

We clarify that DNA damage triggers autophagy by CHK2-FOXK axis, and misregulation of this pathway leads to chemoresistance.
Abstract
Autophagy is an evolutionarily conserved catabolic process, which plays a vital role in removing misfolded proteins and clearing damaged organelles to maintain internal environment homeostasis. Here, we uncovered the checkpoint kinase 2 (CHK2)–FOXK (FOXK1 and FOXK2) axis playing an important role in DNA damage–mediated autophagy at the transcriptional regulation layer. Mechanistically, following DNA damage, CHK2 phosphorylates FOXK and creates a 14-3-3γ binding site, which, in turn, traps FOXK proteins in the cytoplasm. Because FOXK functions as the transcription suppressor of ATGs, DNA damage–mediated FOXKs’ cytoplasmic trapping induces autophagy. In addition, we found that a cancer-derived FOXK mutation induces FOXK hyperphosphorylation and enhances autophagy, resulting in chemoresistance. Cotreatment with cisplatin and chloroquine overcomes the chemoresistance caused by FOXK mutation. Overall, our study highlights a mechanism whereby DNA damage triggers autophagy by increasing autophagy genes via CHK2-FOXK–mediated transcriptional control, and misregulation of this pathway contributes to chemoresistance.
INTRODUCTION
Macroautophagy (hereafter referred to as autophagy) is a self-degradative process that influences vital functions in balancing sources of energy and eliminating harmful metabolic products in the cell, such as misfolded proteins, reactive oxygen species, and broken organelles, in response to various stressors (1–3). Accumulating evidence has demonstrated that autophagy is induced by DNA damage and is required for several functional outcomes of DNA damage response (DDR) signaling, such as DNA repair, senescence, and cytokine secretion, which, in turn, play an important role in maintaining genomic stability (1, 4–6). It is well demonstrated that DNA damage–regulated rapid induction of autophagy is mediated by posttranslational modifications (PTMs) such as phosphorylation, ubiquitination, and acetylation (7–9). For example, DNA damage triggers activation of the ULK1/ATG13/FIP200 kinase complex and initiates autophagy through the ataxia-telangiectasia mutated (ATM)–AMPK-TSC2-TORC1 axis (10). ATM-mediated PTEN phosphorylation promotes its nuclear localization and induces autophagy upon DNA damage (11). Besides PTM-mediated rapid induction of autophagy, the regulation of autophagy may also depend on transcriptional or posttranscriptional programs. However, whether DNA damage induces autophagy at the transcriptional or posttranscriptional level remains largely unknown.
FOXK (FOXK1 and FOXK2 collectively termed FOXK) proteins are members of the Forkhead transcriptional family, which play important roles in proliferation, differentiation, and energy metabolism (12, 13). A recent study showed that FOXK proteins are important transcriptional repressors in autophagy and that mTOR (mammalian target of rapamycin) phosphorylates and activates FOXK to limit basal levels of autophagy under nutrient-rich conditions (14).
Here, we demonstrate that FOXK proteins play an important role in transcriptional regulation to link DNA damage and autophagy. DNA damage induces CHK2-mediated FOXK phosphorylation [FOXK1 at Ser130 (S130) and FOXK2 at Ser61 (S61)] and traps FOXK in the cytoplasm through binding with 14-3-3γ. FOXK trapping in the cytoplasm relieves the inhibition on the expression of autophagy-related genes (ATGs) and facilitates autophagy. Overall, we uncovered a new function of the CHK2-FOXK axis in DNA damage–mediated transcriptional control in autophagy, and this mechanism has important implications in cancer therapy.
RESULTS
CHK2 is essential for DNA damage–induced autophagy
Current research has demonstrated that DNA damage induces autophagy through multiple signaling pathways, which is important for maintaining genomic stability (15). Besides well-demonstrated posttranslational modifications in DNA damage–mediated rapid autophagy initiation, how DNA damage activates transcriptional control in autophagy is still not clear. Hence, we treated two cancer cell lines, HEPG2 and A549, with cisplatin or etoposide for 24 hours to induce DNA damage and then examined the outcomes of autophagy. As shown in Fig. 1A and fig. S1C, DNA damage increased endogenous LC3-II levels and decreased p62 levels. We further examined the autophagic flux by using fluorescent protein–tagged LC3 [EGFP (enhanced green fluorescent protein)–mCherry–LC3B]. Both GFP and mCherry fluorescence will exhibit in autophagosomes (yellow puncta), while because of quenching of GFP signal in low pH in autolysosomes, only mCherry fluorescent signal can be detected in autolysosomes (red puncta). As shown in fig. S1 (A and B), the numbers of red and yellow puncta in cells were markedly increased following cisplatin treatment. In addition, in chloroquine (CQ)–treated cells, cisplatin treatment induced further notably increased autophagosomes (yellow puncta), but not the autolysosomes (red puncta), because CQ is able to block the autophagosome-lysosome fusion (fig. S1, A and B) (16, 17). Collectively, these results suggest that DNA damage induces autophagy, which is consistent with previous reports (18–20). We found that both the transcription and protein level of ATGs (MAP1LC3B, ATG5, ATG7, Beclin1, and ULK1) were markedly increased following cisplatin treatment (Fig. 1, B and C, and fig. S1, D and L), indicating that DNA damage may activate transcriptional control in autophagy. To investigate the potential mechanism for this process, we treated HEPG2 and A549 cells with DNA-damaging agents and various inhibitors, which target key kinases in the DDR pathway, and then examined the outcome of autophagy. As shown in Fig. 1 (D to G) and fig. S1 (E to K), ATM (Ku55933) and CHK2 (BML-227) inhibitors, but not ataxia telangiectasia and Rad3-related (ATR) (VX-970) and CHK1 (CCT245737) inhibitors, markedly decreased transcription of DNA damage–induced ATGs. Because CHK2 is downstream of ATM in the DDR pathway, we next generated CHK2-depleted cells to further confirm the role of CHK2 in DNA damage–induced autophagy. As shown in Fig. 1 (H to J), depletion of CHK2 significantly decreased LC3-II levels and both red-only and yellow puncta induced by DNA damage. Reconstituting CHK2 in CHK2-depleted cells rescued these phenomena. Together, these findings suggest that the ATM-CHK2 axis is important for DNA damage–mediated transcriptional control of autophagy.
Fig. 1. CHK2 regulates DNA damage–induced autophagy.
