

Abstract
Several neurodegenerative disorders like amyotrophic lateral sclerosis (ALS) and spinocerebellar ataxia (SCA) are caused by non‐coding nucleotide repeat expansions. Different pathogenic mechanisms may underlie these non‐coding repeat expansion disorders. While gain‐of‐function mechanisms, such as toxicity associated with expression of repeat RNA or toxicity associated with repeat‐associated non‐ATG (RAN) products, are most frequently connected with these disorders, loss‐of‐function mechanisms have also been implicated. We review the different pathways that have been linked to non‐coding repeat expansion disorders such as C9ORF72‐linked ALS/frontotemporal dementia (FTD), myotonic dystrophy, fragile X tremor/ataxia syndrome (FXTAS), SCA, and Huntington's disease‐like 2. We discuss modes of RNA toxicity focusing on the identity and the interacting partners of the toxic RNA species. Using the C9ORF72 ALS/FTD paradigm, we further explore the efforts and different methods used to disentangle RNA vs. RAN toxicity. Overall, we conclude that there is ample evidence for a role of RNA toxicity in non‐coding repeat expansion diseases.
Keywords: C9ORF72 ALS/FTD, non‐coding repeat expansion disorders, RNA toxicity
This review comprehensively delineates pathogenic mechanisms underlying various neurodegenerative disorders with special focus on disentangling toxicity associated with RNA species or RAN (repeat‐associated non‐ATG) products.

Glossary
- ALS
amyotrophic lateral sclerosis
- ASO
antisense oligonucleotide
- DPR
dipeptide repeat protein
- ELISA
enzyme‐linked immunosorbent assay
- FTD
frontotemporal dementia
- FTLD
frontotemporal lobe degeneration
- FXTAS
fragile X tremor ataxia syndrome
- HDL‐2
Huntington's disease‐like 2
- hnRNP
heterogeneous nuclear ribonucleoprotein
- HRE
hexanucleotide repeat expansion
- iMNs
induced motor neurons
- mRNP
messenger ribonucleoprotein
- PET
positron emission tomography
- RAN
repeat‐associated non‐ATG
- RBP
RNA‐binding protein
- rRBP
repeat RNA‐binding protein
- SCA
spinocerebellar ataxia
- UPS
ubiquitin‐proteasome system
- UTR
untranslated region
Introduction
Non‐coding repeat expansion disorders
Several neurodegenerative disorders are caused by a non‐coding repeat expansion and are referred to as “non‐coding repeat expansion disorders”. So far, ten non‐coding repeat expansion disorders have been described (Table 1). Most of them are adult‐onset disorders and are phenotypically characterized by variable syndromes that include ataxia, cognitive dysfunction, motor neuron symptoms, and extra‐neuronal involvement (Table 1). The most frequent clinical syndromes are myotonic dystrophy, amyotrophic lateral sclerosis (ALS)/frontotemporal dementia (FTD), and spinocerebellar ataxia (SCA; Table 1). The repeat expansion can be located in the promoter region, the 5′UTR (untranslated region), an intron, an alternate exon, or the 3′UTR of the respective gene (Table 1). The repeat sequence is variable, ranging from trinucleotide to hexanucleotide repeats, and is in general characterized by a high GC content (except SCA10 and SCA31). The range of the repeat length in healthy individuals normally does not exceed 30 repeats (Table 1). However, the range of the (unambiguously) pathogenic repeat lengths is highly variable and can be subdivided into three classes. First, repeats in SCA12 and Huntington's disease‐like 2 (HDL‐2) are usually not longer than 100 repeats (Margolis et al, 2004; Dong et al, 2015). Second, repeats generally do not exceed a few hundred (± 200) in fragile X tremor ataxia syndrome (FXTAS; O'Donnell & Warren, 2002) although longer repeats are associated with fragile X syndrome due to FMR1 loss of function (Verkerk et al, 1991). Third, repeats in all other diseases are mostly in the range of many hundreds up to a few thousands (Table 1). A clear length–phenotype correlation (i.e., more aggressive phenotype with increasing repeat length) has only been described in myotonic dystrophy types 1 and 2 (Udd & Krahe, 2012).
Table 1.
Non‐coding repeat expansion disorders
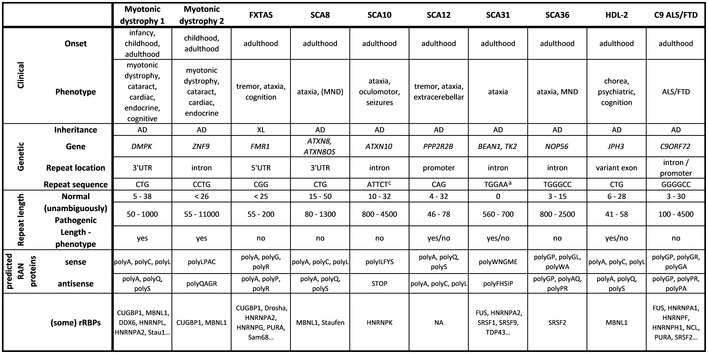
Overview of key features of all non‐coding repeat expansion disorders. Clinical features include age at onset (i.e., the main life phase(s)) and phenotypic presentation(s). Genetic features include inheritance pattern, gene containing the repeat expansion, location of the repeat in the respective gene, and sequence of the repeat. Data regarding repeat length include repeat length in healthy individuals, unambiguously pathogenic repeat lengths, and correlation between repeat length and phenotype. For each disease, all theoretical RAN proteins are described, both in sense and in antisense direction. Regarding possible mechanisms, rRBPs implicated in the disease are listed.
Abbreviations: AD, autosomal dominant; FXTAS, fragile X tremor ataxia syndrome; HDL, Huntington disease‐like; MND, motor neuron degeneration; rRBPs, repeat RNA‐binding proteins; SCA, spinocerebellar ataxia; XL, X‐linked.
Complex pentanucleotide (TAGAA, TAAAA, TAAAATAGAA).
“alteration” of function.
Impurity of repeat (associated with seizures).
Three possible mechanisms
Three non‐mutually exclusive mechanisms have been linked to the pathogenesis of non‐coding repeat expansion disorders (Fig 1; example is given for C9ORF72 ALS/FTD (C9 ALS/FTD)). Repeat RNA can cause toxicity by directly interacting with repeat RNA‐binding proteins and thereby compromising their normal function (Miller et al, 2000). This is referred to as “RNA toxicity”. The repeat RNA might also induce toxicity indirectly by being translated into toxic proteins (Zu et al, 2011). This translation occurs in a non‐ATG‐dependent way called “repeat‐associated non‐ATG” (RAN) translation and generates RAN proteins (Zu et al, 2011). This type of toxicity is called “RAN toxicity”. Depending on the repeat type (i.e., tri‐, penta‐, or hexanucleotide) and the specific repeat code, the RAN proteins consist of iterations of one, two, four, or five amino acids (Table 1). RAN proteins consisting of two amino acid repeats are called “dipeptide repeat proteins” (DPRs; Mackenzie et al, 2013). The presence of the repeat expansion might also lead to loss of function of the respective protein. This can be caused by decreased transcription initiation (e.g., epigenetic alterations), defective transcription (e.g., abortion), or increased mRNA degradation of the host gene (Todd et al, 2010; Haeusler et al, 2014). In the next chapter, we will give an overview of the current state of evidence regarding which mechanism is at play (RNA toxicity, RAN toxicity, and loss of function) in C9 ALS/FTD and the other non‐coding repeat expansion disorders.
Figure 1. Three possible pathogenic mechanisms of non‐coding repeat expansion disorders—example given for C9ORF72 ALS/FTD .
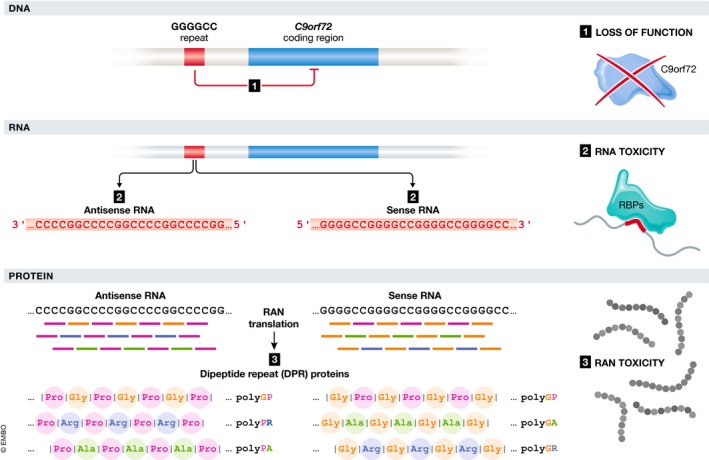
First, the repeat expansion might interfere with the normal transcription of the C9ORF72 gene, leading to loss of function of the C9orf72 protein. Second, repeat‐containing mRNAs might bind to various RNA‐binding proteins, hence disturbing their normal function. This is called “RNA toxicity”. Third, the repeat RNA itself might unconventionally be translated into peculiar toxic RAN peptides. This is called “RAN toxicity”.
