

Structural studies of dARC reveal a Gag exapted from a transposable element and an ancient relationship to orthoretroviruses.
Abstract
The tetrapod neuronal protein ARC and its Drosophila melanogaster homolog, dARC1, have important but differing roles in neuronal development. Both are thought to originate through exaptation of ancient Ty3/Gypsy retrotransposon Gag, with their novel function relying on an original capacity for self-assembly and encapsidation of nucleic acids. Here, we present the crystal structure of dARC1 CA and examine the relationship between dARC1, mammalian ARC, and the CA protein of circulating retroviruses. We show that while the overall architecture is highly related to that of orthoretroviral and spumaretroviral CA, there are substantial deviations in both amino- and carboxyl-terminal domains, potentially affecting recruitment of partner proteins and particle assembly. The degree of sequence and structural divergence suggests that Ty3/Gypsy Gag has been exapted on two separate occasions and that, although mammalian ARC and dARC1 share functional similarity, the structures have undergone different adaptations after appropriation into the tetrapod and insect genomes.
INTRODUCTION
Activity-regulated cytoskeleton-associated protein (ARC) is an immediate early gene product induced in response to high levels of synaptic activity and is directed to neuronal synapses through signaling sequences in its 3′ untranslated region (1). Mammalian ARC (mam-ARC) is essential for neuronal plasticity and is involved in memory (2) acting as a regulator of AMPA receptors (AMPARs) (3, 4). ARC has also been implicated in neurological disorders, including Alzheimer’s disease (5), fragile X syndrome (6), and schizophrenia (7, 8). In Drosophila melanogaster, two homologs of mam-ARC are expressed: dARC1 and dARC2 (9). dARC1 is present at neuromuscular junctions and, along with its mRNA, has been implicated in regulating the behavioral starvation response but is not involved in synaptic plasticity (10). Therefore, comparing the structural and functional properties of mam-ARC and dARC1 might lead to a better understanding of cognition and memory consolidation.
The ARC gene is thought to be derived from the gag gene of a Ty3/Gypsy retrotransposon (11) that, subsequent to genomic insertion, has been repurposed to perform an advantageous function to the host (12). This connection between ARC and retrotransposons was made when sequence alignments revealed that the ARC proteins shared sequence similarity with the Gag protein of retroviruses or retrotransposons (11). These data also suggested that ARC is evolutionarily related to the Ty3/Gypsy family of retrotransposons. Further evidence came from crystal structures of two α-helical domains from Rattus norvegicus ARC (rARC) (13), which revealed that rARC N- and C-terminal capsid (CA) domains were structurally homologous to the N- and C-terminal CA domains of both Orthoretrovirinae (13) and Spumaretrovirinae (14). Further phylogenetic analysis revealed that, despite mam-ARC and dARC1 seemingly providing related functions in the host, dARC1 and the tetrapod ARCs most likely arose from separate lineages of Ty3/Gypsy, because dARC1 clustered with insect Ty3/Gypsy retrotransposons and tetrapod ARCs clustered with fish Ty3/Gypsy retrotransposons (12).
The relevance of ARC’s retrotransposon origin to its function in synaptic plasticity was not immediately obvious until the recent observation that mam-ARC and dARC1 can self-assemble into particles and package RNA for potential transfer between cells (9, 12), similarly to retrotransposons and retroviruses (15, 16). In D. melanogaster, it is proposed that dARC1 expressed at neuromuscular junction presynaptic boutons assembles into particles that encapsidate dARC1 mRNA. Loaded particles might then be packaged and released as extracellular vesicles for intercellular transfer to the postsynapse, where mRNA release and translation can take place (9, 12). Similarly, mam-ARC can also encapsidate ARC mRNA into particles, allowing transfer from donor to recipient neurons, where ARC mRNA can be translated (12).
Because both dARC1 and mam-ARC are able to form CA-like particles (9, 12), it seems likely that they share a degree of structural similarity. To date, crystal structures of the individual domains from rARC have been determined (13), along with the solution nuclear magnetic resonance (NMR) structure of the rARC CA (17). Here, we report two crystal structures of the entire CA region of dARC1 at 1.7 and 2.3 Å and consider these structures in comparison to those of rARC and retroviral CA. dARC1 comprises two α-helical domains with a fold related to that observed in the CA-NTD and CA-CTD of orthoretroviral and spumaretroviral CA. However, we observe significant divergence in the NTD of dARC1, where an extended hydrophobic strand that packs against α1 and α3 of the core fold replaces the N-terminal β hairpin and helix α1 found in orthoretroviral CAs. In the rARC structure, this hydrophobic strand is replaced by peptides from the binding partners Ca2+/calmodulin-dependent protein kinase 2A (CamK2A) and transmembrane AMPAR regulatory protein γ2 (TARPγ2) and may represent a functional adaptation for the recruitment of partner proteins. We also show that dARC1 uses the same CTD-CTD interface required for the assembly of retroviral CA into mature particles and propose that this obligate dimer represents a building block for dARC1 particle assembly. Further examination of the relationship between dARC1, mam-ARC, and Gag from Ty retrotransposon families reveals that, although dARC1 and mam-ARC are functional orthologs, the structural divergence in dARC1 and mam-ARC CA domains is consistent with the notion of Ty3/Gypsy Gag exaptation on two separate occasions. We suggest that they may have undergone different adaptations after appropriation into the tetrapod and insect genomes.
RESULTS
Structures of dARC1 CA
We determined the crystal structure of the CA domain region of dARC1, residues S39 to N205 (dARC1 CA), using single-wavelength anomalous diffraction (SAD) and crystals of Se-Met substituted protein. The structure was determined in both an orthorhombic and a hexagonal crystal form. The orthorhombic crystals diffracted to higher resolution, allowing the structure to be refined to a final resolution of 1.7 Å with an R factor of 18.1% and a free R factor of 21.3%. Details of data collection, phasing, and refinement are presented in table S1. The asymmetric unit (ASU) contains two chains, each containing an α-helical N-terminal (CA-NTD) and C-terminal domain (CA-CTD) (Fig. 1A). The chains are arranged in a dimer with a distinct U-shape reminiscent of a glacial trough (Fig. 1A, right). The CA-CTDs form the base of the trough and pack together to form a homodimer interface, and the CA-NTDs form the sides of the trough and are separated by ~45 Å. Inspection of each domain reveals that the CA-NTD is made up from an extended N-terminal strand and a four-helix core (α1 to α4), and the CA-CTD comprises a further five α-helix bundle (α5 to α9) (Fig. 1B, i and ii). The tertiary folds of each domain are particularly similar and can be superimposed with a root mean square deviation (RMSD) of 2.2 Å over 49 Cα atoms (Fig. 1C). Moreover, it can be seen that the dARC1 N-terminal β strand is topologically equivalent to α5 in the CTD, while NTD α1 to α4 are equivalent to CTD α6 to α9. This strong similarity of dARC1 CA domains provides further evidence for the notion that tandem domains of CA arose as the result of a gene duplication event (14). The hexagonal crystal form was independently solved and refined to a resolution of 2.3 Å and reveals an almost identical dimeric ASU that aligns with an RMSD of only 0.247 Å over 133 Cα pairs (fig. S1, A to C). Both structures appear especially stable around the CTD-mediated dimeric interface and, when aligned through their CTDs, show only small differences in the positioning of NTDs with respect to the CTDs (fig. S1D).
Fig. 1. Crystal structure of the dARC1 CA domain.
