

Abstract
TGF‐β signaling pathway plays a key role in breast cancer metastasis. Recent studies suggest that TGF‐β regulates tumor progression and invasion not only via transcriptional regulation, but also via translational regulation. Using both bioinformatics and experimental tools, we identified a micropeptide CIP2A‐BP encoded by LINC00665, whose translation was downregulated by TGF‐β in breast cancer cell lines. Using TNBC cell lines, we showed that TGF‐β‐activated Smad signaling pathway induced the expression of translation inhibitory protein 4E‐BP1, which inhibited eukaryote translation initiation factor elF4E, leading to reduced translation of CIP2A‐BP from LINC00665. CIP2A‐BP directly binds tumor oncogene CIP2A to replace PP2A's B56γ subunit, thus releasing PP2A activity, which inhibits PI3K/AKT/NFκB pathway, resulting in decreased expression levels of MMP‐2, MMP‐9, and Snail. Downregulation of CIP2A‐BP in TNBC patients was significantly associated with metastasis and poor overall survival. In the MMTV‐PyMT model, either introducing CIP2A‐BP gene or direct injection of CIP2A‐BP micropeptide significantly reduced lung metastases and improved overall survival. In conclusion, we provide evidence that CIP2A‐BP is both a prognostic marker and a novel therapeutic target for TNBC.
Keywords: CIP2A‐BP, invasion and metastasis, PI3K/AKT/NFκB pathways, TNBC
Subject Categories: Cancer, Protein Biosynthesis & Quality Control, RNA Biology
Long non‐coding RNA‐translated micropeptide CIP2A‐BP impairs oncogenic CIP2A‐PI3K‐AKT signaling but is controlled by TGF‐β in the malignant state.
Introduction
Triple‐negative breast cancer (TNBC) is a subtype of breast cancer accounting for 15% of all breast cancers. TNBC is negative for the expression of estrogen receptor (ER) and progesterone receptor (PR) and lacks HER2 amplification or overexpression. It is usually associated with more aggressive tumor behaviors, including younger age of disease onset, shorter time to relapse, and higher risk for local and distant metastasis, therefore with poor overall survival compared to other subtypes of breast cancer (Gluz et al, 2009; Lara‐Medina et al, 2011; Sharma, 2016). Currently, there are no effective targeted therapies available for TNBC.
TGF‐β signal transduction pathway plays an important role in development and growth of normal cells. Alteration in TGF‐β signal pathway has been reported in tumorigenesis, tumor invasion, and metastasis (Akhurst & Hata, 2012). In breast cancer, TGF‐β treatment can induce epithelial–mesenchymal transition (EMT) and induce cancer stem cells whereas TGF‐β inhibition augments chemotherapy against TNBC (Bhola et al, 2013).
Long non‐coding RNAs (lncRNA) are a type of RNA longer than 200nt, which normally do not encode proteins, and participate in normal cellular function and disease development by regulation of transcription or translation (Wapinski & Chang, 2011; Li et al, 2014; Sun & Kraus, 2015). More recent studies suggest that some of these lncRNAs encode biologically active micropeptides which participate in a variety of cellular activities (Anderson et al, 2015; Cai et al, 2017; Matsumoto et al, 2017). Previous studies have shown that TGF‐β treatment can directly regulate lncRNA at transcription level (Richards et al, 2015); however, little is known whether TGF‐β treatment can directly influence lncRNA at translation level.
In this study, we first screened for potential lncRNAs that encoded micropeptides which were regulated by TGF‐β treatment. Subsequently, we determined the clinical utility of lncRNA‐encoded micropeptide. Finally, we determined the function and mechanism of lncRNA‐encoded micropeptide in TNBC invasion and metastasis.
Results
LINC00665 is regulated by TGF‐β at translational level
In order to identify lncRNAs regulated by TGF‐β at translational level, we obtained both Ribo‐seq and RNA‐seq datasets (GSE59817) of a normal human mammary epithelial cell line MCF‐10A before and after TGF‐β treatment from GEO database. Using bioinformatics analysis, we identified 14 lncRNA candidates that showed alterations on translation (Ribo‐seq) but not on transcription (RNA‐seq) after TGF‐β treatment. Subsequently, these lncRNAs were further analyzed by polysome profiling and quantitative PCR (qPCR) in MCF‐10A and MDA‐MB‐231 cells (Appendix Fig S1), and only LINC00665 remained changed at translation level but unchanged at transcription level.
LINC00665 is an annotated human lncRNA (NR_038278.1). Both fluorescence in situ hybridization (FISH) and nuclear/cytoplasm fractionation indicated that LINC00665 mainly resides in cytoplasm (Appendix Fig S2A and B). Because of its ribosome occupancy (Fig 1A), we tested the hypothesis that it might encode proteins or peptides. We performed both polysome profiling analysis and qPCR on LINC00665 in MCF‐10A as well as three TNBC cell lines (Hs578T, BT549, and MDA‐MB‐231) before and after TGF‐β treatment. In all cell lines, we observed that TGF‐β treatment reduced LINC00665‐polysome complex level (Fig 1B), but not LINC00665 RNA level (Fig 1C). Northern blot analysis of MCF‐10A and MDA‐MB‐231 cell lines further confirmed that TGF‐β treatment had no effect on LINC00665 RNA level (Appendix Fig S2C).
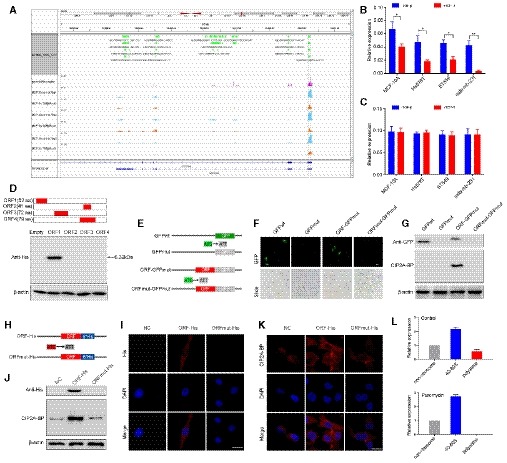
Figure 1. LINC00665 encodes a novel micropeptide and is regulated by TGF‐β at the translational level.
-
ARibosome occupancy map at the LINC00665 locus. MCF‐10A cells were either mock‐treated or treated with TGF‐β, and ribosome profiling on three biological replicates was shown.
-
BRelative levels of CIP2A‐BP were determined by polysome profiling and qRT–PCR in MCF‐10A and TNBC cells either mock‐treated or treated with TGF‐β.
-
CRelative levels of LINC00665 were determined by qRT–PCR in MCF‐10A and TNBC cells either mock‐treated or treated with TGF‐β.
-
DUpper: putative ORFs in LINC00665. Lower: The ORFs were constructed into pcDNA3.1 vector and transfected to MCF‐10A cells for 24 h. The ORFs‐His fusion proteins were determined by Western blot with anti‐His antibody.
-
EDiagram of the GFP fusion constructs. The start codon ATGGTG of the GFP (GFPwt) gene is mutated to ATTGTT (GFPmut). The start codon ATG of the LINC00665 ORF is mutated to ATT.
-
F, GThe indicated constructs were transfected into MCF‐10A cells for 24 h; then, the GFP fluorescence was detected using fluorescence microscope (F); and fusion protein levels were determined by Western blot with anti‐GFP and anti‐CIP2A‐BP antibodies, respectively (G).
-
HDiagram of the His fusion constructs. The start codon ATG of the LINC00665 ORF is mutated to ATT.
-
I–KThe indicated constructs were stably expressed in MDA‐MB‐231 cells; micropeptide CIP2A‐BP was immunostained using anti‐His (I) and anti‐CIP2A‐BP antibodies (K), respectively; and CIP2A‐BP–His fusion protein levels were determined by Western blotting with anti‐His and CIP2A‐BP antibodies (J).
-
LMCF‐10A cells incubated in the absence or presence of puromycin (200 μM) were fractionated on sucrose gradients, and distribution of CIP2A‐BP transcripts was quantified by qPCR.
LINC00665 could potentially encode four open reading frames (ORFs) by in silico analysis. To determine the translational potential of these ORFs, we generated c‐terminal His‐tag fusion protein for each ORF and transfected into MCF‐10A cell. Western blot analysis indicated that only LINC0665‐ORF1 translated into a micropeptide (Fig 1D). This ORF is located on chromosome 19 from 36,313,061 to 36,331,718, within the first exon of LINC00665 encoding a 52‐amino acid peptide (5.5KDa). This is consistent with ribosome occupancy data from GWIPS‐viz database (Fig 1A). We name this micropeptide CIP2A binding peptide (CIP2A‐BP).
To determine whether the starting codon of CIP2A‐BP was functional, we generated an expression construct (ORF‐GFPmut) by fusing GFPmut ORF (in which the starting codon ATGGTG was mutated to ATTGTT) to the c‐terminus of CIP2A‐BP and transfected into MCF‐10A cells (Fig 1E). GFP fusion protein was detected in ORF‐GFPmut transfected MCF‐10A cells (Fig 1F). Western blot using anti‐GFP antibody further confirmed the presence of CIP2A‐BP‐GFP (Fig 1G). However, no GFP fusion protein was detected when the starting codon in CIP2A‐BP was mutated (ORFmut‐GFPmut), nor by Western blot using anti‐GFP antibody (Fig 1F and G). To rule out fusion protein property change due to relative large size of GFP, we further confirmed the functionality of CIP2A‐BP start codon using His‐tag fusion protein constructs in MDA‐MB‐231 cells (Fig 1H–J).
Expression of endogenous CIP2A‐BP
To determine whether CIP2A‐BP was produced endogenously, we first performed polysome profiling in MCF‐10A cell lysate. The mRNA‐protein particles (mRNPs) were separated into three groups: non‐ribosome (mRNPs without any ribosome), 40S‐80S (mRNPs associated with ribosome but not being translated), and polysome (mRNPs being actively translated), and the presence of CIP2A‐BP RNA in each fraction was determined using qRT–PCR (Appendix Fig S1C). We found that CIP2A‐BP RNA was enriched in polysome fraction: CIP2A‐BP RNA enrichment in 40S‐80S and polysome fractions was further reduced by TGF‐β treatment, but completely abolished in polysome fractions by puromycin treatment (Fig 1L and Appendix Fig S1C).
