

This Outlook discusses Cho et al.’s finding showing that CDK7 directly phosphorylates YAP/TAZ/Yki in the nucleus, protecting them from ubiquitination and degradation, in a manner independent of the Hippo cascade and from CDK7 basal transcriptional effects.
Keywords: CDK3, CRL4, Cul4, DCAF12, Hippo, Taz, Yap, Yki, cancerorgan size
Abstract
Inhibition of CDK7 is a promising strategy for cancer therapy. CDK7 so far has been understood mainly in the context of Pol II-driven transcription. However, how are the roles of CDK7 in the “basal” transcriptional machinery reconciled with the function of CDK7 as inducer of specific transcriptional programs in tumor cells? In this issue of Genes & Development, Cho and colleagues (pp. 53–71) advance in this direction, demonstrating that attenuation of CDK7 fosters the oncogenic activity of the YAP/TAZ/Yki coactivators. CDK7 directly phosphorylates YAP/TAZ/Yki in the nucleus, protecting them from ubiquitination and degradation, in a manner independent from the Hippo cascade and independent from CDK7 basal transcriptional functions.
Tumorigenesis is the product of dysregulated gene expression programs controlling proliferation, fate plasticity, and epithelial–stromal interactions (Bradner et al. 2017). Thus, a bewildering array of genetic and epigenetic events must eventually converge on the basal transcriptional machinery, generating “addictions” that may be targeted therapeutically. This may be particularly important for tumor types lacking targeted therapies, such as triple-negative breast cancer (TNBC), or for tumor stages for which definitive driver mutations may not exist, such as metastasis. That said, a key unanswered question is in fact the molecular nature of these transcriptional addictions: by what signaling cascades and what transcriptional effectors?
One popular model posits that tumor cells set high demands on RNA polymerase II (Pol II) itself and its basal transcriptional machinery, warranting the disproportionally high levels of gene expression thought to be required to support the enhanced proliferation and high metabolic demands that typify cancer cells (Bradner et al. 2017). The corollary of this model is that general transcription factors may represent cancer “Achilles’ heels” in plain sight and, as such, targets of anti-cancer therapies. A paradigm example is the vulnerability of certain tumors to a “transcriptional therapy” mediated by drugs targeting the BRD4 coactivator, bridging Pol II to histone acetylation at promoters and enhancers (Shi and Vakoc 2014). The function of cyclin-dependent kinase7 (CDK7) seems another poster child of this model: This kinase is a subunit of TFIIH, directly phosphorylating the Pol II C-terminal domain and supporting Pol II function throughout its transcriptional cycle, from promoter clearance to termination (Fisher 2019). Intriguingly, small molecule inhibitors of CDK7 (CDK7-i), such as THZ1, have been shown to be effective in inhibiting the growth of leukemia and different solid tumors, including TNBC, leading to activation of a key set of genes (Wang et al. 2015; Fisher 2019). In these studies, the effectiveness of CDK7-i has been readily attributed to the well-established functions of CDK7 in boosting Pol II transcription. However, such a “basal transcriptional machinery” model suffers from some inconsistencies: If CDK7 fuels the cancer phenotype through Pol II, how is cancer-specific regulation of gene expression achieved? Tumor cells indeed express their unique and peculiar set of genes, and not just a much higher dosage of the same genes expressed by normal cells. Moreover, there is no reason to think that CDK7 inhibitors would be effective only toward highly expressed genes and not lowly expressed genes equally essential for cancer growth. Consistently, CDK7 inhibitors are also effective against inflammatory conditions (Fisher 2019), a scenario again speaking in favor of great selectivity in target gene activation by such “general” transcription cofactors.
In this issue of Genes & Development, Cho et al. (2020) make advances concerning these outstanding issues by proposing a radically different model for CDK7 function that indeed explains how this kinase connects to cancer-specific gene expression programs. They discovered that CDK7 in fact fosters the activity of YAP/TAZ, potent oncogenic transcriptional activators well known for their role in cellular mechanotransduction and Hippo signaling (Totaro et al. 2018). This function of CDK7 is independent from its basal roles in transcription and cell cycle progression and can be genetically uncoupled from TFIIH.