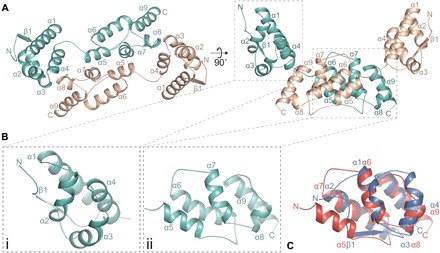
(A) Cartoon representation of the dARC1 CA dimer. The N-terminal extended β strand and α helices are numbered sequentially from the N terminus to the C terminus. Monomer A is colored cyan, and monomer B is colored wheat. The right-hand panel is a view at 90° relative to the left-hand panel. (B) Close-up cartoon representations of dARC1 CA-NTD (left) and dARC CA-CTD (right) showing the helical topology of each domain. (C) Three-dimensional (3D) Cα structural alignment of dARC1 CA-NTD (blue cartoon) with dARC1 CA-CTD (red cartoon), with secondary structure elements labeled.
dARC1 CA dimer interface
The dARC1 CA-CTD monomer consists of a five-helix core comprising α5 (residues A125 to Q134), α6 (residues I143 to Q156), α7 (residues E164 to L171), α8 (residues I177 to H182), and α9 (residues F191 to N204). The dimer interface is located between CA-CTDs, where the outer surfaces of α5 and α7 pack against α5′ and α7′ of the opposing monomer (Fig. 2A). The homodimer interface encompasses 768 Å2 of the buried surface and is defined by numerous intermolecular interactions. The interface is largely hydrophobic with contributions from side-chain packing of the Y126, Y129, M130, F133, L170, F172, and L174 hydrophobic and aromatic residues that are exposed on α5 and α7 and form a continuous apolar network with Y129 and F133 at its center (Fig. 2A, left). This is apparent in the analysis of the dARC1 CA surface hydrophobicity profile, which reveals a distinct apolar patch that locates to the center of the CA-CTD homodimer interface (fig. S2A). In addition, at the periphery of the interface, there is also a salt bridge between R161 on the α6-α7 connecting loop with D169 at the C terminus of α7, providing further stabilization (Fig. 2A, right). The number and hydrophobic nature of interactions within the homodimer interface suggest that the dimer constitutes a relatively stable or obligate structure.
Fig. 2. dARC1 CA dimer interface and solution conformation.
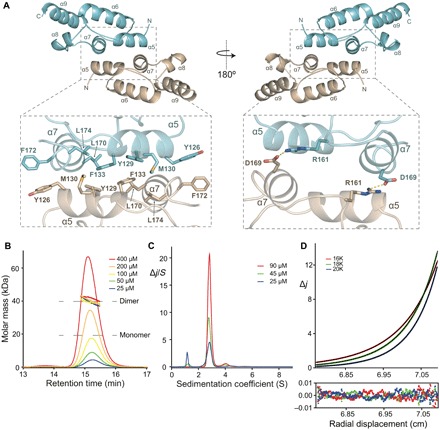
(A) Cartoon representation of dARC1 CA-CTD dimer. α Helices are numbered sequentially from the N terminus to the C terminus. Monomer A is colored cyan, and monomer B is colored wheat. The right-hand panel is a view at 180° relative to the left-hand panel. Insets: Close-up views of molecular details of interactions at the dARC1 dimer interface. Residues that make interactions are shown in stick representation colored by atom type. Salt-bridge interactions between R161 and D169 are shown as dashed lines. (B) SEC-MALLS analysis of dARC1 CA. The sample loading concentrations were 400 μM (8 mg/ml) (red), 200 μM (4 mg/ml) (orange), 100 μM (2 mg/ml) (yellow), 50 μM (1 mg/ml) (green), and 25 μM (0.5 mg/ml) (blue). The differential refractive index is plotted against column retention time, and the molar mass, determined at 1-s intervals throughout the elution of each peak, is plotted as points. The dARC1 CA monomer and dimer molecular mass are indicated with the gray dashed lines. (C) C(S) distributions derived from sedimentation velocity data recorded from dARC1 CA at 25 μM (blue), 50 μM (green), and 100 μM (red). The curves represent the distribution of the sedimentation coefficients that best fit the sedimentation data (ƒ/ƒ0 = 1.41). (D) Multispeed sedimentation equilibrium profile determined from interference data collected on dARC1 CA at 70 μM. Data were recorded at the three speeds indicated. The solid lines represent the global best fit to the data using a single-species model (Mw = 38.9 ± 1 kDa). The lower panel shows the residuals to the fit.
Self-association of dARC1 CA
Given the unexpected nature of the dimer observed in the crystal structure, the solution molecular mass, conformation, and self-association properties of dARC1 CA were examined using a variety of solution hydrodynamic methods. Initial assessment by size exclusion chromatography–coupled multi-angle laser light scattering (SEC-MALLS) was performed with protein concentrations ranging from 25 to 400 μM that yielded an invariant solution molecular weight of 40.0 kDa for dARC1 CA (Fig. 2B). By comparison, the dARC1 CA sequence-derived molecular weight is 19.6 kDa. Given this value, together with the lack of a concentration dependency of the molecular weight, it is apparent that dARC1 CA also forms strong dimers in solution. To confirm and better analyze dARC1 CA oligomerization, we measured the hydrodynamic properties using sedimentation velocity (SV-AUC) and sedimentation equilibrium (SE-AUC) analytical ultracentrifugation. A summary of the experimental parameters, molecular weights derived from these data, and statistics relating to the quality of fits are shown in table S2. Analysis of the sedimentation velocity data for dARC1 CA using both discrete component and the C(S) continuous sedimentation coefficient distribution function (Fig. 2C) revealed a predominant single species with S20,w of 2.92 ± 0.03 S and no significant concentration dependency of the sedimentation coefficient over the range measured (25 to 90 μM). These data show that dARC1 CA comprises a single stable 2.92 S species with a molecular weight derived from either the C(S) function or discrete component analysis (S20,w/D20,w) of 38 kDa (table S2), consistent with a dARC1 CA dimer. The frictional ratio (f/fo) obtained from the analysis of the sedimentation coefficients is 1.41 (table S2), suggesting that the solution dimer has an elongated conformation and is consistent with the U-shaped conformation observed in the crystal structures. Moreover, analysis of the crystal structure using HYDROpro (18) gives calculated S20,w and D20,w values in close agreement with that observed in solution (table S2), supporting the idea that the dimer observed in the crystal structures is wholly representative of the solution conformation. To further ascertain the affinity of dARC1 CA self-association, multispeed SE-AUC studies at varying protein concentration were carried out and typical equilibrium distributions for dARC1 CA are presented in Fig. 2D. Analysis of individual gradient profiles showed no concentration dependency of the molecular weight, and so, all the data were fitted globally with a single ideal molecular species model, producing a weight-averaged molecular weight of 38.9 kDa (table S2). The lack of any concentration dependency precludes any analysis of homodimer affinity but confirms that dARC1 CA forms a stable dimeric structure that has the expected properties of the dimer we observe in the crystal structure.
Attempts to mildly disrupt the central apolar network by introduction of an F133A mutation had no effect on dimerization when assessed by SEC-MALLS (fig. S2B). More aggressive mutations F133A + Y129A and F133A + R161A resulted in complete loss of protein solubility and an inability to purify the constructs, further suggesting that, in dARC1 CA, homodimerization is a requirement for protein folding/structural integrity and likely forms a key building block of dARC1 particle assembly. Analysis of the electrostatic surface potential of the dimeric structure reveals a differential distribution of charge, where the surface of the glacial trough has a net negative charge that spreads across both domains of each dARC1, and the underside where the C-terminus projects has a more positively charged character (fig. S2C), suggesting that, upon assembly, dARC1 particles would have a negatively charged exterior and a more positively charged interior where nucleic acid is contained.
Comparison with mam-ARC CA structure
Given that mam-ARC and dARC1 share functional similarities, we assessed the relationship between rARC and dARC1 by comparing the dARC1 structure with the individual domains from rARC. Overall, the alignments are excellent, reflecting the evolutionary relationship, but there are significant differences between dARC1 and rARC in both their NTDs and CTDs.
There are two crystal structures of the rARC NTD in complex with peptide ligands [Protein Data Bank (PDB): 4X3H and 4X3I] (13) and a recent solution NMR structure [6GSE; (17)] of the entire rARC CA domain that resolves the NTD in an apo form. Superficially, the dARC1 CA-NTD aligns well with all available structures of the rARC CA-NTD, with DALI Z scores of 8 to 10 and RMSDs between 1.5 and 1.9 Å (Fig. 3A).
Fig. 3. Comparison of dARC1 and rARC CA-NTD structures.