Mechanisms in C9 ALS/FTD
Amyotrophic lateral sclerosis (ALS) is an adult‐onset neurodegenerative disease characterized by progressive degeneration of upper and lower motor neurons (Swinnen & Robberecht, 2014). Clinically, patients present with painless subacute focal muscle weakness. The disease is rapidly progressive, generally leading to death in 3–5 years after symptom onset, and is unfortunately still incurable (Swinnen & Robberecht, 2014). The genetic landscape of ALS has been redrawn significantly in recent years. In most (± 90%) cases, ALS does not run in the family and is hence called “sporadic ALS” (sALS). The remainder (± 10%) of ALS patients, however, has an affected first degree, which is called “familial ALS” (fALS). In the latter, a monogenetic cause is evidently suspected. Indeed, such monogenetic mutation is identified in the majority (± 70%) of patients, with C9ORF72, FUS, TARDBP, and SOD1 being the most frequent ones (Renton et al, 2014). Surprisingly, a monogenetic cause is identified in ± 10% of sALS patients, most likely reflecting both de novo mutations and incomplete penetrance of mutations. Frontotemporal dementia (FTD) is the clinical dementia syndrome caused by frontotemporal lobe degeneration (FTLD) and is the second most common dementia after Alzheimer's disease (AD) in patients younger than 65 years (Olney et al, 2017).
Amyotrophic lateral sclerosis and FTD are considered to constitute the extremes of a disease spectrum (Swinnen & Robberecht, 2014). The most frequent cause of ALS/FTD is a repeat expansion in the C9ORF72 gene (DeJesus‐Hernandez et al, 2011; Renton et al, 2011, 2014). Its structure at the DNA, RNA, and protein level is depicted in Fig 2. Post‐mortem examinations of C9 ALS/FTD cases reveal TDP‐43 pathology (Mackenzie et al, 2014; Saberi et al, 2015), reminiscent of sporadic ALS cases, but also RAN proteins (i.e., dipeptide repeat proteins (DPRs)) (Zu et al, 2013) and RNA foci (DeJesus‐Hernandez et al, 2011).
Figure 2. C9ORF72 gene structure, transcription, and translation.
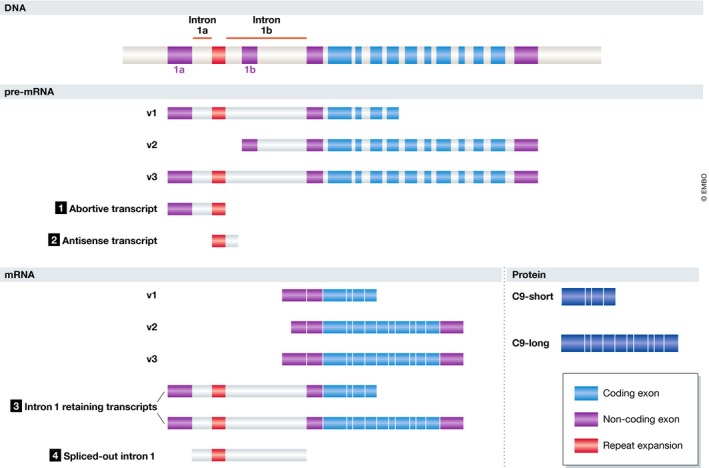
Four potentially pathogenic RNA species can be discerned. (1) At the pre‐mRNA level, transcription of v1 and v3 might stall at the repeat region, resulting in the generation of abortive transcripts. (2) Transcription of the repeat region in the antisense direction generates antisense transcripts. (3) Ineffective splicing of intron 1 in transcripts v1 and v3 might result in intron 1‐retaining transcripts. (4) Effective splicing of intron 1 in transcripts v1 and v3 might generate repeat‐containing spliced‐out intron 1.
C9 ALS/FTD is mainly a gain‐of‐function disease
C9 ALS/FTD is considered to be mainly driven by a gain‐of‐function mechanism, based on several observations. First, patients homozygous for the repeat expansion do not have an excessively aggressive clinical nor pathological phenotype which would have been expected in case of a loss‐of‐function mechanism (Cooper‐Knock et al, 2013; Fratta et al, 2013). Second, while there is one study reporting a C9ORF72 coding mutation (Liu et al, 2016), none were found in a large cohort of ALS patients (Harms et al, 2013). Third, C9ORF72 promoter hypermethylation, associated with gene silencing, is neuroprotective as observed using cross‐sectional and longitudinal neuroimaging data (McMillan et al, 2015). Fourth, several in vitro observations are not in line with a loss‐of‐function hypothesis. Most importantly, C9ORF72 transcript‐directed antisense oligonucleotide (ASO) treatment resulting in decreased or dysfunctional C9ORF72 transcripts rescued the phenotype (e.g., glutamate‐induced cell death (Donnelly et al, 2013) and transcriptional changes (Sareen et al, 2013), cfr. Table 5) in patient‐derived induced motor neurons (iMNs). Furthermore, ASO‐mediated C9ORF72 knockdown has no effect in control iMNs and neuronal primary cultures (Sareen et al, 2013; Sellier et al, 2016). Fifth, none of the C9orf72 knockout murine models develop a neurodegenerative phenotype (Lagier‐Tourenne et al, 2013; Koppers et al, 2015; Atanasio et al, 2016; Burberry et al, 2016; Jiang et al, 2016; O'Rourke et al, 2016; Sudria‐Lopez et al, 2016). Altogether, these data support the conclusion that C9ORF72 loss‐of‐function is not the main pathogenic driver suggesting mainly a gain‐of‐function mechanism; i.e., RNA and/or RAN toxicity.
RNA toxicity in C9 ALS/FTD
The exact nature of the repeat RNA present in RNA foci is still unclear. Four RNA species can be proposed (Fig 2). At the pre‐mRNA level, transcription of transcripts v1 and v3 might stall at the repeat region, resulting in the generation of abortive transcripts. Transcription of the repeat region in the antisense direction also generates antisense transcripts. Ineffective splicing of intron 1 in transcripts v1 and v3 might result in intron 1‐retaining transcripts. Finally, effective splicing of intron 1 in transcripts v1 and v3 might generate repeat‐containing spliced‐out intron 1. In general, repeat RNA is thought to form RNA foci that contain a cluster of repeat RNAs in complex with several RNA‐binding proteins (Kumar et al, 2017). RNA foci are not restricted to neurons and are also found in astrocytes, microglia, and oligodendrocytes (Mizielinska et al, 2013). While RNA foci are mostly intranuclear, cytoplasmic RNA foci as well as RNA foci at the edge of the nucleus have also been observed (Mizielinska et al, 2013; Cooper‐Knock et al, 2015b). RNA foci do not follow a rostrocaudal anatomical distribution as they are equally prevalent in the frontal cortex and in the spinal cord (DeJesus‐Hernandez et al, 2011, 2017).
RAN (DPR) toxicity in C9 ALS/FTD
Both sense DPRs (GA, GR, GP) and antisense DPRs (PR, PA, GP) are formed with sense DPRs being more abundant than antisense and GA being the most frequently observed one (Mori et al, 2013a; Mackenzie et al, 2015). DPRs are exclusively present in neurons and are mainly detected as cytoplasmic aggregates (Ash et al, 2013; Mackenzie et al, 2013). All DPRs have a similar anatomical distribution and are most abundant in cortical and cerebellar regions and almost absent in brainstem and spinal cord (Davidson et al, 2016; Mackenzie et al, 2015).
The toxic potential of the different DPRs has been examined comprehensively both in in vitro and in vivo disease models (Table 2). The potential mechanisms of this DPR toxicity have recently been reviewed (Freibaum & Taylor, 2017). Altogether, these data indicate that the arginine‐rich DPRs can be highly toxic, at least in overexpression systems. Data also support the notion that GA can be toxic, while GP and PA are probably harmless (at least in the currently available disease models). Despite these in vitro and in vivo findings, it remains to be determined whether DPRs contribute to the pathogenesis of C9 ALS/FTD in humans. One should note that obtaining post‐mortem support for DPR toxicity might be difficult as toxic DPR species might kill vulnerable motor neurons, hence leaving no trace to be uncovered. However, recent post‐mortem data favor an association between DPRs and pathology as GR aggregates correlate with neurodegeneration and even colocalize with phospho‐TDP‐43, albeit with some variability (Saberi et al, 2018; Sakae et al, 2018). Moreover, whereas DPR load generally does not correlate with clinical severity (Gendron et al, 2015), cerebellar GP levels inversely correlate with cognitive scores and GA burden is inversely related with age at onset (Davidson et al, 2014). Nevertheless, several post‐mortem observations are difficult to reconcile with DPR toxicity being the main culprit. Anatomical distribution of DPR aggregation post‐mortem does not obviously correlate with neurodegeneration. In short, DPR load is highest in unaffected tissue (i.e., cerebellum) and lowest in affected tissue (i.e., spinal motor neurons; Gomez‐Deza et al, 2015; Mackenzie et al, 2015). Moreover, coexistence of DPR and TDP‐43 aggregates in a given cell is very rare (Mackenzie et al, 2013; Davidson et al, 2014). In addition, their predominant appearance in disease models is not in line with post‐mortem findings (Mackenzie et al, 2015), especially the supposed nucleolar localization of GR and PR in disease models (Kwon et al, 2014; Wen et al, 2014). Nevertheless, the in vitro and in vivo models where GA forms cytoplasmic aggregates recapitulate post‐mortem findings in C9 ALS/FTD patients (Mackenzie et al, 2015). Altogether and despite the DPR toxicity observed in both in vitro and in vivo systems, its pathogenic involvement in ALS is still an open question.
Table 2.
