

mDia1/3 facilitate T cell receptor signaling via F-actin polymerization and are critical for T cell activation and development.
Abstract
The mechanism by which the cytosolic protein Zap70 physically interacts with and phosphorylates its substrate, the transmembrane protein LAT, upon T cell receptor (TCR) stimulation remains largely obscure. In this study, we found that the pharmacological inhibition of formins, a major class of actin nucleators, suppressed LAT phosphorylation by Zap70, despite TCR stimulation–dependent phosphorylation of Zap70 remaining intact. High-resolution imaging and three-dimensional image reconstruction revealed that localization of phosphorylated Zap70 to the immune synapse (IS) and subsequent LAT phosphorylation are critically dependent on formin-mediated actin polymerization. Using knockout mice, we identify mDia1 and mDia3, which are highly expressed in T cells and which localize to the IS upon TCR activation, as the critical formins mediating this process. Our findings therefore describe previously unsuspected roles for mDia1 and mDia3 in the spatiotemporal control of Zap70-dependent LAT phosphorylation at the IS through regulation of filamentous actin, and underscore their physiological importance in TCR signaling.
INTRODUCTION
T cells are activated by the T cell receptor (TCR) upon its recognition of the cognate major histocompatibility complex (MHC)–associated peptide antigen, presented on the surface of antigen-presenting cells (APCs). Resultant TCR signaling is critical for T cell development and activation (1). It is known that TCR activation triggers phosphorylation of proximal signaling molecules and induces the assembly of multimolecular signaling complexes (2). Because the TCR has no intrinsic enzymatic activity, the tyrosine kinase Lck initiates TCR signaling, conveying signals to the cytoplasmic protein kinase Zap70 (3) and then to the single transmembrane adaptor protein LAT (4). It has previously been shown that Zap70 phosphorylates tyrosine residues including Y171 and Y191 in the cytosolic tail of LAT upon TCR stimulation (5). However, although Zap70 and LAT appear to localize in close proximity to one another by diffraction-limited imaging, they are found to be spatially segregated at the subdiffraction scale in T cells both before and after TCR activation (6, 7). Therefore, the mechanism by which phosphorylated Zap70 physically interacts with, and subsequently phosphorylates, LAT remains a central question in immunology. Although several different mechanisms have been previously proposed, such as vesicular trafficking (8) or Lck-dependent bridging (9), it is possible that the efficient phosphorylation of LAT at the cell membrane by cytoplasmic Zap70 is a complex process requiring additional layers of regulation at the molecular level.
In the physiological setting, T cells are activated upon the formation of the immune synapse (IS) with an APC. The IS is a specialized contact interface between T cells and APCs that forms within seconds following the initial recognition of cognate peptide-MHC presented on the surface of APCs (10). Following the initial contact and triggering of TCR signaling, T cells undergo notable changes in their morphology that are driven by the actin cytoskeleton (11, 12). The robust spreading of the IS has previously been demonstrated to occur downstream of LAT phosphorylation (13). Phosphorylated LAT acts as a scaffold for several signaling molecules including the signal-transducing adapter molecule SLP76, forming a signaling complex known as the LAT signalosome (4). SLP76 recruits additional cytoskeleton regulators, such as Vav1 and Nck, and can therefore modify filamentous actin (F-actin) dynamics downstream of LAT (14). In addition, it has been proposed that, together, LAT and SLP76 may interact with phosphatidylinositol 3-kinase (PI3K) and activate the Dock2/Elmo-Rac signaling axis (15), although no obvious abnormalities in proximal TCR signaling have been associated with the suppression of this pathway. These studies demonstrate that F-actin, induced downstream of phosphorylated LAT, can facilitate IS spreading independently of TCR signaling (15). However, F-actin has also been shown to be critical for proximal TCR signaling in the IS, because total suppression of F-actin polymerization abolishes TCR signaling (16). These findings suggest that an F-actin component that is regulated independently of phosphorylated LAT may play a critical role in proximal TCR signaling. However, how such F-actin is regulated and how it participates in LAT activation remains largely unknown.
F-actin dynamics in living cells is regulated by a wide variety of actin regulatory proteins (17). Actin polymerization in mammalian cells is catalyzed not only by the Arp2/3 complex but also by formin-family proteins such as mDia (18). Although formins have been reported to be involved in events downstream of TCR signaling such as MTOC (microtubule-organizing center) repositioning (19) and TCR microcluster centralization (20) during IS formation, the role of formins in proximal TCR signaling per se remains unclear. Furthermore, at least 15 formins (21), many with potentially overlapping functions, are expressed in mammalian cells, and their contribution to TCR signaling and IS organization remains largely uncharacterized. In this work, we reveal previously unsuspected roles for specific formins in proximal TCR signaling, elucidating the underlying molecular mechanism and demonstrating their physiological function.
RESULTS
Formin activity is indispensable for Zap70-mediated LAT phosphorylation and F-actin polymerization in TCR-stimulated naïve CD8 T cells cultured in suspension
To investigate the role of formins in TCR-proximal signaling events in naïve CD8 T cells, we examined the effects of the formin inhibitor SMIFH2 (22) on cells cultured in suspension. We stimulated naïve CD8 T cells with anti-CD3 and anti-CD28 antibodies in the presence or absence of 30 μM SMIFH2 and analyzed the phosphorylation of the key downstream signaling molecules Zap70, LAT, and SLP76 by Western blotting (Fig. 1A). TCR stimulation in control cells induced sustained phosphorylation (up to 10 min) of Zap70 [Y319], while phosphorylation of LAT [Y191] and SLP76 [Y128] was transient, demonstrating peak phosphorylation at 1.5 to 3 min following TCR stimulation, and declining thereafter [Fig. 1, B (left) and C (left)]. Phosphorylation of Zap70 [Y319] was largely intact in SMIFH2-treated cells, while phosphorylation of LAT [Y191] and SLP76 [Y128] was strongly suppressed upon inhibition of formin activity [Fig. 1, B (right) and C (right)], suggesting that the actin polymerizing activity of formins is required for the phosphorylation of LAT and SLP76.
Fig. 1. Impaired TCR signaling and F-actin polymerization in TCR-stimulated naïve CD8 T cells treated with the formin inhibitor SMIFH2.
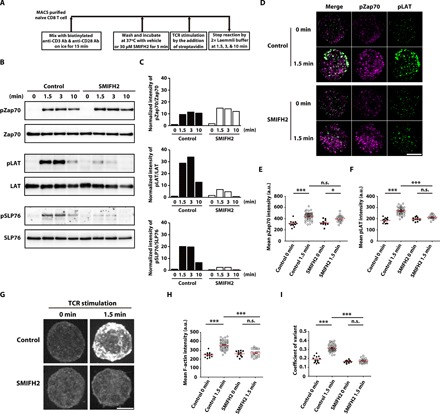
(A) Scheme of the experiment protocol. (B) Western blotting analysis of total levels and phosphorylated (p-) levels of Zap70 [Y319], LAT [Y191], and SLP76 [Y128] in lysates of naïve CD8 T cells unstimulated (0) or stimulated for 1.5, 3, or 10 min with anti-CD3 (10 μg/ml) and anti-CD28 (2 μg/ml) antibodies in the absence or presence of 30 μM SMIFH2 in suspension. Data are representative of three independent experiments. (C) Densitometric quantification of the Western blotting data in (B). The level of phosphorylated protein relative to total protein was normalized to the levels observed in unstimulated control cells. (D) Spinning disk confocal imaging of pZap70 [Y319] and pLAT [Y171] immunofluorescence in naïve CD8 T cells unstimulated (0 min) or stimulated for 1.5 min with anti-CD3 (10 μg/ml) and anti-CD28 (2 μg/ml) antibodies in the absence or presence of 30 μM SMIFH2 in suspension. Image stacks are shown. Scale bar, 5 μm. Quantification of pZap70 [Y319] (E) and pLAT [Y171] (F) staining intensity in unstimulated (0 min) or TCR-stimulated (1.5 min) naïve CD8 T cells untreated (control) or treated with 30 μM SMIFH2. Each data point represents a single cell. Data represent means ± SEM. n.s. = not significant, *P < 0.05, and ***P < 0.001 [one-way analysis of variance (ANOVA) with post hoc test]. (G) Spinning disk confocal imaging of F-actin staining in TCR-stimulated naïve CD8 T cells cultured in suspension in the absence (control) or presence of 30 μM SMIFH2. Image stacks are shown. Scale bar, 5 μm. Quantification of mean F-actin staining intensity (H) and the coefficient of variant of F-actin staining intensity (I). Each data point represents a single cell. Data represent means ± SEM. n.s., not significant, ***P < 0.001 (one-way ANOVA with post hoc test).
We next performed immunocytochemistry for the phosphorylated forms of Zap70 (pZap70 [Y319]) and LAT (pLAT [Y171]) and examined their spatial localization by spinning disk confocal microscopy. In control cells, TCR stimulation induced strong punctate signals for both pZap70 [Y319] and pLAT [Y171] at 1.5 min [Fig. 1, D (top), E (left), and F (left)], indicating that these phosphorylated proteins formed microclusters. In SMIFH2-treated cells, although the number and intensity of pZap70 microclusters were comparable to those in control cells [Fig. 1, D (bottom) and E (right)], we found that the fluorescence signals for pLAT [Y171] were significantly reduced in SMIFH2-treated cells [Fig. 1, D (bottom) and F (right)]. These results suggested that formin activity is required for pZap70-dependent phosphorylation of LAT, even though Zap70 phosphorylation itself is not dependent on formin activity.