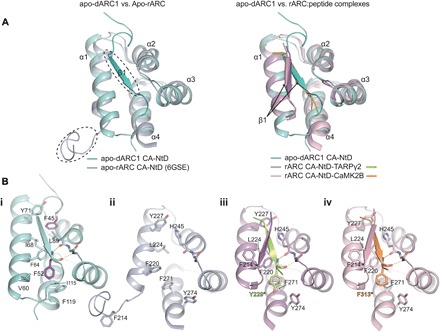
(A) Left: 3D structural alignment of dARC1 CA-NTD (teal cartoon) and apo-rARC CA-NTD (PDB: 6GSE; lilac cartoon). Secondary structure elements are labeled. Circled are the ordered N-terminal β strand of dARC1 and the disordered N-terminal strand of apo-rARC. Right: 3D structural alignment of dARC1 CA-NTD and the peptide-complex structures of rARC CA-NTDs (PDB: 4X3H and 4X3I). The protein backbones are shown in cartoon representation, colored according to the legend. Secondary structure elements are labeled. The arrow indicates the different positioning of the extended N-terminal β strand between the dARC1 and rARC structures. (B, i to iv) Individual views of the structures presented in (A): (i) apo-dARC1, (ii) apo-rARC, (iii) rARC-TARPγ2, and (iv) rARC-CaMK2B. Residues that constitute the hydrophobic NTD cleft are shown in stick format, colored by atom type. In each structure, the side chains of the aromatic residues buried in the interface (F45 and F52, dARC1 CA-NTD; Y229*, rARC CA-NTD–TARPγ2; F313*, rARC CA-NTD–CaMK2B) are colored purple, yellow, and orange, respectively. The conserved main-chain hydrogen bonding interactions between the backbone amide and carbonyl of F52 with the carbonyl of L89 and the amide of Y91 (dARC1), of Y229 with the carbonyl of H245 and the amide of N247 (rARC CA-NTD–TARPγ2), and of F313 (rARC CA-NTD–CaMK2B) with the carbonyl of H245 and the amide of N247 are shown as dashed lines.
Examination of the dARC1 CA-NTD reveals an N-terminal extended strand (NT-strand), residues G43 to R56, with a short β configuration that packs against the core of the NTD. The NT-strand makes many interactions with the apolar and aromatic side chains that extend from α1, α2, and α4, burying 803 Å2 of surface in the interface [Fig. 3, A and B (i), and fig. S3A], and the same configuration is observed in all four instances of the NTDs that we see in our two crystal structures (fig. S3B). The NT-strand residues are highly conserved in dARC genes across Drosophilidae but not with the mam-ARCs (fig. S3C). In particular, two highly conserved aromatic residues, F45 and F52, are entirely buried, surrounded by the conserved side chains of F64, L89, I115, and F119, and act to anchor the NT-strand into the hydrophobic α1-to-α4 cleft of the CA-NTD. In addition, there is a main-chain interaction between the backbone amide and carbonyl of F52 with the carbonyl of L89 and the amide of Y91 that further stabilizes the conformation of the NT-strand [Fig. 3B (i) and fig. S3A].
In apo-rARC CA-NTD (6GSE), the helical core aligns very well with the corresponding region of dARC1 (RMSD = 1.45 Å). However, here, the rARC NT-strand residues D210 to E216 have a disordered conformation (Fig. 3, A and B, ii), and the α1-to-α4 hydrophobic cleft, which in dARC1 contains the native NT-strand, is unoccupied in rARC, suggesting that there is a functional divergence for the NT-strand between the dARC1 and mam-ARC families. This notion is supported by the inspection of the rARC CA-NTD–TARPγ2 and CA NTD–CaMK2B complexes (4X3H and 4X3I), where the α1-to-α4 cleft of rARC is now occupied by the bound TARPγ2- or CaMK2B-derived peptides (Fig. 3B, iii and iv), and the bound peptides adopt the same extended β configuration as the native NT-strand in the dARC1 structure (fig. S3D) and bury a comparable amount of surface, 772 and 641 Å, respectively. Moreover, both bound peptides contain an aromatic residue equivalent to dARC1 F52, Y229 in TARPγ2, and F313 in CaMK2B that packs into the core of rARC CA-NTD and makes an identical main-chain interaction with the backbone carbonyl of H245 and the amide of N247 as that observed between the backbone amide and carbonyl of F52 with the carbonyl of L89 and the amide of Y91 in dARC1 (fig. S3D). In these peptide-complex structures, the rARC NT-strand, D210 to E216, that is disordered in the apo structure now adopts a parallel β configuration to pack against the bound peptides (Fig. 3B, iii and iv), and it is possible that the propensity to form this stabilizing β configuration has been selected for. This notion is supported by the inspection of the dARC and mam-Arc multiple sequence alignment (fig. S3C) that reveals a conserved “TQIF” motif in Amniota that retains β-branched residues, favored in β structure, at the T and I position. This motif is not present in amphibians or in Latimeria chalumnae Gypsy2, the closest known relative to the transposon from which tetrapod ARC was exapted, suggesting that this feature, and possibly peptide binding ability, arose within Amniota.
The structures of dARC1 CA-CTD and rARC CA-CTD (PDB: 4X3X) also superimpose well (RMSD = 2.7 Å). However, the CTD of the apo-rARC CA NMR structure more closely matched the structure of dARC1 CA-CTD (RMSD = 2.2 Å), with all five helices overlaying (Fig. 4A). However, in contrast to our solution studies of dARC1 (Fig. 2, A to C, and fig. S2A), the rARC CA domain was monomeric in solution, even at the high concentrations under which NMR was performed (17).
Fig. 4. Comparison of the dARC1 and rARC CA-CTD structures.
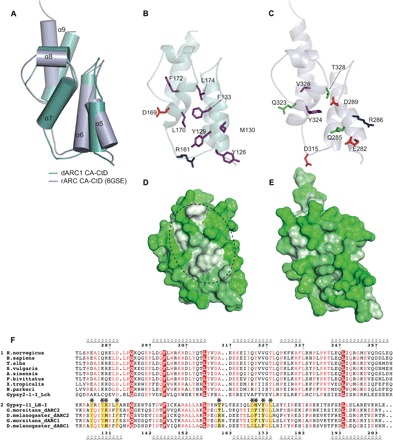
(A) 3D structural alignment of dARC1 CA-CTD and rARC CA-CTD from apo-rARC (PDB: 6GSE). The structures are shown in cartoon, with equivalent helices labeled and shown as cylinders. dARC1 is colored cyan, and rARC is colored light blue. (B and C) Details of the CA-CTD homodimer interfaces. Cartoon representations of the protein backbone of dARC1 CA-CTD (B) and rARC CA-CTD (C) are shown, colored as in (A). The view is of one monomer looking into the dimer interface. Residues that make interactions in dARC1 CA and their equivalents in rARC are shown in stick representation, color-coded by residue type (purple, hydrophobic/aromatic; green, polar; red, acidic; blue, basic). (D and E) Hydrophobic surface representations of (B) and (C), respectively. Circled in (D) is a distinct hydrophobic patch on the surface of dARC1 CA-CTD, which is absent in rARC. (F) Multiple sequence alignment of ARC, dARC1, and dARC2 CA-CTDs and parent retrotransposon sequences. Group 1 contains tetrapod ARC (tARC) sequences and the closely related Latimeria chalumnae (L. ch) Gypsy2 transposon. Top: Secondary structure of rARC; numbers according to the rARC (R. norvegicus) sequence. Group 2 contains dARC1, dARC2, and closely related Linepithema humile (L. h) Gypsy11 retrotransposon. Bottom: Secondary structure of dARC1; numbers according to the dARC1 (D. melanogaster) sequence. Red box and white text represent invariant residues shared between groups. Red text represents residues conserved within a group. Asterisks mark the residues at the dARC1 CTD dimer interface and their equivalents in tARCs, as shown in (B) and (C).