Downstream from oncogenic signaling, YAP/TAZ activation is widespread and pervasive in a vast array of human solid malignancies and their murine models, promoting proliferation, gain-of-stemness attributes, and chemoresistance (Zanconato et al. 2019). The relevance of YAP/TAZ in cancer cells is in stark contrast to their dispensability in healthy tissues and normal physiology. Targeting YAP/TAZ thus promises to display a broad therapeutic window effective against tumor cells while sparing normal cells from the toxicities typically associated with chemotherapy, and is indeed an area of intense investigation. Thus, the work of Cho et al. (2020) suggests that CDK7 inhibitors may indeed serve as anti-YAP/TAZ drugs and could explain why CDK7 represents a selective vulnerability of cancer cells.
Cho et al. (2020) started their investigation taking advantage of Drosophila genetics, screening by RNAi for modifiers of tissue overgrowth caused by overexpression of Yki, the fly homolog of mammalian YAP/TAZ. After identifying CDK7, they first validated the requirement for Yki function of cyclin H and Mat1, factors that regulate CDK7 substrate recognition, but not of Xpd1, which normally bridges the CDK7/cyclin H/Mat1 complex to TFIIH. Next, the investigators extended their findings in mouse models carrying liver-specific conditional inactivation of the Hippo kinases (e.g., MST1/2). These deficiencies cause YAP/TAZ nuclear localization, leading to aberrant proliferation of liver cells, conversion of hepatocytes into progenitor (oval) cells, and overall increased liver size (Zhou et al. 2009). Remarkably, treatment with the CDK7 inhibitor THZ1 alleviated these YAP/TAZ-driven responses. Thus, CDK7 regulates YAP/TAZ in a manner ostensibly independent of Hippo signaling. This conclusion is corroborated by results in Drosophila, where the investigators failed to detect any epistatic relationships between CDK7 and Hippo mutants, and with active Yki mutants insensitive to Hippo regulation remaining sensitive to CDK7 knockdown.
Mechanistically, CDK7 acts in the nucleus by directly phosphorylating YAP in an evolutionary conserved serine, corresponding to human S128 (i.e., S169 in Drosophila Yki), as visualized in vivo with phospho-specific antibodies and reproduced in vitro with purified proteins (Fig. 1). Supporting in vivo the relevance of CDK7–YAP/TAZ/Yki phosphorylation, a TNBC cell line expressing a YAP S128D mutant—i.e., mimicking CDK7 phosphorylation—once injected in recipient mice generates tumors that are refractory to pharmacological CDK7 inhibition. Curiously, the activating phosphorylation in S128 is in stark contrast to the repressive phosphorylation mediated in the preceding residue, YAP S127, by the cardinal Hippo kinase LATS/Wts (Totaro et al. 2018).
Figure 1.
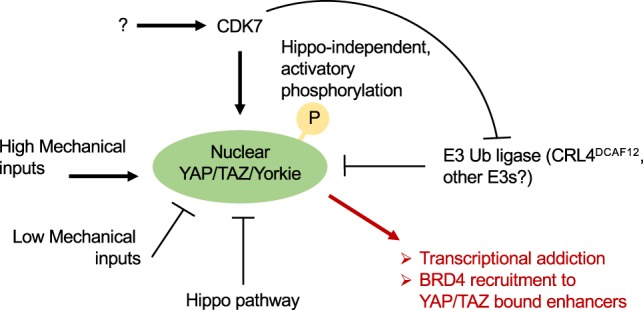
Nuclear localization of YAP/TAZ/Yki is inhibited by Hippo signaling and by conditions associated with reduced mechanical signaling, such as adhesion to a soft extracellular matrix and to a small adhesive area, and by postconfluent cell density. Nuclear localization of YAP/TAZ/Yki is favored by a spread cell shape, adhesion to a stiff extracellular matrix, or other forms of mechanical strains. In this issue of Genes & Development, Cho et al. (2020) propose that nuclear YAP/TAZ/Yki requires activation by CDK7, that directly phosphorylates YAP/TAZ/Yki in an Hippo-independent manner. This step prevents YAP/TAZ/Yki degradation by CRL4DCAF12. CDK7 thus promotes nuclear YAP/TAZ/Yki activity, leading to their association with cognate enhancers, recruitment of BRD4 to the same cis-regulatory elements, and activation of specific gene expression programs. See the text for details.