To investigate the effects of formin inhibition on changes in F-actin organization downstream of TCR activation, T cells were stained with fluorescent phalloidin and observed by spinning disk confocal microscopy. We found that TCR stimulation for 1.5 min significantly increased mean F-actin intensity in control cells, suggesting that actin polymerization is strongly induced [Fig. 1, G (top) and H (left)] downstream of TCR activation. In contrast, we found that such TCR-induced F-actin polymerization was strongly suppressed in SMIFH2-treated naïve CD8 T cells [Fig. 1, G (bottom) and H (right)]. From quantitative analysis of the coefficient of variant, we also detected a larger variation in F-actin intensity in stimulated control cells than in unstimulated cells (Fig. 1I). Therefore, F-actin was not only polymerized but also significantly remodeled upon TCR stimulation. These results suggested that TCR stimulation induces actin polymerization and remodeling in a formin-dependent manner.
Formin inhibition impairs IS spreading and subsequent TCR microcluster centralization in naïve CD8 T cells stimulated on a stimulating lipid bilayer
We next used the stimulating lipid bilayer (SLB) system to mimic the physiological IS (23) and investigated the role of formin-dependent actin polymerization and remodeling upon TCR stimulation in the context of IS organization. MHC loaded with the agonist peptide SL8 (antigenic peptide “SIINFEKL” of the chicken ovalbumin) was incorporated into the planar lipid bilayer system. To examine the dynamics of TCR microclusters and IS formation, we placed naïve CD8 OT-I T cells stained with Alexa Fluor 568–conjugated anti-TCRβ antibody onto the SLB in the presence or absence of 10 μM SMIFH2, and then performed live imaging with total internal reflection fluorescence (TIRF) microscopy (Fig. 2A). In control cells, sporadic TCR microclusters began to appear at the contact site and then underwent relatively slow movement during the early attachment phase [Fig. 2B, top (from 0 to 20 s), and movie S1]. Subsequently, robust spreading of the plasma membrane was induced and new TCR microclusters continuously formed at the edge of the IS during the spreading phase (20 to 40 s after contact). Initially, these new TCR microclusters moved radially outward in conjunction with IS spreading [Fig. 2B, top (after 20 s), and movie S1]. When IS spreading was maximal (at ~40 s), TCR microclusters at the edge of the IS then underwent a rapid centripetal movement [Fig. 2B, top (after 40 s), and movie S1] to form a central supramolecular activation center (cSMAC). This represented the cSMAC formation phase (Fig. 2B, top, white arrow). In SMIFH2-treated cells, however, while the initial formation of TCR microclusters at the contact site was similar to that observed for control cells, their movement was slower (Fig. 2B, bottom, and movie S2). Moreover, the rapid membrane spreading of the IS was largely abolished (Fig. 2B, bottom, and movie S2) and TCR microclusters in SMIFH2-treated cells failed to coalesce to form a cSMAC (Fig. 2B, bottom, and movie S2). Tracking of all TCR microclusters in these images revealed a clear difference in TCR dynamics between control and SMIFH2-treated cells (Fig. 2C). We noted that this finding was consistent with a previous report that the centralization of TCR microclusters and cSMAC formation in Jurkat T cells stimulated on SLBs is regulated by formin (20). Quantification of IS area versus time showed that SMIFH2 treatment suppressed the rate of IS spreading on the SLB (Fig. 2D). Quantification of TCR microclusters further revealed that, although the number (Fig. 2E) and speed (Fig. 2F) of TCR microclusters were similar at early time points following cell attachment to SLBs, they were significantly decreased in SMIFH2-treated cells during the spreading phase of IS formation when compared with control cells. Together, these results suggested that IS spreading and subsequent TCR microcluster formation and movement are dependent on formin activity.
Fig. 2. IS spreading and TCR microcluster centralization are impaired in SMIFH2-treated naïve CD8 T cells.
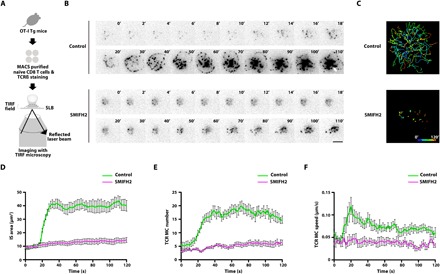
(A) Scheme of experiment design for TIRF live imaging of TCR microclusters in naïve CD8 OT-I T cells stimulated on SLBs. (B) TIRF images of TCRβ-stained naïve CD8 OT-I T cells stimulated on an SLB in the absence (control) or presence of 10 μM SMIFH2. Time is relative to the onset of IS spreading at 20 s. The white arrow indicates cSMAC. Scale bar, 5 μm. (C) Track plot of microcluster trajectories in (B). Plots show the paths taken by individual TCR microclusters over time (path pseudocolor represents time as indicated in the key). IS area (D), TCR microcluster (MC) number (E), and TCR microcluster speed (F) as imaged in (B) are plotted against time. n = 9 for control cells and n = 8 for SMIFH2-treated cells. Error bars represent SEM.
The above experiments examined the effects of formin inhibition from the beginning of IS formation. To further assess the effects of formin inhibition on subsequent events after initial contact and TCR microcluster formation, we imaged TCR microclusters by TIRF live microscopy and treated cells with SMIFH2 before IS spreading, after IS spreading, and after cSMAC formation. We found that the addition of SMIFH2 soon after initial TCR microcluster formation (but before IS spreading) impaired subsequent rapid IS spreading and TCR microcluster centralization (fig. S1, A and B, and movie S3). However, the addition of SMIFH2 after IS spreading had no apparent effect on TCR microcluster centralization and cSMAC formation (fig. S1, C and D, and movie S4). Furthermore, the addition of SMIFH2 after cSMAC formation had no apparent effect on cSMAC stability (fig. S1, E and F, and movie S5). Therefore, formin activity is indispensable during the early phase before IS spreading, but not after IS spreading and cSMAC formation.
Formin-dependent actin polymerization controls phosphorylated Zap70 localization to the IS and facilitates LAT phosphorylation
We next sought to compare the dynamics of F-actin and TCR microclusters within the IS between control and SMIFH2-treated cells. To enable live cell imaging of these structures, we used electroporation to introduce mRNA encoding LifeAct-EGFP (enhanced green fluorescent protein), a probe for F-actin (24), into OT-I T cell blasts, and then labeled surface TCRβ with fluorescently conjugated antibody. We then seeded these cells onto SLBs and subjected them to live-cell TIRF imaging. In control cells, polymerized F-actin first appeared at the site of T cell–SLB contact together with initial TCR microclusters [Fig. 3A, top (0 to 10 s), and movie S6]. Upon IS spreading (at 20 s), F-actin was observed to intensify at the periphery of the IS to form a ring-like structure with an F-actin–poor region at the center, toward which the TCR microclusters flowed to form the cSMAC [Fig. 3A, top (after 20 s), and movie S6]. In contrast, in SMIFH2-treated cells, the colocalization of F-actin with TCR microclusters during the initial contact phase was strongly suppressed (Fig. 3A, bottom, and movie S7). Moreover, at later time points, F-actin was apparently disorganized throughout the IS of SMIFH2-treated cells and the formation of the peripheral F-actin ring was impaired (Fig. 3A, bottom, and movie S7). Consequently, SMIFH2-treated cells did not form a single central F-actin–poor region as observed in control cells, and the centralization of TCR microclusters was disrupted (Fig. 3A, bottom, and movie S7). Consistent with this finding, stacked TIRF images for SMIFH2-treated cells (used to show temporal changes in F-actin dynamics at the T cell–SLB interface within a single composite image) revealed impaired F-actin polymerization upon contact with the SLB and the subsequent sporadic formation of disorganized F-actin structures, the intensity of which fluctuated over time (Fig. 3B, bottom). This contrasted the relatively stable and peripherally restricted F-actin observed in TCR-stimulated control cells (Fig. 3B, top).
Fig. 3. TCR stimulation–dependent F-actin polymerization and subsequent peripheral F-actin ring formation at the IS are dependent on formin activity.
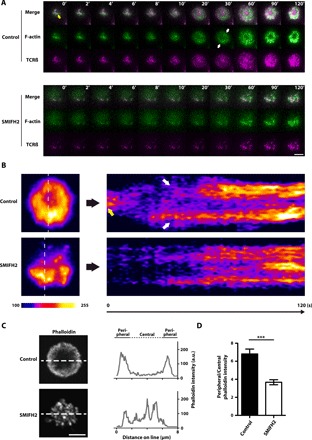
(A) Representative dual-color TIRF images of LifeAct-EGFP and TCR microclusters in CD8 OT-I T blasts cultured on SLBs in the absence (control) or presence of 10 μM SMIFH2. Time is relative time to the commencement of IS spreading at 10 s post-SLB attachment. Scale bar, 5 μm. (B) Time course of dynamic F-actin localization in TCR-stimulated control and SMIFH2-treated CD8 OT-I T blasts. TIRF images of LifeAct-EGFP over 2 min shown in (A) were used to generate the stacked images on the left. The upper and lower pseudocolored images correspond to control and SMIFH2-treated naïve CD8 OT-I T cells, respectively, stimulated on an SLB. Image intensities were scaled to a normalized LifeAct-EGFP intensity and range from 100 to 255 [arbitrary units (a.u.)]. Images in the right panel show linescan profiles of LifeAct-EGFP intensity over time for the dotted lines in the corresponding images on the left. The yellow arrow in (A) and (B) indicates polymerized F-actin colocalized with TCR microclusters at the contact point, and white arrows in (A) and (B) indicate the peripheral F-actin ring of the control CD8 OT-I T blast. (C) Left: Representative confocal images (single plane at the IS level) of F-actin (phalloidin) in control or 10 μM SMIFH2–treated naïve CD8 OT-I T cells stimulated on an SLB and then fixed at 10 min. Scale bar, 5 μm. Right: Linescan profiles of the signal intensities of phalloidin along the dotted white lines shown in the corresponding images on the left. (D) Ratiometric analysis of peripheral versus central F-actin intensity (phalloidin staining fluorescence intensity) within the IS of control and 10 μM SMIFH2–treated naïve CD8 OT-I T cells stimulated on an SLB and then fixed at 10 min. “Central” is defined as the central circular area from the cell origin to the half-radius, while “Peripheral” is defined as the remaining area. n = 23 for control cells and n = 22 for SMIFH2-treated cells. Data represent means ± SEM. ***P < 0.001 (Student’s t test).