In dARC1, a large proportion of the CTD dimer interface results from the packing of hydrophobic side chains projecting from helices 5 and 7 (Fig. 2A). However, upon comparison of the external α5/α7 surfaces of dARC1 and rARC (Fig. 4, B and C), it is apparent that the exposed Y126, Y129, M130, F133, L170, F172, and L174 side chains that are responsible for the hydrophobic character of the dARC1 dimer interface are not conserved in rARC and are replaced by E282, Q285, R286, D289, Y324, V326, and T328 in rARC. Therefore, the hydrophobic patch present on the surface of dARC1 is not evident in the same surface on rARC (Fig. 4, D and E). In addition, R161 and D169, which make a salt bridge interaction in the dARC1 interface, are also not conserved, being replaced by D315 and Q323 in rARC (Fig. 4, B and C). These sequence differences are also apparent throughout the entire dARC and mam-ARC families. Hence, there is strong sequence conservation of residues that constitute the core fold of the CA-CTD across both dARC and mam-ARCs, but the hydrophobic CA-CTD dimer interface residues are only present in the dARC lineage (Fig. 4F). Together, these data reveal that, while tertiary structure topology of dARC1 and rARC CA-CTDs is conserved, there are substantial differences in the character of the surface that is presented around α5 to α7; in dARC1, the hydrophobic nature of this surface drives the formation of a strong CTD dimer, whereas in rARC, the more polar nature of this surface may explain why the protein is monomeric in solution. Given these differences, although there is strong evidence for the assembly of both dARC1 and mam-ARC into CA-like particles (9, 12), it seems likely that if dARC1 and mam-ARC use the α5/α7 interface in a particle assembly pathway, the interface may be substantially weaker for mam-ARC.
Fig. 5. Structural similarity with ortho- and spumaretroviral CA.
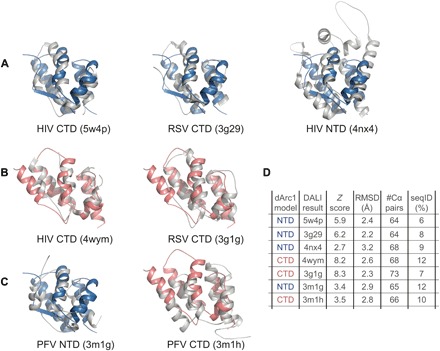
(A) Pairwise DALI 3D Cα structural alignment of dARC1 CA-NTD with HIV CA-CTD (left), RSV CA-CTD (middle), and HIV-NTD (right). In each panel, the cartoon of the dARC1 CA-NTD backbone is shown in blue, and the backbone of the aligned structures is shown in gray. (B) Pairwise 3D Cα structural alignment of dARC1 CA-CTD with HIV CA-CTD (left) and RSV CA-CTD (right). In each panel, the cartoon of the dARC1 CA-CTD backbone is shown in red, and the backbone of the aligned structures is shown in gray. (C) Pairwise 3D Cα structural alignment of dARC1 CA-NTD with prototypic foamy virus (PFV) CA-NTD (left) and dARC1 CA-CTD with PFV CA-CTD (right). (D) DALI Z scores, RMSD, number of aligned residues, and sequence identities for 3D Cα alignments.
Retroviral CA domain structures are related to both dARC1 CA domains
The topology of the α-helical two-domain fold of dARC1 is highly reminiscent of retroviral CA structures. Interrogation of the PDB database with dARC1 CA using the DALI alignment/search engine (19) produced an overwhelming number of matches to Gag proteins (87%, Z score ≥ 5.0) and identified rARC, together with many orthoretroviral and spumaretroviral CA-NTD and CA-CTD structures. Alignments with CA-NTDs and CA-CTDs from HIV CA, Rous sarcoma virus (RSV) CA, and prototypic foamy virus CA (PFV) are presented in Fig. 5. The best structural alignments to dARC1-NTD were with retroviral Gag CA-CTD structures rather than with Gag CA-NTD structures (Fig. 5, A and D), indicating that the dARC1 CA-NTD is more closely related to the orthoretroviral CA-CTD than to the orthoretroviral CA-NTD. Alignments with dARC1-CTD also had the best structural alignment with orthoretroviral Gag CA-CTD structures (Fig. 5, B and D), perhaps not unexpected given the observation of close resemblance of dARC1 CA-NTD to dARC1 CA-CTD (Fig. 1B, iii). Alignments with PFV CA-NTD and CA-CTD were also found (Fig. 5, C and D); although not as significant as with the orthoretroviral CA, these data support previous observations of a relationship of spumaretroviral Gag with mam-ARC (14).
These data provide evidence for a structural conservation between orthoretroviral CA and ARC proteins, and the weaker alignments observed with orthoretroviral CA-NTDs suggest that orthoretroviral CA-NTDs have undergone much more structural divergence than has occurred in the Ty3 family or ARC proteins. Moreover, these data further support the previously proposed idea that a duplication of a CA-CTD progenitor first gave rise to double domain ancestors and that subsequent divergence of domains resulted in spumaretroviral, orthoretroviral, and Metaviridae-derived proteins, such as ARC, that are found presently (14, 20).
The dARC1 CTD dimer is an ancient assembly interface conserved in orthoretroviridae
Given the existence of the dARC1 CA dimer and the distant relationship with orthoretroviral CA, we next looked to see whether the dimer interface was conserved between dARC1 and the CTD dimers of HIV-1 CA and RSV CA that are known to be essential for CA assembly in orthoretroviruses. For these comparisons, the interhexamer CA CTD-CTD dimers observed in HIV-1 and RSV CA-hexamer crystal structures (21, 22) were used, as these most closely relate to those observed in cryo-electron microscopy (cEM) studies of whole CA assemblies (22, 23). Cartoon representations of the dARC1, HIV-1, and RSV CA-CTD dimers are shown in Fig. 6 (A to C). In each, the domain arrangement that presents the dimer interface is the same, and this is also seen in the CA-CTD dimer of native Ty3 particles visualized by cEM (24), but with some repositioning of the CA-NTDs (fig. S4). The structures have been aligned to find the best Cα alignment over the entire dimer (HIV, RMSD = 2.8 Å over 117 Cα; RSV, RMSD = 3.1 Å over 101 Cα) (Fig. 6, D and E), and it is apparent that each interface is made up from interactions between residues on CTD helices α5 and α7 of dARC1, which correspond to α7′ and α8 in the orthoretroviral CA-CTD structures. Notably, in the orthoretroviruses, α7′ is reduced to a single turn, and the monomers are rotated with respect to each other. Therefore, in dARC1, residues on α5 and α7 contribute equally to the interface, while in the orthoretroviruses, α8 contributes more to the interface than does α7′. This combination of the larger contribution of α5 in dARC1, together with the rotation and displacement of CA-CTDs seen in the orthoretroviruses, has the effect of reducing the surface area that is buried at the interface from 768 Å2 in dARC1 to 452 Å2 in HIV-1. Notably, the homodimer affinity for orthoretroviral CA-CTD dimers is much weaker than the dARC1 dimer. Equilibrium dissociation constants ranging between 10 and 20 μM have been reported for HIV-1 (25, 26), and CA-CTD dimerization is undetectable for other genera (27–29). Nevertheless, given the domain organization and the similarity in character of the orthoretroviral and dARC1 CA-CTD dimers, we suggest that this interface is a key building block of CA assembly, retained in dARC1 and conserved from Ty3/Gypsy transposable elements to orthoretroviridae.
Fig. 6. Comparison with retroviral CA-CTD dimers.
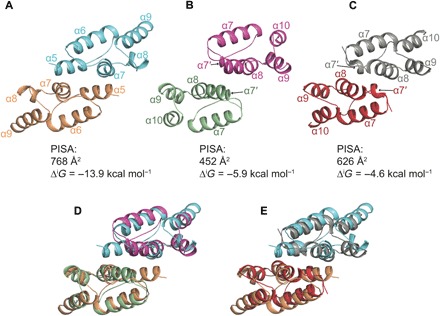
(A to C) Cartoon representations of CA-CTD dimers. (A) dARC1 is colored cyan and wheat. (B) HIV-1 is colored magenta and pale green (PDB: 2XFX). (C) RSV is colored gray and red (PDB: 3G21). The orthoretroviral structures are aligned with respect to the dARC1 dimer. CTD helices α5 to α9 are labeled in the dARC1 structure, and the equivalent α7′ to α10 are labeled in the orthoretroviral structures. The buried surface area (Å2) and free energy of interaction (ΔiG) of each interface, calculated in PDBePISA, are displayed below each structure. (D and E) Structural alignment of dARC1 CA with HIV-1 CA and RSV CA dimers, respectively. Protein backbones are colored as in (A) to (C).