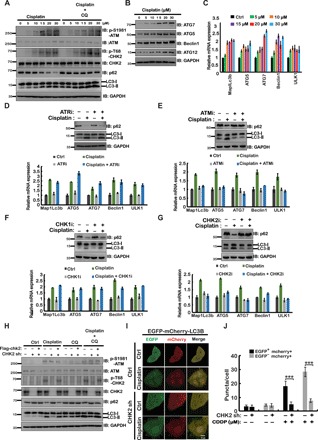
(A) HEPG2 cells were treated with various concentrations of cisplatin (24 hours) and/or chloroquine (CQ) (2 hours, 50 μM) as indicated. Western blot was performed with the indicated antibodies. GAPDH, glyceraldehyde-3-phosphate dehydrogenase. (B) HEPG2 cells were treated with the indicated concentrations of cisplatin. After 24 hours, cells were lysed, and Western blot was performed with the indicated antibodies. IB, immunoblotting. (C) Cells from (B) were subjected to mRNA extraction, and quantitative polymerase chain reaction (qPCR) assay was performed. The expression of ATGs was examined. aa, amino acid. (D to G) HEPG2 cells were treated with 20 μM cisplatin and/or several DDR inhibitors as indicated, 100 nM ATR inhibitor (VX-970) (D), 10 μM ATM inhibitor (Ku55933) (E), 10 μM CHK1 inhibitor (CCT245737) (F), or 10 μM CHK2 inhibitor (BML-227) (G). Top: After 24 hours, cells were lysed, and Western blot was performed with the indicated antibodies. Bottom: The mRNA was extracted from cells and subjected to quantitative reverse transcription PCR (qRT-PCR) to examine the expression of several key ATGs. (H) CHK2-depleted cells were transiently transfected with Flag-CHK2 and treated with vehicle or 20 μM cisplatin for 24 hours. Before harvesting, cells were treated with or without CQ for 2 hours as indicated. Western blot was performed with the indicated antibodies. (I) CHK2-depleted HEPG2 cells or control HEPG2 cells stably expressing EGFP-mCherry-LC3B were treated with vehicle or cisplatin (CDDP) for 24 hours. Green (EGFP) and red (mCherry) fluorescence were analyzed by confocal microscopy (40×). Representative images are shown. Scale bar, 10 μm. (J) Quantification of the data in (I). ***P < 0.001. Statistical analyses were performed using Student’s t test.
CHK2 interacts with FOXK
We next investigated the mechanisms underlying CHK2-mediated regulation of DNA damage–induced autophagy. We used Flag-tagged CHK2 as the bait to perform tandem affinity purification and mass spectrometry analysis. We identified FOXK2 as a binding partner of CHK2 (data not shown). Because a previous study showed that FOXK proteins function as transcriptional suppressors in ATG expression, we were interested in investigating whether CHK2 regulates autophagy through FOXK proteins. We first performed a coimmunoprecipitation assay to confirm the binding between CHK2 and FOXK proteins. As shown in fig. S2A, immunoprecipitation of endogenous CHK2 pulled down FOXK proteins (FOXK1 and FOXK2). The interaction between CHK2 and FOXK was confirmed using reciprocal coimmunoprecipitation assay (Fig. 2, A and B). In addition, we tried to detect whether there is an interaction between CHK1 and FOXK. As shown in fig. S2B, CHK1 is unable to bind with FOXK. Furthermore, bacterially expressed glutathione S-transferase (GST)–FOXK proteins were able to pull down CHK2 (fig. S2, C and D). We found that the interaction between CHK2 and FOXK proteins was markedly increased following DNA damage (Fig. 2, A and B). In addition, treatment with both ATM inhibitor and phosphatase impaired the DNA damage–induced CHK2-FOXK binding (fig. S2, E and F), suggesting that this interaction is phosphorylation dependent. Furthermore, we found that full-length CHK2, but not CHK2 deleted of the SCD domain (SQ/TQ cluster domain containing multiple S/T sites phosphorylated by ATM), interacted with FOXK proteins in cells (Fig. 2C). In addition, the CHK2 T68A mutation abolished the interaction between CHK2 and FOXK (fig. S2G). These data suggest that the phosphorylation of CHK2 at Thr68 by ATM is important for CHK2-FOXK interaction. Moreover, bacterially expressed full-length GST-FOXK and forkhead-associated (FHA) domain, but not the delta-FHA form of FOXK, pull down CHK2 in cells treated with cisplatin (Fig. 2D and fig. S2H). Together, these results suggest that CHK2 interacts with FOXK, and ATM-mediated CHK2 phosphorylation at Thr68 is essential for its binding to the FHA domain of FOXK following DNA damage (fig. S2I).
Fig. 2. CHK2 interacts with FOXK in vivo and in vitro, and CHK2 regulates DNA damage–induced autophagy though FOXK.
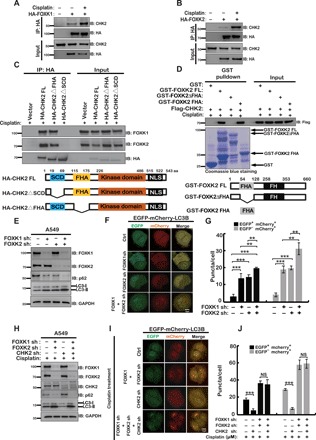
(A and B) Human embryonic kidney (HEK) 293T cells transfected with HA-FOXK1 (A) or HA-FOXK2 (B) were treated vehicle or 20-μM cisplatin for 24 hours and purified using anti–HA-agarose beads. The immunoprecipitates were then blotted with the indicated antibodies. (C) HEK293T cells transfected with HA-tagged WT CHK2 (FL), FHA deletion mutant of CHK2 (ΔFHA), or SCD deletion mutant of CHK2 (ΔSCD) and treated with 20 μM cisplatin before harvest were lysed, and then cell lysates were subjected to immunoprecipitation with anti–HA-agarose beads. The immunoprecipitates were then blotted with the indicated antibodies. The schema below depicts the various constructs of CHK2 used in this experiment. (D) HEK293T cells were transfected with Flag-CHK2 and treated with 20-μM cisplatin before harvest, and then cell lysates were incubated with Sepharose coupled with GST, GST-FOXK2, GST-FOXK2△FHA, or GST-FOXK2 FHA. After washing, proteins bound to Sepharose were blotted with indicated antibodies. The schema below depicts the various constructs of FOXK2 used in this experiment. (E) A549 cells stably expressing the indicated FOXK constructs were lysed. Western blot was performed with the indicated antibodies. (F) EGFP-mCherry-LC3B and the indicated constructs were stably expressed in HEPG2 cells. Green (EGFP) and red (mCherry) fluorescence were analyzed by confocal microscopy (40×). Representative images are shown. Scale bar, 10 μm. (G) Quantification of the data in (F). **P < 0.01 and ***P < 0.001. Statistical analyses were performed using Student’s t test. NS stands for no significant change. (H) A549 cells stably expressing the indicated constructs were treated with cisplatin for 24 hours. Western blot was performed with the indicated antibodies. (I) EGFP-mCherry-LC3B and the indicated constructs were stably expressed in HEPG2 cells. Cells were treated with cisplatin for 24 hours. Green and red fluorescence were analyzed by confocal microscopy (40×). Representative images are shown. Scale bar, 10 μm. (J) Quantification of the data in (I). ***P < 0.001. Statistical analyses were performed using Student’s t test.
CHK2 regulates autophagy through FOXK
Because it has been previously reported that FOXK plays important roles in regulating autophagy (14), we next examined whether CHK2 regulates DNA damage–induced autophagy through FOXK. We depleted FOXK1 and FOXK2 individually or simultaneously in HEPG2 and A549 cells and then examined the levels of autophagy by assessing the p62 protein level, LC3-II level, and LC3B puncta formation. As shown in fig. S3 (A and B), depletion of FOXK1 or FOXK2 individually resulted in a significant increase in autophagy, as demonstrated by a reduction in p62 protein level and an increase in LC3-II level. Double knockdown of FOXK1 and FOXK2 showed a higher autophagic level (Fig. 2, E to G, and fig. S3C). Furthermore, we found that depletion of CHK2 markedly decreased DNA damage–induced autophagy in control cells but not FOXK-depleted cells (Fig. 2, H to J, and fig. S3D). Together, these results suggest that CHK2 regulates DNA damage–induced autophagy through FOXK.