Our findings from experiments performed using fixed cell specimens supported the live-cell imaging results; while a peripheral F-actin ring was present in control naïve CD8 OT-I T cells 10 min after stimulation on SLBs (Fig. 3C, top), F-actin was localized throughout the IS in SMIFH2-treated cells (Fig. 3C, bottom). Quantification of the F-actin distribution within the IS demonstrated significant differences between control and SMIFH2-treated naïve CD8 OT-I T cells (Fig. 3D). Together, these results suggest that the actin nucleation and polymerization activity of formins is required for initial F-actin polymerization upon TCR stimulation, as well as the subsequent spatiotemporal remodeling of F-actin at the IS periphery.
Next, we examined the relationship between formin-dependent polymerized F-actin and pZap70 localization in the IS of naïve CD8 OT-I T cells stimulated for 1.5 min on SLBs using confocal microscopy and three-dimensional (3D) image reconstruction. We found that, while most of the pZap70 [Y319] signal was observed beneath the peripheral F-actin ring of the IS in control cells [Fig. 4, A (top) and B (top), and movie S8], the pZap70 [Y319] signal was distributed throughout the cytoplasm, with no obvious accumulation at the IS in SMIFH2-treated cells [Fig. 4, A (bottom) and B (bottom), and movie S9]. Quantitative analysis of fluorescence intensity revealed that the overall intensity of pZap70 [Y319] staining was not significantly different between control and SMIFH2-treated cells (Fig. 4C). Together, these results suggest that the localization of pZap70 [Y319] to the IS is dependent on formin-mediated actin polymerization. In addition to pZap70 staining, we also examined pLAT [Y171] staining together with F-actin in naïve CD8 OT-I T cells stimulated for 1.5 min on SLBs. In control cells, a strong pLAT [Y171] signal was observed beneath the peripheral F-actin ring of the IS [Fig. 4, D (top) and E (top), and movie S10]. In contrast, we found that, while pLAT localization at the IS was still observed in SMIFH2-treated cells, the intensity and size of the signal were significantly attenuated when compared with controls [Fig. 4, D (bottom), E (bottom), and F, and movie S11]. Therefore, we conclude that formin-dependent polymerized F-actin facilitates LAT phosphorylation by pZap70 at the IS.
Fig. 4. Formin-dependent F-actin is critical for LAT phosphorylation by pZap70 at the IS.
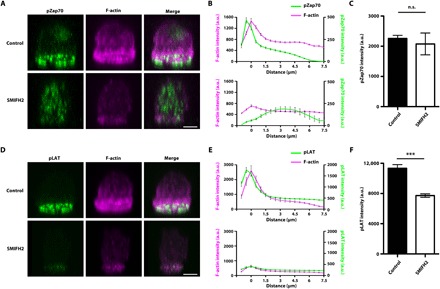
3D reconstruction of confocal image stack of control and 10 μM SMIFH2–treated naïve CD8 OT-I T cells stimulated on SLBs and stained for F-actin and pZap70 [Y319] (A) or F-actin and pLAT [Y171] (D). Scale bars, 3 μm. Quantification of staining intensity of F-actin together with pZap70 (B) or pLAT (E) in the images from each confocal plane (0.5 μm per slice). The distance is relative to the plane of maximum F-actin staining intensity. Quantification of pZap70 (C) and pLAT (F) staining intensity per cell. For pZap70 immunocytochemistry experiment, n = 39 for control cells and n = 21 for SMIFH2-treated cells. For pLAT immunocytochemistry experiment, n = 15 for control cells and n = 26 for SMIFH2-treated cells. Data represent means ± SEM. n.s. = not significant, ***P < 0.001 (Student’s t test).
The formins mDia1 and mDia3 are required for TCR signaling–dependent activation of T cells
mDia1, mDia2, and mDia3 are a group of formin proteins that can be activated by Rho guanosine triphosphatases (GTPases) (25). Given previous reports that mDia1 is strongly expressed in T cells (19, 20), we examined the expression of these three mDia isoforms in naïve CD8 T cells by quantitative reverse transcription polymerase chain reaction (qRT-PCR). We found that, in addition to mDia1, mDia3 was also expressed in naïve CD8 T cells, albeit at a lower level (Fig. 5A). We then isolated naïve CD8 T cells from knockout (KO) mice for each of these isoforms to examine the roles of mDia1 and mDia3 in TCR stimulation–dependent Zap70 and LAT phosphorylation. We were unable to detect any significant difference in pZap70 [Y319] or pLAT [Y191] levels between TCR-stimulated wild-type (WT) and mDia1 KO (fig. S2A), or WT and mDia3 KO, naïve CD8 T cells treated in suspension (fig. S2B). Given previous findings from our group, as well as others, on the functional redundancy between mDia1 and mDia3 (26–30), we suspected that mDia1 and mDia3 may have overlapping functions in TCR signaling, and thus may be able to functionally compensate for one another. We therefore used the UBC-creERT2 tamoxifen-inducible system to generate mDia1 and mDia3 conditional double-KO (cDKO) naïve CD8 T cells (Fig. 5B). mDia1/mDia3 protein expression and loss of these protein expressions were confirmed by immunoblotting of lysates from control and mDia1/3 cDKO naïve CD8 T cells, respectively (Fig. 5C). We then examined pZap70 [Y319] and pLAT [Y191] levels upon TCR stimulation and found that pLAT [Y191] was strongly suppressed in TCR-stimulated mDia1/3 cDKO naïve CD8 T cells (Fig. 5, D and E), in a similar manner to cells treated with SMIFH2 (Fig. 1, B and C). It is noteworthy that we also found that TCR stimulation–dependent interleukin-2 (IL-2) production and cell proliferation were strongly impaired in mDia1/3 cDKO naïve CD8 T cells (Fig. 5, F and G). Therefore, we concluded that, together, mDia1 and mDia3 are critical formins in the regulation of TCR signaling through Zap70-dependent LAT phosphorylation, and therefore play indispensable roles in T cell activation.
Fig. 5. mDia1 and mDia3 are expressed in naïve CD8 T cells and are critical for proximal TCR signaling and T cell activation.
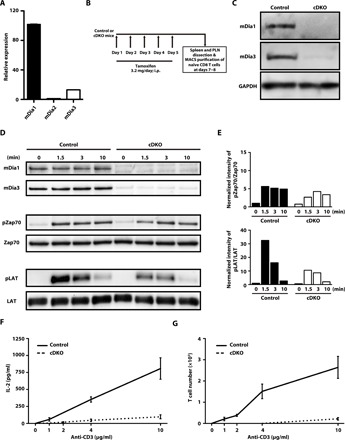
(A) Relative expression of the mDia isoforms in naïve CD8 T cells as determined by quantitative reverse transcription polymerase chain reaction (qRT-PCR). mDia1 and mDia3 levels are normalized to that of mDia2. Bars represent means ± SEM (n = 3). (B) Scheme of the experimental schedule for the conditional KO of mDia1 and mDia3 in UBC-creERT2 × mDia1/3 double-floxed mice by tamoxifen injection. Mice received intraperitoneal (i.p.) tamoxifen injections consecutively for 5 days. Spleen and peripheral lymph nodes were isolated at 2 or 3 days after the last tamoxifen injection and naïve CD8 T cells were then purified by magnetic-activated cell sorting (MACS). (C) Representative Western blotting data of mDia1 and mDia3 protein levels in the lysates of control mDia1/3 double-floxed and tamoxifen-induced mDia1/3 conditional DKO (cDKO) naïve CD8 T cells. (D) Western blotting analysis of mDia1 and mDia3 protein levels and total and phosphorylated protein levels of pZap70 [Y319] and pLAT [Y191] in the lysates of control mDia1/3 double-floxed and tamoxifen-induced mDia1/3 cDKO naïve CD8 T cells unstimulated (0) or stimulated for 1.5, 3, or 10 min with anti-CD3 (10 μg/ml) and anti-CD28 (2 μg/ml) antibodies. Data are representative of three independent experiments. (E) Densitometric quantification of the Western blotting data in (D). The value of the phosphorylated form relative to total protein was normalized to that obtained for unstimulated control cells. (F) ELISA analysis of IL-2 levels secreted by control mDia1/3 double-floxed and mDia1/3 cDKO naïve CD8 T cells after stimulation for 48 hours in vitro without (0) or with anti-CD3 antibody (1, 2, 4, or 10 μg/ml) and anti-CD28 antibody (2 μg/ml). n = 4 for each group. Error bars represent SD. (G) Anti-CD3/CD28–induced proliferation of naïve CD8 T cells from control mDia1/3 double-floxed and mDia1/3 cDKO mice. Naïve CD8 T cells were seeded in 96-well plates coated with anti-CD3 antibody (0, 1, 2, 4, or 10 μg/ml) and in the presence of soluble anti-CD28 antibody (2 μg/ml). Cells were cultured for 48 hours and then assayed for proliferation. n = 4. Error bars represent SD.