To further confirm the presence of CIP2A‐BP peptide, we generated antibody against CIP2A‐BP. The CIP2A‐BP fusion protein was detected in the ORF‐GFPmut and ORF‐His transfected cells using the anti‐CIP2A‐BP antibody, but no fusion protein expression was detected in the cells transfected with the other constructs (Fig 1G, J and K). To determine the specificity of CIP2A‐BP antibody, we showed that the level of CIP2A‐BP protein was significantly lower in LINC00665 shRNA transfected MCF‐10A and MDA‐MB‐231 cells (Appendix Fig S3A–C).
Immunofluorescence analysis indicated that CIP2A‐BP was present in MCF‐10A and MDA‐MB‐231 cells, and TGF‐β treatment significantly reduced CIP2A‐BP expression (Fig 2A). To investigate whether CIP2A‐BP plays a role in tumorigenesis of TNBC, we determined CIP2A‐BP peptide level by Western blot in MCF‐10A and three TNBC cell lines (Hs578T, BT549, and MDA‐MB‐231). CIP2A‐BP expression was lower in TNBC cell lines than in MCF‐10A cell line, and TGF‐β treatment reduced CIP2A‐BP expression in all four cell lines (Fig 2B). To rule out the possibility that CIP2A‐BP was processed from a larger protein precursor, we showed that CIP2A‐BP translation‐blocking antisense oligo blocked the expression of CIP2A‐BP (Fig 2C).
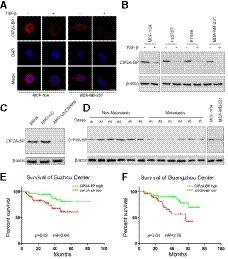
Figure 2. Micropeptide CIP2A‐BP is endogenously expressed, and downregulation is associated with poor prognosis in TNBC patients.
-
AMicropeptide CIP2A‐BP was immunostained with anti‐CIP2A‐BP antibody in MCF‐10A and MDA‐MB‐231 cells either mock‐treated or treated with TGF‐β.
-
BMicropeptide CIP2A‐BP levels were determined by Western blot with anti‐CIP2A‐BP antibody in MCF‐10A and TNBC cells either mock‐treated or treated with TGF‐β.
-
CMCF‐10A cells were first transfected with anti‐LINC00665 translation‐blocking antisense oligo, and then, micropeptide CIP2A‐BP was detected.
-
DMicropeptide CIP2A‐BP levels were detected by Western blot with anti‐CIP2A‐BP antibody in the indicated non‐metastatic TNBC tissues (non‐metastasis) and metastatic TNBC tissues (metastasis). Micropeptide CIP2A‐BP expression in MCF‐10A cells was used as the positive control.
-
E, FKaplan–Meier overall survival curves for TNBC patients with high or low micropeptide CIP2A‐BP expression in Suzhou cohort (discovery set, n = 112) and Guangzhou cohort (validation set, n = 105).
Low expression of CIP2A‐BP was associated with poor survival in triple‐negative breast cancer patients
Because CIP2A‐BP peptide level was lower in TNBC cell lines than in MCF‐10A cells, we determined whether CIP2A‐BP peptide level differed in TNBC patients by metastasis status, and showed that CIP2A‐BP level was lower in patients with metastasis than in patients without metastasis (Fig 2D). However, the transcript of LINC00665 level had no significant difference between metastatic and non‐metastatic patients (Appendix Fig S4A). Next, we determined whether CIP2A‐BP level was associated with overall survival among TNBC patients. Using median expression level CIP2A‐BP among TNBC patients, we separated TNBC patients into two different groups: patients with high CIP2A‐BP (relative expression level>median expression level) and patients with low CIP2A‐BP (relative expression level ≤ median expression level), in both the Suzhou cohort and the Guangzhou cohort. Using the log‐rank test and Kaplan–Meier survival curves, we showed that patients with low CIP2A‐BP expression had significantly lower OS than patients with high CIP2A‐BP expression in both Suzhou cohort [median survival time (MST): 29 versus 43 months, log rank P = 0.02, hazard ratio (HR) = 2.64] and Guangzhou cohort (MST: 26 versus 42 months, log rank P = 0.01, HR = 2.76) (Fig 2E and F). Interestingly, in the Suzhou cohort (MST: 34 versus 39 months, log rank P = 0.58, HR = 1.28) and Guangzhou cohort (MST: 32 versus 38 months, log rank P = 0.68, HR = 1.18), we found that there was no significant difference in OS between patients with low and high expression of LINC00665 transcript (Appendix Fig S4B and C).
Generation of CIP2A‐BP knockout breast cancer cell lines using CRISP/Cas9 system
To investigate whether CIP2A‐BP micropeptide was involved in TNBC progression, we knocked out CIP2A‐BP in two TNBC cell lines (Hs578T and MDA‐MB‐231) using CRISPR/Cas9‐mediated homologous recombination technique. After a series of screenings, two independent knockout cell lines (Hs578TKO and MDA‐MB‐231KO) were obtained. Sequence analysis revealed that both cell lines contained ATG codon mutations which prevented translation of CIP2A‐BP (Appendix Fig S5A). To verify the properties of these two knockout cell lines, we determined the expression of LINC00665 by qPCR, CIP2A‐BP translation by polysome profiling, and CIP2A‐BP expression by Western blot analysis. We showed that translation of CIP2A‐BP was decreased but transcription of LINC00665 was not affected (Appendix Fig S5B–D).
Micropeptide CIP2A‐BP, not LINC00665, inhibits migration and invasion of TNBC cells
To determine whether CIP2A‐BP plays a role in the progression of TNBC, we conducted Transwell and wound‐healing assays, using TNBC cells with CIP2A‐BP overexpression or knocked out (Appendix Fig S5E and F). we showed that overexpression of His‐tagged LINC00665 ORF in TNBC cell lines Hs578T and MDA‐MB‐231 reduced both migration and invasion in Transwell assays (Fig 3A and B), as well as migration in wound‐healing scratch assay (Fig 3E and F). On the contrary, CIP2A‐BP knockout in Hs578T and MDA‐MB‐231 cell lines increased migration and invasion in Transwell assays (Fig 3C and D), and migration in wound‐healing scratch assay (Fig 3E and F). Such increase in migration and invasion could be reversed by overexpression of LINC00665 ORF in Hs578TKO and MDA‐MB‐231KO cell lines (Fig 3C–F). To investigate whether transcript of LINC00665 is functional in the progression of TNBC, we knocked down LINC00665 in wild‐type MDA‐MB‐231 and MDA‐MB‐231KO cells. We showed that LINC00665 knockdown promoted migration and invasion in wild‐type MDA‐MB‐231 cell, but not in MDA‐MB‐231KO cell (Appendix Fig S4D–I). These results indicated that CIP2A‐BP, not LINC00665 transcript, played an important role in TNBC migration and invasion.
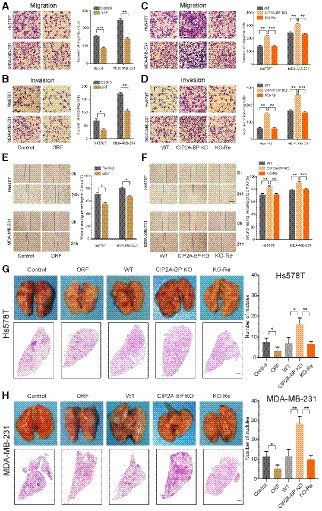
Figure 3. The micropeptide CIP2A‐BP suppresses migration and invasion of triple‐negative breast cancer cells in vitro and in vivo .
-
A, BMigration and invasion of TNBC cells transfected with LINC00665 ORF and control lentiviruses for 48 h. Right panel is the quantification of TNBC cells that migrated through the membrane without the Matrigel (upper) and invaded through Matrigel‐coated membrane (lower).
-
C, DMigration and invasion of wild‐type and CIP2A‐BP knockout TNBC cells and CIP2A‐BP re‐expressed Hs578TKO and MDA‐MB‐231KO cells. Right panel is the quantification of cells that migrated through the membrane without the Matrigel (upper) and invaded through Matrigel‐coated membrane (lower).
-
EWound‐healing assay on Hs578T and MDA‐MB‐231 cells transfected with LINC00665 ORF expressing and control lentiviruses. The wound edges were photographed at the indicated time points after wounding. Right panel is the quantification of the relative wound‐healing area.
-
FWild‐type and CIP2A‐BP knockout TNBC cells and CIP2A‐BP re‐expressed Hs578TKO and MDA‐MB‐231KO were seeded on cell culture inserts for wound‐healing assay. The wound edges were photographed at the indicated time points after wounding. Right panel is the quantification of the relative wound‐healing area.
-
G, HMacroscopic and histological analysis of the lungs of nude mice injected with the indicated Hs578T (G) and MDA‐MB‐231 cells (H) (1.0 × 105–106 cells/mouse) via tail vein; lung metastatic nodules were visualized 8 weeks post‐transplantation (5 mice per group). Right panel is the quantification of pulmonary metastases.
We next determined the effect of LINC00665 ORF overexpression and CIP2A‐BP knockout on tumor metastasis using mouse TNBC lung metastasis xenograft models. We showed that LINC00665 ORF overexpression of TNBC cell lines generated lower number of lung metastatic nodules compared with wild‐type TNBC cell lines, while CIP2A‐BP knockout TNBC cell lines generated higher number of lung metastatic nodules (Fig 3G and H). These results suggested that LINC00665‐encoded micropeptide CIP2A‐BP functioned as a suppressor for cell migration and invasion in TNBC.