DISCUSSION
dARC1 CA structures
Our crystal structures demonstrate that the central region of dARC1 contains two largely α-helical domains that, despite the lack of sequence conservation, have the same predominantly α-helical folds observed in the structures of CA domains from the ortho- and spumaretroviruses. A more detailed inspection of dARC1 CA-NTD and CA-CTD reveals that they comprise four- and five-helix bundles, respectively, with a topology that aligns well with the arrangement of secondary structure elements observed in orthoretroviral CA NTDs and CTDs (Fig. 5). However, it is apparent that both the ARC CA-NTD and CA-CTD are much more closely related to the orthoretroviral CA-CTDs than they are to orthoretroviral CA-NTDs (Fig. 5), consistent with our previous notion that an ancient domain duplication was a key event during retrotransposon evolution (14). Notably, orthoretroviral CA-NTDs contain an extra N-terminal β hairpin and an additional two helices compared to the ARCs and the CA domains of Ty3/Gypsy transposons (fig. S4) (24). This suggests that unique aspects of the retroviral life cycle might be driving specific changes in the structure of the retroviral CA-NTD. One such pressure might be associated with the process of maturation that follows retrovirus budding from the cell. Maturation involves proteolytic cleavage of immature viral cores, followed by CA reassembly to yield mature virions and although it is proposed that dARC1 and mam-ARC transport mRNA between cells, it is thought likely that particles are packaged into extracellular vesicles for cell-to-cell transfer (9, 12). Similarly, maturation events do not occur in Ty3 elements, which also do not bud from the cell and have Gag that assembles directly into mature forms (24). The absence of maturation also characterizes spumaviruses, and it was observed previously that the CA NTD–equivalent region of PFV Gag showed greater similarity to rARC than to orthoretroviral CA (14).
Structural differences between insect and mam-ARCs
Our three-dimensional (3D) superimpositions have demonstrated that there is a large degree of structural conservation between the dARC1 and mam-ARC CA structures. However, despite this strong similarity, two regions of distinct differences between the dARC1 and rARC structure are apparent. The first region concerns the ARC CA-NTD and the interaction with potential binding partners; the second region concerns the putative dimerization domain of the CTD.
Functionally important interactions between mam-ARC and a variety of neuronal proteins, including the TARPγ2 and CaMK2B proteins, as well as the NMDA (N-methyl-d-aspartate) receptor, have been defined (13, 17). However, no such interactions have been reported for dARC1. In the rARC structures with bound TARPγ2 or CaMK2B peptides, the disordered N-terminal region of rARC seen in the apo structure now forms a short parallel β sheet, with the bound peptide stabilizing the peptide binding within a hydrophobic cleft on rARC. It is apparent that the conformation of these rARC-bound peptides strongly resembles that of the NT-strand of dARC1 NTD (Fig. 3). Therefore, given the sequence differences in the NT-strand region between the dARC and mam-ARCs (fig. S3C), one notion is that mam-ARC has evolved an N-terminal strand that no longer binds into the CA-NTD hydrophobic cleft but has gained the ability to promote the binding of synaptic protein ligands, perhaps acting as a sensor of synaptic stimuli. This sensing property might then contribute control to a functional role for ARC based on assembly and mRNA trafficking.
There are also significant differences between dARC1 and rARC CA-CTD, illustrated in Fig. 4. Overall, our crystal structure of dARC1 and the NMR structure of full-length rARC (17) are very similar, with good overlay in all five helices. However, inspection of the dARC1 surface reveals a substantial hydrophobic patch that is absent in rARC (Fig. 4, D and E). This hydrophobic patch is shared with the orthoretroviruses (25, 30) and seems to be associated with the formation of stable dARC1 dimers, whereas rARC is monomeric. Whether this translates to differences in the stability of assembled particles in vivo remains to be determined; however, it is possible that differences in the physiological roles of dARC1 and mam-ARC may mean that mam-ARC has evolved to require a weaker interface that facilitates disassembly. Alternatively, it is possible that mam-ARC may require a conformational change to facilitate dimerization or uses a completely different assembly mechanism that uses other surfaces of the molecule.
ARC particle assembly
The observation that residues at the dARC1 CA-CTD interface are not conserved between the insect and mam-ARC lineages suggests the possibility that, although mam-ARC particles have been observed in vitro and in cells, their mode of assembly may not use an obligate CA-CTD dimer as a building block. This type of observation has been made with orthoretroviruses that assemble through a combination of NTD-NTD, NTD-CTD, and CTD-CTD interactions to form the viral CA shell, where the relative contribution that different types of CA interaction make to the overall formation of the viral core varies depending on the retroviral genera. For instance, in lentiviruses, it is apparent that CA assembly requires a strong intrinsic CTD-CTD dimeric interaction (25, 30). However, more generally, CA shell formation requires three types of interaction: intrahexamer NTD-NTD self-association (30–33), intrahexamer NTD-CTD interactions between adjacent CA monomers (30, 34, 35), and interhexamer CTD-CTD interactions (25, 30). Therefore, it is entirely possible that, in dARC1 and mam-ARC particles, the relative contributions of each type of interface may also differ.
ARC exaptation
Mam-ARC and dARC1 appear to have different biological properties. However, it remains to be determined whether these differences result from the capture of two different Ty3/Gypsy elements or they reflect evolutionary adaptations. Perhaps the best studied example of the appropriation of retroelement encoded genes by mammalian hosts is the case of syncytin, a fusagenic protein essential for proper placenta formation (36). It is evident that syncytin capture appears to have occurred on multiple independent occasions, involving envelope proteins from different retroviruses (37, 38), resulting in placentae with subtly different morphologies (39). Determining whether this is also the case with the ARC genes, as well as their close relatives in the mammalian genome (11), will require further characterization of existing retrotransposon elements using structural methods not reliant on the comparative similarities in related nucleic acid sequences that have disappeared with the passage of time.
MATERIALS AND METHODS
Protein expression and purification
dARC1 residues S39 to N205 were determined to represent the CA domain according to multiple sequence alignment and secondary structural analysis performed in ClustalX (40) and Psipred (41). An Escherichia coli codon-optimized complementary DNA (cDNA) for D. melanogaster dARC1 (UniProt, Q7K1U0) was synthesized (GeneArt), and the relevant sequence was polymerase chain reaction–amplified and subcloned into a pET22b plasmid (Novagen). The resulting construct comprised residues 39 to 205 of dARC1, with an N-terminal Met and a C-terminal PLEHHHHHH His-tag extension. Proteins were expressed in E. coli strain BL21 (DE3) grown in LB broth by induction of log-phase cultures with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) and incubated overnight at 20°C. Cells were pelleted and resuspended in 50 mM tris-HCl, 150 mM NaCl, 10 mM imidazole, 5 mM MgCl2, and 1 mM dithiothreitol (pH 8.0), supplemented with lysozyme (1 mg/ml; Sigma-Aldrich), deoxyribonuclease (DNase) I (10 μg/ml; Sigma-Aldrich), and one Protease Inhibitor cocktail tablet (EDTA-free, Pierce) per 40 ml of buffer. Cells were lysed using an EmulsiFlex-C5 homogenizer (Avestin), and dARC1 CA was captured from clarified lysate using immobilized metal ion affinity on a 5-ml Ni2+-NTA superflow column (Qiagen). Bound dARC1 CA was eluted in nonreducing buffer (50 mM tris-HCl, 150 mM NaCl, and 300 mM imidazole), and carboxypeptidase A (CPA; Sigma-Aldrich, C9268) was added at a ratio of ~100 mg of dARC1 per mg of CPA. The resulting mixture was incubated overnight at 4°C to allow digestion of the C-terminal His-tag. The CPA was inactivated by the addition of TCEP-HCl [tris (2-carboxyethyl) phosphine hydrochloride] to 2 mM. dARC1 CA was further purified by size exclusion chromatography using a Superdex 75 (26/60) (GE Healthcare) column, equilibrated in 20 mM tris-HCl, 150 mM NaCl, and 1 mM TCEP (pH 8.0). Purified protein eluted in a single peak. Selenomethionine derivative protein was produced using an identical procedure, but with Methionine auxotroph E. coli B834 (DE3) cells, grown in selenomethionine medium (Molecular Dimensions, Newmarket, United Kingdom), used to express the protein. Electrospray-ionization mass spectrometry was used to confirm the identity of dARC1 and, where applicable, selenomethionine incorporation. It also confirmed that the N-terminal Met had been processed and that the His-tag had been completely digested, leaving the motif “PLE” at the C terminus. Protein was concentrated by centrifugal ultrafiltration (Vivaspin; molecular weight cutoff, 10 kDa), then snap-frozen, and stored at −80°C. Protein concentrations were determined by ultraviolet-visible absorbance spectroscopy using an extinction coefficient at 280 nm derived from the tyrosine and tryptophan content.