mDia1 and mDia3 localize to the IS periphery and are required for IS spreading, peripheral F-actin ring formation, and TCR microcluster centralization
Given the importance of mDia1/3 in proximal TCR signaling, we next used immunocytochemistry in conjunction with TIRF microscopy to examine and compare the endogenous localization of mDia1 and TCRβ in fixed naïve CD8 T cells stimulated on SLBs. We found that mDia1 was concentrated at the periphery of the spreading IS (Fig. 6A, left). At later stages of spreading, mDia1 was almost completely absent within the central area of the IS where TCR microclusters centralize to form the cSMAC (Fig. 6A, right). Furthermore, we performed TIRF live imaging of EGFP-mDia1 and TCRβ in OT-I T cell blasts and found that mDia1 initially colocalized with TCR microclusters within the IS upon cell contact with the SLB but then concentrated at the periphery upon IS spreading (Fig. 6B and movie S12). Notably, newly formed TCR microclusters that appeared at the edge of the spreading IS moved centrally, across the peripheral mDia1 ring, and eventually reached the center area to form the cSMAC. TIRF live imaging of EGFP-mDia3 and TCRβ in OT-I T cell blasts revealed similar localization dynamics for mDia3 within the IS (fig. S3 and movie S13). Together, these results suggest that mDia1 and mDia3 initially localize to TCR microclusters but then form a ring at the periphery of the spreading IS.
Fig. 6. Peripheral F-actin ring formation is impaired in the IS of mDia1/3-deficient naïve CD8 T cells.
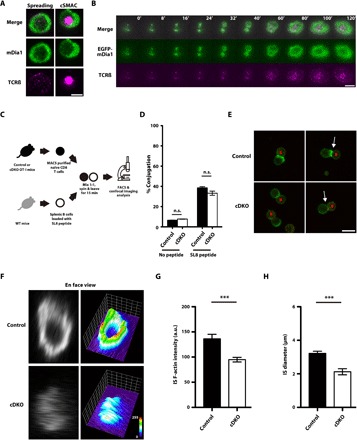
(A) Representative TIRF images of TCRβ and mDia1 in the IS of a naïve CD8 OT-I T cell fixed at 3 min after stimulation on an SLB. Scale bar, 5 μm. (B) Representative TIRF images of TCR microclusters together with EGFP-mDia1 in a CD8 OT-I T blast stimulated on an SLB. Time is relative to initial cell-SLB contact. Scale bar, 5 μm. (C) Scheme of the experimental design for the cell conjugation assay for control OT-I or mDia1/3 cDKO naïve CD8 OT-I T cells cocultured with SL8 peptide–loaded splenic B cells. Naïve CD8 OT-I T cells from control mice or tamoxifen-induced mDia1/3 cDKO mice and SL8 peptide–loaded splenic B cells were mixed at a one-to-one ratio, centrifuged, and then incubated for 15 min. After fixation, conjugated cells were stained with phalloidin. Half of the stained cells were then analyzed by FACS, while the remaining cells were subjected to F-actin staining and imaging by confocal microscopy. (D) Percentage of cell conjugates from coculture assays, as determined by FACS. Data are from three independent experiments. Bars represent means ± SEM. n.s. = not significant (one-way ANOVA with post hoc test). (E) Representative confocal image stacks of cell conjugates from control (upper) and mDia1/3 cDKO (lower) naïve CD8 OT-I T cells cultured with SL8 peptide–pulsed splenic B cells. Cells were labeled with anti-B220 antibody (red) and phalloidin (green) and imaged with a confocal microscope. White arrows indicate the cell conjugates used for the reconstructed en face views shown in (F). B, B cell. Scale bar, 10 μm. (F) Reconstructed en face views of F-actin at the IS in control and mDia1/3 cDKO naïve CD8 OT-I T cells (left) and surface plots of the corresponding en face view (right). Quantification of IS F-actin intensity (G) and IS diameter (H) in conjugates of control and mDia1/3 cDKO naïve CD8 OT-I T cells. n = 11 for control cells and n = 13 for mDia1/3 cDKO cells. Bars represent means ± SEM. ***P < 0.001 (Student’s t test).
Next, we assessed the impact of mDia1/3 loss on IS formation between naïve CD8 OT-I T cells and SL8-pulsed splenic B cells using a conjugation assay (Fig. 6C). Although fluorescence-activated cell sorting (FACS) analysis revealed that the formation of cell conjugates was not significantly affected (Fig. 6D), confocal imaging analysis revealed that the IS of mDia1/3 cDKO cells was smaller and that F-actin staining at the IS interface was reduced when compared with controls (Fig. 6E). Confocal image stacks and the reconstructed en face view of the IS revealed a strong signal for polymerized F-actin at the periphery and an F-actin–devoid space at the center in control cells (Fig. 6F, top), a finding that was consistent with previous reports (31, 32). Conversely, the polymerized F-actin signal was weaker in the IS of mDia1/3 cDKO cells (Fig. 6F, bottom left). Impaired peripheral F-actin ring formation in the IS of mDia1/3 cDKO naïve CD8 OT-I T cells was further confirmed by surface plot analysis of the en face view (Fig. 6F, bottom right). Quantification of image data confirmed the significant reduction in F-actin intensity at the IS interface of cell conjugates formed using mDia1/3 cDKO naïve CD8 OT-I T cells (Fig. 6G) and also an associated decrease in IS diameter (Fig. 6H). Together, these results suggest that mDia1/3 is indispensable for both IS peripheral F-actin ring formation and IS spreading. Notably, live-cell imaging of TCR microcluster dynamics in mDia1/3 cDKO naïve CD8 OT-I T cells stimulated on SLBs revealed that loss of mDia1/3 impairs IS spreading, TCR microcluster centralization, and cSMAC formation (fig. S4 and movies S14 and S15) in a manner similar to that observed for SMIFH2-treated cells (Fig. 2B and movies S1 and S2).
mDia1/3 are indispensable for the positive selection of T cells
To explore the physiological significance of mDia1/3 in T cell function, we investigated the phenotype of conventional mDia1/3 double-KO (DKO) mice (26). We found that these mice exhibited T cell lymphopenia (fig. S5, A to D). Histological analysis of the thymus revealed that the cortex, the region where CD4 and CD8 double-positive (DP) thymocytes reside, was larger and the medulla, the region where CD4 single-positive (SP) and CD8 SP thymocytes reside, was smaller in mDia1/3 DKO mice when compared with control WT mice (fig. S5E). We therefore performed FACS analysis of CD4 and CD8 surface expression on thymocytes and found significantly higher proportions of DP and CD4+CD8int DP thymocytes and significantly lower proportions of CD4 SP and CD8 SP thymocytes in the thymus of mDia1/3 DKO mice when compared with WT controls (fig. S5, F and G). This phenotype was not observed in mDia1 KO mice (fig. S6, A and B) or mDia3 KO mice (fig. S6, C and D). These findings therefore suggested that, together, mDia1 and mDia3 are required for the positive selection of T cells in the thymus. The impaired positive selection phenotype associated with mDia1/3 DKO is intrinsic to developing T cells because bone marrow transfer of mDia1/3 DKO cells to irradiated WT mice (fig. S7) and lck-cre–driven conditional deletion of mDia1/3 mimicked the impaired positive selection phenotype (fig. S8). To exclude the possibility that the polyclonal TCR repertoire may complicate the interpretation of these results, we also assessed the effects of mDia1/3 deficiency on positive selection in a defined antigen-specific TCR background, OT-I and OT-II, and obtained similar results (fig. S9). These results demonstrate the critical and redundant role of mDia1 and mDia3 in the positive selection of T cells, a process dependent on TCR signaling (1).
Last, we confirmed the impact of mDia1 and mDia3 double deficiency on IS spreading, TCR microcluster dynamics, and F-actin assembly in thymocytes by TIRF imaging of cells stimulated on SLBs. Similar to naïve CD8 OT-I T cells (Fig. 2B, top, and fig. S4A, top), control OT-I thymocytes rapidly spread and that TCR microclusters continuously formed at the cell membrane edge (fig. S10A, top, black arrows). Subsequently, when IS spreading reached the maximum, TCR microclusters moved toward the cell center to form a cSMAC (fig. S10A, top, white arrow, and movie S16). On the other hand, in mDia1/3 cDKO (lck-cre) OT-I thymocytes, while the initial formation of TCR microclusters at the contact site was unaffected, their movement was significantly slower when compared with controls (fig. S10A, bottom, and movie S17). Moreover, rapid membrane spreading associated with IS maturation was largely abolished and TCR microclusters in mDia1/3 cDKO (lck-cre) OT-I thymocytes failed to coalesce to form a cSMAC (fig. S10A, bottom, and movie S17). Quantitative analysis of image data revealed that the TCR microcluster number and the speed of TCR microcluster movement during IS formation were significantly reduced in mDia1/3 cDKO (lck-cre) OT-I thymocytes when compared with controls (fig. S10B). Consistent with these findings, an analysis of fixed samples revealed that, after seeding on SLBs at 10 min, while 71.8 ± 3.7% of TCRβ-positive control OT-I thymocytes formed a cSMAC, cSMAC formation was seen in only 35.6 ± 11.2% of TCRβ-positive mDia1/3 cDKO (lck-cre) OT-I thymocytes at the same time point (fig. S10C). In addition, mDia1/3 cDKO (lck-cre) OT-I thymocytes fixed 3 min after stimulation on SLBs also showed reduced IS area (fig. S10D). Analysis of F-actin and TCRβ staining in cells fixed 10 min after contact with SLBs revealed a peripheral F-actin ring surrounding the cSMAC in control OT-I thymocytes (fig. S10E, top). In contrast, F-actin was localized throughout the IS and cSMAC formation was impaired in mDia1/3 cDKO (lck-cre) OT-I thymocytes (fig. S10E, bottom). Quantitative analysis of the F-actin intensity distribution within the IS confirmed a significant reduction in peripheral F-actin in the IS of mDia1/3 cDKO (lck-cre) OT-I thymocytes when compared with controls (fig. S10, F and G). Together, these results suggest that mDia1/3, through their regulation of F-actin, are critical for IS spreading, TCR microcluster dynamics, and cSMAC formation also in thymocytes.