TGF‐β inhibits expression of micropeptide CIP2A‐BP through 4E‐BP1
Because our data suggest that TGF‐β treatment significantly reduced the level of CIP2A‐BP and TGF‐β activates the Smad signal transduction pathway to regulate translation through 4E‐BP1 (Schmierer & Hill, 2007; Azar et al, 2009), we tested the hypothesis that TGF‐β treatment reduced CIP2A‐BP translation through 4E‐BP1. First, we showed that TGF‐β treatment activated epithelial mesenchymal transition (EMT) and Smad signaling pathway in TNBC cell lines. After 48 h of TGF‐β treatment of TNBC cell lines Hs578T and MDA‐MB‐231, we detected the downregulation of E‐cadherin and upregulation of N‐cadherin. In addition, we also detected increased level of p‐Smad2 and p‐Smad3 (Fig 4A). Finally, we detected upregulation of 4E‐BP1.
Figure 4. TGF‐β inhibits expression of micropeptide CIP2A‐BP through 4E‐BP1.
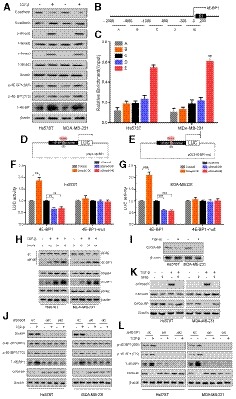
-
AImmunoblotting analysis of expression of the indicated proteins in TNBC cells cultured with or without TGF‐β.
-
BDesign of initial primer sets for chromatin immunoprecipitation assay.
-
CChromatin immunoprecipitation showing Smad4 occupancy at the 4E‐BP1 locus in TNBC cells. Co‐precipitated DNA was analyzed for amplicons A–E by qPCR. Values represent the enrichment of bound protein fractions relative to input.
-
D, EA diagram of luciferase reporter constructs with wild‐type and mutant 4E‐BP1 promoter.
-
F, GLuciferase reporter assay was performed in Hs578T (F) and MDA‐MB‐231 (G) cells following co‐transfection with wild‐type or mutant 4E‐BP1 promoter fragment and Smad4 overexpressed, or knocked down and respective controls for 36 h and treatment with TGF‐β for 24 h. The reporter constructs were expressing the luciferase gene under 4E‐BP1 promoter segment or 4E‐BP1 promoter deleted 0 to +200 region.
-
HRegulation of 4E‐BP1 expression and binding to eIF4E by TGF‐β. TNBC cells were treated with TGF‐β, and equal amounts of proteins were subjected to immunoprecipitation and/or examined by immunoblotting analysis.
-
IImmunoblotting analysis of expression of micropeptide CIP2A‐BP and β‐actin in TNBC cells cultured with or without TGF‐β.
-
JSmad4 siRNA and corresponding control transfected TNBC cells were cultured with or without TGF‐β. The indicated proteins were determined by immunoblotting analysis.
-
KTNBC cells were pretreated with specific antagonist against Smad3 (SIS3, 3 μM) for 1 h and then cultured with or without TGF‐β. The indicated proteins were determined by immunoblotting analysis.
-
M4E‐BP1 siRNA and corresponding control transfected TNBC cells were cultured with or without TGF‐β. The indicated proteins were determined by immunoblotting analysis.
Next, we showed that Smad4 was the transcription factor for 4E‐BP1 in TNBC cells. Chromatin immunoprecipitation (CHIP) experiment showed that Smad4 bound to the 4E‐BP1 promoter adjacent to exon 1 (−300/+200) (Fig 4B and C), which contained a potential Smad4 binding element (SBE: CAGCCAGA) (Azar et al, 2009, 2013). Using both wild‐type and mutant 4E‐BP1 promoter luciferase reporter plasmids (pGL3‐4E‐BP1) (Fig 4D and E), we showed that with wild‐type 4E‐BP1 promoter reporter, the luciferase activity was significantly upregulated in Smad4 overexpressed cells and downregulated by Smad4 knockdown after 24‐h treatment with TGF‐β (Fig 4F and G). Such changes were completely abolished when mutant 4E‐BP1 promoter luciferase reporter was used. We further showed that TGF‐β/Smad4 signaling pathway also significantly increased endogenous 4E‐BP1 mRNA level (Appendix Fig S6A and B). These results suggested that Smad4 is the transcription factor for 4E‐BP1.
Because 4E‐BP1 inhibits translation through binding to eukaryote translation initiation factor‐4F (eIF4F) (Bjornsti & Houghton, 2004; Richter & Sonenberg, 2005), we determined whether TGF‐β treatment in TNBC cell lines increased binding of 4E‐BP1 to eIF4E. Co‐IP experiment indicated that the amount of elF4E‐associated 4E‐BP1 increased in a time‐dependent manner after TGF‐β treatment of TNBC cell lines of Hs578T and MDA‐MB‐231 (Fig 4H). This was correlated with decreased level of CIP2A‐BP after TGF‐β treatment (Fig 4I). And further research has found that 4E‐BP1 could inhibit the translation of multiple genes (Appendix Fig S7A–C). In summary, our data indicate that activation of TGF‐β/Smad pathway leads to increased expression of 4E‐BP1, which reduced expression of CIP2A‐BP through directly binding to eIF4E.
It has been reported that mTOR/4E‐BP1 signaling pathway regulates protein translation through hypophosphorylated 4E‐BP1 (Wang et al, 2019), and we determined whether 4E‐BP1 phosphorylation level influenced on LINC00665 translation. When we treated the triple‐negative breast cancer cells with mTOR inhibitor, 4E‐BP1 phosphorylation and translation of micropeptide CIP2A‐BP were significantly reduced without TGF‐β stimulation, but translation of micropeptide CIP2A‐BP was not affected with TGF‐β stimulation (Appendix Fig S7D). Our results indicated that under high level of TGF‐β, LINC00665 translation was insensitive to regulation by mTOR signaling pathway.
To further confirm that downregulation of CIP2A‐BP in TNBC was a direct consequence of activation of TGF‐β/Smad signaling pathway, we found that downregulation of CIP2A‐BP by TGF‐β treatment could be reversed by siRNA knockdown of Smad4 (Fig 4J and Appendix Fig S6C), siRNA knockdown of 4E‐BP1 (Fig 4L and Appendix Fig S6C), or inhibition of Smad3 phosphorylation by Smad3 phosphorylation inhibitor (SIS3) (Fig 4K).
CIP2A‐BP competes with PP2A subunit B56γ but not B56α to bind to CIP2A
Because CIP2A‐BP lacks homology to known proteins, we first sought to identify CIP2A‐BP interacting proteins using both co‐IP and mass spectrometry (MS) analysis. A total 12 candidate proteins were identified (Fig 5A). Western blot analysis of top six candidate proteins indicated that only CIP2A binds to CIP2A‐BP (Fig 5B). Co‐IP experiment also indicated that CIP2A binds to CIP2A‐BP (Fig 5C). We also confirmed that endogenous CIP2A interacted with endogenous CIP2A‐BP (Fig 5D). We further confirmed the interaction between CIP2A‐BP and CIP2A in the presence of RNase A, and the results indicate the interaction of CIP2A‐BP with CIP2A was RNA independent (Fig 5E).
Figure 5. Micropeptide CIP2A‐BP interacts with the region of CIP2A which is required for interaction with PP2A subunit B56γ.
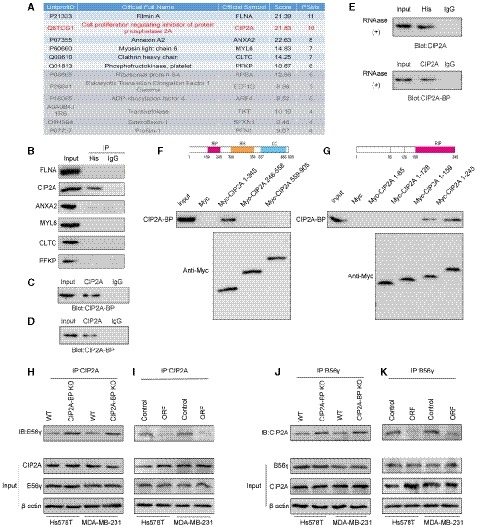
-
AMass spectrometric analysis of co‐immunoprecipitation experiments.
-
BLINC00665 ORF‐His plasmid was transfected into MDA‐MB‐231 cells. Whole‐cell lysates were subjected to co‐immunoprecipitation using anti‐His antibody and immunoblot analysis with CIP2A antibody to verify that CIP2A bound to CIP2A‐BP.
-
CLINC00665 ORF‐His plasmid was transfected into MDA‐MB‐231 cells. Whole‐cell lysates were subjected to co‐immunoprecipitation by CIP2A antibody and immunoblot analysis with CIP2A‐BP antibodies to verify that CIP2A‐BP bound to CIP2A.
-
DEndogenous CIP2A‐BP and CIP2A were co‐immunoprecipitated in MCF‐10A cell line.
-
ELINC00665 ORF‐His plasmid was transfected into MDA‐MB‐231 cells. Whole‐cell lysates were treated with 30 mg/ml RNase A for 30 min, the indicated complexes were co‐immunoprecipitated by anti‐His or anti‐CIP2A antibody, and CIP2A or micropeptide CIP2A‐BP was detected.
-
FUpper: a diagram of the domain structure of full length CIP2A protein. Red: RIP = required for interaction with PPP2R5C; orange: RH = required for homodimerization; blue, CC = coiled coil. Lower: Co‐immunoprecipitation‐coupled Western blot analysis assays revealed the interaction of CIP2A‐BP with CIP2A via the N‐terminus domain of CIP2A.
-
GUpper: A diagram of the domain structure of CIP2A protein fragment (1–245). Red: RIP = Required for Interaction with PPP2R5C. Lower: Co‐immunoprecipitation‐coupled Western blot analysis assays revealed the interaction of CIP2A‐BP with CIP2A via the N‐terminus domain of CIP2A (amino acid 159–245).
-
H–KCo‐immunoprecipitation assays showed that the interaction between CIP2A and B56γ in TNBC cells was affected by CIP2A‐BP expression.
CIP2A (cancerous inhibitor of PP2A cancer inhibitory factor) is an oncogene that promotes tumor progression through inhibiting PP2A and promoting AKT, MYC, and E2F1 activities (Junttila et al, 2007; Khanna et al, 2013; Janghorban et al, 2014).
To identify the binding site for CIP2A‐BP, we generated MYC‐tagged CIP2A truncated protein fragments. Using co‐IP and Western blot analysis, we located the CIP2A‐BP binding site at the N‐terminus domain of CIP2A (amino acid 159–245) (Fig 5F and G).