CHK2 phosphorylates FOXK (FOXK1 at S130 and FOXK2 at S61) in vivo and in vitro
Because CHK2 functions as one of the key kinases in the DDR pathway, it is possible that CHK2 is able to phosphorylate FOXK. To test this hypothesis, we used purified wild-type (WT) CHK2 or CHK2 kinase dead (KD) mutant and FOXK proteins (GST-FOXK1 and GST-FOXK2) to perform phosphorylation assay in a cell-free system. As shown in Fig. 3 (A and B), we found that FOXK proteins displayed an evident mobility shift upon incubation with WT CHK2, but not the CHK2 KD, mutant, in Phos-tag gels. It has been well established that RXXS/T is the consensus phosphorylation motif targeted by CHK2 kinase (21, 22). After analyzing the sequences of FOXK proteins, we found that S130 on FOXK1 and S61 on FOXK2 are potential CHK2 phosphorylation sites, and they are conserved across multiple species (fig. S4A). In addition, the sequence around the S130 on FOXK1 is also highly conserved, similar to that of the S61 on FOXK2 (fig. S4A). We purified GST-FOXK WT and FOXK SA (FOXK1 S130A and FOXK2 S61A) mutant proteins and performed in vitro phosphorylation assays. As shown in Fig. 3 (C and D), GST-FOXK WT, but not the SA mutant, was phosphorylated by CHK2. Moreover, we generated an antibody specifically recognizing FOXK1 pS130 and FOXK2 pS61 and examined FOXK phosphorylation in cells and in vitro. As shown in Fig. 3 (E and F), CHK2 phosphorylated FOXK WT, but not the SA mutants in vitro. In addition, cisplatin induced phosphorylation of FOXK1 at S130 and FOXK2 at S61 in cells (Fig. 3, G and H). However, depletion of CHK2 or treating cells with phosphatase abolished the cisplatin-induced FOXK phosphorylation (Fig. 3, I and J). Together, these results suggest that CHK2 specifically phosphorylates FOXK1 at S130 and FOXK2 at S61 in response to DNA damage.
Fig. 3. CHK2 phosphorylates FOXK in response to DNA damage in vivo and in vitro.
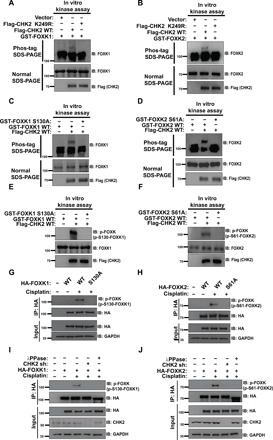
(A and B) Purified Flag-CHK2 WT or Flag-CHK2 K249R proteins from HEK293T were incubated with GST-FOXK1 (A) or GST-FOXK2 (B) in CHK2 kinase buffer at 30°C for 30 min. The samples were separated by Phos-tag SDS–polyacrylamide gel electrophoresis (PAGE) gels or normal SDS-PAGE gels as indicated. Phosphorylation was examined using indicated antibodies. (C and D) Purified Flag-CHK2 WT from HEK293T cells was incubated with GST-FOXK1 WT or GST-FOXK1 S130A (C) and GST-FOXK2 WT or GST-FOXK2 S61A (D) in CHK2 kinase buffer at 30°C for 30 min. The samples were separated by Phos-tag SDS-PAGE gels or normal SDS-PAGE gels as indicated. Phosphorylation was examined by probing the blots with the indicated antibodies. (E and F) Samples from (C) to (F) were separated by normal SDS-PAGE gels. Phosphorylation was examined by probing blots with the indicated antibodies (p-FOXK antibody is able to recognize both phosphorylation of FOXK1 at S130 and FOXK2 at S61). (G and H) HEK293T cells were transfected with HA-FOXK1 (G) or HA-FOXK2 (H) and then treated with vehicle or 20 μM cisplatin. HA-FOXK proteins were purified using anti–HA-agarose beads, and proteins bound on Sepharose were blotted with indicated antibodies. (I and J) HA-FOXK1 (I) or HA-FOXK2 (J) were transiently transfected into control cells or CHK2-depleted HEK293T cells. These cells were subsequently treated with vehicle or 20 μM cisplatin for 24 hours. Cells were lysed and purified using anti–HA-agarose beads. One of the samples was additionally treated with λPPase as indicated. The immunoprecipitates were then blotted with the indicated antibodies.
Phosphorylation of FOXK is essential for their translocation from the nucleus to the cytoplasm
Next, we studied the functional significance of FOXK phosphorylation in response to DNA damage. We first examined the cellular localization of FOXK proteins. As shown in Fig. 4 (A to D), cisplatin treatment induced translocation of FOXK proteins from the nucleus to the cytoplasm in cells expressing WT FOXK, but not the SA mutants. Furthermore, depletion of CHK2 markedly decreased FOXK translocation to the cytoplasm in response to DNA damage (Fig. 4, E to J). Moreover, phosphorylated species of FOXK (FOXK1 at S130, FOXK2 at S61) exclusively remained in the cytoplasmic fraction (Fig. 4, I and J). In addition, CHK2 inhibitor treatment also blocked FOXK translocation from the nucleus to the cytoplasm in response to DNA damage (Fig. 4, K and L, and fig. S4, B to E). Together, these results suggest that FOXK phosphorylation by CHK2 is important for its nuclear export following DNA damage.
Fig. 4. CHK2-induced FOXK translocation from nucleus to cytoplasm in response to DNA damage is dependent on FOXK phosphorylation (FOXK1 S130 and FOXK2 S61).
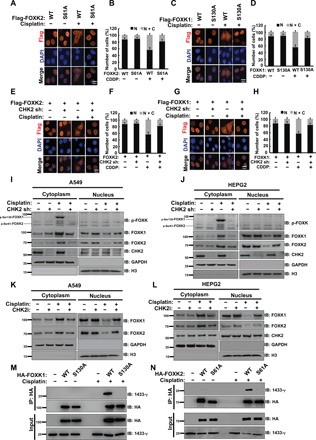
(A) HEPG2 cells were transiently transfected with HA-FOXK2 WT or HA-FOXK2 S61A plasmid. Twenty-four hours after transfection, cells were treated with or without 20 μM cisplatin (CDDP). Representative images are shown. Scale bar, 10 μm. DAPI, 4′,6-diamidino-2-phenylindole. (B) Quantification of at least 100 cells from (A) viewed in five to eight random fields from n = 3 independent experiments. N: nucleus; C: cytoplasm. (C) HEPG2 cells were transiently transfected with HA-FOXK1 WT or HA-FOXK1 S130A plasmid. Twenty-four hours after transfection, cells were treated with or without 20 μM cisplatin (CDDP). Representative images are shown. Scale bar, 10 μm. (D) Quantification of at least 100 cells from (C) viewed in five to eight random fields from n = 3 independent experiments. (E to H) HA-FOXK2 WT (E) or HA-FOXK1 WT (G) plasmid was transfected into HEPG2 control cells or cells depleted CHK2. Twenty-four hours after transfection, cells were treated with or without 20 μM cisplatin (CDDP). Representative images are shown. Scale bar, 10 μm. Quantification of at least 100 cells from (E), (F), (G), or (H) viewed in five to eight random fields from n = 3 independent experiments is shown. (I and J) Western blot analysis was performed to assess endogenous FOXK cellular localization in A549 cells (I) or HEPG2 cells (J) transfected with control or CHK2 shRNA and treated with vehicle or 20 μM cisplatin for 24 hours. (K and L) Western blot analysis was performed to assess endogenous FOXK cellular localization in A549 (K) or HEPG2 (L) cells treated with CHK2 inhibitors and/or 20 μM cisplatin for 24 hours. (M and N) HEK293T cells transfected with HA-FOXK1 WT or HA-FOXK1 S130A (M) or HA-FOXK2 WT or HA-FOXK2 S61A (N) were treated with vehicle or 20 μM cisplatin for 24 hours and purified using anti–HA-agarose beads. The immunoprecipitates were then blotted with the indicated antibodies.