DISCUSSION
Stimulation of T cells with cognate antigen rapidly induces phosphorylation of proximal components of the TCR signaling complex. In this study, we started by examining the effects of formin inhibition on the phosphorylation kinetics of the key proximal TCR signaling molecules Zap70, LAT, and SLP76 in naïve CD8 T cells TCR-stimulated in suspension. We found that, while Zap70 phosphorylation was relatively sustained, the phosphorylation of the Zap70 substrates LAT and SLP76 was transient and dependent upon the activity of formins. It has previously been shown that two distinct pools of pLAT exist in TCR-stimulated T cells, a plasma membrane pLAT pool and a vesicular pLAT pool (4, 8). Recently, it has also been demonstrated that, while the plasma membrane pool of LAT is rapidly phosphorylated, the vesicular pool is phosphorylated at later time point following TCR activation (33). Because formin inhibition suppressed LAT phosphorylation from the earliest time points examined, we concluded that phosphorylation of the membrane pool of LAT occurs in a formin-dependent manner. However, formin activity is dispensable for Zap70 phosphorylation. Zap70 phosphorylation has previously been shown to be regulated by its catch and release from the TCR (34), and its TCR dwell time has been shown to determine its kinase activity independently of LAT (35). Our results therefore suggest that formin-dependent F-actin polymerization and subsequent F-actin remodeling are not involved in the interaction between Zap70 and the TCR. It should be noted that a previous study has reported that formin activity is critical for TCR signaling from the Zap70 level (20). Although we did not address the reason for this discrepancy experimentally, this may be because of technical difference between the studies, such as the cell types used; our study used naïve CD8 T cells, while the previous study used Jurkat T cells (20). It should also be noted that in this previous study, the authors examined the level of Zap70 and LAT phosphorylation in cell conjugate assays using APCs. Given that F-actin has previously been implicated in APC-mediated TCR signaling (36), it is possible that formin inhibition in APCs may also indirectly affect TCR signaling.
In our study, we also found a correlation between Zap70-mediated LAT phosphorylation and formin-dependent F-actin polymerization in TCR-stimulated naïve CD8 T cells in suspension. Our results suggest the possibility that formin-dependent F-actin networks interact with pZap70 and pLAT upon TCR stimulation. We further dissected the role of formin-dependent F-actin in the regulation of pZap70 and pLAT in the context of the IS using an SLB system. Using this system, we observed that pZap70 is localized beneath the peripheral F-actin ring in the IS of stimulated control T cells. However, treatment with a formin inhibitor suppressed the formation of this peripheral F-actin ring and impaired pZap70 localization to the IS. Consistent with this finding, phosphorylated LAT, which also localizes to the IS periphery in control cells, was significantly diminished in the IS of formin inhibitor–treated T cells. Together, these results suggest that pZap70-dependent LAT phosphorylation at the IS is regulated by formin-dependent F-actin assembly. We speculate that the recently proposed mechanism whereby Lck served as a bridge between pZap70 and pLAT (9) may occur subsequently to the formin-mediated transient and physical coupling of pZap70 and LAT that we report here to further stabilize the complex and therefore promote efficient LAT phosphorylation.
It should also be noted that we found formin activity to be critical for IS spreading and TCR microcluster formation during the early stages of IS formation in naïve CD8 T cells, but found it to be dispensable at later stages, after IS spreading or cSMAC formation. Given that IS spreading upon TCR stimulation occurs downstream of LAT phosphorylation (37), impaired IS spreading and the subsequent impaired TCR microcluster formation are likely to be secondary consequences of suppressed LAT phosphorylation upon formin inhibition. Although the TCR microcluster number was reduced, total cellular levels of Zap70 phosphorylation were not significantly affected. Our 3D image reconstruction analysis revealed that although pZap70 levels at the IS of SMIFH2-treated naïve CD8 T cells were reduced, this was not a consequence of impaired protein phosphorylation, but rather because the phosphorylated protein was mislocalized to the cytoplasm. Given the catch and release mechanisms for the regulation of phosphorylated Zap70 at the TCR (34), it is possible that pZap70 may be released into the cytoplasm in the absence of formin-dependent polymerized F-actin at the IS.
A previous study into the role of the formin and Rho effector, mDia1, in TCR signaling and IS formation in Jurkat T cells concluded that this protein had no effect on TCR signaling and the TCR activation–dependent formation of F-actin–rich structures (19). Consistent with these findings, we found that naïve CD8 T cells from mDia1 KO mice did not exhibit an impaired proximal TCR signaling phenotype. However, given that previous studies from our group (26–29) and others (30) have demonstrated functionally redundant roles between mDia1 and mDia3 isoforms in the polymerization of F-actin, we further investigated potential T cell phenotypes associated with loss of both mDia1 and mDia3. We found that Zap70-dependent LAT phosphorylation was significantly impaired in TCR stimulated naïve CD8 T cells from mDia1/3 cDKO mice in suspension, a finding that was similar to that observed for SMIFH2-treated cells. Loss of mDia1/3 in naïve CD8 T cells strongly suppressed TCR stimulation–dependent IL-2 production and cell proliferation, suggesting a role of mDia1/3 in T cell activation. Using the SLB system, we have shown that mDia1 initially colocalizes with early TCR microclusters but then concentrates at the IS periphery, which is the site where the formin-dependent peripheral F-actin ring subsequently forms. Furthermore, using a cell conjugation assay, we demonstrated that loss of mDia1/3 in naïve CD8 T cells impaired peripheral F-actin ring formation and IS spreading in a similar manner to that observed in SMIFH2-treated cells. We also found that thymocytes from mice deficient in mDia1/3 showed impaired IS spreading and TCR microcluster centralization on SLBs. Notably, the positive selection of these cells in vivo was also impaired, suggesting that mDia1/3 is critical for TCR signaling in the physiological context. It has previously been shown that thymocytes expressing a constitutively active form of RhoA, an upstream regulator of mDia1/3 (38), demonstrate augmented positive selection (39). On the basis of this previous report and our findings, we speculate that RhoA may exert its effects on thymocyte positive selection through mDia1/3. The underlying molecular mechanism controlling mDia1/3 activation upon TCR stimulation warrants future investigation.
In this study, we have combined pharmacological inhibition of formins, genetic manipulation of mDia1/3, an SLB system, and high-resolution imaging in conjunction with 3D image reconstruction to reveal an indispensable yet hitherto unrecognized role for the formins, mDia1 and mDia3, in Zap70-dependent LAT phosphorylation at the IS. Our study therefore advances a new conceptual model in which F-actin spatiotemporally regulates proximal TCR signaling, providing an important framework for further dissection of the role of F-actin in T cell development and activation.
MATERIALS AND METHODS
Study design
The aim of this study was to reveal the function of formin-mediated F-actin assembly in proximal TCR signaling using mouse naïve CD8 T cells and mouse CD8 T blasts. All animal experiments were conducted in accordance with the U.S. National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of Kyoto University Graduate School of Medicine and University of California, San Francisco. WT mice were used as the control for experiments with mDia1 KO, mDia3 KO, and mDia1/3 DKO mice. mDia1/3 double-floxed mice were used as the control for experiments with mDia1/3 double-floxed × UBC-creERT2 mice (mDia1/3 cDKO mice). mDia1/3 double-floxed × OT-I mice were used as the control for experiments with mDia1/3 double-floxed × UBC-creERT2 × OT-I mice. Lck-cre transgenic mice were used as the control for experiments with mDia1/3 double-floxed × lck-cre mice [mDia1/3 cDKO (lck-cre) mice]. mDia1/3 double-floxed × OT-I mice and mDia1/3 double-floxed × OT-II mice were used as the control for experiments with mDia1/3 double-floxed × lck-cre × OT-I mice and mDia1/3 double-floxed × lck-cre × OT-II mice, respectively. Age- and gender-matched mice of the same strain were used in each experiment. Experimental replicates were indicated in the figure legends. Randomization was not performed in the animal experiments of this study.
Mice
mDia1 KO mice, mDia3 KO mice, and mDia1/3 DKO mice have been described previously (26). In brief, mDia1 KO mice and mDia3 KO mice (each lacking exon 1 of the WT gene) were generated by homologous recombination and backcrossed for more than 10 generations to C57BL/6N mice. mDia1/3 DKO mice were generated as follows: mDia1 heterozygous males were crossed with mDia3 heterozygous females. Among the offspring, mDia1 heterozygous/mDia3 hemizygous male mice were then mated with mDia3 heterozygous female mice, and the resultant male and female mice of mDia1 heterozygous/mDia3 homozygous genotype were then intercrossed to generate mDia1/3 DKO mice. mDia1/3 double-floxed mice were generated by crossing mDia1 floxed mice with mDia3 floxed mice (28) backcrossed to C57BL/6N mice for more than 10 generations. To delete mDia1 and mDia3 in the T cell–lineage, mDia1/3 double-floxed mice were mated with transgenic mice expressing Cre recombinase under the control of lck proximal promoter. Mice with transgenic expression of lck-cre, UBC-creERT2, OT-I, and OT-II were from the Jackson Laboratory. CD45.1-congenic C57BL/6J mice for bone marrow transplantation experiments were also obtained from the Jackson Laboratory. Tamoxifen (T-5648, Sigma) was dissolved in ethanol and then diluted in corn oil (C8267, Sigma) to a final concentration of 20 mg/ml. For tamoxifen-induced deletion of mDia1/3, tamoxifen was intraperitoneally injected into mDia1/3 double-floxed × UBC-creERT2 mice or mDia1/3 double floxed × UBC-creERT2 × OT-I mice at a dose of 3.2 mg/day for five consecutive days.