Because previous report has shown that PP2A's subunits B56γ and B56α also bind to the N‐terminus of CIP2A (amino acid 159–245) to maintain the stability of CIP2A dimer (Wang et al, 2017; Wu et al, 2017), we hypothesized that CIP2A‐BP and these two subunits might compete for the same CIP2A binding site. CIP2A‐BP knockout increased the binding of B56γ for CIP2A (Fig 5H and J), while CIP2A‐BP overexpression decreased binding of B56γ for CIP2A (Fig 5I and K). These results indicated that CIP2A‐BP competes for B56γ's binding site on CIP2A. On the contrary, CIP2A‐BP has no effect on B56α's binding to CIP2A (Appendix Fig S8A). But such binding did not affect the stability of CIP2A dimer (Appendix Fig S8B).
CIP2A‐BP binds to CIP2A to inhibit migration and invasion of TNBC through CIP2A‐mediated PP2A inhibition of PI3K/AKT/NFκB Pathway
Activated AKT plays an important role in tumorigenesis and metastasis through phosphorylation of downstream NFκB (Ozes et al, 1999; Zheng et al, 2012; Yang et al, 2019). To investigate CIP2A‐BP's role in the PI3K/AKT/NFκB pathway, we analyzed activities of PI3K/AKT/NFκB pathway using CIP2A‐BP overexpression and knockdown cell lines. CIP2A‐BP knockdown increased AKT Thr308, Ser473 and IκBα Ser32 phosphorylation, while CIP2A‐BP overexpression reduced phosphorylation of these three sites and further affected the expression of MMP2, MMP9, and Snail (Fig 6A). After TGF‐β treatment, CIP2A‐BP knockdown did not affect phosphorylation of in PI3K/AKT/NFκB signaling pathway, while CIP2A‐BP overexpression downregulated phosphorylation of in PI3K/AKT/NFκB signaling pathway (Appendix Fig S9A). In addition, we confirmed that transcription of MMP2, MMP9, and Snail was increased, but E‐cadherin was decreased, after activation of PI3K/AKT/NF‐kB signaling pathway (Appendix Fig S10). We also determined PP2A activity in CIP2A‐BP overexpression and knockdown cell lines. CIP2A‐BP knockdown significantly decreased PP2A activity, while CIP2A‐BP overexpression increased PP2A activity (Fig 6B). This was consistent with immunohistochemistry analysis on clinical samples that high CIP2A‐BP expression was significantly associated with downregulation of p‐AKT (Fig 6C). These results suggest that CIP2A‐BP can effectively inhibit activation of PI3K/AKT/NFκB pathway, and expression of downstream targets, including MMP2, MMP9, and Snail.
Figure 6. Micropeptide CIP2A‐BP inhibits the activation of the PI3K/AKT/NFκB pathway by increasing PP2A activity.
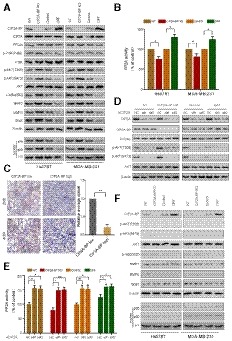
- The expression levels of the indicated proteins were detected by immunoblotting analysis in CIP2A‐BP KO, LINC00665 ORF overexpressed (OE) and respective controls of TNBC cells.
- PP2A activity was measured using a protein phosphatase assay kit. PP2A activity assay was performed to evaluate PP2A activity in CIP2A‐BP KO, LINC00665 ORF overexpressed (OE) and respective control of TNBC cells.
- Immunohistochemistry (IHC) staining of p‐AKT from TNBC primary tumor (n = 20) with high or low micropeptide CIP2A‐BP expression. Lower panels show higher‐magnification images of insets in upper panels. Right panel is the quantification of IHC staining p‐AKT.
- MDA‐MB‐231 cells were transfected with anti‐CIP2A siRNAs. Whole‐cell lysates of these cells were subjected to immunoblot analysis with CIP2A, CIP2A‐BP, PP2Ac, p‐AKT, AKT, and β‐actin antibodies.
- CIP2A‐BP KO, LINC00665 ORF overexpressed (OE) and respective control MDA‐MB‐231 cells were transfected with anti‐CIP2A siRNAs. PP2A activity assay was performed to evaluate PP2A activity in the indicated cells.
- The indicated cells were pretreated with specific antagonist against AKT (MK‐2206, 3 μM) for 1 h. Then, whole lysates of these cells were subjected to immunoblot analysis.
To further confirm that CIP2A‐BP functions through CIP2A/PP2A and its downstream PI3K/AKT/NFκB signaling pathway, we knocked down CIP2A expression in CIP2A‐BP overexpression and CIP2A‐BP knockdown cell lines (Appendix Fig S6C). We showed that CIP2A knockdown completely blocked CIP2A‐BP's effect on AKT Thr308 and Ser473 phosphorylation (Fig 6D and Appendix Fig S9B), as well as CIP2A‐BP's effect on PP2A activity (Fig 6E and Appendix Fig S9C).
Finally, we showed that AKT phosphorylation inhibitor treatment completely abolished Ser/Thr phosphorylation of both AKT and IκBα (Fig 6F and Appendix Fig S11A). Our results indicate that CIP2A‐BP can effectively inhibit activation of PI3K/AKT/NFκB pathway.
Micropeptide CIP2A‐BP inhibited breast cancer metastasis and invasion in MMTV‐PyMT mouse model
In order to investigate the effect of micropeptide CIP2A‐BP on breast cancer metastasis and invasion, we first introduced CIP2A‐BP into C57BL/6 mice and then mated with MMTV‐PyMT mice to generate MMTV‐PyMT; CIP2A‐BP +/+ mice (Fluck & Schaffhausen, 2009). We showed that compared to MMTV‐PyMT mice, MMTV‐PyMT; CIP2A‐BP +/+ mice had less lung metastasis (Fig 7A). Interestingly, we showed that primary tumor in MMTV‐PyMT;CIP2A‐BP +/+ mice had lower p‐AKT expression compared to MMTV‐PyMT mice (Fig 7B). Using MMTV‐PyMT mice model, we further investigated the effect of exogenous CIP2A‐BP treatment on lung metastasis (Appendix Fig S11B). We showed that mammary pad injection of CIP2A‐BP significantly reduced the number of lung metastasis loci (Fig 7C). At the same time, we showed that exogenous CIP2A‐BP treatment significantly reduced p‐AKT level of primary tumor (Fig 7D). In summary, we showed that CIP2A‐BP inhibited AKT phosphorylation in primary tumor and lung metastasis in MMTV‐PyMT mouse model.
Figure 7. Micropeptide CIP2A‐BP inhibits metastasis and invasion of breast cancer in the MMTV‐PyMT mouse model and acts as a potential anticancer peptide in vivo .
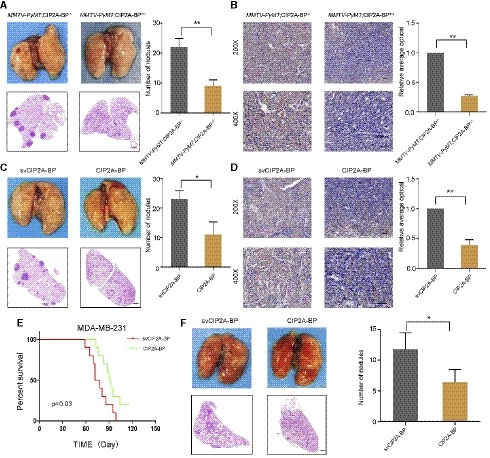
- Macroscopic and histological analysis of the lungs of 140 days old MMTV‐PyMT mice; lung metastatic nodules were visualized 8 weeks post‐transplantation (5 mice per group). Right panel is the quantification of pulmonary metastases.
- Immunohistochemistry (IHC) staining of p‐AKT of mammary tumors from MMTV‐PyMT;CIP2A‐BP +/+ or MMTV‐PyMT;CIP2A‐BP −/− mice. Lower panels show higher‐magnification images of insets in upper panels. Right panel is the quantification of IHC staining p‐AKT.
- MMTV‐PyMT mice were injected with CIP2A‐BP or svCIP2A‐BP via mammary fat pad. Macroscopic and histological analysis of the lungs of 140‐day‐old MMTV‐PyMT mice; lung metastatic nodules were visualized 8 weeks post‐transplantation (5 mice per group). Right panel is the quantification of pulmonary metastases.
- Representative IHC images of p‐AKT of mammary tumors from MMTV‐PyMT mice that were injected with CIP2A‐BP or svCIP2A‐BP via mammary fat pad; lower panels show higher‐magnification images of insets in upper panels. Right panel is the quantification of IHC staining p‐AKT.
- Kaplan–Meier survival curves of nude mice transplanted with MDA‐MB‐231 cells and injected with CIP2A‐BP or svCIP2A‐BP (10 mice per group). The horizontal line indicates the time after the mice were injected with the cells via the tail vein.
- Macroscopic and histological analysis of the lungs of nude mice injected with MDA‐MB‐231 cells and CIP2A‐BP or svCIP2A‐BP via tail vein; lung metastatic nodules were visualized 8 weeks post‐transplantation (5 mice per group). Right panel is the quantification of pulmonary metastases.
Micropeptide CIP2A‐BP is a potential anti‐tumor peptide
Since anti‐tumor peptides have been reported before and important role of micropeptide CIP2A‐BP in TNBC (Sookraj et al, 2010; Jiao et al, 2014), we determined whether micropeptide CIP2A‐BP has anti‐tumor activity. In order to determine the in vivo anti‐tumor activity of micropeptide, we directly injected CIP2A‐BP and MDA‐MB‐231 cells into mice through tail vein (Appendix Fig S11C). CIP2A‐BP was injected again 1 week later. We showed that mice with CIP2A‐BP injection significantly improved survival compared to control mice (Fig 7E). Further, tissue HE staining indicated that CIP2A‐BP also significantly reduced the number of lung metastases (Fig 7F). These results indicated that CIP2A‐BP can effectively suppress breast cancer metastasis and invasion, therefore improve overall survival.