We further checked the sequence around the FOXK phosphorylation sites and found that it fits the predicated consensus sequence of the 14-3-3 binding site (fig. S4F). Given that 14-3-3 binds to phosphorylated proteins and traps them in the cytoplasm (23–25), we speculated that phosphorylated FOXK might interact with the 14-3-3 protein. Coimmunoprecipitation assays were performed to detect the binding between FOXK and the 14-3-3 protein. As shown in Fig. 4 (M and N), WT but not the SA mutants of FOXK proteins bound to 14-3-3γ in response to cisplatin treatment. Moreover, specific small interfering RNA (siRNA)–mediated depletion of 14-3-3γ in cells decreased cisplatin-induced autophagy (fig. S5A). In addition, treatment with R18 (an inhibitor that is able to block 14-3-3 proteins from binding with phosphorylated protein) also decreased autophagy induced by cisplatin (fig. S5B). Together, these findings indicate that FOXK phosphorylation by CHK2 induces FOXK binding to the 14-3-3 protein, which, in turn, traps FOXK in the cytoplasm and induces autophagy following DNA damage.
FOXK phosphorylation is key for DNA damage–induced autophagy
A previous study showed that FOXK proteins negatively regulate autophagy by suppressing transcription of ATGs (14). We next examined whether FOXK phosphorylation by CHK2 is important for the regulation of autophagy via transcriptional control. We reconstituted WT and SA mutants of FOXK (FOXK1 and FOXK2) in cells depleted of endogenous FOXK1 and FOXK2 and examined transcription of ATGs following DNA damage. As shown in Fig. 5A, transcripts of ATGs were markedly increased following cisplatin treatment in cells expressing WT FOXK but not the SA mutants. Furthermore, cells expressing WT FOXK but not the SA mutants showed enhanced LC3-II level and increased red and yellow LC3 puncta following DNA damage (Fig. 5, B to E, and fig. S5C). We also used electron microscopy to examine autophagy in cells expressing WT FOXK and the SA mutants. As shown in Fig. 5F, cells expressing WT FOXK showed vastly increased number of autophagic vacuoles following DNA damage compared with those expressing the SA mutants. Together, these results indicate that FOXK phosphorylation by CHK2 plays an important role in DNA damage–induced autophagy via transcriptional regulation.
Fig. 5. FOXK phosphorylation (FOXK1 S130 and FOXK2 S61) is important for DNA damage–induced autophagy.
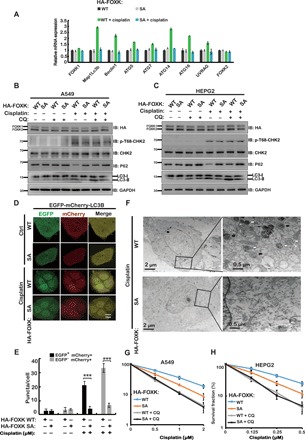
(A) Cells stably expressing FOXK WT or FOXK SA in endogenous FOXK-depleted cells were treated with vehicle or 20 μM cisplatin for 24 hours. The expression of ATGs was analyzed by quantitative PCR (RT-qPCR). (B and C) A549 (B) or HEPG2 (C) cells depleted of endogenous FOXK and stably expressing HA-FOXK WT or HA-FOXK SA were treated with 20 μM cisplatin and/or CQ as indicated. Western blot analysis was performed with the indicated antibodies. (D) HEPG2 cells were depleted of endogenous FOXK and rescued with FOXK WT or FOXK SA. Cells were transfected to stably express EGFP-mCherry-LC3B and treated with 10 μM cisplatin for 24 hours. Green (EGFP) and red (mCherry) fluorescence were analyzed by confocal microscopy (40×). Representative images are shown. Scale bar, 10 μm. (E) The numbers of red and green puncta per cell in (D) were quantified in at least 100 cells in five to eight random areas for each experiment, and data are presented as means ± SD. ***P < 0.001. Statistical analyses were performed using Student’s t test. (F) Cells depleted of endogenous FOXK and stably expressing FOXK WT or FOXK SA were treated with 20 μM cisplatin for 24 hours, harvested, and subjected to transmission electron microscopy analysis. The right panels show enlarged regions of the left panels. The white arrows indicate autophagosomes, and the black arrows indicate autolysosome. (G) Cells from (B) were used to perform colony formation as indicated. (H) Cells from (C) were used to perform colony formation as indicated.
Although autophagy has controversial roles (prosurvival or prodeath) in cancer (26, 27), accumulating evidence suggests that inhibition of autophagy is a promising strategy to overcome drug resistance in cancer therapy (5, 28). Hence, we examined the role of FOXK phosphorylation in chemoresponse. As shown in Fig. 5 (G and H), cells reconstituted with the FOXK SA mutants were hypersensitive to cisplatin treatment. However, combination of autophagy inhibitor CQ with cisplatin efficiently killed cells expressing either the WT or the SA mutant (Fig. 5, G and H). These results suggest that FOXK phosphorylation leads to chemoresistance in cancer cells by modulating autophagy.
Cancer-derived FOXK mutations enhances DNA damage–induced autophagy and drug resistance by affecting FOXK phosphorylation
Recent studies suggest that autophagy is used by tumor cells to escape from radiation- or chemotherapy-induced cell death. Here, we demonstrated that FOXK phosphorylation plays an important role in DNA damage–induced autophagy, which may cause chemoresistance in cancer (5, 29, 30). We were interested in evaluating the clinical relevance of FOXK phosphorylation in cancer. We first checked the FOXK proteins in The Cancer Genome Atlas database (www.cbioportal.org). We found several cancer-derived FOXK mutation sites in the vicinity of the FOXK phosphorylation sites (fig. S6A). We next examined whether these mutations affect FOXK phosphorylation and cytoplasmic localization in response to DNA damage. As shown in Fig. 6A, the FOXK1 N128K mutant showed hyperphosphorylation following DNA damage compared with FOXK1 WT and the I125M mutant. We observed a similar phenomenon for the N59K mutation on FOXK2 (Fig. 6B). However, the FOXK NK (FOXK1 N128K and FOXK2 N59K) mutants did not further promote the interaction between CHK2 and FOXK (fig. S6, B and C). In addition, CHK2 inhibitors (BML-227) abolished phosphorylation of FOXK in both FOXK WT and NK mutation cells (fig. S6, D and E). We speculated that FOXK NK mutation (FOXK1 N128K and FOXK2 N59K) might change the conformation of FOXK so that CHK2 is more readily able to phosphorylate FOXK (FOXK1 at S130 and FOXK2 at S61). Furthermore, FOXK NK mutations facilitated their cytoplasmic localization compared with FOXK WT or FOXK SA mutations upon DNA damage (fig. S6, F to K). We hypothesized that hyperphosphorylation may lead to further activation of autophagy in response to DNA damage. To test this hypothesis, we depleted cells of FOXK1/2, reconstituted these cells with WT FOXK1/2 or NK mutants, and examined DNA damage–induced autophagy. As shown in Fig. 6C and fig. S7 (A to D), compared with cells expressing WT FOXK1/2, cells expressing the NK mutants showed higher LC3-II levels and increased red and yellow LC3 puncta following DNA damage. Together, these results suggest that cancer-associated mutant FOXK is hyperphosphorylated following DNA damage, which, in turn, elevates DNA damage–induced autophagy. We next examined the role of the cancer-derived FOXK mutants in chemoresponse. As shown in Fig. 6D and fig. S7E, compared with WT cells, cells expressing the NK mutants were resistant to cisplatin treatment in vitro. However, combination of CQ and cisplatin treatment overcame the chemoresistance observed in the NK mutants (Fig. 6D and fig. S7E). Next, we further confirmed the role of cancer-derived FOXK mutants in response to chemotherapy in vivo. As shown in Fig. 6 (E and F) and fig. S7F, cancer-derived FOXK mutation did not affect cancer cell growth in vivo. However, cancer cells expressing the FOXK NK mutants were resistant to cisplatin treatment in a xenograft model. On the other hand, CQ treatment overcame chemoresistance of cells expressing the FOXK NK mutants in vivo (Fig. 6 (E and F) and fig. S7F). Together, our results demonstrate that cancer-derived FOXK mutations enhance DNA damage–induced autophagy and chemoresistance in cancers by affecting FOXK phosphorylation. Our study also implies that FOXK mutation status and FOXK phosphorylation status may be prognostic biomarkers of chemoresponse in cancer, and combination chemotherapy and autophagy inhibitor would help overcome chemoresistance in cancers with misregulation of the CHK2-FOXK axis.