Protein crystallization and structure determination
dARC1 CA was crystallized using sitting drop vapor diffusion at 18°C using Swissci MRC two-drop trays (Molecular Dimensions), with drops set using a Mosquito LCP robot with a humidity chamber (TTP Labtech). Native protein was initially concentrated to 20 mg/ml. Typically, drops were 200 to 300 nl, made by mixing protein:mother liquor in a 3:1 or 1:1 ratio, with a 75-μl reservoir. Initial crystal hits were obtained using the Structure Screen 1&2 (Molecular Dimensions) under a condition containing 4.3 M NaCl and 0.1 M Hepes (pH 7.5). Two crystal forms could be observed in these conditions: thin rods, which had a primitive orthorhombic (oP) lattice, and hexagonal disks or trapezoidal prisms, which had a primitive hexagonal (hP) lattice. Datasets were collected for these native crystals, but they could not be solved by molecular replacement methods. SeMet dARC1 CA was crystallized under conditions that optimized protein concentration, NaCl concentration, and pH. The best crystals grew in 300- to 400-nl drops set with protein at 12.5 to 16 mg/ml, with mother liquor NaCl ranging between 2.8 and 3.3 M. Rods were ~400 μm × 30 μm × 30 μm, and hexagons/trapezoids were ~130 μm across and up to 30 μm thick. Crystals were harvested using MiTeGen lithographic loops. The best cryoprotection was achieved using sodium malonate mixed into mother liquor to a concentration of 1.6 M. This was added directly to the drop, or crystals were bathed in this solution before flash freezing in liquid nitrogen.
Data collection and structure determination
Data were collected at the tunable SLS beamline PXIII. For the orthorhombic crystal form, a peak dataset was collected to 2.06 Å (see table S1). Data were processed by the SLS GoPy pipeline in P212121 using XDS (42) and showed significant anomalous signal to 2.82 Å. The resultant dataset was solved using SAD methods with Phenix (43), and despite a relatively low Figure of Merit (FOM), the experimental map was readily interpretable and it was possible to almost completely autobuild an initial structure with BUCCANEER (44). A higher-resolution (1.55 Å) dataset was collected at a non-anomalous, low-energy remote wavelength (table S1). This dataset was processed using the Xia2 (45) pipeline, DIALS (46) for indexing and integration, and AIMLESS (47) for scaling and merging. This dataset was initially used for refinement to 1.7 Å and manual model building in COOT (48). It was evident that the data were anisotropic and that they might benefit from anisotropic correction. Diffraction images were reprocessed using the autoPROC pipeline (49), XDS, POINTLESS (50), AIMLESS, and STARANISO (http://staraniso.globalphasing.org/cgi-bin/staraniso.cgi). This dataset was used for further refinement of the model, and there was an improvement in map quality, and in agreement between model and data. For the hexagonal crystal form, a highly redundant peak dataset was collected to 2.14 Å. This was processed using the Xia2 pipeline, DIALS for indexing and integration, and AIMLESS for scaling and merging, showing significant anomalous signal to 2.59 Å, in P6122. This dataset was solved using SAD methods in Phenix. Again, the experimental map was readily interpretable, and it was possible to almost completely autobuild an initial structure with BUCCANEER. Refinement and model building were carried out in Phenix and COOT, respectively. Anomalous signal was very strong in this dataset, and so, Friedel pairs were treated separately during refinement. MolProbity (51) and PDB_REDO (52) were used to monitor and assess model geometry. Details of data collection, phasing, and structure refinement statistics are presented in table S1.
Size exclusion chromatography–coupled multi-angle laser light scattering
SEC-MALLS was used to determine the molar mass of dARC CA. Samples ranging from 25 to 400 μM were applied in a volume of 100 μl to a Superdex INCREASE 200 10/300 GL column equilibrated in 20 mM tris-HCl, 150 mM NaCl, 0.5 mM TCEP, and 3 mM NaN3 (pH 8.0) at a flow rate of 1.0 ml/min. The scattered light intensity and the protein concentration of the column eluate were recorded using a DAWN HELEOS laser photometer and an OPTILAB-rEX differential refractometer, respectively. The weight-averaged molecular mass of material contained in chromatographic peaks was determined from the combined data from both detectors using the ASTRA software version 6.0.3 (Wyatt Technology Corp., Santa Barbara, CA).
Analytical ultracentrifugation
Sedimentation velocity experiments were performed in a Beckman Optima Xl-I analytical ultracentrifuge using conventional aluminum double-sector centerpieces and sapphire windows. Solvent density and the protein partial specific volumes were determined as described (53). Before centrifugation, dARC1 CA samples were prepared by exhaustive dialysis against the buffer blank solution, 20 mM tris-HCl (pH 8), 150 mM NaCl, and 0.5 mM TCEP (tris buffer). Samples (420 μl) and buffer blanks (426 μl) were loaded into the cells, and centrifugation was performed at 50,000 rpm and 293 K in an An50-Ti rotor. Interference data were acquired at time intervals of 180 s at varying sample concentrations (25, 50, and 100 μM). Data recorded from moving boundaries were analyzed in terms of the size distribution functions C(S) using the program Sedfit (54).
Sedimentation equilibrium experiments were performed in a Beckman Optima XL-I analytical ultracentrifuge using aluminum double-sector centerpieces in an An-50 Ti rotor. Before centrifugation, samples were dialyzed exhaustively against the buffer blank (tris buffer). Samples (150 μl) and buffer blanks (160 μl) were loaded into the cells, and after centrifugation for 30 hours, interference data were collected at 2 hourly intervals until no further change in the profiles was observed. The rotor speed was then increased, and the procedure was repeated. Data were collected on samples of different concentrations of dARC1 CA (25, 50, and 70 μM) at three speeds, and the program SEDPHAT (55) was used to determine weight-averaged molecular masses by nonlinear fitting of individual multispeed equilibrium profiles to a single-species ideal solution model. Inspection of these data revealed that the molecular mass of dARC1 CA showed no significant concentration dependency, and so, global fitting incorporating the data from multiple speeds and multiple sample concentrations was applied to extract a final weight-averaged molecular mass.
Structure analysis and alignments
Molecular interfaces were analyzed using the EBI protein structure interface analysis service PDBePISA (www.ebi.ac.uk/pdbe/pisa). Electrostatic surface potential and the surface hydrophobicity/hydrophilicity distribution of the dARC1 CA dimer were calculated with APBS (56) and using the pymol script (https://pymolwiki.org/index.php/Color_h), respectively. The DALI comparison server (http://ekhidna2.biocenter.helsinki.fi/dali) was used to search for and align structural homologs from the PDB.
Sequence alignment
Amino acid alignments were produced with MAFFT v7.271 (57), within tcoffee v11.00.8cbe486 (58), weighting alignments using three-state secondary-structure predictions produced with RaptorX Property v1.02 (59). Alignment images were produced with ESPript (60).