Discussion
In the current study, we identified a long non‐coding RNA LINC00665 that encodes a micropeptide, CIP2A‐BP. Translation of this peptide is downregulated by TGF‐β treatment. Mechanistically, we showed that in TNBC cell lines TGF‐β‐activated Smad signaling pathway induced the expression of translation inhibitory protein 4E‐BP1, which inhibited eukaryote translation initiation factor elF4E, leading to reduced translation of CIP2A‐BP from LINC00665. In TNBC, expression of CIP2A‐BP is downregulated which is associated with tumor invasion and metastasis as well as poor overall survival. In vitro knockdown and overexpression studies confirmed that CIP2A‐BP, but not LINC00665 transcript, acts as a tumor suppressor gene in breast cancer development and progression. CIP2A‐BP directly binds tumor oncogene CIP2A to replace PP2A's B56γ subunit, thus releasing PP2A activity that inhibits activation of PI3K/AKT/NFκB pathway, and further affected the expression of downstream targets, including MMP2, MMP9, and Snail. In MMTV‐PyMT mouse model of human breast cancer, we demonstrate that either introducing CIP2A‐BP gene or direct injection of CIP2A‐BP micropeptide significantly reduces lung metastases and improves overall survival. In summary, we showed that in TNBC, TGF‐β downregulates the translation of CIP2A‐BP to induce tumor invasion and metastasis, through CIP2A/PP2A and PI3K/AKT/NFκB signaling pathways.
Increased TGF‐β expression is a hallmark of cancer progression, contributing to tumor invasion and metastasis (Akhurst & Hata, 2012; Oshimori et al, 2015; Xu et al, 2015). Many studies indicate that TGF‐β signaling pathway induces epithelial mesenchymal transition (EMT) through Smad‐mediated transcriptional regulation (Meyer et al, 2011; Hata & Chen, 2016; Zhang, 2017). Remarkably, it has also been shown that TGF‐β can directly regulate 4E‐BP1 expression via Smad4 (Azar et al, 2009). Specifically, TGF‐β activates Smad signaling pathway and recruits Smad4 into nucleus to act as a transcription factor to induce the expression of translation inhibitor protein 4E‐BP1. In eukaryotic cells, translation initiation factor eIF‐4E is required for CAP‐dependent translation. eIF‐4E interacts with eIF4G to allow ribosomal binding to the 5′ end of mRNA. 4E‐BP1 inhibits translation in eukaryotic cells by competitively binding to eIF‐4E through eIF4G binding site (Richter & Sonenberg, 2005; Peter et al, 2015; Sekiyama et al, 2015). Our data suggest that TGF‐β regulates CIP2A‐BP translation through Smad pathway. In TNBC cells, we showed that TGF‐β treatment can activate Smad signaling pathway; Smad4 directly induced expression of 4E‐BP1, which binds to eIF4E and leads to reduced translation of CIP2A‐BP from LINC00665. It should be noted that TGF‐β acts as an oncogene in late stage tumor, and we hypothesize that TGF‐β induces metastasis in TNBC in part through regulating CIP2A‐BP translation.
In our study, CIP2A‐BP acts as a tumor suppressor in TNBC by binding and inhibiting CIP2A. CIP2A stands for cancerous inhibitor of protein phosphatase 2A, also referred to KIAA1524 or P90 autoantigen. CIP2A promotes tumor cell proliferation and tumor growth through inhibiting tumor suppressor PP2A (Junttila et al, 2007; Puustinen & Jaattela, 2014). PP2A is a major serine/threonine phosphatase in eukaryotic cells. It is a heterotrimeric protein complex, comprised of scaffolding subunit A, regulatory subunit B, and catalytic subunit C. Each subunit has a variety of isoforms and splice variants, resulting in more than 80 distinct combinations of PP2A holoenzyme, which can specifically target different substrates and lead to different effects (Petritsch et al, 2000; Lechward et al, 2001; Andrabi et al, 2007; Sablina et al, 2010; Rodgers et al, 2011). Inactivation of PP2A occurs in up to 90% of breast cancer, either through PP2A phosphorylation or overexpression of its endogenous inhibitors SET or CIP2A. PP2A inhibition is associated with poor response to therapies (Anazawa et al, 2005; Sangodkar et al, 2016).
Protein kinase AKT is a major regulator of eukaryote's signaling networks (Sever & Brugge, 2015; Manning & Toker, 2017). Activated AKT stimulates the downstream signaling molecule NFκB through phosphorylation (Ozes et al, 1999; Yang et al, 2019). Many studies have shown that activated AKT can increase the expression levels of MMP2, MMP9, and Snail by activating the NFkB pathway through phosphorylation of IκB kinase (IKK), leading to IκB degradation and NFκB nuclear translocation, thus promoting the process of metastasis and invasion of tumors (Julien et al, 2007; Sarkar et al, 2008; Muscella et al, 2017). Our results indicate that CIP2A‐BP can release PP2A through competitive binding with PP2A, inhibit the phosphorylation of AKT, and then inhibit the activation of NFκB, resulting in the decrease of the expression levels of MMP2, MMP9, and Snail, thus inhibiting metastasis and invasion in triple‐negative breast cancer cells.
It should be noted that in normal cells, mTOR acts downstream of AKT pathway and influences 4E‐BP1 phosphorylation and CIP2A‐BP translation. However, in TNBC cells, high level of TGF‐β disrupts the dynamics among CIP2A‐AKT‐mTOR‐4E‐BP1 to promote tumor progression.
Mechanistically, we showed that CIP2A‐BP competes with PP2A's subunit B56γ to bind to CIP2A. In TNBC cells, downregulation of CIP2A‐BP frees CIP2A to bind to PP2A through B56γ, while CIP2A‐BP overexpression binds to CIP2A, thus releasing PP2A. Because PP2A acts as a tumor suppressor gene, CIP2A acts as an oncogene through inhibiting PP2A function; therefore, CIP2A‐BP acts as a tumor suppressor gene in TNBC. We further confirmed that in TNBC, CIP2A‐BP inhibits tumor metastasis through blocking activation of PI3K/AKT/NFκB signaling pathway, thereby resulting in decreased expression levels of MMP2, MMP9, and Snail.
Recent reports suggest that many lncRNAs modulate biological pathways and processes through encoding and translating micropeptides. For example, LINC00961 encodes SPAR peptide that negatively regulates mTORC1 activation and muscle regeneration (Matsumoto et al, 2017); LncRNA HOXB‐AS3 encodes a 53‐aa peptide, which suppresses colon cancer growth (Huang et al, 2017); LncRNA‐Six1 encodes a 7.26 kDa micropeptide that activates Six1 gene to promote cell proliferation and muscle growth (Cai et al, 2017); myoregulin is a regulator of skeletal muscle physiology encoded by a skeletal muscle‐specific LncRNA (Anderson et al, 2015). Our data indicate that LINC0065 encodes a functional micropeptide CIP2A‐BP that acts as a tumor suppressor gene in TNBC.
Since we showed that CIP2A‐BP can effectively inhibit TNBC cells metastasis and invasion both in vitro and in vivo, we next examined whether direct injection of CIP2A‐BP could also inhibit tumor invasion and metastasis. Interestingly, our data indicate that direct injection of CIP2A‐BP can significantly reduce lung metastasis in mouse mammary tumor model and significantly increase progression‐free survival. This is similar to super‐TDU peptide targeting YAP‐TEADs pathway to inhibit gastric cancer growth (Jiao et al, 2014); macrocyclic peptide decreasing c‐Myc protein levels to reduce prostate cancer cell growth (Mukhopadhyay et al, 2017); and PNC‐27 peptide inducing lysis of breast cancer cells (Sookraj et al, 2010). Our data add to existing literature on anti‐tumor peptides.
In summary, our clinical experiments show that CIP2A‐BP expression is significantly lower among TNBC patients with metastasis when compared to patients without metastasis, and low expression of CIP2A‐BP is associated with poor overall survival. Our data suggest that CIP2A‐BP expression could be used as a prognosis marker among TNBC patients. Our mechanistic experiments show that CIP2A‐BP functions downstream of TGF‐β signaling pathway. CIP2A‐BP's competitive binding to CIP2A releases PP2A from inhibition by CIP2A, which inactivates PI3K/AKT/NFκB pathway, resulting in decreased expression levels of MMP2, MMP9, and Snail, thus inhibiting breast cancer invasion and metastasis. Overexpression of CIP2A‐BP or direct injection of CIP2A‐BP peptide significantly reduces breast cancer lung metastasis and increases overall survival in a mouse mammary gland malignancy model. We conclude that CIP2A‐BP represents a potential therapeutic candidate to treat TNBC metastasis through augmenting PP2A activity.
Materials and Methods
Human study subjects
A total of 112 tissue samples of fresh TNBC were obtained from patients in eastern China who underwent tylectomies at the Affiliated Hospitals of Soochow University (Suzhou). Another 105 samples of fresh TNBC were collected from patients in southern China at the Cancer Hospitals affiliated with Guangzhou Medical University. None of the patients received anticancer treatment before surgery, including chemotherapy or radiotherapy. The clinical characteristics of the patients are listed in Appendix Table S1. The Medical Ethics Committees of Soochow University and Guangzhou Medical College approved this study.
Cell culture and treatments
All cell lines were purchased from Procell Life Science & Technology Co., Ltd. These cell lines were all characterized by DNA finger printing analysis and passaged < 6 months in this study. BT549 cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS); HEK293Tand MDA‐MB‐231 cells were grown in DMEM medium supplemented with 10% fetal bovine serum (FBS); MCF‐10A cells were grown in DMEM/F12 medium supplemented with 5% horse serum plus growth additive; and Hs578T cells were grown in DMEM medium supplemented with 10% fetal bovine serum (FBS) plus 0.01 mg/ml bovine insulin. All cell lines were grown in penicillin‐/streptomycin‐containing medium, at 37°C in a humidified atmosphere with 5% CO2.
Hs578T and MDA‐MB‐231 cells were infected with the lentivirus expresses the indicated LINC00665 ORF constructs, and these cells were then selected with G418 at 800 μg/ml (Gibco). MCF‐10A, MDA‐MB‐231, and MDA‐MB‐231 KO cells were transfected with the indicated plasmids with Lipofectamine 2000 for 48 h. The lentiviruses and plasmids used in this study are listed in the Appendix Table S2.