Cell preparation and stimulation
Thymocytes from thymus and lymphocytes from spleen and lymph nodes were isolated by mechanical disruption between the frosted ends of glass slides (S2215, Matsunami) and filtrated through a 70-μm nylon cell strainer (352350, BD Falcon). Naïve CD8 T cells from control or tamoxifen-induced mDia1/3 conditional DKO mice were isolated using a negative selection MACS isolation kit (130-096-543, Miltenyi Biotec). For the stimulation of naïve CD8 T cells in suspension, cells were incubated at 37°C under 5% CO2 for 2 hours in RPMI 1640 medium (R-8758, Sigma) containing 10% fetal calf serum (12483-020, Gibco), resuspended in phosphate-buffered saline (PBS), incubated with biotin-conjugated anti-mouse CD3ε antibody (553059, BD Pharmingen) and anti-mouse CD28 antibody (102102, BioLegend) on ice for 15 min, washed, and then stimulated by cross-linking with streptavidin (43-4301, Invitrogen) at 37°C.
Immunocytochemistry (in suspension)
For immunocytochemistry of naïve CD8 T cells in suspension, cells were fixed with 4% paraformaldehyde (PFA) in phosphate buffer for 15 min, washed with PBS, and allowed to settle for 45 min onto poly-l-lysine (P6282, Sigma)–coated coverslips (Matsunami) at 37°C. Cells were then washed with PBS, permeabilized with 0.3% Triton-X100 in PBS for 10 min, and blocked with 1% bovine serum albumin in PBS for 45 min at room temperature. The cells were then incubated with indicated primary antibodies at 4°C overnight, washed, and stained with secondary antibodies for 45 min at room temperature. For F-actin staining, cells were incubated with fluorescent dye–labeled phalloidin diluted in 0.3% Triton-X100/PBS for 20 min at room temperature. Cells were finally washed with PBS and mounted onto slides with ProLong Diamond Antifade reagent (P36970, Life Technologies). Fluorescence images were acquired with an SD-OSR IX83 inverted microscope (Olympus) equipped with a 100× numerical aperture (NA) 1.4, UPLSAPO oil immersion objective (Olympus) and Yokogawa W1 spinning disk unit (Yokogawa) controlled by MetaMorph software (Universal Imaging). Quantification of F-actin, pZap70 [Y319], and pLAT [Y171] staining intensity was performed using ImageJ software. Coefficient of variant of F-actin staining intensity was calculated as the ratio of the SD and average staining intensity.
Imaging of the IS on SLBs
TIRF imaging was performed as previously described (23). An Olympus IX-81 invert microscope equipped with a laser TIRF slider (Olympus) was used for imaging of naïve CD8 OT-I T cells and CD8 OT-I T blasts. For TIRF microscopy, a 100× NA 1.4 PlanApo objective lens was used. For two-color TIRF imaging, a G-base, two-channel simultaneous imaging system (G-Angstrom) with a 560-nm longpass dichroic filter and 525-nm/50-nm and 605-nm/70-nm bandpass emission filters was used to split the camera field into two image channels for simultaneous imaging of EGFP and Alexa Fluor 568 fluorescence. Images were acquired with an iXon Ultra 888 EMCCD (Andor) and MetaMorph software (Universal Imaging). For imaging of OT-I thymocytes, a Zeiss Axiovert 200M equipped with a laser TIRF slider and a 100× NA 1.45 Plan-Fluar objective lens was used. Images were acquired with an Evolve EMCCD (Photometrics) and MetaMorph software (Universal Imaging).
For labeling of surface TCRs, 2 × 106 OT-I thymocytes, naïve CD8 OT-I T cells, or CD8 OT-I T blasts were stained for 30 min on ice with 1 μg of Alexa Fluor 568–labeled H57-597 anti-TCRβ antibody (Bio X Cell) in 0.05 ml of complete phenol red–free RPMI medium. For imaging of cells stimulated on bilayers, 1 × 105 cells in 0.1 ml of complete phenol red–free RPMI medium were added to the PBS (0.25 ml) overlaying the bilayer. In TIRF time-lapse imaging experiments, cells undergoing initial spreading on bilayers were located, and TIRF images were acquired for 2 to 5 min at intervals of 1 or 2 s for naïve CD8 OT-I T cells and CD8 OT-I T blasts and for 2 to 5 min at intervals of 0.25 or 0.5 s for OT-I thymocytes. The exposure length was 100 to 200 ms. Imaris software was used for tracking and quantitative analysis of TCR microcluster dynamics. In immunocytochemistry of experiment of cells stimulated on SLBs, cells were fixed with 4% PFA and then blocked and stained according to standard protocols. TIRF images of fixed cells were acquired on the same microscope used for live-cell imaging. Confocal images of fixed cells on SLBs were acquired with a laser scanning confocal imaging system (Zeiss LSM710) using a 63×, NA 1.4 oil immersion objective lens. Line scanning analysis of F-actin staining within a single confocal plane corresponding to the IS was conducted using ImageJ software (NIH). 3D reconstruction of confocal image stacks was performed using Imaris software (Bitplane). Quantification of the percentage of cSMAC was performed by visual inspection of the TIRF images. Quantification of IS area and staining intensity in the images of each confocal plane was conducted using ImageJ software (NIH).
mRNA electroporation
For electroporation, 1 μg of LifeAct-EGFP mRNA (TriLink), 1 μg of EGFP-mDia1 mRNA (TriLink), or 1 μg of EGFP-mDia3 mRNA (TriLink) was mixed with 5 × 105 CD8 OT-I T blasts (TCR-stimulated with SL8 peptide–loaded splenocytes for 48 hours) in 10 μl of T buffer (Thermo Fisher Scientific). Cells were then loaded in 10-μl tips and electroporated with the Neon Transfection System (Invitrogen) according to the manufacturer’s protocol with three pulses of 1400 V for 10 ms.
Conjugation assay
Naïve CD8 T cells were isolated from control or mDia1/3 cDKO OT-I mice using a negative selection MACS purification kit (130-096-543, Miltenyi Biotec). B cells from WT mice were isolated using MACS beads (18954, Stemcell Technologies), incubated with SL8 peptide (1 μg/ml) (Sigma) for 30 min at 37°C, and then surface-stained with phycoerythrin (PE)–conjugated anti-B220 antibody (12-0452-83, eBioscience). For cell conjugation assays, equal volumes of naïve CD8 OT-I T cells and SL8 peptide–pulsed B cells were mixed and incubated at 37°C for 15 min. Conjugated cells were then fixed with 0.5% PFA for FACS analysis using the LSR Fortessa System (BD Biosciences). The percentage of conjugates within the culture was analyzed using FlowJo software (Tree Star Inc.). For imaging of the F-actin ring, cell conjugates were fixed with 4% PFA, permeabilized, stained with Alexa Fluor 488-conjugated phalloidin, and then imaged with a Leica SP5 laser confocal microscope equipped with 100× NA 1.4 HCX PL APO CS objective lens (Leica). 3D reconstruction of stacked confocal images and en face view images were generated by Volocity software (PerkinElmer). Surface plots of 3D reconstructed images were generated using ImageJ software (NIH). Quantification of IS F-actin intensity and IS diameter was performed using ImageJ software (NIH).
Statistical analyses
Quantitative data were reported as means ± SEM or means ± SD. Excel (Microsoft) and Prism (GraphPad Software) were used for statistical analyses. The Student’s t test or one-way ANOVA with post hoc test was used for the evaluation of significance as specified in the figure legends. The sample number (N) refers to the number of biological replicates. n.s. = not significant (P > 0.05), *P < 0.05, **P < 0.01, and ***P < 0.001.
Supplementary Material
Acknowledgments
We thank J. Monypenny for critical reading of the manuscript, P. Beemiller for advice on experiments using the SLB system, A. Nomachi and M. Madhu for technical assistance, and K. Naruo for assistance with animal experiments. We are also grateful to T. Arai for secretarial assistance. Funding: This work was supported by a grant from the Ono Medical Research Foundation (203170600055 to D.T.), a grant from the Singapore National Research Foundation Fellowship (NRF-NRFF-2011-04 to P.K.), grants from Ministry of Education Academic Research Fund Tier 2 (MOE-T2-1-124 and MOE-T2-1-045 to P.K.), a grant from MBI seed funding (P.K.), grants from the US National Institutes of Health (R01 AI052116 to M.F.K.) and Grants-in-Aid for Scientific Research from MEXT of the Japanese Government (25860188 and 15K18986 to D.T., 19H01020 to N.W. and 17H01531 to S.N.). Author contributions: D.T., M.F.K., and S.N. designed this study. D.T. and Y.K. carried out most of the experiments including statistical analysis with the exception at the following. Y.N. performed imaging experiments with the spinning disk confocal microscope. K.T. and T.I. generated genetically engineered mice. S.K. and C.T. contributed to mRNA electroporation experiments. T.H. supervised FACS experiments. P.K. and N.W. supervised imaging experiments. D.T. and S.N. wrote the manuscript. All authors contributed to editing of the manuscript. Competing interests: D.T. and S.N. were supported by the Coordination Fund from JST and Astellas Pharm Inc. SN is a scientific advisor to Astellas Pharma Inc. The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/1/eaay2432/DC1
Supplementary Materials and Methods
Fig. S1. Effects of formin inhibition after initial TCR microcluster formation in naïve CD8 OT-I T cells stimulated on SLBs.
