, 10 mM HEPES, 1% N2, 2% B27 (all from Life Technologies), 50 ng ml−1 epidermal growth factor (EGF) (R&D Systems), 100 ng ml−1 fibroblast growth factor-(FGF)-10 (PeproTech), 1 mM N-acetyl-cysteine, 10 μM Y-27632, 10 mM nicotinamide (all from Sigma-Aldrich), 1 μM TGFβi (A83–01), 10 nM gastrin (both from Tocris) and penicillin/streptomycin. Organoids were maintained by replacing culture media every 3–4 days and passaging every 5 to 7 days at a ratio of 1:3. To passage, organoids were split by mechanical shearing using a flame-polished glass pipette, collected by centrifugation and re-embedded in fresh Matrigel.
Generation and maintenance of human gastric organoids.
All experiments conducted using human tissue were approved by the Ethics Committees at the University Health Network (protocol no. 15–9437) or Cincinnati Children’s Hospital Medical Center (protocol no. 00002988). Gastric tissue sections from patients undergoing pancreatico-duodenectomy (Whipple procedure) or sleeve gastrectomy were used to culture human gastric organoids. Tissue samples were washed in PBS and cut into 2 mm pieces. Pancreatico-duodenectomy samples were incubated in 5 mM EDTA in PBS at 4 °C with rocking for 2–3 h. Glands were extracted by mechanical shaking in 55 mM d-sorbitol, 44 mM sucrose in PBS. Sleeve gastrectomy samples were prepared as described previously46. Briefly, glands were released by incubation in a solution containing Clostridium histolyticum collagenase (1 mg ml−1) and BSA (2 mg ml−1) for 30 min at 37 °C. The glands were collected by centrifugation at 500g for 10 min and embedded in Matrigel. After Matrigel polymerization, embedded glands were overlaid with organoid media (described above). Organoids were maintained by replacing culture media every 2–3 days and passaging (described above) every 7 to 10 days at a ratio of 1:2.
Generation of gastric epithelial organoid monolayers.
To form monolayers, 5–7-day-old organoids were collected, sheared briefly and washed once with Advanced DMEM/F12 containing 2 mM l-glutamine, 10 mM HEPES and penicillin/streptomycin. Organoid fragments were resuspended in organoid media (described above) and seeded onto collagen-coated plates or coverslips coated with poly-l-lysine (Sigma-Aldrich) or diluted Matrigel (BD Bioscience). After 24 h, organoid medium was replaced with 2D medium consisting of Advanced DMEM/ F12, 10% FBS, 1% N2, 2% B27, 10 mM nicotinamide, 50 ng ml−1 EGF, 10 μM Y-27632 and 1 μM TGFβi. Every two days, organoid monolayers were washed twice with PBS and overlaid with fresh 2D media.
Infection of murine gastric organoids.
Infections were conducted using epithelial organoid monolayers and were performed when organoids had spread and formed monolayers with confluency >60%. Cells were washed twice with PBS and overlaid with fresh 2D media, before starting the infection. Monolayers were infected with SS1 or SS1 s1i1 H. pylori at a multiplicity of infection of 1:100 and allowed to invade for ~16 h. After invasion, cells were washed three times with PBS and overlaid with Advanced DMEM/F12 media containing 100 μg ml−1 gentamicin for 1 h to kill extracellular bacteria. Monolayers were then washed three times with PBS and overlaid with 2D media containing 10 μg ml−1 gentamicin for an additional 3 h. When indicated, ML-SA1 (20 μM) or DMSO was added for the last 3 h. To release intracellular bacteria, monolayers were washed three times with PBS and incubated with 0.1% saponin made up in Advanced DMEM/F12 media for 15 min at 37 °C. Bacteria were quantified in triplicate by serial dilution and plating on Columbia 5% blood agar plates.
Infection, VacA and ML-SA1 treatment of human gastric organoids.
For imaging, treatments were carried out using epithelial organoid monolayers. Monolayers were infected overnight with VacA- or VacA+ H. pylori followed by 4 h ML-SA1 or DMSO administration. Alternatively, VacA+ or VacA− culture supernatants were added to cells for 16 h, followed by 3 h with DMSO or ML-SA1 and coverslips fixed with formaldehyde. Western blot analysis was performed on 3D organoids treated with VacA+ or VacA− culture supernatant for 16 h, followed by 5 h with DMSO or ML-SA1. The Matrigel was dissociated from the organoids by incubating with Dispase (Corning) for ~20 min at 37 °C. After Matrigel digestion, Dispase was inactivated using 5 mM EDTA. Organoids were then centrifuged at 500g for 5 min, washed once with PBS, and lysed in RIPA buffer for 30–60 min on ice.
Statistical analysis.