Cells were treated with TGF‐β (10 ng/ml) for 48 h (R&D Systems) to activate TGF‐β signaling pathway. To inhibit specific signaling pathways, cells were pretreated with vehicle (DMSO), 1 μM PP242 (Selleck Chemicals), 3 μM SIS3 (Selleck Chemicals), and 3 μM MK‐2206 (Selleck Chemicals) for 1 h at 37°C prior to the experiments.
Animal breeding and treatments
Female nude mice of 6–8 weeks of age were purchased from Shanghai Laboratory Animal Center at the Chinese Academy of Sciences (Shanghai, China). MMTV‐PyMT mice were obtained from Zhongjun Dong’ lab at Institute for Immunology and School of Medicine, Tsinghua University. All mice experiments were carried out in accordance with guidelines approved by the Laboratory Animal Center of Soochow University.
Hs578T and MDA‐MB‐231 cells lines stably expressing LINC00665 ORF and CIP2A‐BP knockout cell lines (Hs578T KO and MDA‐MB‐231 KO) were used. For experimental metastasis, tumor cells (1.0 × 105–106) were re‐suspended in 100 μl PBS and injected into the tail vein of nude mice. The metastatic foci in the lungs were directly visualized in randomly selected fields 8 weeks after implantation.
For tumor metastasis experiments, on day 1, both MDA‐MB‐231 cells (1.0 × 105–106) and CIP2A‐BP (300 μg/ml) were injected into the tail vein of nude mice. On day 7, CIP2A‐BP (15 mg/kg) was re‐injected via the tail vein. The metastatic foci in the lungs were directly visualized in randomly selected fields 8 weeks after implantation. CIP2A‐BP (15 mg/kg) was also injected into the mammary fat pad of 8‐week‐old MMTV‐PyMT mice. On day 7 after injection, CIP2A‐BP (15 mg/kg) was injected weekly for additional 5 weeks for each mice group (5 mice per group). The metastatic foci in the lungs were directly visualized in randomly selected fields 12 weeks after implantation.
Generation of MMTV‐PyMT;CIP2A‐BP +/+ mice
We generated a CIP2A‐BP overexpressing mouse models using the gene‐targeting construct. A single copy of the CIP2A‐BP cDNA was inserted downstream to the STOP cassette through homologous recombination into the ROSA26 (R26) locus, so the exogenous CIP2A‐BP cDNA is transcribed under the control of R26 promoter allowing a reproducible and stable overexpression of CIP2A‐BP. This gene‐targeting construct was electroporated into ES cells, and microinjection of the recombinant ES clones led to the generation of chimeric animal.
To analyze the in vivo consequences of CIP2A‐BP overexpression, conditional KI animals were crossed with the CMV‐Cre strain in order to obtain CIP2A‐BP +/− heterozygous mice. CIP2A‐BP +/+ homozygous animals were generated by intercrossing heterozygous mice. Finally, CIP2A‐BP +/+ mice were crossed with C57BL/6 MMTV‐PyMT mice to generate MMTV‐PyMT;CIP2A‐BP +/+ on the C57BL/6 background.
Ribosome profiling and RNA sequencing data analysis
The ribosome profiling and RNA sequencing data of TGF‐β‐treated MCF‐10A cells (GSE59817) were obtained from GEO database. For ribosome profiling data, the raw reads were preprocessed by cutadapt software, and then, the filtered reads were aligned to Genome Reference Consortium Human Build 37 (GRCh37) using TopHat2 algorithm. After alignment to the human genome, the gene expression levels were calculated by Cufflinks, and the differentially expressed genes were calculated by Cuffdiff. Finally, the genes which FDR < 0.05 were defined as differentially expressed genes. For RNA sequencing data, the reads were aligned to Genome Reference Consortium Human Build 37 (GRCh37) using TopHat2 algorithm. After alignment to the human genome, the gene expression levels were calculated by Cufflinks, and the differentially expressed genes were calculated by Cuffdiff. Finally, the genes which FDR < 0.05 were defined as differentially expressed genes.
Plasmid constructs
To generate eGFP fusion protein constructs with the LINC00665 ORF (ORF‐GFPmut), the LINC00665 ORF sequences were amplified using RT–PCR and cloned into a pEGFP‐N1 vector in which the GFP start codon (ATGGTG) was mutated to ATTGTT (pGFPmut) (Clontech). A mutant (ORFmut‐GFPmut) in which the LINC00665 ORF start codon was mutated to ATT in the LINC00665 ORF‐GFPmut vector was generated using a QuikChange Site‐Directed Mutagenesis Kit (Agilent Technologies).
To generate His fusion protein constructs with the LINC00665 ORF (ORF‐His), the LINC00665 ORF sequence was amplified and cloned into the pcDNA3.1(+) vector (Invitrogen). A mutation construct (ORFmut‐His) in which the ORF start codon was mutated to ATT in the LINC00665 ORF‐His vector was generated using a QuikChange Site‐Directed Mutagenesis Kit (Agilent Technologies).
Lentiviral production and transduction
A 177 bp sequence of LINC00665 ORF‐His was synthesized by Genewiz (Beijing, China) and then cloned into the lentiviral expression vector pLVX‐IRES‐neo (Clontech Laboratories Inc.). To produce lentivirus containing CIP2A‐BP targeting sequence, 293T cells were co‐transfected with the vector described above and the lentiviral vector packaging system using Lipofectamine 2000. Infectious lentiviruses were collected at 48 and 72 h after transfection and filtered through 0.45‐μm PVDF filters. We used empty plenty‐CMV‐IRES‐Puromycin vector to generate negative control lentiviruses. Recombinant lentiviruses were concentrated by centrifugation. The virus‐containing pellet was dissolved in DMEM, and aliquots were stored at −80°C until use. Hs578T, MDA‐MB‐231, Hs578TKO, and MDA‐MB‐231KO cells were infected with concentrated virus in the presence of polybrene (Sigma‐Aldrich). The supernatant was replaced with complete culture medium after 24 h, followed by selection with 800 μg/ml G418 (Gibco), and the expression of CIP2A‐BP in infected cells was verified by qRT–PCR, and the drug‐resistant cell populations were used for subsequent studies.
Immunofluorescence staining
MCF‐10A cells were transfected with LINC00665 ORF‐GFPmut and ORFmut‐GFPmut vectors for 24 h, and GFP fluorescence was directly visualized and recorded. MDA‐MB‐231 cells stably expressing LINC00665 ORF‐His, and ORFmut‐His, were plated on glass coverslips. These cells were fixed with 4% paraformaldehyde, permeabilized with 0.1% Triton X‐100, incubated with anti‐His (sc‐53073, Santa Cruz) or anti‐CIP2A‐BP (N/A, This paper) antibodies, and subsequently incubated with the corresponding Cy3‐conjugated secondary IgG antibodies. Cellular nuclei were stained with DAPI.
Anti‐CIP2A‐BP antibody preparation
Peptide synthesis and anti‐CIP2A‐BP antibody preparation were prepared as described (Yu et al, 2017) with some modifications. Briefly, a BSA and OVA‐coupled peptide ESWPFASTGGKLA‐Cys was synthesized, and polyclonal antibodies against the LINC00665 peptide were obtained from inoculated rabbits. Antibodies were purified using affinity chromatography on columns containing the corresponding peptides.
Antibodies and Western blotting
Cells and tissue were collected and lysed in cell/tissue lysis buffer for Western and IP (Beyotime Institute of Biotechnology). Proteins were separated on SDS–polyacrylamide gel and transferred to nitrocellulose membrane. Immunoblotting of the membranes was performed using the primary antibodies are listed in Appendix Table S3.
RNA extraction and qRT–PCR
Total RNA was isolated from MCF‐10A, TNBC using the RNA Isolater Total RNA Extraction Reagent (Vazyme). First‐strand cDNA was synthesized with the Superscript II‐reverse transcriptase kit (Invitrogen, Carlsbad). All qRT–PCR primers are listed in Appendix Table S4.
Northern blot analysis
LINC00665 northern blot was performed using a Roche DIG Northern Starter Kit (Roche) according to the manufacturer's instructions. A total of 10 μg of RNA from each sample was subjected to formaldehyde gel electrophoresis and transferred to a Biodyne Nylon membrane (Pall). The PCR primers used to generate the northern blot probe were listed in Appendix Table S5.
Migration and invasion assays
The ability of the cells to migrate and invade was assessed using Corning Transwell insert chambers with pores 8 μm in size (Corning) and a BioCoat Matrigel Invasion Chamber (BD Biosciences), respectively. Approximately 1.0 × 104 (migration assay) or 2.0 × 105 (invasion assay) transfected cells in 200 μl of serum‐free DMEM medium were seeded in the upper well; the chambers were then incubated with medium plus 20% fetal bovine serum for 48 h at 37°C to allow the cells to migrate to the lower well. The cells that had migrated or invaded through the membrane were fixed in methanol, stained with crystal violet (Invitrogen), imaged, and counted.
Wound‐healing assay
Cells were seeded on cell culture inserts (ibidi GmbH, Inc., Munchen, Germany) in 35‐mm dishes and incubated at 37°C in 5% CO2. After 24 h, the culture inserts were removed and medium was added. Wound healing within the gap was observed at different time points, and wound closure was evaluated in five random fields using an inverted microscope. The gap was analyzed using ImageJ software. And each experiment was repeated in triplicate.
Polysome profiling analysis
Polysome profiling was performed to measure the translation of CIP2A‐BP monitored by qRT–PCR. We performed polysome profiling following the procedure described before (Gandin et al, 2014). The primers for qRT–PCR were listed in Appendix Table S4.
Rna‐fish
We performed RNA‐FISH experiments using the lncRNA FISH Probe Mix (RiboBio) according to the manufacturer's instructions.
Cell fractionation of RNA
To determine the cellular localization of LINC00665, cytosolic and nuclear fractions were collected from MCF‐10A and MDA‐MB‐231 cells as per the manufacturer's instructions for the Nuclear/cytoplasmic Isolation Kit (Biovision).