How can YAP/TAZ/Yki phosphorylation by CDK7 induce their transcriptional responses? Cho et al. (2020) propose that this step protects nuclear YAP/TAZ/Yki from ubiquitination mediated by the E3 ubiquitin ligase CRL4DCAF12 followed by their clearance through proteasomal degradation (Fig. 1). What is less clear in this mechanistic segment of the study is the in vivo relevance of CRL4DCAF12, as Drosophila CRL4DCAF12 inactivation is, by itself, inconsequential for tissue growth. There are some plausible technical and biological explanations for this discrepancy; for example, CRL4DCAF12 knockdown may leave sufficient protein for Yki degradation. In addition, other E3 ligases of the DCAF family may overlap and compensate for loss of CRL4DCAF12. Thus, additional experiments are required to firmly establish mechanisms of and players in a nuclear YAP/TAZ/Yki degradation. Irrespective of these considerations, Cho et al. (2020) shed light on the so far underexplored regulations of YAP/TAZ/Yki in the nucleus, adding a positive, oncogenic regulatory tier mediated by CDK7, potentially balancing nuclear YAP/TAZ inactivation by sequestration into ARID1A-containing SWI/SNF tumor-suppressive complexes (Chang et al. 2018).
Previous work noted how the effects of CDK7 inhibitors on gene expression correlated with transcriptional regulation of genes controlled by superenhancers; the latter represent a restricted set of enhancer regions bound by the BRD4 coactivator and able to drive high-level of target gene transcription (Wang et al. 2015; Bradner et al. 2017). The mechanisms of the intriguing CDK7–superenhancer correlation have remained obscure thus far, but may be potentially connected to the present CDK7–YAP axis. Recent work has indeed revealed that BRD4-mediated transcriptional addiction in different cancer cells is also dependent on YAP/TAZ, which recruits BRD4 at high levels at YAP/TAZ-bound enhancers and superenhancers (Zanconato et al. 2018). Thus, a potential unifying scenario may be one in which CDK7 would sustain nuclear YAP/TAZ/Yki, securing their binding at cognate enhancer sites, in turn leading to BRD4 accrual; this would lead to Pol II recruitment of specific and YAP/TAZ-addicted gene expression programs. Clearly, this possibility remains speculative.
In sum, Cho et al. (2020) highlight a number of potential novelties in the regulation of YAP/TAZ/Yki, shed light on the mechanisms of CDK7 transcriptional addictions in cancer cells, and open exciting scenarios for future investigations.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.335562.119.
References
- Bradner JE, Hnisz D, Young RA. 2017. Transcriptional addiction in cancer. Cell 168: 629–643. 10.1016/j.cell.2016.12.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Azzolin L, Di Biagio D, Zanconato F, Battilana G, Lucon Xiccato R, Aragona M, Giulitti S, Panciera T, Gandin A, et al. 2018. The SWI/SNF complex is a mechanoregulated inhibitor of YAP and TAZ. Nature 563: 265–269. 10.1038/s41586-018-0658-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YS, Li S, Wang X, Zhu J, Zhuo S, Han Y, Yue T, Yang Y, Jiang J. 2020. CDK7 regulates organ size and tumor growth by safeguarding the Hippo pathway effector Yki/Yap/Taz in the nucleus. Genes Dev 10.1101/gad.333146.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher RP. 2019. Cdk7: a kinase at the core of transcription and in the crosshairs of cancer drug discovery. Transcription 10: 47–56. 10.1080/21541264.2018.1553483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Vakoc CR. 2014. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell 54: 728–736. 10.1016/j.molcel.2014.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Totaro A, Panciera T, Piccolo S. 2018. YAP/TAZ upstream signals and downstream responses. Nat Cell Biol 20: 888–899. 10.1038/s41556-018-0142-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Zhang T, Kwiatkowski N, Abraham BJ, Lee TI, Xie S, Yuzugullu H, Von T, Li H, Lin Z, et al. 2015. CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell 163: 174–186. 10.1016/j.cell.2015.08.063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato F, Battilana G, Forcato M, Filippi L, Azzolin L, Manfrin A, Quaranta E, Di Biagio D, Sigismondo G, Guzzardo V, et al. 2018. Transcriptional addiction in cancer cells is mediated by YAP/TAZ through BRD4. Nat Med 24: 1599–1610. 10.1038/s41591-018-0158-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato F, Cordenonsi M, Piccolo S. 2019. YAP and TAZ: a signalling hub of the tumour microenvironment. Nat Rev Cancer 19: 454–464. 10.1038/s41568-019-0168-y [DOI] [PubMed] [Google Scholar]
- Zhou D, Conrad C, Xia F, Park JS, Payer B, Yin Y, Lauwers GY, Thasler W, Lee JT, Avruch J, et al. 2009. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer Cell 16: 425–438. 10.1016/j.ccr.2009.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]