In vitro and in vivo toxicity of individual DPRs
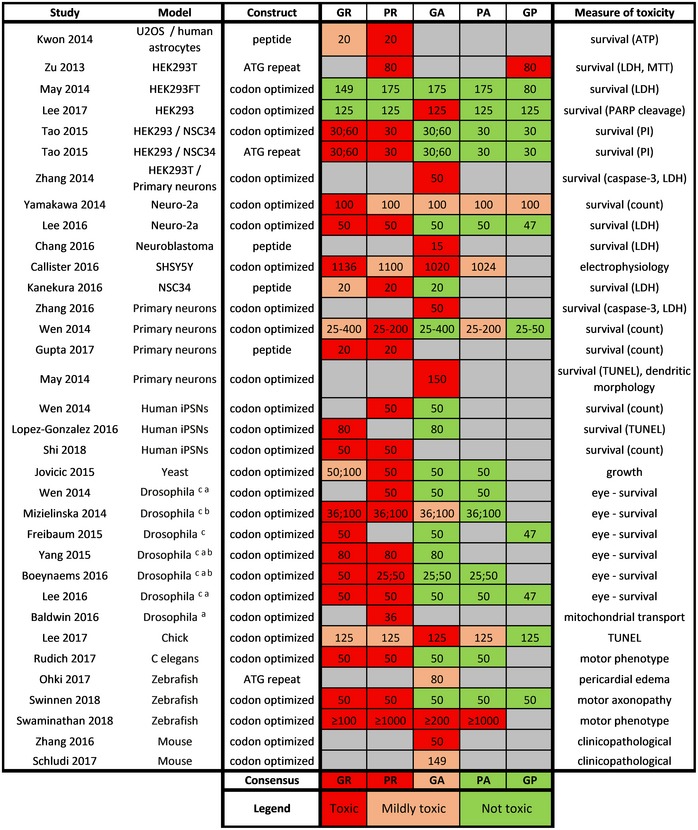
Numbers indicate the repeat lengths used.
Abbreviations: ATP, adenosine triphosphate; LDH, lactate dehydrogenase; MTT, 3‐(4,5‐dimethylthiazol‐2‐yl)‐2,5‐diphenyltetrazolium bromide; PI, propidium iodide; TUNEL, terminal deoxynucleotidyl transferase dUTP nick end labeling.
Motor neuron driver (OK371 or D42).
Pan‐neuronal driver (elav or tubulin).
Eye driver (GMR).
Loss of function in C9 ALS/FTD
Despite C9 ALS/FTD being considered as mainly having a gain‐of‐function disease mechanism, some data indicate that C9ORF72 loss‐of‐function might contribute to disease pathogenesis and could enhance the gain‐of‐function mechanisms. In C9 ALS/FTD post‐mortem brain tissue, C9ORF72 transcript levels are decreased by 50% (DeJesus‐Hernandez et al, 2011; Gijselinck et al, 2012; van Blitterswijk et al, 2015). Similarly, decreased levels of the long C9orf72 protein isoform have been observed in the frontal and temporal cortex of C9 ALS/FTD patients (Waite et al, 2014; Xiao et al, 2015; Saberi et al, 2018). Knockdown of C9ORF72 in in vitro models is associated with autophagic dysfunction, including p62 accumulation, perinuclear clustering of swollen lysosomes, and TDP‐43 aggregation (Sellier et al, 2016; Webster et al, 2016; Yang et al, 2016; Amick & Ferguson, 2017; Aoki et al, 2017). In patient‐derived cells, the glutamate hypersensitivity phenotype is rescued by C9ORF72 overexpression as well as being recapitulated by C9ORF72 knockout in control cells (Shi et al, 2018). The mechanism of C9ORF72 loss of function as well as its contribution to disease pathogenesis has recently been reviewed in detail (Balendra & Isaacs, 2018). Essentially, C9ORF72 loss‐of‐function might contribute to pathology via its role in autophagy (Balendra & Isaacs, 2018; Webster et al, 2018).
Mechanisms in other repeat expansion diseases
Myotonic dystrophy type 1
Myotonic dystrophy type 1 is caused by CTG repeats in the 3′ UTR of DMPK and is mainly driven by RNA toxicity. CUG repeat RNA adopts a stable hairpin conformation (Tian et al, 2000) that forms nuclear RNA foci (Taneja et al, 1995; Davis et al, 1997). The RNA foci can sequester muscleblind‐like (MBNL) proteins (Miller et al, 2000; Mankodi et al, 2001) leading to an imbalance between MBNL proteins and CUGBP1 (Lin et al, 2006; Kuyumcu‐Martinez et al, 2007). This imbalance causes altered splicing of several mRNAs (e.g., the insulin receptor IR2, the chloride channel CLC2, and the cardiac troponin cTNNT2) in a tissue‐dependent manner (Philips et al, 1998; Charlet‐B et al, 2002; Mankodi et al, 2002; Fugier et al, 2011) explaining the various multisystemic phenotypic features. Missplicing has been confirmed in patient tissue and correlates with clinical features (Savkur et al, 2001; Fugier et al, 2011; Freyermuth et al, 2016), and MBNL1 dysfunction is regarded as the key mechanism involved in myotonic dystrophy 1. Observations in several mouse models [i.e., Mbnl1 knockout (Kanadia et al, 2003), Mbnl2 knockout (Hao et al, 2008), Cugbp1 overexpressing (Ho et al, 2005; Ward et al, 2010), and (CUG)n expressing (Mahadevan et al, 2006)] are consistent with this view. However, RNA toxicity might encompass more than missplicing alone. Several additional modes of action of CUG repeat RNA toxicity have been proposed, including miRNA misprocessing (Perbellini et al, 2011), transcriptional dysregulation (Botta et al, 2007), global translational inhibition through stress granule induction (Onishi et al, 2008; Huichalaf et al, 2010), and use of alternative polyadenylation sites (Batra et al, 2014). In addition to RNA toxicity, RAN toxicity has been suggested as well. While polyQ, derived from antisense CAG repeat RNA, has been found in patient material (Zu et al, 2011), its pathogenic contribution is still not clear. DMPK loss of function is unlikely given the absence of a clear relevant phenotype in Dmpk knockout mice (Jansen et al, 1996; Reddy et al, 1996).
Myotonic dystrophy type 2
Myotonic dystrophy type 2 is caused by CCTG repeats in the intron of ZNF9 and resembles myotonic dystrophy type 1 in many regards. As a consequence, the underlying mechanism is believed to be very similar as well. Essentially, the CCUG repeat RNA leads to an MBNL‐CUGBP1 imbalance (Salisbury et al, 2009; Jones et al, 2011), making RNA toxicity the prevailing mechanism. However, as both sense (LPAC) and antisense (QAGR) RAN peptides are present in post‐mortem tissue and display in vitro toxicity (Zu et al, 2017), they might contribute to certain aspects of the disease as well. Additionally, CNBP loss of function might also play a role, as Cnbp‐deficient mice develop key features of myotonic dystrophy (Chen et al, 2007).
Fragile X tremor ataxia syndrome
FXTAS is caused by CGG repeats in the 5′ UTR of the FMR1 gene. Pathological hallmarks of FXTAS consist of Purkinje cell loss and intranuclear ubiquitin‐positive inclusions containing a polyglycine RAN peptide (Buijsen et al, 2014; Boivin et al, 2018). Loss of function is excluded as patients with Fragile X syndrome, caused by FMR1 loss‐of‐function due to very long (> 200) CGG repeats, do not develop any FXTAS features (Boivin et al, 2018). Moreover, FMR1 mRNA levels are even increased in FXTAS patients (Kenneson et al, 2001; Allen et al, 2004) and expression of CGG repeat RNA induces in vitro and in vivo neurotoxicity (Jin et al, 2003; Willemsen et al, 2003; Hukema et al, 2014), suggesting a primary gain‐of‐function mechanism. The CGG repeat RNA is able to adopt secondary structures (i.e., G‐quadruplexes, duplexes and hairpins; Malgowska et al, 2014), which might compromise the function of various RNA‐binding proteins like Pur‐alpha (Jin et al, 2007), hnRNPA2/B1 (Sofola et al, 2007), CUGBP1 (Sofola et al, 2007), Sam68 (Sellier et al, 2010), and Drosha‐DGCR8 (Sellier et al, 2013). The observation that overexpression of most of these proteins can rescue the phenotype in CGG Drosophila (Jin et al, 2007; Sofola et al, 2007; Sellier et al, 2013) supports a functional role of these proteins in FXTAS pathogenesis. However, RNA toxicity seems to be insufficient to explain FXTAS pathogenesis because of the following three observations. First, the repeat size is relatively short, compared to (mainly) RNA toxicity driven diseases (e.g., myotonic dystrophy—cfr. Table 1). Second, the large ubiquitin‐positive intranuclear inclusions in FXTAS are reminiscent of aggregates typically seen in protein‐mediated neurodegenerative disorders (e.g., Huntington's disease). Third, the toxicity of CGG constructs in Drosophila and mouse models seems to depend on FMRpolyG production (Todd et al, 2013; Sellier et al, 2017) suggesting a contribution of RAN toxicity. FMRpolyG has been found in patient‐derived cells (Sellier et al, 2017), mouse models (Hukema et al, 2015; Sellier et al, 2017) and in post‐mortem tissue (Todd et al, 2013; Buijsen et al, 2014), where it colocalizes with the ubiquitin‐positive intranuclear inclusions (Todd et al, 2013). In several models, FMRpolyG displays a length‐dependent propensity to aggregate in the nucleus (Todd et al, 2013; Sellier et al, 2017), and it is suggested to be neurotoxic by disturbing the ubiquitin‐proteasome system (UPS) and the nuclear lamina structure (Oh et al, 2015; Sellier et al, 2017). Interestingly, antisense RAN proteins have also been observed in patient material (Krans et al, 2016). However, their pathogenic contribution has not yet been characterized.