Co‐immunoprecipitation and mass spectrometry
Whole‐cell lysates were prepared using lysis buffer (20 mM Tris (pH 7.5). Co‐immunoprecipitation assay was performed using Pierce™ Co‐Immunoprecipitation Kit (Thermo Scientific) according to the manufacturer's instructions. The lysates were applied to columns containing 10 μg of immobilized antibodies covalently linked to an amine‐active resin and incubated overnight at 4°C. Then, the co‐immunoprecipitate was eluted and analyzed by SDS–PAGE or mass spectrometry along with the controls. Co‐immunoprecipitation assays were performed using the antibodies are listed in Appendix Table S3.
Chromatin immunoprecipitation
ChIP assays were performed with an EZ‐ChIP kit (Millipore, Bedford, MA) according to the manufacturer's instructions. Chromatin from TNBC cells was immunoprecipitated with antibodies against Smad4, p65, Snail, and IgG control. The primers for ChIP were listed in Appendix Table S6.
RNA interference
Small interfering RNA (siRNA) targeting the Smad4, 4E‐BP1, and CIP2A gene and non‐targeting siRNA control were purchased from GenePharma. Using Lipofectamine 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer's protocol, TNBC cells were transfected with 75 nM siRNA. Also, 48 h after transfection the cells were then harvested. The siRNA used in this study is listed in Appendix Table S7.
Construction of reporter plasmids and dual‐luciferase reporter assay
Fragments of the human 4E‐BP1 promoter were synthesized and then inserted into the pGL3‐basic vector (Promega) (termed the pGL3‐4E‐BP1 promoter). Another plasmid was the deficient version, in which the binding motif was deleted from the pGL3‐4E‐BP1 promoter (termed the pGL3‐4E‐BP1‐mut promoter).
The promoter activity of 4E‐BP1 was determined using the dual‐luciferase reporter assay kit (Promega) according to the manufacturer's instructions. TNBC cells were seeded in 24‐well plates (1 × 105 cells per well) and cultured to 60–70% confluence before transfection. Then, cells were transfected with 800 ng of the reporter plasmids described above using Lipofectamine 2000 (Invitrogen). Twenty‐four hours post‐transfection, cells were treated with TGF‐β (10 ng/μl) for 48 h. The cells were collected using 100 μl passive buffer, and Renilla luciferase activity was detected using the Dual‐Luciferase Reporter Assay System (Promega), and a TD‐20/20 illuminometer (Turner Biosystems). The construction of other reporter gene plasmids used in this article is consistent with the above description. The promoter activity was repeated three times in parallel for statistical analysis.
Production of CIP2A‐BP KO cells by the CRISPR system
The gRNA sequence designed specifically for the ORF of CIP2A‐BP start codon inserted to the Cas9/gRNA (puro‐GFP) vector (VK001‐02, ViewSolid BioTech) was 5′‐GGCCACGTGCCTGCCGGACA‐3′. The constructed targeting vector was subsequently transfected into the TNBC cells using Lipofectamine 2000 (Thermo Fisher Scientific), after which the cells were cultured under puromycin drug selection (2 μg/ml). Single‐cell clones were selected and evaluated using the T7 endonuclease assay to detect mutation. Next, PCR amplification was performed using purified genomic DNA as a template and primers designed for sites near the start codon of CIP2A‐BP. Then, the obtained PCR products were subcloned into pUCm‐T vector (Sangon Biotech (Shanghai) Co., Ltd.). Lastly, the constructed clones were selected and sequenced using a 3100 Genetic Analyzer (ABI) to confirm the presence of mutation in the target sequence.
PP2A activity assay
The phosphatase activity of PP2A was detected by a commercial PP2A immunoprecipitation phosphatase assay kit (Millipore, Billerica, MA, USA) according to the manufacturer's instructions. Briefly, 100 μg protein extracts and PP2A were immunoprecipitated using 4 μg of PP2A antibody and 25 μl Protein A agarose slurry, both supplied by the kit. After 2 h of incubation in constant rocking, samples were washed three times with TBS 1× followed by one additional wash with a ser/thr assay buffer also provided by the kit. Next, 60 μl of a diluted phosphopeptide at 750 μM and 40 μl of ser/thr assay buffer were added, and the mix was incubated for 5 min at 30°C in a shaking incubator, and then, 25 μl of the mix was transferred into each well of a 96‐well plate. Each measurement was performed in triplicates. 100 μl of Malachite Green Detection Solution was added, and the mix was incubated for 15 min at room temperature. Absorbance at 650 nm was used to calculate the amount of phosphate released (pmol). To avoid variability due to differences in the amounts of immunoprecipitated protein between samples, the phosphatase activities were normalized to the amount of PP2A immunoprecipitated, as detected and quantified by immunoblot analysis for each treatment group.
Immunohistochemistry (IHC)
Paraffin‐embedded samples were sectioned at 4 μm thickness. Antigen retrieval was performed by a pressure cooker for 15–20 min in 0.01 M citrate buffer (pH 6.0) to remove aldehyde links formed during initial tissue fixation. Specimens were incubated with antibodies specific for p‐AKT (ab8933, Abcam) and CIP2A‐BP overnight at 4°C, and immunodetection was performed on the following day using DAB (Dako) according to the manufacturer's instructions.
Statistics
All experiments were repeated for at least three times unless stated in the figure legend. Two‐tailed paired Student's t‐tests were applied for comparisons between two groups. Survival curves were obtained using the Kaplan–Meier method and compared using the log‐rank test. Statistical analyses were performed using Prism 7 software. The data are presented as the mean ± SD except where stated otherwise. The differences with *P < 0.05, **P < 0.01, or ***P < 0.001 were considered statistically significant.
Study approval
Ethical consent was given by Soochow University Committee for Ethical Review of Research Involving Human Subjects. The use of human TNBC cancer tissue specimens was evaluated and approved by the Ethical Committee of the Soochow University and Guangzhou Medical University, and written informed consent was obtained from all participants or their appropriate surrogates. All animal studies were conducted with the approval of Soochow University Institutional Animal Care and Use Committee and were performed in accordance with established guidelines.
Author contributions
YZ and BG: study concept and design, analysis and interpretation of data, drafting of the manuscript, statistical analysis, obtained funding, and study supervision. XZ, LZ, SW, JD, and JL: acquisition of data, analysis and interpretation of data, drafting of the manuscript, and statistical analysis. RW, SZ, FL, and YW: acquisition of data.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Review Process File
Acknowledgements
This work was supported by the National Scientific Foundation of China grants 81772544, 81972649, and 81472630; Science Foundation for Distinguished Young Scholars in Jiangsu (BK20160008); A Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions; National Key R&D Program of China (2016YFC1302100); and the Program for Guangdong Introducing Innovative and Entrepreneurial Teams (2017ZT07S096).
The EMBO Journal (2020) 39: e102190
Data availability
All data used in this study can be downloaded from the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) with the following accession number: GSE59817.
References
- Akhurst RJ, Hata A (2012) Targeting the TGFbeta signalling pathway in disease. Nat Rev Drug Discov 11: 790–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anazawa Y, Nakagawa H, Furihara M, Ashida S, Tamura K, Yoshioka H, Shuin T, Fujioka T, Katagiri T, Nakamura Y (2005) PCOTH, a novel gene overexpressed in prostate cancers, promotes prostate cancer cell growth through phosphorylation of oncoprotein TAF‐Ibeta/SET. Cancer Res 65: 4578–4586 [DOI] [PubMed] [Google Scholar]
- Anderson DM, Anderson KM, Chang CL, Makarewich CA, Nelson BR, McAnally JR, Kasaragod P, Shelton JM, Liou J, Bassel‐Duby R et al (2015) A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell 160: 595–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrabi S, Gjoerup OV, Kean JA, Roberts TM, Schaffhausen B (2007) Protein phosphatase 2A regulates life and death decisions via Akt in a context‐dependent manner. Proc Natl Acad Sci USA 104: 19011–19016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azar R, Alard A, Susini C, Bousquet C, Pyronnet S (2009) 4E‐BP1 is a target of Smad4 essential for TGFbeta‐mediated inhibition of cell proliferation. EMBO J 28: 3514–3522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azar R, Lasfargues C, Bousquet C, Pyronnet S (2013) Contribution of HIF‐1alpha in 4E‐BP1 gene expression. Mol Cancer Res 11: 54–61 [DOI] [PubMed] [Google Scholar]
- Bhola NE, Balko JM, Dugger TC, Kuba MG, Sanchez V, Sanders M, Stanford J, Cook RS, Arteaga CL (2013) TGF‐beta inhibition enhances chemotherapy action against triple‐negative breast cancer. J Clin Invest 123: 1348–1358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjornsti MA, Houghton PJ (2004) Lost in translation: dysregulation of cap‐dependent translation and cancer. Cancer Cell 5: 519–523 [DOI] [PubMed] [Google Scholar]
- Cai B, Li Z, Ma M, Wang Z, Han P, Abdalla BA, Nie Q, Zhang X (2017) LncRNA‐Six1 encodes a micropeptide to activate Six1 in Cis and is involved in cell proliferation and muscle growth. Front Physiol 8: 230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fluck MM, Schaffhausen BS (2009) Lessons in signaling and tumorigenesis from polyomavirus middle T antigen. Microbiol Mol Biol Rev 73: 542–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandin V, Sikstrom K, Alain T, Morita M, McLaughlan S, Larsson O, Topisirovic I (2014) Polysome fractionation and analysis of mammalian translatomes on a genome‐wide scale. J Vis Exp 87: e51455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gluz O, Liedtke C, Gottschalk N, Pusztai L, Nitz U, Harbeck N (2009) Triple‐negative breast cancer–current status and future directions. Ann Oncol 20: 1913–1927 [DOI] [PubMed] [Google Scholar]