Fig. 6. Two novel point mutations on FOXK (FOXK1 N128K and FOXK2 N59K) are important for FOXK function in DNA damage–induced autophagy and response to chemotherapy.
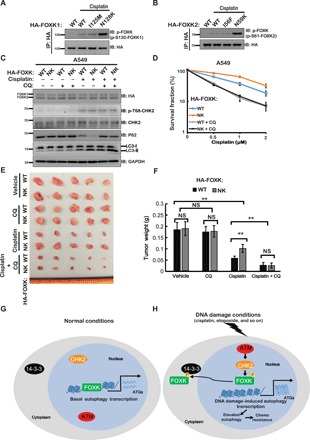
(A and B) HEK293T cells transfected with indicated FOXK1 (A) or FOXK2 (B) constructs were treated with vehicle or 20 μM cisplatin for 24 hours and purified using anti–HA-agarose beads. The immunoprecipitates were then blotted with the indicated antibodies. (C) A549 cells depleted of endogenous FOXK and stably expressing HA-FOXK WT or HA-FOXK NK were treated with 20 μM cisplatin and/or CQ as indicated. Western blot was performed with the indicated antibodies. (D) Cells from (C) were used to perform colony formation as indicated. (E and F) Tumor growth assay for FOXK WT or FOXK NK cells treated with vehicle, CQ, cisplatin, or cisplatin together with CQ. Tumor images were acquired as shown in (E), and weights (F) were measured. n = 5; data points in (F) represent mean tumor weight ± SD. **P < 0.01. Statistical analyses were performed using Student’s t test. (G) Under normal conditions, FOXK localizes to the nucleus as a transcriptional repressor to control the expression of ATGs. (H) Upon DNA damage conditions (caused by agents such as cisplatin and etoposide), ATM activates CHK2, which further directly phosphorylates FOXK (FOXK1 at S130 and FOXK2 at S61) and creates a 14-3-3γ binding site, which, in turn, traps FOXK proteins in the cytoplasm. Because FOXK functions as the transcriptional suppressor of ATGs, DNA damage–mediated FOXKs’ cytoplasmic trapping induces expression of ATGs and facilitates autophagy in response to DNA damage. Moreover, DNA damage–induced autophagy results in chemoresistance.
DISCUSSION
DDR and autophagy are two important processes for maintaining cellular and organismal homeostasis. Recent studies suggest that autophagy is activated by DNA damage, which, in turn, regulates DNA repair, senescence, and chemoresponse in cancer (1, 4–6). Acute regulation of autophagy in response to DNA damage by protein posttranslational modification is well defined (7, 31). For example, DNA damage initiates autophagy through ATM-mediated phosphorylation of AMPK, which activates TSC2 to inhibit mTORC1 (mTOR complex 1), which, in turn, triggers activation of the ULK1/ATG13/FIP200 kinase complex (32). ATM also phosphorylates PTEN to promote its nuclear localization and induces autophagy upon DNA damage (11). We found DNA damage still induced autophagy in AMPK−/− or PTEN-deficient cells (data not shown), suggesting there may be other mechanisms contributing to DNA damage–induced autophagy. We observed that DNA damage markedly up-regulated the transcription of ATGs, suggesting that, besides posttranslational modification, transcriptional control may be another important mechanism for DNA damage–mediated autophagy. A previous study showed that the ATR-CHK1 axis responds to ultraviolet/methyl methanesulfonate (inducing DNA single-strand breaks)–induced autophagy by regulating the RhoB-TSC2-mTOC1 axis (33). However, we demonstrated here that the ATM-CHK2, but not the ATR-CHK1, axis is responsible for the cisplatin- or etoposide-induced autophagy.
In this study, we revealed the molecular mechanism of DNA damage–induced transcriptional control of autophagy. We demonstrated that ATM and CHK2 are the key factors in this process. By performing protein purification, we identified the new CHK2-binding proteins FOXK proteins, which have been previously reported to negatively regulate ATG transcription. We further demonstrated that following DNA damage, ATM phosphorylates CHK2 and promotes its binding to the FHA domain of FOXK proteins. CHK2, in turn, phosphorylates FOXK and creates a 14-3-3 binding site to trap FOXK proteins in the cytoplasm. Last, we found that ATM/CHK2-mediated FOXK nuclear export is a novel mechanism that activates transcription of ATGs following DNA damage (Fig. 6, G and H). Because a previous paper showed that FOXK played an important role in starvation-induced autophagy (14), we sought to verify whether starvation also activates the ATM-CHK2-FOXK pathway. We found that both starvation and cisplatin treatment induced autophagy, but only cisplatin activated the ATM-CHK2-FOXK pathway (fig. S8, A and B). Another study showed that the Rad53 (CHK2 ortholog in yeast)–Rph1/KDM4 axis was involved in the regulation of genotoxin-induced autophagy in yeast (34). However, in this study, knockdown of KDM4a could not rescue cisplatin-induced autophagy in CHK2-depleted cells, suggesting that KDM4a may not be involved in CHK2-mediated autophagy activation in human cells (fig. S8C).
DNA damage–induced autophagy is one the mechanisms used by tumor cells to resist radiochemotherapy. In addition, the combination of autophagy inhibitor and genotoxic therapy has been proven to overcome radio- and chemoresistance both in in vitro and in vivo experimental mouse models (5, 29, 30). We demonstrated that DNA damage–induced FOXK phosphorylation activates autophagy, which, in turn, leads to drug resistance in cancer cells. Compared with WT FOXK, the FOXK phosphorylation–inactive mutant continually represses transcription of ATGs, in turn sensitizing cancer cells to chemotherapy. We characterized some cancer-derived FOXK mutations and showed that they are hyperphosphorylated, resulting in hyperactive autophagy in cancer cells and chemoresistance. Combination treatment with CQ (autophagy inhibitor) and cisplatin is able to overcome the chemoresistance in the cells expressing cancer-derived FOXK mutants.