Fig. S2. TCR signaling in naïve CD8 T cells from mDia1 KO or mDia3 KO mice.
Fig. S3. Dynamic localization of EGFP-mDia3 in the IS.
Fig. S4. IS spreading and TCR microcluster centralization are impaired in mDia1/3 cDKO naïve CD8 OT-I T cells.
Fig. S5. T cell lymphopenia results from impaired positive selection in mDia1/3 DKO mice.
Fig. S6. T cell development in mDia1 KO mice and mDia3 KO mice.
Fig. S7. Impaired T cell development phenotypes in mDia1/3 DKO are cell intrinsic.
Fig. S8. Conditional deletion of mDia1 and mDia3 in the T cell lineage.
Fig. S9. mDia1/3 deficiency impaired thymocyte-positive selection in OT-I and OT-II transgenic mice.
Fig. S10. Impaired TCR microcluster centralization and peripheral F-actin ring formation following loss of mDia1/3 in OT-I thymocytes.
Movie S1. TIRF live imaging of TCR microcluster dynamics in a control naïve CD8 OT-I T cell stimulated on a SLB (2 s per frame; 60 frames).
Movie S2. TIRF live imaging of TCR microcluster dynamics in a 10 μM SMIFH2–treated naïve CD8 OT-I T cell stimulated on an SLB (2 s per frame; 60 frames).
Movie S3. TIRF live imaging of TCR microcluster dynamics in a naïve CD8 OT-I T cell stimulated on an SLB and treated with 15 μM SMIFH2 at 10 s after initial microcluster formation (2 s per frame; 75 frames).
Movie S4. TIRF live imaging of TCR microcluster dynamics in a naïve CD8 OT-I T cell stimulated on an SLB and treated with 15 μM SMIFH2 at 20 s after initial microcluster formation (2 s per frame; 75 frames).
Movie S5. TIRF live imaging of cSMAC dynamics in a naïve CD8 OT-I T cell stimulated on an SLB for 10 min and treated with 15 μM SMIFH2 (2 s per frame; 160 frames).
Movie S6. TIRF live imaging of TCR microcluster and LifeAct-EGFP dynamics in a control CD8 OT-I T blast stimulated on an SLB (2 s per frame; 60 frames).
Movie S7. TIRF live imaging of TCR microcluster and LifeAct-EGFP dynamics in a 10 μM SMIFH2–treated CD8 OT-I T blast stimulated on an SLB (2 s per frame; 60 frames).
Movie S8. 3D reconstruction of a confocal image stack of pZap70 and F-actin staining in a control naïve CD8 OT-I T cell stimulated on an SLB for 1.5 min.
Movie S9. 3D reconstruction of a confocal image stack of pZap70 and F-actin staining in a 10 μM SMIFH2–treated naïve CD8 OT-I T cell stimulated on an SLB for 1.5 min.
Movie S10. 3D reconstruction of a confocal image stack of pLAT and F-actin staining in a control naïve CD8 OT-I T cell stimulated on an SLB for 1.5 min.
Movie S11. 3D reconstruction of a confocal image stack of pLAT and F-actin staining in a 10 μM SMIFH2–treated naïve CD8 OT-I T cell stimulated on an SLB for 1.5 min.
Movie S12. TIRF live imaging of TCR microcluster and EGFP-mDia1 dynamics in a CD8 OT-I T blast stimulated on an SLB (2 s per frame; 60 frames).
Movie S13. TIRF live imaging of TCR microcluster and EGFP-mDia3 dynamics in a CD8 OT-I T blast stimulated on an SLB (2 s per frame; 60 frames).
Movie S14. TIRF live imaging of TCR microcluster dynamics in a control naïve CD8 OT-I T cell stimulated on an SLB (2 s per frame; 60 frames).
Movie S15. TIRF live imaging of TCR microcluster dynamics in an mDia1/3 cDKO naïve CD8 OT-I T cell stimulated on an SLB (2 s per frame; 60 frames).
Movie S16. TIRF live imaging of TCR microcluster dynamics in a control OT-I thymocyte stimulated on an SLB (0.5 s per frame; 240 frames).
Movie S17. TIRF live imaging of TCR microcluster dynamics in an mDia1/3 cDKO (lck-cre) OT-I thymocyte stimulated on an SLB (0.5 s per frame; 240 frames).
REFERENCES AND NOTES
- 1.Gaud G., Lesourne R., Love P. E., Regulatory mechanisms in T cell receptor signalling. Nat. Rev. Immunol. 18, 485–497 (2018). [DOI] [PubMed] [Google Scholar]
- 2.Malissen B., Grégoire C., Malissen M., Roncagalli R., Integrative biology of T cell activation. Nat. Immunol. 15, 790–797 (2014). [DOI] [PubMed] [Google Scholar]
- 3.Au-Yeung B. B., Shah N. H., Shen L., Weiss A., ZAP-70 in signaling, biology, and disease. Annu. Rev. Immunol. 36, 127–156 (2018). [DOI] [PubMed] [Google Scholar]
- 4.Balagopalan L., Kortum R. L., Coussens N. P., Barr V. A., Samelson L. E., The linker for activation of T cells (LAT) signaling hub: From signaling complexes to microclusters. J. Biol. Chem. 290, 26422–26429 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang W., Sloan-Lancaster J., Kitchen J., Trible R. P., Samelson L. E., LAT: The ZAP-70 tyrosine kinase substrate that links T cell receptor to cellular activation. Cell 92, 83–92 (1998). [DOI] [PubMed] [Google Scholar]
- 6.Lillemeier B. F., Mörtelmaier M. A., Forstner M. B., Huppa J. B., Groves J. T., Davis M. M., TCR and Lat are expressed on separate protein islands on T cell membranes and concatenate during activation. Nat. Immunol. 11, 90–96 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yi J., Balagopalan L., Nguyen T., McIntire K. M., Samelson L. E., TCR microclusters form spatially segregated domains and sequentially assemble in calcium-dependent kinetic steps. Nat. Commun. 10, 277 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Larghi P., Williamson D. J., Carpier J.-M., Dogniaux S., Chemin K., Bohineust A., Danglot L., Gaus K., Galli T., Hivroz C., VAMP7 controls T cell activation by regulating the recruitment and phosphorylation of vesicular Lat at TCR-activation sites. Nat. Immunol. 14, 723–731 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Lo W.-L., Shah N. H., Ahsan N., Horkova V., Stepanek O., Salomon A. R., Kuriyan J., Weiss A., Lck promotes Zap70-dependent LAT phosphorylation by bridging Zap70 to LAT. Nat. Immunol. 19, 733–741 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dustin M. L., Chakraborty A. K., Shaw A. S., Understanding the structure and function of the immunological synapse. Cold Spring Harb. Perspect. Biol. 2, a002311 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ritter A. T., Angus K. L., Griffiths G. M., The role of the cytoskeleton at the immunological synapse. Immunol. Rev. 256, 107–117 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumari S., Curado S., Mayya V., Dustin M. L., T cell antigen receptor activation and actin cytoskeleton remodeling. Biochim. Biophys. Acta 1838, 546–556 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bunnell S. C., Kapoor V., Trible R. P., Zhang W., Samelson L. E., Dynamic actin polymerization drives T cell receptor–induced spreading: A role for the signal transduction adaptor LAT. Immunity 14, 315–329 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Jordan M. S., Koretzky G. A., Coordination of receptor signaling in multiple hematopoietic cell lineages by the adaptor protein SLP-76. Cold Spring Harb. Perspect. Biol. 2, a002501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Le Floc'h A., Tanaka Y., Bantilan N. S., Voisinne G., Altan-Bonnet G., Fukui Y., Huse M., Annular PIP3 accumulation controls actin architecture and modulates cytotoxicity at the immunological synapse. J. Exp. Med. 210, 2721–2737 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Varma R., Campi G., Yokosuka T., Saito T., Dustin M. L., T cell receptor-proximal signals are sustained in peripheral microclusters and terminated in the central supramolecular activation cluster. Immunity 25, 117–127 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pollard T. D., Actin and actin-binding proteins. Cold Spring Harb. Perspect. Biol. 8, a018226 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Campellone K. G., Welch M. D., A nucleator arms race: Cellular control of actin assembly. Nat. Rev. Mol. Cell Biol. 11, 237–251 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gomez T. S., Kumar K., Medeiros R. B., Shimizu Y., Leibson P. J., Billadeau D. D., Formins regulate the actin-related protein 2/3 complex-independent polarization of the centrosome to the immunological synapse. Immunity 26, 177–190 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murugesan S., Hong J., Yi J., Li D., Beach J. R., Shao L., Meinhardt J., Madison G., Wu X., Betzig E., Hammer J. A., Formin-generated actomyosin arcs propel T cell receptor microcluster movement at the immune synapse. J. Cell Biol. 215, 383–399 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kühn S., Geyer M., Formins as effector proteins of Rho GTPases. Small GTPases 5, e29513 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rizvi S. A., Neidt E. M., Cui J., Feiger Z., Skau C. T., Gardel M. L., Kozmin S. A., Kovar D. R., Identification and characterization of a small molecule inhibitor of formin-mediated actin assembly. Chem. Biol. 16, 1158–1168 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beemiller P., Jacobelli J., Krummel M. F., Integration of the movement of signaling microclusters with cellular motility in immunological synapses. Nat. Immunol. 13, 787–795 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Riedl J., Crevenna A. H., Kessenbrock K., Yu J. H., Neukirchen D., Bista M., Bradke F., Jenne D., Holak T. A., Werb Z., Sixt M., Wedlich-Soldner R., Lifeact: A versatile marker to visualize F-actin. Nat. Methods 5, 605–607 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thumkeo D., Watanabe S., Narumiya S., Physiological roles of Rho and Rho effectors in mammals. Eur. J. Cell Biol. 92, 303–315 (2013). [DOI] [PubMed] [Google Scholar]