For mouse experiments, no specific blinding method was used. The sample size (n) of each experimental group is described in each corresponding figure legend. GraphPad Prism software was used for all statistical analyses. Quantitative data displayed as histograms are expressed as means + standard error of the mean (represented as error bars). A two-tailed unpaired Student’s t-test with equal s.d., or one-way analysis of variance with multiple comparisons (Tukey correction) was used for comparisons among groups. Statistical significance was set at P < 0.05.
Supplementary Material
Acknowledgements
We thank S. Slaugenhaupt for the Trpml1-deficient mice, S. Blanke for the purified VacA toxin and anti-VacA polyclonal antibody and J. Atherton for the H. pylori SS1 strains. This work was supported by the Canadian Institutes of Health Research (CIHR), the Canadian Association of Gastroenterology (CAG) and the North American Society for Paediatric Gastroenterology, Hepatology and Nutrition Foundation. L.K.G. was supported by a CIHR/CAG/Canadian Crohn’s and Colitis and CIHR/CAG/AbbVie Pharmaceuticals Canada Fellowship.
Footnotes
Competing interests
The authors declare no competing interests.
Reprints and permissions information is available at www.nature.com/reprints.
Additional information
Supplementary information is available for this paper at https://doi.org/10.1038/s41564–019-0441–6.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Data availability
Material used in this study is readily available from the authors or from the commercial source. AGS cells and H. pylori 60190 strain are available from the corresponding author.
References
- 1.Mégraud F, Bessède E. & Varon C. Helicobacter pylori infection and gastric carcinoma. Clin. Microbiol. Infect 21, 984–990 (2015). [DOI] [PubMed] [Google Scholar]
- 2.Hooi JKY et al. Global prevalence of Helicobacter pylori infection: systematic review and meta-analysis. Gastroenterology 153, 420–429 (2017). [DOI] [PubMed] [Google Scholar]
- 3.IARC Helicobacter pylori Working Group. Helicobacter pylori eradication as a strategy for preventing gastric cancer IARC Working Group Reports, No. 8 (WHO Press, 2014). [Google Scholar]
- 4.Tacconelli E. et al. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis 18, 318–327 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Foegeding NJ, Caston RR, McClain MS, Ohi MD & Cover TL An overview of Helicobacter pylori VacA toxin biology. Toxins 8, 173 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Terebiznik MR et al. Helicobacter pylori VacA toxin promotes bacterial intracellular survival in gastric epithelial cells. Infect. Immun 74, 6599–6614 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Terebiznik MR et al. Effect of Helicobacter pylori’s vacuolating cytotoxin on the autophagy pathway in gastric epithelial cells. Autophagy 5, 370–379 (2009). [DOI] [PubMed] [Google Scholar]
- 8.Raju D. et al. Vacuolating cytotoxin and variants in Atg16L1 that disrupt autophagy promote Helicobacter pylori infection in humans. Gastroenterology 142, 1160–1171 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Galluzzi L. et al. Autophagy in malignant transformation and cancer progression. EMBO J. 34, 856–880 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Waller-Evans H. & Lloyd-Evans E. Regulation of TRPML1 function. Biochem. Soc. Trans 43, 442–446 (2015). [DOI] [PubMed] [Google Scholar]
- 11.Venkatachalam K, Wong CO & Zhu MX The role of TRPMLs in endolysosomal trafficking and function. Cell Calcium 58, 48–56 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng X, Shen D, Samie M. & Xu H. Mucolipins: intracellular TRPML1–3 channels. FEBS Lett. 584, 2013–2021 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dong X. et al. PI(3,5)P2 controls membrane trafficking by direct activation of mucolipin Ca2+ release channels in the endolysosome. Nat. Commun 1, 38 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shen D. et al. Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat. Commun 3, 731 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schiffmann R. et al. Constitutive achlorhydria in mucolipidosis type IV. Proc. Natl Acad. Sci. USA 95, 1207–1212 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Venugopal B. et al. Neurologic, gastric, and opthalmologic pathologies in a murine model of mucolipidosis type IV. Am. J. Hum. Genet 81, 1070–1083 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vergarajauregui S, Connelly PS, Daniels MP & Puertollano R. Autophagic dysfunction in mucolipidosis type IV patients. Hum. Mol. Genet 17, 2723–2737 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chandra M. et al. A role for the Ca2+ channel TRPML1 in gastric acid secretion, based on analysis of knockout mice. Gastroenterology 140, 857–867 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miller A. et al. Mucolipidosis type IV protein TRPML1-dependent lysosome formation. Traffic 16, 284–297 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Lee A. et al. A standardized mouse model of Helicobacter pylori infection: introducing the Syndey strain. Gastroenterology 122, 1386–1397 (1997). [DOI] [PubMed] [Google Scholar]