Overall, our study identifies a novel mechanism linking DNA damage and autophagy that sheds light on the potential therapeutic targeting of the CHK2-FOXK axis in dysregulated autophagy-related disease. Moreover, our study implies that FOXK phosphorylation status may be able to serve as a biomarker for combination chemotherapy and autophagy inhibitor treatment in cancer.
MATERIALS AND METHODS
Cell culture, plasmids, reagents, and antibodies
Human embryonic kidney (HEK) 293T cells and the human cancer cell lines A549 and Hepg2 were purchased from the American Type Culture Collection. The medical genome facility of Mayo Clinic in Rochester, Minnesota, confirmed the identities of all the cell lines. Cells were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and were transfected with polyetherimide or Lipofectamine 2000 (Thermo Fisher Scientific). For inducing DNA damage, cells were treated with 10 or 20 μM cisplatin or etoposide for 24 hours. Flag–hemagglutinin (HA)–FOXK2 and Flag-HA-FOXK1 were subcloned using plvx3 vector (Clontech). FOXK1 S130A, I125M, and N128K and FOXK2 S61A, I56M, and N59k constructs were generated by a two-step mutation method (35, 36). pBABE-puro mCherry-EGFP-LC3B (#22418) plasmids were purchased from Addgene.
The anti–glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody was purchased from Proteintech (60004-1-lg). Anti-FOXK1 antibody (sc-134550) was purchased from Santa Cruz Biotechnology, and anti-FOXK2 (A301-730A) antibody was purchased from Bethyl Laboratories. Anti–phospho-FOXK (p-S130-FOXK1 and p-S61-FOXK2) antibody was generated by GenScript. Anti-CHK2 (05-649) antibody was purchased from Millipore. Anti-Flag (F1804) and anti-HA (F9658) antibodies, R18 (SML0108-1MG), and cisplatin (P4394-25MG) were purchased from Sigma-Aldrich. p62/Sqstm1 (MBL PM045) and H3 (Proteintech, no. 17168-1-AP) antibodies were used in this study. Anti-KDM4a (ab24545) antibody was purchased from Sigma-Aldrich. Anti–p-S1981-ATM (5883T), anti-ATM (2873T), anti-CHK1 (2360S), anti–p-T68-CHK2 (2197T), and anti-LC3B (2775s) antibodies were purchased from Cell Signaling Technology.
Immunofluorescence staining
Immunofluorescence staining was performed per standard procedures. Briefly, Hepg2 cells were seeded and transfected with HA-FOXK WT (HA-FOXK2 and HA-FOXK1) and HA-FOXK mutants (HA-FOXK1 S130A, HA-FOXK1 N128K, HA-FOXK2 S61A, and HA-FOXK2 N59K) in six-well plates containing coverslips. After treatment with appropriate agents, cells were fixed using 4% paraformaldehyde for 20 min at room temperature, washed three times in 1× phosphate-buffered saline (PBS), and then extracted with 0.5% Triton X-100 solution for 5 min. After blocking with PBS containing 1% goat serum albumin, cells were incubated with the indicated primary antibodies for 1 hour at 37°C. After that, cells were washed three times using 1× PBS and incubated with Alexa Fluor 488– or Alexa Fluor 594–conjugated second primary antibodies (Thermo Fisher Scientific) for 1 hour at room temperature. Last, cells were counterstained with 4′,6-diamidino-2-phenylindole (100 ng/ml) for 5 min to visualize nuclear DNA. The coverslips were mounted onto glass slides with antifade solution and visualized under a Leica Eclipse E800 fluorescence microscope with a 40× objective lens (numerical aperture, 1.30).
RNA extraction, reverse transcription, and real-time PCR
RNA samples were extracted with RNAiso Plus reagent (Takara), and reverse transcription was performed according to the manufacturer’s instruction. Real-time polymerase chain reaction (PCR) was performed using Power SYBR Green PCR Master Mix (Takara). The 2−ΔΔCt method was used for quantification of gene expression. GAPDH was used as control.
Lentivirus packaging and infection
FOXK1 short hairpin RNAs (shRNAs) (SHCLNV-NM_001037165), FOXK2 shRNAs (SHCLNV-NM_004514), and CHK2 shRNAs (SHCLND-NM_007194) were purchased from Sigma-Aldrich. Lentivirus FOXK and CHK2 shRNAs were made according to the protocol shown on the Sigma website (www.sigmaaldrich.com/): FOXK1 shRNA #1: GCTGCTATGAAGACAGGATTA; FOXK1 shRNA #2: GTCGCTCTATCACAAAGAAGA; FOXK2 shRNA #1: CCCGAGCACAAACATCAAGAT; FOXK2 shRNA #2: AGCGGAGACATGTGGAATTAG; CHK2 shRNA #1: GAACAGATAAATACCGAACAT; CHK2 shRNA #2: ACTCCGTGGTTTGAACACGAA.
Briefly, plko.1 lentiviral or plvx3 constructs and packaging plasmids (pMD2G and pSPAX2) were used to package virus into HEK293T cells. Viral supernatant was collected two times (24 and 48 hour) after the cotransfection of lentiviral vectors and packaging plasmids (pMD2G and pSPAX2). Hepg2 or 293T cells were infected with viral supernatant with the addition of polybrene (8 μg/ml; Sigma-Aldrich), and stable cells were selected with media containing puromycin (2 μg/ml; Sigma-Aldrich). The stable cell lines were determined by immunoblotting.
Coimmunoprecipitation and Western blotting
For transient transfection and coimmunoprecipitation assays, constructs encoding Flag-tagged CHK2 or HA-tagged FOXK constructs were transiently cotransfected into HEK293T cells. The transfected cells were lysed with NETN buffer [20 mM tris-HCl (pH 8.0), 100 mM NaCl, 1 mM EDTA, and 0.5% NP-40] containing 1× protease inhibitors on ice for 25 min. After removal of cell debris by centrifugation at 12,000 rpm for 10 min, the soluble fractions were collected and incubated with 3× Flag beads for 4 hours at 4°C. Beads were washed three times with NETN buffer, boiled in 1× SDS loading buffer for 5 min, and resolved on SDS–polyacrylamide gel electrophoresis (PAGE). For endogenous immunoprecipitation assay, the cells were solubilized in NETN lysis buffer supplemented with protease and phosphatase inhibitors following standard procedure. After removal of cell debris by centrifugation, the soluble fractions were collected. One milligram of the whole-cell extract was then incubated with 25 μl of a 1:1 slurry of protein A–Sepharose coupled with 2 μg of the indicated antibodies for 2 hours at 4°C. The Sepharose beads were washed three times with NETN buffer, boiled in 1× SDS loading buffer, and resolved on SDS-PAGE. Membranes were blocked in 5% milk in TBST (tris-buffered saline with Tween 20) buffer and then probed with antibodies as indicated.
Phosphatase assay
FOXK immunoprecipitates were washed with 1× NETN buffer five times and incubated in a total of 50 μl containing 400-U λ protein phosphatase (New England BioLabs) and 1× mM MnCl2 for 1 hour at 30°C. Immunoprecipitates were then washed three times with 1× NETN to remove nonbinding proteins.