- 26.Shinohara R., Thumkeo D., Kamijo H., Kaneko N., Sawamoto K., Watanabe K., Takebayashi H., Kiyonari H., Ishizaki T., Furuyashiki T., Narumiya S., A role for mDia, a Rho-regulated actin nucleator, in tangential migration of interneuron precursors. Nat. Neurosci. 15, 373–380 (2012). [DOI] [PubMed] [Google Scholar]
- 27.Thumkeo D., Shinohara R., Watanabe K., Takebayashi H., Toyoda Y., Tohyama K., Ishizaki T., Furuyashiki T., Narumiya S., Deficiency of mDia, an actin nucleator, disrupts integrity of neuroepithelium and causes periventricular dysplasia. PLOS ONE 6, e25465 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deguchi Y., Harada M., Shinohara R., Lazarus M., Cherasse Y., Urade Y., Yamada D., Sekiguchi M., Watanabe D., Furuyashiki T., Narumiya S., mDia and ROCK mediate actin-dependent presynaptic remodeling regulating synaptic efficacy and anxiety. Cell Rep. 17, 2405–2417 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Sakamoto S., Thumkeo D., Ohta H., Zhang Z., Huang S., Kanchanawong P., Fuu T., Watanabe S., Shimada K., Fujihara Y., Yoshida S., Ikawa M., Watanabe N., Saitou M., Narumiya S., mDia1/3 generate cortical F-actin meshwork in Sertoli cells that is continuous with contractile F-actin bundles and indispensable for spermatogenesis and male fertility. PLOS Biol. 16, e2004874 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Litschko C., Brühmann S., Csiszár A., Stephan T., Dimchev V., Damiano-Guercio J., Junemann A., Körber S., Winterhoff M., Nordholz B., Ramalingam N., Peckham M., Rottner K., Merkel R., Faix J., Functional integrity of the contractile actin cortex is safeguarded by multiple Diaphanous-related formins. Proc. Natl. Acad. Sci. U.S.A. 116, 3594–3603 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jenkins M. R., Stinchcombe J. C., Au-Yeung B. B., Asano Y., Ritter A. T., Weiss A., Griffiths G. M., Distinct structural and catalytic roles for Zap70 in formation of the immunological synapse in CTL. eLife 3, e01310 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kumari S., Depoil D., Martinelli R., Judokusumo E., Carmona G., Gertler F. B., Kam L. C., Carman C. V., Burkhardt J. K., Irvine D. J., Dustin M. L., Actin foci facilitate activation of the phospholipase C-γ in primary T lymphocytes via the WASP pathway. eLife 4, e04953 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Balagopalan L., Yi J., Nguyen T., McIntire K. M., Harned A. S., Narayan K., Samelson L. E., Plasma membrane LAT activation precedes vesicular recruitment defining two phases of early T-cell activation. Nat. Commun. 9, 2013 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Katz Z. B., Novotná L., Blount A., Lillemeier B. F., A cycle of Zap70 kinase activation and release from the TCR amplifies and disperses antigenic stimuli. Nat. Immunol. 18, 86–95 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Klammt C., Novotná L., Li D. T., Wolf M., Blount A., Zhang K., Fitchett J. R., Lillemeier B. F., T cell receptor dwell times control the kinase activity of Zap70. Nat. Immunol. 16, 961–969 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Comrie W. A., Li S., Boyle S., Burkhardt J. K., The dendritic cell cytoskeleton promotes T cell adhesion and activation by constraining ICAM-1 mobility. J. Cell Biol. 208, 457–473 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bunnell S. C., Hong D. I., Kardon J. R., Yamazaki T., McGlade C. J., Barr V. A., Samelson L. E., T cell receptor ligation induces the formation of dynamically regulated signaling assemblies. J. Cell Biol. 158, 1263–1275 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Watanabe N., Madaule P., Reid T., Ishizaki T., Watanabe G., Kakizuka A., Saito Y., Nakao K., Jockusch B. M., Narumiya S., p140mDia, a mammalian homolog of Drosophila diaphanous, is a target protein for Rho small GTPase and is a ligand for profilin. EMBO J. 16, 3044–3056 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Corre I., Gomez M., Vielkind S., Cantrell D. A., Analysis of thymocyte development reveals that the GTPase RhoA is a positive regulator of T cell receptor responses in vivo. J. Exp. Med. 194, 903–914 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/1/eaay2432/DC1
Supplementary Materials and Methods
Fig. S1. Effects of formin inhibition after initial TCR microcluster formation in naïve CD8 OT-I T cells stimulated on SLBs.
Fig. S2. TCR signaling in naïve CD8 T cells from mDia1 KO or mDia3 KO mice.
Fig. S3. Dynamic localization of EGFP-mDia3 in the IS.
Fig. S4. IS spreading and TCR microcluster centralization are impaired in mDia1/3 cDKO naïve CD8 OT-I T cells.
Fig. S5. T cell lymphopenia results from impaired positive selection in mDia1/3 DKO mice.
Fig. S6. T cell development in mDia1 KO mice and mDia3 KO mice.
Fig. S7. Impaired T cell development phenotypes in mDia1/3 DKO are cell intrinsic.
Fig. S8. Conditional deletion of mDia1 and mDia3 in the T cell lineage.
Fig. S9. mDia1/3 deficiency impaired thymocyte-positive selection in OT-I and OT-II transgenic mice.
Fig. S10. Impaired TCR microcluster centralization and peripheral F-actin ring formation following loss of mDia1/3 in OT-I thymocytes.
Movie S1. TIRF live imaging of TCR microcluster dynamics in a control naïve CD8 OT-I T cell stimulated on a SLB (2 s per frame; 60 frames).
Movie S2. TIRF live imaging of TCR microcluster dynamics in a 10 μM SMIFH2–treated naïve CD8 OT-I T cell stimulated on an SLB (2 s per frame; 60 frames).
Movie S3. TIRF live imaging of TCR microcluster dynamics in a naïve CD8 OT-I T cell stimulated on an SLB and treated with 15 μM SMIFH2 at 10 s after initial microcluster formation (2 s per frame; 75 frames).
Movie S4. TIRF live imaging of TCR microcluster dynamics in a naïve CD8 OT-I T cell stimulated on an SLB and treated with 15 μM SMIFH2 at 20 s after initial microcluster formation (2 s per frame; 75 frames).
Movie S5. TIRF live imaging of cSMAC dynamics in a naïve CD8 OT-I T cell stimulated on an SLB for 10 min and treated with 15 μM SMIFH2 (2 s per frame; 160 frames).
Movie S6. TIRF live imaging of TCR microcluster and LifeAct-EGFP dynamics in a control CD8 OT-I T blast stimulated on an SLB (2 s per frame; 60 frames).
Movie S7. TIRF live imaging of TCR microcluster and LifeAct-EGFP dynamics in a 10 μM SMIFH2–treated CD8 OT-I T blast stimulated on an SLB (2 s per frame; 60 frames).
Movie S8. 3D reconstruction of a confocal image stack of pZap70 and F-actin staining in a control naïve CD8 OT-I T cell stimulated on an SLB for 1.5 min.
Movie S9. 3D reconstruction of a confocal image stack of pZap70 and F-actin staining in a 10 μM SMIFH2–treated naïve CD8 OT-I T cell stimulated on an SLB for 1.5 min.
Movie S10. 3D reconstruction of a confocal image stack of pLAT and F-actin staining in a control naïve CD8 OT-I T cell stimulated on an SLB for 1.5 min.
Movie S11. 3D reconstruction of a confocal image stack of pLAT and F-actin staining in a 10 μM SMIFH2–treated naïve CD8 OT-I T cell stimulated on an SLB for 1.5 min.
Movie S12. TIRF live imaging of TCR microcluster and EGFP-mDia1 dynamics in a CD8 OT-I T blast stimulated on an SLB (2 s per frame; 60 frames).
Movie S13. TIRF live imaging of TCR microcluster and EGFP-mDia3 dynamics in a CD8 OT-I T blast stimulated on an SLB (2 s per frame; 60 frames).
Movie S14. TIRF live imaging of TCR microcluster dynamics in a control naïve CD8 OT-I T cell stimulated on an SLB (2 s per frame; 60 frames).
Movie S15. TIRF live imaging of TCR microcluster dynamics in an mDia1/3 cDKO naïve CD8 OT-I T cell stimulated on an SLB (2 s per frame; 60 frames).
Movie S16. TIRF live imaging of TCR microcluster dynamics in a control OT-I thymocyte stimulated on an SLB (0.5 s per frame; 240 frames).
Movie S17. TIRF live imaging of TCR microcluster dynamics in an mDia1/3 cDKO (lck-cre) OT-I thymocyte stimulated on an SLB (0.5 s per frame; 240 frames).