- Hata A, Chen YG (2016) TGF‐beta signaling from receptors to Smads. Cold Spring Harb Perspect Biol 8: a022061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JZ, Chen M, Chen D, Gao XC, Zhu S, Huang H, Hu M, Zhu H, Yan GR (2017) A peptide encoded by a putative lncRNA HOXB‐AS3 suppresses colon cancer growth. Mol Cell 68: 171–184 e6 [DOI] [PubMed] [Google Scholar]
- Janghorban M, Farrell AS, Allen‐Petersen BL, Pelz C, Daniel CJ, Oddo J, Langer EM, Christensen DJ, Sears RC (2014) Targeting c‐MYC by antagonizing PP2A inhibitors in breast cancer. Proc Natl Acad Sci USA 111: 9157–9162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao S, Wang H, Shi Z, Dong A, Zhang W, Song X, He F, Wang Y, Zhang Z, Wang W et al (2014) A peptide mimicking VGLL4 function acts as a YAP antagonist therapy against gastric cancer. Cancer Cell 25: 166–180 [DOI] [PubMed] [Google Scholar]
- Julien S, Puig I, Caretti E, Bonaventure J, Nelles L, van Roy F, Dargemont C, de Herreros AG, Bellacosa A, Larue L (2007) Activation of NF‐kappaB by Akt upregulates Snail expression and induces epithelium mesenchyme transition. Oncogene 26: 7445–7456 [DOI] [PubMed] [Google Scholar]
- Junttila MR, Puustinen P, Niemela M, Ahola R, Arnold H, Bottzauw T, Ala‐aho R, Nielsen C, Ivaska J, Taya Y et al (2007) CIP2A inhibits PP2A in human malignancies. Cell 130: 51–62 [DOI] [PubMed] [Google Scholar]
- Khanna A, Pimanda JE, Westermarck J (2013) Cancerous inhibitor of protein phosphatase 2A, an emerging human oncoprotein and a potential cancer therapy target. Cancer Res 73: 6548–6553 [DOI] [PubMed] [Google Scholar]
- Lara‐Medina F, Perez‐Sanchez V, Saavedra‐Perez D, Blake‐Cerda M, Arce C, Motola‐Kuba D, Villarreal‐Garza C, Gonzalez‐Angulo AM, Bargallo E, Aguilar JL et al (2011) Triple‐negative breast cancer in Hispanic patients: high prevalence, poor prognosis, and association with menopausal status, body mass index, and parity. Cancer 117: 3658–3669 [DOI] [PubMed] [Google Scholar]
- Lechward K, Awotunde OS, Swiatek W, Muszynska G (2001) Protein phosphatase 2A: variety of forms and diversity of functions. Acta Biochim Pol 48: 921–933 [PubMed] [Google Scholar]
- Li W, Zheng J, Deng J, You Y, Wu H, Li N, Lu J, Zhou Y (2014) Increased levels of the long intergenic non‐protein coding RNA POU3F3 promote DNA methylation in esophageal squamous cell carcinoma cells. Gastroenterology 146: 1714–26 e5 [DOI] [PubMed] [Google Scholar]
- Manning BD, Toker A (2017) AKT/PKB signaling: navigating the network. Cell 169: 381–405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto A, Pasut A, Matsumoto M, Yamashita R, Fung J, Monteleone E, Saghatelian A, Nakayama KI, Clohessy JG, Pandolfi PP (2017) mTORC1 and muscle regeneration are regulated by the LINC00961‐encoded SPAR polypeptide. Nature 541: 228–232 [DOI] [PubMed] [Google Scholar]
- Meyer C, Godoy P, Bachmann A, Liu Y, Barzan D, Ilkavets I, Maier P, Herskind C, Hengstler JG, Dooley S (2011) Distinct role of endocytosis for Smad and non‐Smad TGF‐beta signaling regulation in hepatocytes. J Hepatol 55: 369–378 [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay A, Hanold LE, Thayele Purayil H, Gisemba SA, Senadheera SN, Aldrich JV (2017) Macrocyclic peptides decrease c‐Myc protein levels and reduce prostate cancer cell growth. Cancer Biol Ther 18: 571–583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muscella A, Vetrugno C, Marsigliante S (2017) CCL20 promotes migration and invasiveness of human cancerous breast epithelial cells in primary culture. Mol Carcinog 56: 2461–2473 [DOI] [PubMed] [Google Scholar]
- Oshimori N, Oristian D, Fuchs E (2015) TGF‐beta promotes heterogeneity and drug resistance in squamous cell carcinoma. Cell 160: 963–976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB (1999) NF‐kappaB activation by tumour necrosis factor requires the Akt serine‐threonine kinase. Nature 401: 82–85 [DOI] [PubMed] [Google Scholar]
- Peter D, Igreja C, Weber R, Wohlbold L, Weiler C, Ebertsch L, Weichenrieder O, Izaurralde E (2015) Molecular architecture of 4E‐BP translational inhibitors bound to eIF4E. Mol Cell 57: 1074–1087 [DOI] [PubMed] [Google Scholar]
- Petritsch C, Beug H, Balmain A, Oft M (2000) TGF‐beta inhibits p70 S6 kinase via protein phosphatase 2A to induce G(1) arrest. Genes Dev 14: 3093–3101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puustinen P, Jaattela M (2014) KIAA1524/CIP2A promotes cancer growth by coordinating the activities of MTORC1 and MYC. Autophagy 10: 1352–1354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards EJ, Zhang G, Li ZP, Permuth‐Wey J, Challa S, Li Y, Kong W, Dan S, Bui MM, Coppola D et al (2015) Long non‐coding RNAs (LncRNA) regulated by transforming growth factor (TGF) beta: LncRNA‐hit‐mediated TGFbeta‐induced epithelial to mesenchymal transition in mammary epithelia. J Biol Chem 290: 6857–6867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter JD, Sonenberg N (2005) Regulation of cap‐dependent translation by eIF4E inhibitory proteins. Nature 433: 477–480 [DOI] [PubMed] [Google Scholar]
- Rodgers JT, Vogel RO, Puigserver P (2011) Clk2 and B56beta mediate insulin‐regulated assembly of the PP2A phosphatase holoenzyme complex on Akt. Mol Cell 41: 471–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sablina AA, Hector M, Colpaert N, Hahn WC (2010) Identification of PP2A complexes and pathways involved in cell transformation. Cancer Res 70: 10474–10484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangodkar J, Farrington CC, McClinch K, Galsky MD, Kastrinsky DB, Narla G (2016) All roads lead to PP2A: exploiting the therapeutic potential of this phosphatase. FEBS J 283: 1004–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar FH, Li Y, Wang Z, Kong D (2008) NF‐kappaB signaling pathway and its therapeutic implications in human diseases. Int Rev Immunol 27: 293–319 [DOI] [PubMed] [Google Scholar]
- Schmierer B, Hill CS (2007) TGFbeta‐SMAD signal transduction: molecular specificity and functional flexibility. Nat Rev Mol Cell Biol 8: 970–982 [DOI] [PubMed] [Google Scholar]
- Sekiyama N, Arthanari H, Papadopoulos E, Rodriguez‐Mias RA, Wagner G, Leger‐Abraham M (2015) Molecular mechanism of the dual activity of 4EGI‐1: dissociating eIF4G from eIF4E but stabilizing the binding of unphosphorylated 4E‐BP1. Proc Natl Acad Sci USA 112: E4036–E4045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sever R, Brugge JS (2015) Signal transduction in cancer. Cold Spring Harb Perspect Med 5: a006098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P (2016) Biology and management of patients with triple‐negative breast cancer. Oncologist 21: 1050–1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sookraj KA, Bowne WB, Adler V, Sarafraz‐Yazdi E, Michl J, Pincus MR (2010) The anticancer peptide, PNC‐27, induces tumor cell lysis as the intact peptide. Cancer Chemother Pharmacol 66: 325–331 [DOI] [PubMed] [Google Scholar]
- Sun M, Kraus WL (2015) From discovery to function: the expanding roles of long noncoding RNAs in physiology and disease. Endocr Rev 36: 25–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Okkeri J, Pavic K, Wang Z, Kauko O, Halonen T, Sarek G, Ojala PM, Rao Z, Xu W et al (2017) Oncoprotein CIP2A is stabilized via interaction with tumor suppressor PP2A/B56. EMBO Rep 18: 437–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Feng X, Molinolo AA, Martin D, Vitale‐Cross L, Nohata N, Ando M, Wahba A, Amornphimoltham P, Wu X et al (2019) 4E‐BP1 is a tumor suppressor protein reactivated by mTOR inhibition in head and neck cancer. Can Res 79: 1438–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wapinski O, Chang HY (2011) Long noncoding RNAs and human disease. Trends Cell Biol 21: 354–361 [DOI] [PubMed] [Google Scholar]
- Wu CG, Chen H, Guo F, Yadav VK, McIlwain SJ, Rowse M, Choudhary A, Lin Z, Li Y, Gu T et al (2017) PP2A‐B’ holoenzyme substrate recognition, regulation and role in cytokinesis. Cell Discov 3: 17027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Acharya S, Sahin O, Zhang Q, Saito Y, Yao J, Wang H, Li P, Zhang L, Lowery FJ et al (2015) 14‐3‐3zeta turns TGF‐beta's function from tumor suppressor to metastasis promoter in breast cancer by contextual changes of Smad partners from p53 to Gli2. Cancer Cell 27: 177–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Hu X, Mo YY (2019) Acidosis promotes tumorigenesis by activating AKT/NF‐kappaB signaling. Cancer Metastasis Rev 38: 179–188 [DOI] [PubMed] [Google Scholar]
- Yu X, Abdullahi AY, Wu S, Pan W, Shi X, Hu W, Tan L, Li K, Wang Z, Li G (2017) Prokaryotic expression of alpha‐13 giardin gene and its intracellular localization in giardia lamblia. Biomed Res Int 2017: 1603264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YE (2017) Non‐Smad signaling pathways of the TGF‐beta family. Cold Spring Harb Perspect Biol 9: a02212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Jiang L, Zhang L, Yang L, Deng J, You Y, Li N, Wu H, Li W, Lu J et al (2012) Functional genetic variations in the IL‐23 receptor gene are associated with risk of breast, lung and nasopharyngeal cancer in Chinese populations. Carcinogenesis 33: 2409–2416 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Review Process File
Data Availability Statement
All data used in this study can be downloaded from the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) with the following accession number: GSE59817.