In vitro phosphorylation assay
Bacterial expression constructs (pGEX-4T-2) containing the indicated genes were transformed into Escherichia coli DH5α. Cells were induced to express protein using 0.5 mM IPTG (isopropyl-β-d-thiogalactopyranoside) at 18°C with 180 rpm rotation overnight. Cells were resuspended in PBS containing 0.5% Triton X-100 and 2 mM β-mercaptoethanol, followed by ultrasonication. The proteins were purified using glutathione beads according to the manufacturer’s protocol (Amersham Biosciences). Purified bacterially expressed GST-FOXK proteins were used to perform CHK2 kinase assay under cell-free conditions. Flag-CHK2 proteins were prepared from the HEK293T cells, purified using Flag beads, and subsequently eluded using 3× Flag peptide. For the in vitro phosphorylation assay, GST-FOXK WT proteins and GST-FOXK SA mutant proteins were incubated with Flag-CHK2 in kinase buffer [10 mM Hepes (pH 7.5), 10 mM MgCl2, 10 mM MnCl2, and 1 mM dithiothreitol, which was added immediately before use] at 30°C for 30 min. Next, 5× SDS loading buffer was added to the reaction and boiled for 5 min at 100°C. Samples were separated by SDS-PAGE and immunoblotted with the indicated antibodies.
GST pulldown assay
As mentioned above, GST fusion proteins were prepared following standard protocol. For in vitro binding assays, various GST fusion proteins bound to the glutathione Sepharose beads were incubated with cell lysates, which contained Flag-CHK2. After washing, the bound proteins were separated by SDS-PAGE and immunoblotted with the indicated antibodies.
Colony formation assay
HEPG2 or A549 cells (500 to 2000) were seeded in triplicate in each well of six-well plates. For chemotherapy sensitivity assay, after 1 day, cells were treated with cisplatin at the indicated doses and left for 10 to 14 days in the incubator to allow colony formation. Colonies were stained with Giemsa and quantified. Results were normalized to plating efficiencies.
Tumor xenograft
Stably expressing HA-FOXK WT or HA-FOXK NK A549 cells, which had been depleted endogenous FOXK using specific shRNAs and reconstituted with siRNA-resistant HA-FOXK WT or HA-FOXK NK, were injected subcutaneously and bilaterally into the flanks of 6-week-old female athymic nude Ncr nu/nu (National Cancer Institute/National Institutes of Health) mice using 18-gauge needles. Each mouse received two injections of a 200-μl mixture of 2 × 106 cells with 30% growth factor reduced Matrigel (BD Biosciences). Mice bearing tumors of 150 to 200 mm3 were randomly assigned into four groups: vehicle control group (saline), CQ group (60 mg kg−1), cisplatin group (3 mg kg−1), and cisplatin (3 mg kg−1), together with CQ group (60 mg kg−1). The treated mice were intraperitoneally injected two times per week. Tumor volume was subsequently measured every 3 days using calipers, and tumor volume was calculated using the formula L × W2. Mice were euthanized for tumor dissection on day 28 after the start of treatment. Data were analyzed using Student’s t test. A P value <0.5 was considered significant. Mice were subjected to euthanasia if they displayed pain or distress, such as lethargy, lying down, not eating or drinking, weight loss greater than 10% body weight, or difficulty breathing. According to the blinding procedures, two people as a group performed all the mice experiments. One person performed the experiments, and another one totally blinded to the experiment group measured the tumor volume and weight and analyzed the data.
Statistics
For cell survival and xenograft assay, data are represented as means ± SEM of three independent experiments. For the animal study, data are represented as means ± SEM of five mice. Statistical analyses were performed in GraphPad Prism with the Student’s t test or χ2 test. Statistical significance is represented in the figures by *P < 0.05, **P < 0.01, ***P < 0.001, and NS (not significant).
Supplementary Material
Acknowledgments
Funding: This work was supported by the National Natural Science Foundation of China (91749115 to J.Y., 81572770 to K.L., 81872298 to Y.L., and 81802754 to L.L.), the Natural Science Foundation of Jiangxi Province (20181ACB20021 to J.Y.), and the China Scholarship Council (201706260203 to Y.P.C.). Author contributions: J.Y., K.L., and Yuping Chen conceived the project and designed the studies. Yuping Chen and J.W. performed the experiments. Yuping Chen wrote the paper. J.Y., Z.Lo., and S.N. revised the manuscript. G.L., G.Ge., F.Z., P.Y., C.W., Y.L., L.L., W.K., Q.Z., J.H., J.L., C.Z., G.Gu., M.D., X.T., X.G., Z.Li., and Yihan Chen provided the constructional suggestions and help for this paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/1/eaax5819/DC1
Fig. S1. Chk2 is required for DNA damage–induced autophagy.
Fig. S2. CHK2 interacts with FOXK.
Fig. S3. CHK2 regulates autophagy induced by DNA damage via FOXK.
Fig. S4. CHK2 phosphorylates FOXK (FOXK1 S130 and FOXK2 S61) in response to DNA damage and then promotes their cytoplasmic translocation.
Fig. S5. FOXK phosphorylation (FOXK1 at S130 and FOXK2 at S61) is essential for DNA damage–induced autophagy.
Fig. S6. Two novel patient-derived point mutations on FOXK (FOXK1 N128K and FOXK2 N59K) facilitate FOXK cytoplasmic translocation and autophagy and confers chemoresistance in response to DNA damage.
Fig. S7. Two novel point mutations on FOXK (FOXK1 N128K and FOXK2 N59K) are important for FOXK function in response to chemotherapy.
Fig. S8. DNA damage and starvation induce autophagy.
REFERENCES AND NOTES
- 1.Abedin M., Wang D., McDonnell M., Lehmann U., Kelekar A., Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ. 14, 500–510 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Notte A., Leclere L., Michiels C., Autophagy as a mediator of chemotherapy-induced cell death in cancer. Biochem. Pharmacol. 82, 427–434 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Li W.-w., Li J., Bao J.-k., Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 69, 1125–1136 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muñoz-Gámez J. A., Rodríguez-Vargas J. M., Quiles-Pérez R., Aguilar-Quesada R., Martín-Oliva D., de Murcia G., de Murcia J. M., Almendros A., de Almodóvar M. R., Oliver F. J., PARP-1 is involved in autophagy induced by DNA damage. Autophagy 5, 61–74 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Sui X., Chen R., Wang Z., Huang Z., Kong N., Zhang M., Han W., Lou F., Yang J., Zhang Q., Wang X., He C., Pan H., Autophagy and chemotherapy resistance: A promising therapeutic target for cancer treatment. Cell Death Dis. 4, e838 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robert T., Vanoli F., Chiolo I., Shubassi G., Bernstein K. A., Rothstein R., Botrugno O. A., Parazzoli D., Oldani A., Minucci S., Foiani M., HDACs link the DNA damage response, processing of double-strand breaks and autophagy. Nature 471, 74–79 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wani W. Y., Boyer-Guittaut M., Dodson M., Chatham J., Darley-Usmar V., Zhang J., Regulation of autophagy by protein post-translational modification. Lab. Invest. 95, 14–25 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McEwan D. G., Dikic I., The three musketeers of autophagy: Phosphorylation, ubiquitylation and acetylation. Trends Cell Biol. 21, 195–201 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.F. Nazio, E. Maiani, F. Cecconi, in Ubiquitination Governing DNA Repair-Implications in Health and Disease (IntechOpen, 2017).