- 21.Winter JA et al. A role for the vacuolating cytotoxin, VacA, in colonization and Helicobacter pylori-induced metaplasia in the stomach. J. Infect. Dis 210, 954–963 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Salama NR, Otto G, Tompkins L. & Falkow S. Vacuolating cytotoxin of Helicobacter pylori plays a role during colonization in a mouse model of infection. Infect. Immun 69, 730–736 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oertli M. et al. Helicobacter pylori γ-glutamyl transpeptidase and vacuolating cytotoxin promote gastric persistence and immune tolerance. Proc. Natl Acad. Sci. USA 110, 3047–3052 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Veldhuyzen van Zanten SJO, Kolesnikow T, Leung V, O’Rourke JL & Lee A. Gastric transitional zones, areas where Helicobacter treatment fails: results of a treatment trial using the Sydney strain mouse model. Antimicrob. Agents Chemother 47, 2249–2255 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dubois A. & Borén T. Helicobacter pylori is invasive and it may be a facultative intracellular organism. Cell. Microbiol 9, 1108–1116 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Necchi V. et al. Natural history of Helicobacter pylori VacA toxin in human gastric epithelium in vivo: vacuoles and beyond. Sci. Rep 7, 14526 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cao Q. et al. BK channels alleviate lysosomal storage diseases by providing positive feedback regulation of lysosomal Ca2+ release. Dev. Cell 33, 427–441 (2015). [DOI] [PubMed] [Google Scholar]
- 28.Zhong XZ, Yang Y, Sun X. & Dong X-P Methods for monitoring Ca2+ and ion channels in the lysosome. Cell Calcium 64, 20–28 (2017). [DOI] [PubMed] [Google Scholar]
- 29.Cover TL, Tummuru MKR, Cao P, Thompson SA & Blaser MJ Divergence of genetic sequences for the vacuolating cytotoxin among Helicobacter pylori strains. J. Biol. Chem 269, 10566–10573 (1994). [PubMed] [Google Scholar]
- 30.Patel HK et al. Plasma membrane cholesterol modulates cellular vacuolation induced by the Helicobacter pylori vacuolating cytotoxin. Infect. Immun 70, 4112–4123 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McCartney AJ, Zhang Y. & Weisman LS Phosphatidylinositol 3,5-bisphosphate: low abundance, high significance. BioEssays 36, 52–64 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen C-C et al. A small molecule restores function to TRPML1 mutant isoforms responsible for mucolipidosis type IV. Nat. Commun 5, 1–10 (2014). [DOI] [PubMed] [Google Scholar]
- 33.Javier Pérez-Victoria F, Mardones GA & Bonifacino JS Requirement of the human GARP complex for mannose 6-phosphate-receptor-dependent sorting of cathepsin D to lysosomes. Mol. Biol. Cell 19, 2350–2362 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coutinho MF, Prata MJ & Alves S. Mannose-6-phosphate pathway: a review on its role in lysosomal function and dysfunction. Mol. Genet. Metab 105, 542–550 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Canuel M, Korkidakis A, Konnyu K. & Morales CR Sortilin mediates the lysosomal targeting of cathepsins D and H. Biochem. Biophys. Res. Commun 373, 292–297 (2008). [DOI] [PubMed] [Google Scholar]
- 36.Coutinho MF, Prata MJ & Alves S. A shortcut to the lysosome: the mannose-6-phosphate-independent pathway. Mol. Genet. Metab 107, 257–266 (2012). [DOI] [PubMed] [Google Scholar]
- 37.Necchi V, Sommi P, Ricci V. & Solcia E. In vivo accumulation of Helicobacter pylori products, NOD1, ubiquitinated proteins and proteasome in a novel cytoplasmic structure. PLoS ONE 5, e9716 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Abe K. & Puertollano R. Role of TRP channels in the regulation of the endosomal pathway. Physiology 26, 14–22 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sahoo N. et al. Gastric acid secretion from parietal cells is mediated by a Ca2+ efflux channel in the tubulovesicle. Dev. Cell 41, 262–273 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Remis NN et al. Mucolipin co-deficiency causes accelerated endolysosomal vacuolation of enterocytes and failure-to-thrive from birth to weaning. PLoS Genet. 10, e1004833 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu H. & Ren D. Lysosomal physiology. Annu. Rev. Physiol 77, 57–80 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Y, Wandinger-Ness A, Goldenring JR & Cover TL Clustering and redistribution of late endocytic compartments in response to Helicobacter pylori vacuolating toxin. Mol. Biol. Cell 15, 1946–1959 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Palframan SL, Kwok T. & Gabriel K. Vacuolating cytotoxin A (VacA), a key toxin for Helicobacter pylori pathogenesis. Front. Cell. Infect. Microbiol 2, 1–9 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stauber T. & Jentsch TJ Chloride in vesicular trafficking and function. Annu. Rev. Physiol 75, 453–477 (2013). [DOI] [PubMed] [Google Scholar]
- 45.Kasper D. et al. Loss of the chloride channel ClC-7 leads to lysosomal storage disease and neurodegeneration. EMBO J. 24, 1079–1091 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bertaux-Skeirik N. et al. CD44 plays a functional role in Helicobacter pylori-induced epithelial cell proliferation. PLoS Pathog. 11, e1004663 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.