SCA8
The 3′UTR repeat expansion in SCA8 is bidirectionally transcribed (i.e., (CTG.CAG)n), complicating the quest for the underlying mechanism. In the ATXN8 strand, the CUG repeat RNA is believed to cause RNA toxicity via MBNL1 dysfunction, similar to what is seen in myotonic dystrophy. Supporting this, nuclear CUG RNA foci colocalize with MBNL1 in molecular layer interneurons of SCA8 patients and mouse models, and loss of Mbnl1 exacerbates the phenotype of SCA8 mice (Daughters et al, 2009). Additionally, splicing changes in a target of MBNL1 (i.e., GAT4) have been established in post‐mortem tissue, validating the pathogenic relevance of RNA toxicity (Daughters et al, 2009). In the ATXN8OS strand, the CAG repeat RNA is translated into toxic polyglutamine and intranuclear polyglutamine inclusions have been seen in Purkinje cells and brainstem neurons of SCA8 mice and post‐mortem tissue (Moseley et al, 2006). Moreover, in post‐mortem tissue polyserine derived from the ATXN8OS strand by RAN translation has been discovered in degenerating white matter regions (Ayhan et al, 2018). Loss of function of the host gene(s) seems unlikely, as individuals harboring a genomic deletion in the SCA8 region do not exhibit cerebellar degeneration (Mandrile et al, 2016). Moreover, CTG expression in Drosophila and mouse models leads to neurodegenerative phenotypes further supporting a gain‐of‐function mechanism (Moseley et al, 2006; Tripathi et al, 2016). Therefore, SCA8 seems to be mainly driven by two gain‐of‐function mechanisms, being RNA and RAN toxicity, arising from bidirectional repeat RNAs.
SCA10
SCA10 is caused by ATTCT repeats in the intron of ATXN10, and a gain‐of‐function mechanism has been proposed due to two main observations. First, an ATXN10 loss‐of‐function mechanism is unlikely as ATXN10 transcript levels are unaltered in SCA10 patients (Wakamiya et al, 2006), as heterozygous Atxn10 knockout mice display no abnormalities (Wakamiya et al, 2006) and as loss‐of‐function ATXN10 mutations do not give rise to a SCA10 phenotype in humans (Keren et al, 2010). Second, in vitro and in vivo (mainly mouse) models overexpressing ATTCT repeat constructs exhibit phenotypes resembling SCA10 (White et al, 2010; White et al, 2012). The exact nature of this gain of function is still unclear, but current data suggest RNA toxicity as the prevailing mechanism. The repeat expansion is spliced out and adopts a hairpin structure (Handa et al, 2005; Park et al, 2015) that binds hnRNPK in vitro and forms nuclear and cytoplasmic RNA foci in patient‐derived cells which colocalize with hnRNPK (White et al, 2010). Furthermore, hnRNPK overexpression rescues in vitro ATTCT toxicity (White et al, 2010), indicating that hnRNPK dysfunction is a key factor in SCA10. However, post‐mortem examinations have not been performed yet and RAN peptides (i.e., poly(ILFYS)) have not been assessed leaving the role of RAN toxicity in SCA10 unclear.
SCA12
SCA12 is caused by CAG repeats in the promoter region of PPP2R2B that encodes a subunit of the phosphatase PP2A. The repeat is located in the promoter region of one (of many) protein isoforms, leading to increased promoter activity upon repeat expansion (O'Hearn et al, 2015). Overexpression of PPP2R2B is toxic both in vitro (O'Hearn et al, 2015) and in Drosophila (Wang et al, 2011) suggesting gain‐of‐function toxicity. However, PPP2R2B mRNA and protein levels have not yet been assessed in post‐mortem tissue making the PPP2R2B gain‐of‐function mechanism still hypothetical. The contribution of RNA and RAN toxicity has also not been evaluated in post‐mortem tissue, and suitable disease models are also lacking. Nevertheless, SCA12 is unlikely to be a polyglutamine disease, as polyglutamine inclusions are absent in a post‐mortem case (O'Hearn et al, 2015). The disease phenotypes are also rather mild compared to other polyglutamine diseases.
SCA31
SCA31 is caused by intronic TGGAA repeats in the BEAN1 and TK2 genes. Small nuclear sense RNA foci are present exclusively in Purkinje cells (Niimi et al, 2013) and colocalize with TDP‐43 (Ishiguro et al, 2017). Repeat RNA‐binding proteins include TDP‐43, SRSF1, SRSF9, NONO, Matrin3, and several hnRNPs (Sato et al, 2009; Ishiguro et al, 2017). In vitro and in vivo (Drosophila) expression of TGGAA repeat constructs leads to toxicity that is suppressed by overexpression of the RNA‐binding proteins TDP‐43, hnRNPA2, and FUS, supporting an RNA toxicity gain‐of‐function mechanism (Niimi et al, 2013; Ishiguro et al, 2017). Nevertheless, poly(WNGME) has been detected as granular cytoplasmic inclusions in Purkinje cells and in Drosophila (Ishiguro et al, 2017). Moreover, production of poly(WNGME) in the latter was reduced upon TDP‐43 overexpression (Ishiguro et al, 2017). Therefore, RAN toxicity in SCA31 cannot be excluded at this moment.
SCA36
SCA36 is caused by intronic TGGGCC expansions in NOP56, and sense RNA foci are abundantly present throughout the brain (Liu et al, 2014). Interestingly, antisense RNA foci have not been observed in post‐mortem tissue nor in a mixed neuronal population derived from induced pluripotent stem cells (iPSCs; Matsuzono et al, 2017). SRSF2, an RNA‐binding protein that mainly functions as a splicing factor, binds to UGGGCC repeat RNA and colocalizes with RNA foci in patient‐derived lymphoblasts (Kobayashi et al, 2011). The role of RAN toxicity in SCA36 is difficult to evaluate based on present data. While a first report did not observe any neuronal inclusions of ubiquitin or p62 (Obayashi et al, 2015), these were present in a second case (Liu et al, 2014), predominantly in the inferior olivary nucleus.
Huntington's disease‐like 2
Huntington's disease‐like 2 is caused by CTG repeats in an alternatively spliced exon of JPH3. The disease clinically and pathologically mimics Huntington's disease. In post‐mortem tissue, corticostriatal degeneration and intranuclear ubiquitin‐positive polyglutamine inclusions are evident (Greenstein et al, 2007; Rudnicki et al, 2008). Given what we know about Huntington's disease, RAN toxicity is likely to be the prevailing mechanism in HDL‐2, driven by polyglutamine generated from antisense CAG repeat RNA. This is supported by the observation of antisense transcripts as well as polyglutamine inclusions in an HDL‐2 mouse model (Wilburn et al, 2011). Nevertheless, protein toxicity mediated by polyalanine or polyleucine in the sense direction through canonical translation cannot be excluded. RNA toxicity driven by CUG repeat RNA, supposedly through MBNL1 dysfunction, might also be at play. As RNA foci are present in post‐mortem tissue and as HDL‐2 repeat RNA is toxic in vitro (Rudnicki et al, 2007), more work still needs to be done to investigate the role of RNA toxicity in HDL‐2. Loss of function of the JPH3 gene might also contribute as JPH3 protein levels are decreased in post‐mortem samples and as jph3 knockout mice develop a motor phenotype (Seixas et al, 2012).
Disentangling RNA and RAN toxicity: a Gordian knot in C9 ALS/FTD?
In order to further understand the contribution of RNA and RAN toxicity, we will focus on the C9 ALS/FTD paradigm. Recent research into C9 ALS/FTD has generated many disease models that allow for a more complete evaluation of the contribution of both mechanisms to pathology. Unfortunately, disentangling these two mechanisms in disease models is very difficult (cfr. Box 1). To specifically assess the contribution of RNA toxicity in the pathogenesis of C9 ALS/FTD, several approaches can be employed, each targeting a different step in the pathway leading to RNA toxicity (Fig 3). We will systematically discuss the current state of evidence at these different levels.
Figure 3. Roadmap to prove/disprove RNA toxicity.
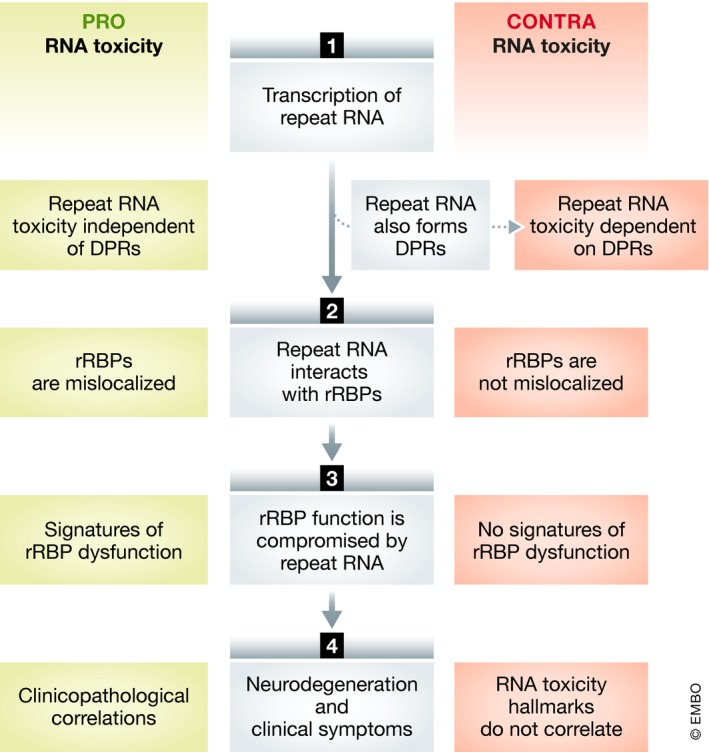
Arguments pro/contra RNA toxicity can be generated at four levels of the presumed pathogenic cascade of RNA toxicity.
Box 1. The enigma of modeling RNA toxicity with “non‐ATG repeat constructs” (with Fig 4).