- 10.Kim J., Kundu M., Viollet B., Guan K.-L., AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 13, 132– 141 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen J.-H., Zhang P., Chen W.-D., Li D.-D., Wu X.-Q., Deng R., Jiao L., Li X., Ji J., Feng G.-K., Zeng Y.-X., Jiang J.-W., Zhu X.-F., ATM-mediated PTEN phosphorylation promotes PTEN nuclear translocation and autophagy in response to DNA-damaging agents in cancer cells. Autophagy 11, 239–252 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hannenhalli S., Kaestner K. H., The evolution of Fox genes and their role in development and disease. Nat. Rev. Genet. 10, 233–240 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Katoh M., Katoh M., Human FOX gene family (Review). Int. J. Oncol. 25, 1495–1500 (2004). [PubMed] [Google Scholar]
- 14.Bowman C. J., Ayer D. E., Dynlacht B. D., Foxk proteins repress the initiation of starvation-induced atrophy and autophagy programs. Nat. Cell Biol. 16, 1202–1214 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodriguez-Rocha H., Garcia-Garcia A., Panayiotidis M. I., Franco R., DNA damage and autophagy. Mutat. Res. 711, 158–166 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Che L., Yang X., Ge C., el-Amouri S. S., Wang Q.-E., Pan D., Herzog T. J., Du C., Loss of BRUCE reduces cellular energy level and induces autophagy by driving activation of the AMPK-ULK1 autophagic initiating axis. PLOS ONE 14, e0216553 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mauthe M., Orhon I., Rocchi C., Zhou X., Luhr M., Hijlkema K.-J., Coppes R. P., Engedal N., Mari M., Reggiori F., Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy 14, 1435–1455 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin J.-F., Lin Y. C., Tsai T. F., Chen H. E., Chou K. Y., Hwang T. I. S., Cisplatin induces protective autophagy through activation of BECN1 in human bladder cancer cells. Drug Des. Devel. Ther. 11, 1517–1533 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Leisching G., Loos B., Botha M., Engelbrecht A.-M., A nontoxic concentration of cisplatin induces autophagy in cervical cancer: Selective cancer cell death with autophagy inhibition as an adjuvant treatment. Int. J. Gynecol. Cancer 25, 380–388 (2015). [DOI] [PubMed] [Google Scholar]
- 20.Xie B.-S., Zhao H.-C., Yao S.-K., Zhuo D.-X., Jin B., Lv D.-C., Wu C.-L., Ma D.-L., Gao C., Shu X.-M., Ai Z.-L., Autophagy inhibition enhances etoposide-induced cell death in human hepatoma G2 cells. Int. J. Mol. Med. 27, 599–606 (2011). [DOI] [PubMed] [Google Scholar]
- 21.Seo G.-J., Kim S. E., Lee Y. M., Lee J. W., Lee J. R., Hahn M. J., Kim S. T., Determination of substrate specificity and putative substrates of Chk2 kinase. Biochem. Biophys. Res. Commun. 304, 339–343 (2003). [DOI] [PubMed] [Google Scholar]
- 22.Zannini L., Delia D., Buscemi G., CHK2 kinase in the DNA damage response and beyond. J. Mol. Cell Biol. 6, 442–457 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu P., Li S., Gan L., Kao T. P., Huang H., A transcription-independent function of FOXO1 in inhibition of androgen-independent activation of the androgen receptor in prostate cancer cells. Cancer Res. 68, 10290–10299 (2008). [DOI] [PubMed] [Google Scholar]
- 24.Tzivion G., Dobson M., Ramakrishnan G., FoxO transcription factors; Regulation by AKT and 14-3-3 proteins. Biochim. Biophys. Acta 1813, 1938–1945 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Fu H., Subramanian R. R., Masters S. C., 14-3-3 proteins: Structure, function, and regulation. Annu. Rev. Pharmacol. Toxicol. 40, 617–647 (2000). [DOI] [PubMed] [Google Scholar]
- 26.Kondo Y., Kanzawa T., Sawaya R., Kondo S., The role of autophagy in cancer development and response to therapy. Nat. Rev. Cancer 5, 726–734 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Inguscio V., Panzarini E., Dini L., Autophagy contributes to the death/survival balance in cancer photodynamic therapy. Cells 1, 464–491 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumar A., Singh U. K., Chaudhary A., Targeting autophagy to overcome drug resistance in cancer therapy. Future Med. Chem. 7, 1535–1542 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Chude C., Amaravadi R., Targeting autophagy in cancer: Update on clinical trials and novel inhibitors. Int. J. Mol. Sci. 18, E1279 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pagotto A., Pilotto G., Mazzoldi E. L., Nicoletto M. O., Frezzini S., Pastò A., Amadori A., Autophagy inhibition reduces chemoresistance and tumorigenic potential of human ovarian cancer stem cells. Cell Death Dis. 8, e2943 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Xie Y., Kang R., Sun X., Zhong M., Huang J., Klionsky D. J., Tang D., Posttranslational modification of autophagy-related proteins in macroautophagy. Autophagy 11, 28–45 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tripathi D. N., Chowdhury R., Trudel L. J., Tee A. R., Slack R. S., Walker C. L., Wogan G. N., Reactive nitrogen species regulate autophagy through ATM-AMPK-TSC2–mediated suppression of mTORC1. Proc. Natl. Acad. Sci. 110, E2950–E2957 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu M., Zeng T., Zhang X., Liu C., Wu Z., Yao L., Xie C., Xia H., Lin Q., Xie L., Zhou D., Deng X., Chan H.-L., Zhao T.-J., Wang H.-R., ATR/Chk1 signaling induces autophagy through sumoylated RhoB-mediated lysosomal translocation of TSC2 after DNA damage. Nat. Commun. 9, 4139 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eapen V. V., Waterman D. P., Bernard A., Schiffmann N., Sayas E., Kamber R., Lemos B., Memisoglu G., Ang J., Mazella A., Chuartzman S. G., Loewith R. J., Schuldiner M., Denic V., Klionsky D. J., Haber J. E., A pathway of targeted autophagy is induced by DNA damage in budding yeast. Proc. Natl. Acad. Sci. U.S.A. 114, E1158–E1167 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Horton R. M., Cai Z., Ho S. N., Pease L. R., Gene splicing by overlap extension: Tailor-made genes using the polymerase chain reaction. Biotechniques 8, 528–535 (1990). [PubMed] [Google Scholar]
- 36.Li H., Havens W. M., Nibert M. L., Ghabrial S. A., RNA sequence determinants of a coupled termination-reinitiation strategy for downstream open reading frame translation in Helminthosporium victoriae virus 190S and other victoriviruses (family Totiviridae). J. Virol. 85, 7343–7352 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/1/eaax5819/DC1
Fig. S1. Chk2 is required for DNA damage–induced autophagy.
Fig. S2. CHK2 interacts with FOXK.
Fig. S3. CHK2 regulates autophagy induced by DNA damage via FOXK.
Fig. S4. CHK2 phosphorylates FOXK (FOXK1 S130 and FOXK2 S61) in response to DNA damage and then promotes their cytoplasmic translocation.
Fig. S5. FOXK phosphorylation (FOXK1 at S130 and FOXK2 at S61) is essential for DNA damage–induced autophagy.
Fig. S6. Two novel patient-derived point mutations on FOXK (FOXK1 N128K and FOXK2 N59K) facilitate FOXK cytoplasmic translocation and autophagy and confers chemoresistance in response to DNA damage.
Fig. S7. Two novel point mutations on FOXK (FOXK1 N128K and FOXK2 N59K) are important for FOXK function in response to chemotherapy.
Fig. S8. DNA damage and starvation induce autophagy.