Supplementary Material
Acknowledgments
We thank the Swiss Light Source for beamtime and the staff of beamline PXIII. Funding: This work was supported by the Francis Crick Institute, which receives its core funding from the Cancer Research UK (FC001162 and FC001178), the UK Medical Research Council (FC001162 and FC001178), and the Wellcome Trust (FC001162 and FC001178), and by the Wellcome Trust (108014/Z/15/Z and 108012/Z/15/Z). Author contributions: M.A.C., S.C.L., and I.A.T. performed experiments. M.A.C., S.C.L., G.R.Y., J.P.S., and I.A.T. contributed to experimental design, data analysis, and manuscript writing. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The coordinates and structure factors for dARC1 CA (S39 to N205) have been deposited in the PDB under accession numbers 6S7X and 6S7Y.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/1/eaay6354/DC1
Table S1. dARC1 CA statistics of data collection, phasing, and refinement.
Table S2. Hydrodynamic parameters of dARC1 CA.
Fig. S1. Crystal structures of dARC1 CA.
Fig. S2. dARC1 CA dimer.
Fig. S3. Comparison of dARC1and mam-ARC CA-NTDs.
Fig. S4. Comparison of dARC1 and Ty3 CA.
REFERENCES AND NOTES
- 1.Dynes J. L., Steward O., Dynamics of bidirectional transport of Arc mRNA in neuronal dendrites. J. Comp. Neurol. 500, 433–447 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Plath N., Ohana O., Dammermann B., Errington M. L., Schmitz D., Gross C., Mao X., Engelsberg A., Mahlke C., Welzl H., Kobalz U., Stawrakakis A., Fernandez E., Waltereit R., Bick-Sander A., Therstappen E., Cooke S. F., Blanquet V., Wurst W., Salmen B., Bosl M. R., Lipp H. P., Grant S. G., Bliss T. V., Wolfer D. P., Kuhl D., Arc/Arg3.1 is essential for the consolidation of synaptic plasticity and memories. Neuron 52, 437–444 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Chowdhury S., Shepherd J. D., Okuno H., Lyford G., Petralia R. S., Plath N., Kuhl D., Huganir R. L., Worley P. F., Arc/Arg3.1 interacts with the endocytic machinery to regulate AMPA receptor trafficking. Neuron 52, 445–459 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shepherd J. D., Rumbaugh G., Wu J., Chowdhury S., Plath N., Kuhl D., Huganir R. L., Worley P. F., Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron 52, 475–484 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu J., Petralia R. S., Kurushima H., Patel H., Jung M. Y., Volk L., Chowdhury S., Shepherd J. D., Dehoff M., Li Y., Kuhl D., Huganir R. L., Price D. L., Scannevin R., Troncoso J. C., Wong P. C., Worley P. F., Arc/Arg3.1 regulates an endosomal pathway essential for activity-dependent beta-amyloid generation. Cell 147, 615–628 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park S., Park J. M., Kim S., Kim J. A., Shepherd J. D., Smith-Hicks C. L., Chowdhury S., Kaufmann W., Kuhl D., Ryazanov A. G., Huganir R. L., Linden D. J., Worley P. F., Elongation factor 2 and fragile X mental retardation protein control the dynamic translation of Arc/Arg3.1 essential for mGluR-LTD. Neuron 59, 70–83 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fromer M., Pocklington A. J., Kavanagh D. H., Williams H. J., Dwyer S., Gormley P., Georgieva L., Rees E., Palta P., Ruderfer D. M., Carrera N., Humphreys I., Johnson J. S., Roussos P., Barker D. D., Banks E., Milanova V., Grant S. G., Hannon E., Rose S. A., Chambert K., Mahajan M., Scolnick E. M., Moran J. L., Kirov G., Palotie A., McCarroll S. A., Holmans P., Sklar P., Owen M. J., Purcell S. M., O'Donovan M. C., De novo mutations in schizophrenia implicate synaptic networks. Nature 506, 179–184 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manago F., Mereu M., Mastwal S., Mastrogiacomo R., Scheggia D., Emanuele M., De Luca M. A., Weinberger D. R., Wang K. H., Papaleo F., Genetic disruption of Arc/Arg3.1 in mice causes alterations in dopamine and neurobehavioral phenotypes related to schizophrenia. Cell Rep. 16, 2116–2128 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ashley J., Cordy B., Lucia D., Fradkin L. G., Budnik V., Thomson T., Retrovirus-like Gag protein Arc1 binds RNA and traffics across synaptic boutons. Cell 172, 262–274.e11 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mattaliano M. D., Montana E. S., Parisky K. M., Littleton J. T., Griffith L. C., The Drosophila ARC homolog regulates behavioral responses to starvation. Mol. Cell. Neurosci. 36, 211–221 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Campillos M., Doerks T., Shah P. K., Bork P., Computational characterization of multiple Gag-like human proteins. Trends Genet. 22, 585–589 (2006). [DOI] [PubMed] [Google Scholar]
- 12.Pastuzyn E. D., Day C. E., Kearns R. B., Kyrke-Smith M., Taibi A. V., McCormick J., Yoder N., Belnap D. M., Erlendsson S., Morado D. R., Briggs J. A. G., Feschotte C., Shepherd J. D., The neuronal gene Arc encodes a repurposed retrotransposon Gag protein that mediates intercellular RNA transfer. Cell 172, 275–288.e18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang W., Wu J., Ward M. D., Yang S., Chuang Y. A., Xiao M., Li R., Leahy D. J., Worley P. F., Structural basis of Arc binding to synaptic proteins: Implications for cognitive disease. Neuron 86, 490–500 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taylor W. R., Stoye J. P., Taylor I. A., A comparative analysis of the foamy and ortho virus capsid structures reveals an ancient domain duplication. BMC Struct. Biol. 17, 3 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Curcio M. J., Lutz S., Lesage P., The Ty1 LTR-retrotransposon of budding yeast, Saccharomyces cerevisiae. Microbiol. Spectr. 3, 1–35 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Freed E. O., HIV-1 assembly, release and maturation. Nat. Rev. Microbiol. 13, 484–496 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nielsen L. D., Pedersen C. P., Erlendsson S., Teilum K., The capsid domain of Arc changes its oligomerization propensity through direct interaction with the NMDA receptor. Structure 27, 1071–1081.e5 (2019). [DOI] [PubMed] [Google Scholar]
- 18.Ortega A., Amoros D., Garcia de la Torre J., Prediction of hydrodynamic and other solution properties of rigid proteins from atomic- and residue-level models. Biophys. J. 101, 892–898 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holm L., Sander C., Dali/FSSP classification of three-dimensional protein folds. Nucleic Acids Res. 25, 231–234 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ball N. J., Nicastro G., Dutta M., Pollard D. J., Goldstone D. C., Sanz-Ramos M., Ramos A., Mullers E., Stirnnagel K., Stanke N., Lindemann D., Stoye J. P., Taylor W. R., Rosenthal P. B., Taylor I. A., Structure of a spumaretrovirus Gag central domain reveals an ancient retroviral capsid. PLOS Pathog. 12, e1005981 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gres A. T., Kirby K. A., KewalRamani V. N., Tanner J. J., Pornillos O., Sarafianos S. G., STRUCTURAL VIROLOGY. X-ray crystal structures of native HIV-1 capsid protein reveal conformational variability. Science 349, 99–103 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bailey G. D., Hyun J. K., Mitra A. K., Kingston R. L., A structural model for the generation of continuous curvature on the surface of a retroviral capsid. J. Mol. Biol. 417, 212–223 (2012). [DOI] [PubMed] [Google Scholar]
- 23.Zhao G., Perilla J. R., Yufenyuy E. L., Meng X., Chen B., Ning J., Ahn J., Gronenborn A. M., Schulten K., Aiken C., Zhang P., Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature 497, 643–646 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dodonova S. O., Prinz S., Bilanchone V., Sandmeyer S., Briggs J. A. G., Structure of the Ty3/Gypsy retrotransposon capsid and the evolution of retroviruses. Proc. Natl. Acad. Sci. U.S.A. 116, 10048–10057 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gamble T. R., Yoo S., Vajdos F. F., von Schwedler U. K., Worthylake D. K., Wang H., McCutcheon J. P., Sundquist W. I., Hill C. P., Structure of the carboxyl-terminal dimerization domain of the HIV-1 capsid protein. Science 278, 849–853 (1997). [DOI] [PubMed] [Google Scholar]