Proving that repeat RNA exerts its toxicity independent of DPR formation is very challenging due to intrinsic methodological limitations of the constructs used to model C9ORF72 gain of function (Fig 4). Theoretically, four different types of constructs can be used, each potentially modeling DPR and/or RNA toxicity (Fig 4). The origin of potential toxicity is clear for “codon‐optimized” and “RNA only” constructs (i.e., DPRs and RNA, respectively). “ATG repeat constructs” cannot be used to model RNA toxicity as DPRs are generated by default, hence obscuring any potential RNA toxicity. For “non‐ATG repeat constructs,” the situation is difficult as DPRs can only be generated by RAN translation and hence are not present by default. Therefore, if no DPRs are detected, toxicity is to be attributed to the repeat RNA itself, indicating RNA toxicity. As such, a good DPR detection approach is important. Three major aspects are important to consider (for a given study, this is covered under the headings ‘Adequate methodology?’ in Tables 3, 4, 5). First, the DPR detection method needs to have a high sensitivity and specificity. Only highly sensitive DPR detection methods (i.e., ELISA or dot blot, opposed to immunohistochemistry or Western blot) have sufficient power to confidently assess DPR presence. Specificity relies on the use of appropriate positive and negative controls. Second, the presence of all possible DPRs should be investigated. Third, (raw) data concerning DPR detection should be provided. Additionally, these models should display C9 ALS/FTD hallmarks in order to claim disease relevance (i.e., RNA foci, TDP‐43 pathology, or motor neuron degeneration).
Figure 4. Constructs used to model C9ORF72 gain‐of‐function toxicity.

First, codon‐optimized DPR constructs generate DPRs but do not have the potential to inflict RNA toxicity since the lack of a repetitive sequence. Therefore, these constructs allow an easy modeling of DPR toxicity. Second, ATG repeat constructs can theoretically induce RNA toxicity but by default also generate DPRs. Therefore, RNA toxicity cannot be investigated with these constructs. Third, repeat constructs lacking an ATG start codon (i.e., “non‐ATG repeat constructs”) can also give rise to RNA toxicity while DPR generation is uncertain since it needs to rely on RAN translation. Therefore, by assessing the presence of DPRs these constructs can be used to assess RNA toxicity. Fourth, so‐called “RNA only” constructs should theoretically only give rise to RNA toxicity as the repeat sequence is regularly interrupted by stop codons interfering with RAN translation.
RNA toxicity in the absence of DPRs in C9ORF72 hexanucleotide disease models
Mammalian cell culture non‐ATG repeat models
Only a minority of in vitro studies have performed a full and complete characterization of hexanucleotide repeat expansion (HRE) models (Table 3). A key problem concerns the DPR detection methodology in many of the studies. Four studies assessed the relationship between toxicity and DPR formation (Wen et al, 2014; Burguete et al, 2015; Hautbergue et al, 2017; Stopford et al, 2017). Two of these studies found repeat toxicity to occur in the absence of detecting DPRs, suggesting an involvement of RNA toxicity (Wen et al, 2014; Burguete et al, 2015). However, the DPR detection methodology might not be optimal in order to draw strong conclusions (low sensitivity with inadequate/absent positive control).
Table 3.
Mammalian cell culture non‐ATG C9ORF72 repeat models
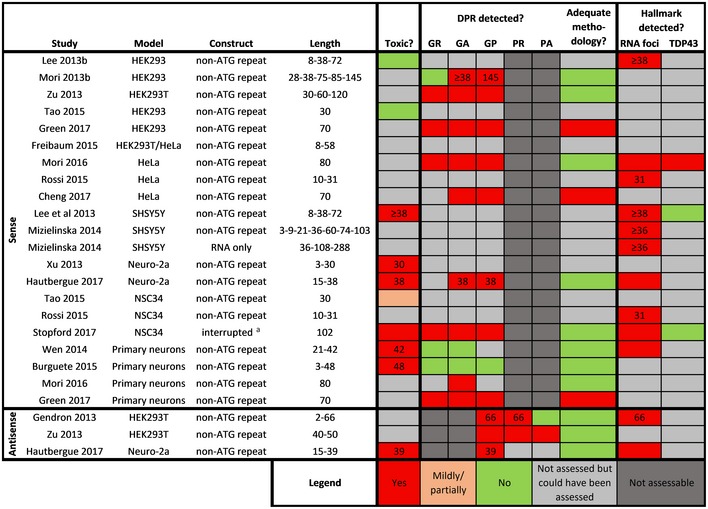
Overview of toxicity, detection of DPRs, and presence of C9ORF72 ALS hallmarks in sense and antisense in vitro models expressing non‐ATG C9ORF72 repeat constructs. If a modality was only found to be present at certain repeat lengths, these are indicated as numbers in the respective boxes.
Non‐stop‐codon interrupted.
In vivo non‐ATG repeat models
Different in vivo HRE models have been characterized also with respect to the presence of DPRs (Table 4). Altogether, three studies found toxicity in the absence of DPR detection, suggestive of RNA toxicity. First, a Drosophila model expressing (GGGGCC)30 in the eye or in adult neurons showed toxicity without detection of GR and GP (Zhang et al, 2015). Unfortunately, a non‐ideal positive control was used (i.e., construct under control of a different promoter and different treatment condition) and the presence of GA was not assessed. In a second study, GGGGCC repeats induced toxicity in a chicken embryo model without obvious presence of DPRs as assessed by immunohistochemistry (Lee et al, 2017). Third, using a zebrafish model we observed GGGGCC repeat RNA to induce motor axonal toxicity in the absence of DPRs (Swinnen et al, 2018). Presence of DPRs was assessed with a quantitative and sensitive immunoassay for GP and GA and with a sensitive dot blot assay for GR and PR. Interestingly, this model also revealed antisense repeat RNA to induce toxicity independent of DPRs (Swinnen et al, 2018).
Table 4.
In vivo non‐ATG C9ORF72 repeat models
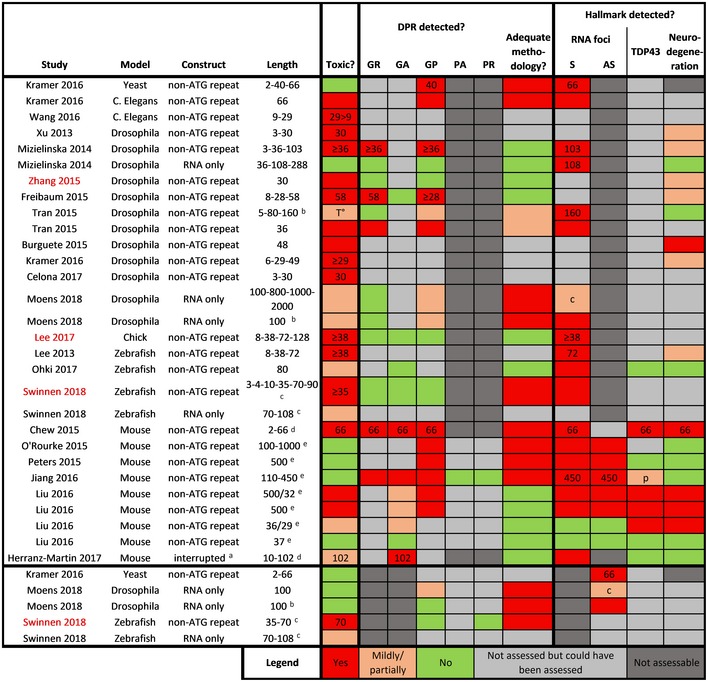
Overview of toxicity, detection of DPRs, and presence of C9 ALS hallmarks in sense and antisense in vivo models expressing non‐ATG C9ORF72 repeat constructs. Studies in red indicate those that found toxicity in the absence of DPRs. If a modality was only found to be present at certain repeat lengths, these are indicated as numbers in the respective boxes.
Abbreviations: c, cytoplasmic; p, increased levels of phospho‐TDP43; T°, only toxic upon higher temperature.
Non‐stop‐codon interrupted.
Intronic.
RNA microinjection.
AAV intracerebroventricular injection.
BAC.
Overall, Drosophila models seem to be very sensitive to arginine‐rich DPR‐induced toxicity (Table 2). As such, the slightest presence of GR or PR generated through RAN translation in repeat expansion Drosophila models might mask potential RNA toxicity.
Box 2. “RNA only constructs” and the search for the holy grail.
Expression constructs harboring the pure GGGGCC repeat sequences (i.e., “ATG repeat” and “non‐ATG repeat”—Fig 4) are easily confounded by the generation of RAN peptides and are therefore not well suited to assess RNA toxicity. Therefore, a GGGGCC repeat construct in which no RAN translation takes place would be the ideal paradigm. Mizielinska et al (2014) have generated “RNA only” constructs that lack an ATG start codon and contain the GGGGCC sequence which is regularly (every 15 repeats) interrupted by stop codons in all reading frames, both in the sense and in the antisense direction. As such, any toxicity arising from these constructs can only be attributed to RNA toxicity. “RNA only” constructs were initially found not to be toxic in Drosophila (Mizielinska et al, 2014), arguing against RNA toxicity. We as well as others, however, found that these constructs can induce (limited) neuronal toxicity in zebrafish and Drosophila (Moens et al, 2018; Swinnen et al, 2018). The limited toxicity could be explained in two ways. First, the “RNA only” repeat RNA might not resemble the physiological situation, as the original repeat sequence is regularly interrupted by non‐repeat sequences. This might disrupt the secondary structure of the repeat RNA and therefore interfere with its interaction with proteins. Second, the absence of toxicity with the “RNA only” constructs might be due to differences in the RNA‐binding protein pool in models versus humans, rendering the former more resistant to RNA toxicity. The observation that knockdown of the Drosophila orthologue of HNRNPH is harmless underscores this view (Moens et al, 2018), especially since HNRNPH knockdown is detrimental in human cells (Lefave et al, 2011). Altogether, “RNA only” constructs have so far not provided conclusive data on the role of RNA toxicity in disease pathogenesis.
Patient‐derived cellular disease models
Disentangling the pathological mechanisms at play in C9 patient‐derived disease models (Table 5) is complex and in addition to potential roles of RNA and DPR toxicity, loss of function also needs to be taken into consideration. Various reported phenotypes have been rescued by ASO‐mediated decrease in C9ORF72 transcript levels (containing the repeat RNA), strongly arguing for a gain‐of‐function mechanism (Donnelly et al, 2013; Sareen et al, 2013; Zhang et al, 2015). Presence of DPRs has often not been assessed, and as discussed above, issues with detecting DPRs complicate the matter regarding the relationship between toxicity and DPRs in C9 cells. In general, DPRs are difficult to detect in C9 cells and have not been found in an aggregated state. Detection of antisense DPRs is even more challenging and PA has never been detected (Westergard et al, 2016). RNA foci, if assessed, were invariably present, hence any correlation with toxicity was absent. There is one study using patient‐derived iMNs supporting RNA toxicity (Donnelly et al, 2013). Treatment of C9 iMNs rescued the observed phenotype and reduced RNA foci but had no effect on GP expression (Donnelly et al, 2013). However, the presence of GR, PR, and GA was not assessed (Donnelly et al, 2013).
Table 5.
Patient‐derived cellular in vitro C9ORF72 models
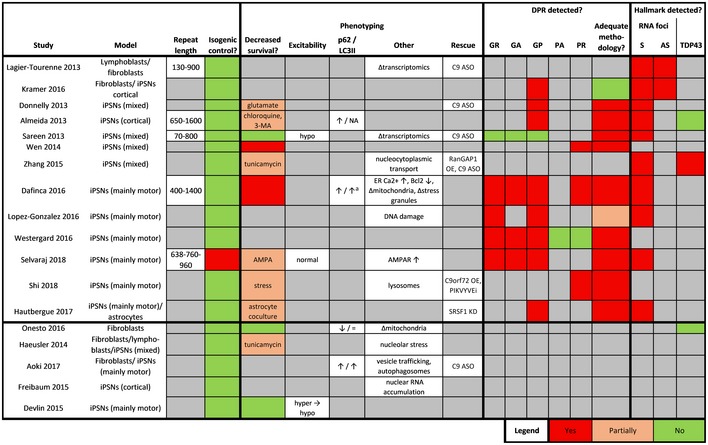
Systematic overview of all studies employing and characterizing patient‐derived in vitro models for C9ORF72 ALS/FTD. For each model, repeat length, use of isogenic control, phenotyping, DPR detection, RNA foci, and TDP43 pathology are reported systematically. Upper part of table contains studies assessing DPR and/or RNA foci presence. Four studies (Burguete et al, 2015; Mori et al, 2016; Niblock et al, 2016; Webster et al, 2016) were excluded because no characterization was performed. If a survival phenotype was observed upon additional treatment, this treatment is indicated.
Abbreviations: AS, antisense; ASO, antisense oligonucleotide; ER, endoplasmic reticulum; iPSNs, induced pluripotent stem cell‐derived neurons; KD, knockdown; NA, not assessed; OE, overexpression; S, sense.
Increased in cortical neurons, not in motor neurons.
While a few in vivo C9ORF72 models have provided some support for a role of RNA toxicity in these models, more work is needed to support this and to dissect DPR from RNA toxicity. When modeling repeat RNA toxicity, the original GGGGCC repeat sequence needs to be used. Therefore, these models have an unavoidable propensity to generate DPRs through RAN translation, making it almost impossible to discriminate RNA toxicity from DPR toxicity.
Mislocalization of repeat RNA‐binding proteins (rRBPs)
In the second step of the pathological cascade of RNA toxicity, the repeat RNA interacts with several RBPs (Fig 3). Demonstrating this interaction in a disease‐relevant context is very challenging. rRBP mislocalization is generally used as a surrogate marker to indicate this interaction, hence providing indirect support for RNA toxicity. This mislocalization consists of a colocalization with RNA foci and/or a subcellular mislocalization. An overview of the involvement of rRBPs is given in Table 6.
Table 6.
Mechanistic involvement of repeat RNA‐binding proteins in C9ORF72 ALS/FTD
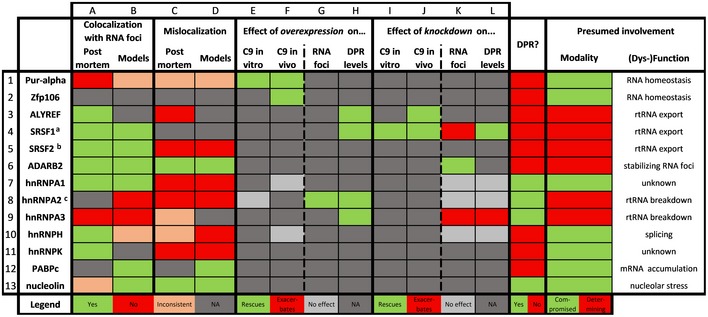
For a subset of RNA‐binding proteins known to bind the C9ORF72 hexanucleotide repeat RNA, their individual involvement in C9ORF72 RNA toxicity is reviewed systematically. Subcellular mislocalization and colocalization with RNA foci in disease models as well as post‐mortem are depicted. The effect of overexpression and/or knockdown of the protein in C9 disease models is reviewed at four levels; effect on the toxicity in in vitro models, effect on the toxicity in in vivo models, effect on RNA foci, effect on DPR levels. For each protein, their possible involvement in DPR toxicity is depicted as well. Finally, based on the current literature, the presumed modality (compromised vs determining) and mechanism of their involvement are listed. Color legends are indicated at the bottom of the table. Abbreviations: NA, not assessed; rtRNA, repeat RNA.
References: 1A (Lee et al, 2013b); 1B (Sareen et al, 2013; O'Rourke et al, 2015; Rossi et al, 2015); 1C, (Lee et al, 2013b; Xu et al, 2013); 1D (Donnelly et al, 2013; Xu et al, 2013; Rossi et al, 2015); 1E (Xu et al, 2013); 1F (Xu et al, 2013; Swinnen et al, 2018); 2F (Celona et al, 2017); 3A (Cooper‐Knock et al, 2014, 2015b); 3C (Cooper‐Knock et al, 2015b); 3H (Hautbergue et al, 2017); 3J, (Freibaum et al, 2015; Hautbergue et al, 2017); 4A (Lee et al, 2013b); 4B (Lee et al, 2013b; Stopford et al, 2017); 4H (Hautbergue et al, 2017); 4I (Hautbergue et al, 2017); 4J (Hautbergue et al, 2017); 4K (Hautbergue et al, 2017); 4L (Hautbergue et al, 2017); 5A (Lee et al, 2013b; Cooper‐Knock et al, 2014, 2015b); 5B (Lee et al, 2013b; Stopford et al, 2017); 5C (Cooper‐Knock et al, 2015b); 5D (Yin et al, 2017); 6A (Donnelly et al, 2013); 6B (Donnelly et al, 2013); 6C (Donnelly et al, 2013); 6D (Donnelly et al, 2013); 6K, (Donnelly et al, 2013); 7A (Cooper‐Knock et al, 2014, 2015b); 7B (Sareen et al, 2013); 7C (Cooper‐Knock et al, 2015b; Fifita et al, 2017); 7D (Donnelly et al, 2013; Yin et al, 2017); 7F (Swinnen et al, 2018); 7K (Mori et al, 2016); 7L (Mori et al, 2016); 8B (Almeida et al, 2013; Sareen et al, 2013; O'Rourke et al, 2015); 8C (Fifita et al, 2017); 8D (Almeida et al, 2013); 8E (Xu et al, 2013); 8G (Mori et al, 2016); 8H (Mori et al, 2016); 8K (Mori et al, 2016); 8L (Mori et al, 2016); 9A (Lee et al, 2013b); 9B (Sareen et al, 2013; O'Rourke et al, 2015); 9C (Lee et al, 2013b; Mori et al, 2013b; Boeynaems et al, 2016; Davidson et al, 2017; Fifita et al, 2017); 9H (Mori et al, 2016); 9K (Mori et al, 2016); 9L (Mori et al, 2016); 10A (Lee et al, 2013b; Cooper‐Knock et al, 2014, 2015b); 10B (Almeida et al, 2013; Lee et al, 2013b; O'Rourke et al, 2015; Rossi et al, 2015; Conlon et al, 2016); 10C (Cooper‐Knock et al, 2015b; Conlon et al, 2016); 10D (Almeida et al, 2013); 10F (Swinnen et al, 2018); 10K (Mori et al, 2016); 10L (Mori et al, 2016); 11A (Cooper‐Knock et al, 2015b); 11C (Cooper‐Knock et al, 2015b); 11D (Haeusler et al, 2014); 12B (Rossi et al, 2015); 12D (Rossi et al, 2015); 13A (Haeusler et al, 2014; Cooper‐Knock et al, 2015b; Stopford et al, 2017); 13B (Stopford et al, 2017); 13C (Cooper‐Knock et al, 2015b); 13D (Haeusler et al, 2014),(O'Rourke et al, 2015).
a.k.a. SF1.
a.k.a. SC35.
a.k.a. hnRNPA2/B1.