- 26.Yoo S., Myszka D. G., Yeh C., McMurray M., Hill C. P., Sundquist W. I., Molecular recognition in the HIV-1 capsid/cyclophilin A complex. J. Mol. Biol. 269, 780–795 (1997). [DOI] [PubMed] [Google Scholar]
- 27.Khorasanizadeh S., Campos-Olivas R., Summers M. F., Solution structure of the capsid protein from the human T-cell leukemia virus type-I. J. Mol. Biol. 291, 491–505 (1999). [DOI] [PubMed] [Google Scholar]
- 28.Mortuza G. B., Goldstone D. C., Pashley C., Haire L. F., Palmarini M., Taylor W. R., Stoye J. P., Taylor I. A., Structure of the capsid amino-terminal domain from the betaretrovirus, Jaagsiekte sheep retrovirus. J. Mol. Biol. 386, 1179–1192 (2009). [DOI] [PubMed] [Google Scholar]
- 29.Kingston R. L., Fitzon-Ostendorp T., Eisenmesser E. Z., Schatz G. W., Vogt V. M., Post C. B., Rossmann M. G., Structure and self-association of the Rous sarcoma virus capsid protein. Structure 8, 617–628 (2000). [DOI] [PubMed] [Google Scholar]
- 30.Ganser-Pornillos B. K., Cheng A., Yeager M., Structure of full-length HIV-1 CA: A model for the mature capsid lattice. Cell 131, 70–79 (2007). [DOI] [PubMed] [Google Scholar]
- 31.Li S., Hill C. P., Sundquist W. I., Finch J. T., Image reconstructions of helical assemblies of the HIV-1 CA protein. Nature 407, 409–413 (2000). [DOI] [PubMed] [Google Scholar]
- 32.Briggs J. A., Wilk T., Welker R., Krausslich H. G., Fuller S. D., Structural organization of authentic, mature HIV-1 virions and cores. EMBO J. 22, 1707–1715 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mortuza G. B., Haire L. F., Stevens A., Smerdon S. J., Stoye J. P., Taylor I. A., High-resolution structure of a retroviral capsid hexameric amino-terminal domain. Nature 431, 481–485 (2004). [DOI] [PubMed] [Google Scholar]
- 34.Lanman J., Lam T. T., Emmett M. R., Marshall A. G., Sakalian M., Prevelige P. E. Jr., Key interactions in HIV-1 maturation identified by hydrogen-deuterium exchange. Nat. Struct. Mol. Biol. 11, 676–677 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Lanman J., Lam T. T., Barnes S., Sakalian M., Emmett M. R., Marshall A. G., Prevelige P. E. Jr., Identification of novel interactions in HIV-1 capsid protein assembly by high-resolution mass spectrometry. J. Mol. Biol. 325, 759–772 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Mi S., Lee X., Li X., Veldman G. M., Finnerty H., Racie L., LaVallie E., Tang X. Y., Edouard P., Howes S., Keith J. C. Jr., McCoy J. M., Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature 403, 785–789 (2000). [DOI] [PubMed] [Google Scholar]
- 37.Funk M., Cornelis G., Vernochet C., Heidmann O., Dupressoir A., Conley A., Glickman S., Heidmann T., Capture of a hyena-specific retroviral envelope gene with placental expression associated in evolution with the unique emergence among carnivorans of hemochorial placentation in Hyaenidae. J. Virol. 93, e01811-18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cornelis G., Heidmann O., Degrelle S. A., Vernochet C., Lavialle C., Letzelter C., Bernard-Stoecklin S., Hassanin A., Mulot B., Guillomot M., Hue I., Heidmann T., Dupressoir A., Captured retroviral envelope syncytin gene associated with the unique placental structure of higher ruminants. Proc. Natl. Acad. Sci. U.S.A. 110, E828–E837 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.P. Wooding, G. Burton, Comparative Placentation: Structures, Functions and Evolution (Springer, 2008). [Google Scholar]
- 40.Larkin M. A., Blackshields G., Brown N. P., Chenna R., McGettigan P. A., McWilliam H., Valentin F., Wallace I. M., Wilm A., Lopez R., Thompson J. D., Gibson T. J., Higgins D. G., Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948 (2007). [DOI] [PubMed] [Google Scholar]
- 41.Jones D. T., Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292, 195–202 (1999). [DOI] [PubMed] [Google Scholar]
- 42.Kabsch W., XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zwart P. H., Afonine P. V., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., McKee E., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Storoni L. C., Terwilliger T. C., Adams P. D., Automated structure solution with the PHENIX suite. Methods Mol. Biol. 426, 419–435 (2008). [DOI] [PubMed] [Google Scholar]
- 44.Cowtan K., The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. D Biol. Crystallogr. 62, 1002–1011 (2006). [DOI] [PubMed] [Google Scholar]
- 45.Winter G., xia2: An expert system for macromolecular crystallography data reduction. J. Appl. Cryst. 43, 186–190 (2009). [Google Scholar]
- 46.Winter G., Waterman D. G., Parkhurst J. M., Brewster A. S., Gildea R. J., Gerstel M., Fuentes-Montero L., Vollmar M., Michels-Clark T., Young I. D., Sauter N. K., Evans G., DIALS: Implementation and evaluation of a new integration package. Acta Crystallogr. D Struct. Biol. 74, 85–97 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Evans P. R., Murshudov G. N., How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Emsley P., Cowtan K., Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 49.Vonrhein C., Flensburg C., Keller P., Sharff A., Smart O., Paciorek W., Womack T., Bricogne G., Data processing and analysis with the autoPROC toolbox. Acta Crystallogr. D Biol. Crystallogr. 67, 293–302 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Evans P., Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 (2006). [DOI] [PubMed] [Google Scholar]
- 51.Chen V. B., Wedell J. R., Wenger R. K., Ulrich E. L., Markley J. L., MolProbity for the masses-of data. J. Biomol. NMR 63, 77–83 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Joosten R. P., Salzemann J., Bloch V., Stockinger H., Berglund A. C., Blanchet C., Bongcam-Rudloff E., Combet C., Da Costa A. L., Deleage G., Diarena M., Fabbretti R., Fettahi G., Flegel V., Gisel A., Kasam V., Kervinen T., Korpelainen E., Mattila K., Pagni M., Reichstadt M., Breton V., Tickle I. J., Vriend G., PDB_REDO: Automated re-refinement of X-ray structure models in the PDB. J. Appl. Cryst. 42, 376–384 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.T. M. Laue, B. D. Shah, T. M. Ridgeway, S. L. Pelletier, in Analytical Ultracentrifugation in Biochemistry and Polymer Science, S. E. Harding, A. J. Rowe, J. C. Horton, Eds. (The Royal Society of Chemistry, 1992), pp. 90–125. [Google Scholar]
- 54.Schuck P., Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys. J. 78, 1606–1619 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Vistica J., Dam J., Balbo A., Yikilmaz E., Mariuzza R. A., Rouault T. A., Schuck P., Sedimentation equilibrium analysis of protein interactions with global implicit mass conservation constraints and systematic noise decomposition. Anal. Biochem. 326, 234–256 (2004). [DOI] [PubMed] [Google Scholar]
- 56.Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A., Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Katoh K., Standley D. M., MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Notredame C., Higgins D. G., Heringa J., T-Coffee: A novel method for fast and accurate multiple sequence alignment. J. Mol. Biol. 302, 205–217 (2000). [DOI] [PubMed] [Google Scholar]
- 59.Wang S., Peng J., Ma J., Xu J., Protein secondary structure prediction using Deep Convolutional Neural Fields. Sci. Rep. 6, 18962 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Robert X., Gouet P., Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 42, W320–W324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/1/eaay6354/DC1
Table S1. dARC1 CA statistics of data collection, phasing, and refinement.
Table S2. Hydrodynamic parameters of dARC1 CA.
Fig. S1. Crystal structures of dARC1 CA.
Fig. S2. dARC1 CA dimer.
Fig. S3. Comparison of dARC1and mam-ARC CA-NTDs.
Fig. S4. Comparison of dARC1 and Ty3 CA.