Mislocalization of rRBPs has been demonstrated in some patient‐derived in vitro models. Most importantly, post‐mortem examination revealed mislocalization of numerous rRBPs. Sense nuclear RNA foci have been shown to contain SRSF1, SRSF2, ALYREF, ADARB2, HNRNPA1, HNRNPH, and HNRNPF (Donnelly et al, 2013; Lee et al, 2013a; Cooper‐Knock et al, 2014). Antisense nuclear RNA foci have been demonstrated to colocalize with SRSF2, ALYREF, HNRNPA1, HNRNPH, and HNRNPK (Cooper‐Knock et al, 2015b). However, some potential limitations of these studies should be mentioned. The colocalization was often not demonstrated in disease‐relevant tissue (i.e., frontal cortex, motor cortex, and spinal cord). Moreover, colocalization data are often conflicting between different studies, probably related to methodological differences. Finally, some DPRs (i.e., GR and PR) also interact and/or colocalize with hnRNPs (Table 6). Therefore, the mislocalization and/or dysfunction of these rRBPs might be related to DPR toxicity.
Importantly, one needs to keep in mind that the absence of rRBP mislocalization or colocalization with RNA foci does not exclude mislocalization as the sensitivity of conventional immunohistochemistry might be too low to detect a (subtle) altered subcellular distribution. This makes validation of rRBP mislocalization in post‐mortem tissue very challenging.
Signatures of rRBP dysfunction
In the third step of the pathological cascade of RNA toxicity, the function of several rRBPs is compromised. Monitoring signatures of this dysfunction might be an indirect proof of RNA toxicity. Given the complexity of this pleiotropic pool of rRBPs, only a limited amount of rRBP dysfunction hallmarks have been identified so far. An increase in splicing errors has been noted in C9ORF72 iMNs (Cooper‐Knock et al, 2015a), which was confirmed in C9 ALS/FTD brains and mainly constituted intron retention events (Prudencio et al, 2015). This is in line with several rRBPs having a cardinal role in RNA splicing. Moreover, a large proportion of the misspliced transcripts in C9 ALS/FTD were targets of HNRNPH and SRSF1, indicating their dysfunction (Prudencio et al, 2015; Conlon et al, 2016). Additionally, transcriptomics in patient‐derived material generally indicates an alteration of genes involved in RNA metabolism thereby circumstantially suggesting rRBP misregulation (Chew et al, 2015; Cooper‐Knock et al, 2015a; Selvaraj et al, 2018). More research will be needed to investigate downstream signatures of rRBP dysfunction. Given the large functional pleiotropy of each of these rRBPs, identifying these disturbances will be challenging.
Clinicopathological correlations
In the final step of the pathological cascade of RNA toxicity, neurodegeneration results in clinical phenotypic manifestations. Demonstrating a correlation between key clinical or pathological features and hallmarks of RNA toxicity would be the best proof of RNA toxicity. Indeed, the burden of sense RNA foci was inversely correlated with age at onset in C9 FTD patients (Mizielinska et al, 2013). Moreover, the burden of sense RNA foci in spinal motor neurons was higher in C9 ALS than in C9 FTD patients, suggesting RNA foci to be correlated with the clinical phenotype (Cooper‐Knock et al, 2014). In addition, the splicing error rate in C9 iMNs and lymphoblastoids was correlated with disease severity (Cooper‐Knock et al, 2015a). Despite these correlations, most clinical and pathological data do not provide a correlation with RNA toxicity hallmarks. Several studies identified no detrimental associations between RNA foci and clinicopathological features (Gendron et al, 2013; Mizielinska et al, 2013; DeJesus‐Hernandez et al, 2017). On the contrary, a higher antisense RNA foci burden was even correlated with a delayed age at onset (DeJesus‐Hernandez et al, 2017). Moreover, RNA foci generally do not follow the pattern of neurodegeneration and TDP‐43 pathology (Gendron et al, 2013; Mizielinska et al, 2013; DeJesus‐Hernandez et al, 2017). Finally, there has been no correlation between RNA foci and toxicity in any of the repeat expansion models developed so far, both in vitro and in vivo (Tables 3, 4, 5).
Mechanisms of RNA toxicity: new insights from C9 ALS/FTD
rRBP loss of function
Using pulldown approaches, the interacting partners of repeat RNA have been studied extensively (Table 7). Classical RNA‐binding proteins (RBPs) constitute the majority of the interactome. These mainly include hnRNPs and mRNA transport proteins. Notably, there is a large variability in the identified proteins between studies likely due to methodological differences (e.g., cell lysate species, probe length, and/or identification method). Additionally, the repeat length was below the presumed threshold of toxicity in most studies (i.e., ± 30 repeats). While it is clear that C9ORF72 repeat RNA interacts with a large number of proteins, the involvement of RNA toxicity as well as its pathological underpinnings is still unclear.
Table 7.
Sense repeat RNA interactome
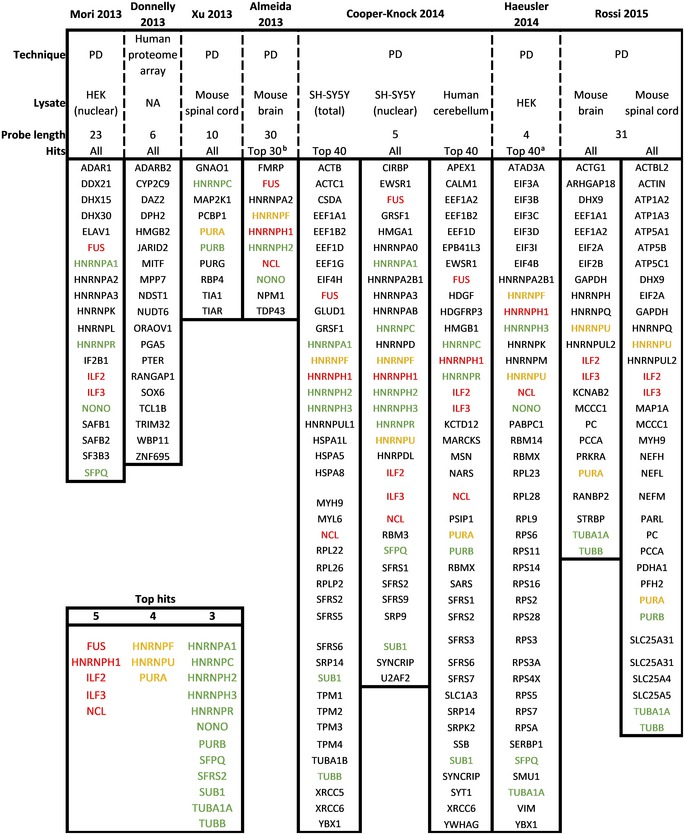
Overview of proteins identified to bind sense (GGGGCC) repeat RNA. In case the list of identified proteins exceeded 40 hits, only the top 40 proteins were included. Box inset left bottom indicates most frequently identified (five times = red, four times = orange, three times = green) proteins.
Abbreviations: NA, not available; PD, pulldown.
Based on “unique peptides”.
Incomplete list in original manuscript.
Research on some of the rRBPs in several C9ORF72 models has corroborated the idea of repeat RNA interfering with their normal function, leading to a loss of function of the rRBP (Table 6). First, overexpression of some of these rRBPs rescued the phenotype induced by the repeat expansion [i.e., Pur‐alpha (Xu et al, 2013; Swinnen et al, 2018) and Zfp106 (Celona et al, 2017)], indicating that repeat RNA might impair their functionality and/or protein level. Additionally, knockdown of these rRBPs in wild‐type models was detrimental (Xu et al, 2013; Celona et al, 2017), suggesting that dysfunction of these rRBPs is harmful. Second, for some rRBPs [Pur‐alpha (Rossi et al, 2015), ADARB2 (Donnelly et al, 2013), nucleolin (Cooper‐Knock et al, 2015b), hnRNPA3 (Boeynaems et al, 2016), and hnRNPH (Conlon et al, 2016)], the repeat expansion altered their subcellular distribution. This indicates that the physical interaction between repeat RNA and rRBPs might lead to a functional sequestration of the latter.
When evaluating the repeat RNA interactome (Table 7), FUS is one of the proteins most consistently shown to bind to repeat RNA. This suggests that FUS dysfunction could contribute to C9 ALS/FTD pathogenesis and hence hints toward convergent mechanisms between FUS ALS and C9ORF72 ALS. In addition, nucleolin is shown in several studies to bind C9 ALS/FTD repeat RNA, suggesting that repeat RNA could directly induce nucleolar stress. This might constitute a convergence point between RNA toxicity and DPR toxicity as DPRs are believed to lead to nucleolar stress (Balendra & Isaacs, 2018). The consistently observed interaction of repeat RNA with ILF2 and ILF3 might also mediate various dysregulations of RNA metabolism. These proteins are known to bind DNA:RNA hybrids and are involved in dynamics of various RNA granules as well as in nucleolar homeostasis and splicing (Shiina & Nakayama, 2014; Nadel et al, 2015; Wandrey et al, 2015). Surprisingly, albeit less consistent, repeat RNA has regularly been reported to bind cytoskeletal proteins (e.g., TUBB and TUBA1A), possibly contributing to axonal dysfunction of motor neurons like has been described for FUS mutations (Guo et al, 2017).
The presumed loss‐of‐function of several rRBPs might lead to disturbances in several cellular processes (Fig 5). Given that many rRBP processes are related to RNA metabolism like splicing (e.g., HNRNPH), nuclear mRNA export (e.g., PABPC), mRNA translation (e.g., ZFP106), cytoplasmic RNA transport (e.g., PURA), and nucleolar stress (e.g., nucleolin), this could lead to cellular stress via different pathways. However, the exact contribution and net effect of each rRBP in RNA toxicity is a puzzle that still needs to be solved.
Figure 5. Processes possibly disturbed by C9ORF72 RNA toxicity.
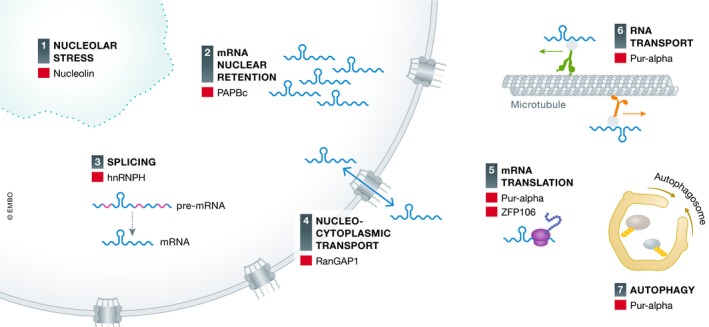
(1) Compromised function of nucleolin (NCL) might induce nucleolar stress. (2) mRNA might be retained in the nucleus due to repeat RNA‐induced nuclear accumulation of mRNA export proteins like PABPC. (3) Splicing might be disturbed due to compromised function of several splicing factors like HNRNPH. (4) Nucleocytoplasmic transport might be directly disturbed by repeat RNA via RanGAP1 dysfunction. (5) Translation of mRNA might be altered due to compromised function of translational factors like Pur‐alpha and ZFP106. (6) Cytoplasmic RNA transport might be disturbed by compromised function of RNA transport factors like Pur‐alpha. (7) Autophagy might be compromised by dysfunction of Pur‐alpha.
rRBPs contribute to repeat RNA localization dynamics
As discussed above, repeat RNA has been shown to induce dysfunction of several rRBPs. Moreover, repeat RNA has also been shown to affect localization of a subset of rRBPs (Table 6). This is illustrated by the intriguing observation that some rRBPs alleviate the repeat toxicity upon knockdown instead of overexpression. Several of these rRBPs are involved in mRNA transport including ALYREF (Hautbergue et al, 2017), SRSF1 (Hautbergue et al, 2017), and FMRP (Burguete et al, 2015). This implies that rRBPs might regulate the subcellular localization of repeat RNA and indirectly contribute to RNA (and DPR) toxicity. Indeed, three independent findings support this interpretation. First, SRSF1 knockdown increased nuclear and decreased cytoplasmic RNA foci (Hautbergue et al, 2017), suggesting that SRSF1 mediates the nuclear export of repeat RNA to the cytoplasm. Similar findings were obtained for ALYREF, another known mRNA export protein (Hautbergue et al, 2017). Second, ADARB2 knockdown reduced the amount of nuclear RNA foci (Donnelly et al, 2013), indicating that it might be involved in the nuclear retention of repeat RNA or that it might have a stabilizing effect on RNA foci. Third, FMRP knockdown decreased the toxicity induced by repeat RNA located in the neurites (Burguete et al, 2015), while it also colocalized with this repeat RNA suggesting that FMRP might be responsible for the transport of repeat RNA to the neurites. These studies support that a subset of rRBPs have the ability to regulate the toxic potential of the repeat RNA by affecting its stability and/or subcellular transport.
Nuclear versus cytoplasmic repeat RNA
In line with the observation that RNA transport factors modify repeat RNA toxicity, the localization of the repeat RNA seems to be crucial for its toxicity. It is still unclear whether the nuclear or the cytoplasmic repeat RNA species are the most toxic ones or whether they both contribute to toxicity. Analysis in post‐mortem samples shows that nuclear RNA foci are more abundant than cytoplasmic ones. However, since nuclear RNA foci show very little to no correlation with neurodegeneration, their pathogenic role is unclear. Increasing evidence supports the notion of cytoplasmic repeat RNA being the main culprit. Cytoplasmic RNA foci have been detected in post‐mortem tissue (Cooper‐Knock et al, 2015b), and sense repeat RNA was found in neurites of C9 iMNs (Burguete et al, 2015). Moreover, the existence of cytoplasmic repeat RNA is a prerequisite for DPR production. As a consequence, the presence of DPRs could be considered as an indirect proof of cytoplasmic repeat RNA. Additionally, decrease of cytoplasmic RNA foci by SRSF1 knockdown was beneficial (Hautbergue et al, 2017). Finally, repeat RNA localized in the neurites was sufficient to cause toxicity (Burguete et al, 2015).
The hypothesis that cytoplasmic repeat RNA is the main culprit still leaves the discussion open between DPR and RNA toxicity, as DPRs are generated from cytoplasmic repeat RNA. Moreover, it is still unclear in which transcriptional context the repeat RNA is generated (Fig 1). In case it is mainly generated from intron 1 retaining transcripts, the repeat RNA should indeed have a high propensity for cytoplasmic localization as it has a polyA tail. In contrast, if it is mainly generated from spliced‐out intron 1 or abortive transcripts, the repeat RNA is more likely to be retained in the nucleus.
RNA foci versus soluble repeat RNA
While RNA foci are an important hallmark of C9 ALS/FTD, their involvement in disease pathogenesis is still elusive. In fact, current post‐mortem data do not support RNA foci as the driver of neurodegeneration. First, similar to DPRs, they do not follow the pattern of neurodegeneration, as RNA foci are equally present in non‐affected regions (e.g., cerebellum and hippocampus) with the highest level in cerebellar Purkinje cells (Mackenzie et al, 2014; Saberi et al, 2015; DeJesus‐Hernandez et al, 2017). Second, the presence of RNA foci does not correlate with TDP‐43 pathology (Mizielinska et al, 2013), even though some correlation in motor neurons between antisense RNA foci and TDP‐43 pathology has been suggested (Cooper‐Knock et al, 2015b). Third, an extensive study identified no detrimental associations between RNA foci and clinical features (DeJesus‐Hernandez et al, 2017). On the contrary, a higher antisense burden of RNA foci was correlated with a delayed age at onset (DeJesus‐Hernandez et al, 2017). Current in vitro and in vivo data further question the disease relevance of RNA foci. Most importantly, neither toxicity nor TDP‐43 pathology is correlated with the presence of RNA foci, both in non‐ATG GGGGCC models (Tables 3 and 4) and in patient‐derived in vitro models (Table 5). Altogether, this evidence seems to be in line with the idea that RNA foci do not contribute significantly to C9 ALS/FTD pathogenesis. As a consequence, repeat RNA not confined in RNA foci (i.e., “soluble repeat RNA”) could be the real perpetrator of RNA toxicity. However, assessing soluble repeat RNA is very challenging and so far only one study has been able to visualize soluble repeat RNA species (Burguete et al, 2015).
Lessons from other non‐coding repeat expansion disorders for RNA toxicity
While the mechanism and exact contribution of RNA toxicity in the other non‐coding repeat expansion disorders is still poorly understood, they can provide important insights into the existence and mechanism of RNA toxicity (in C9 ALS/FTD).
First, analysis in other non‐coding repeat expansion disorders questions the pathogenic role of nuclear RNA foci. Instead of being stable aggregates of repeat RNA sequestering RNA‐binding proteins, work in myotonic dystrophy type 1 supports the notion that RNA foci are highly dynamic structures, with several RNA‐binding proteins themselves being involved in this dynamic process (Lopez‐Morato et al, 2018). As a consequence, nuclear RNA foci might not entirely account for rRBP dysfunction suggesting another mediator of toxicity. Interestingly, cytoplasmic soluble repeat RNA seems a possible candidate. Also in myotonic dystrophy type 1, repeat RNA has been demonstrated to reside in the cytoplasm as well, often adapting a single mRNP conformation (i.e., soluble repeat RNA), opposed to RNA foci (Pettersson et al, 2015). Interestingly, work in FXTAS has shown that the function of several rRBPs (e.g., Pur‐alpha and hnRNPA2) can be altered without them physically being mislocalized (Boivin et al, 2018). Therefore, rRBP mislocalization cannot necessarily always be equated with rRBP dysfunction.
Second, several rRBPs that are able to rescue in models of these diseases have also been implicated in C9 ALS/FTD, suggesting mechanistic commonalities. These include Pur‐alpha (FXTAS; Boivin et al, 2018), hnRNPA2 [FXTAS (Boivin et al, 2018), and SCA31 (Ishiguro et al, 2017)] as well as hnRNPK (SCA10; White et al, 2010). Interestingly, despite commonalities, the pool of involved rRBPs (Table 1) is considerably divergent between different diseases. This might partially underlie the observed pathological and clinical differences.
Third, the repeat sequence in SCA36 (TGGGCC) is strikingly similar to the GGGGCC repeat expansion in C9 ALS/FTD. Moreover, SCA36 and C9 ALS/FTD are the only non‐coding repeat expansion disorders with significant motor neuron involvement (Kobayashi et al, 2011; Ikeda et al, 2012), suggesting that a similar mechanism might be at play. Interestingly, two RAN proteins (GP and PR) are mutual as well as several rRBPs. This observation makes a loss‐of‐function unlikely to be the main mediator of toxicity. Moreover, it indicates that DPR toxicity is insufficient to explain the pathogenesis, as GP has no toxic potential (as described previously) and as PR is only sporadically detected, suggesting an important role for RNA toxicity.
How to disentangle RNA from RAN—Future directions
Deciphering the exact pathogenic code underlying each of the non‐coding repeat expansion disorders is important to identify therapies to halt these aggressive diseases. To maximize the effectiveness of therapies, the exact involvement of each of the three possible mechanisms (RNA, RAN and loss‐of‐function) needs to be uncovered. Assessing whether RNA toxicity, alone or in addition to RAN toxicity, plays a substantial pathogenic role is crucial to predict the effectiveness of RAN directed therapies (e.g., nanobodies). At this moment, a multimodal therapeutic strategy aiming at all three possible mechanisms (or at least the two gain‐of‐function mechanisms) has the best potential of clinical success.
Disease models
As already pointed out, assessment of RNA toxicity in disease models is difficult since “non‐ATG repeat RNA constructs” have the intrinsic propensity to undergo RAN translation (cf. Box 1). Therefore, both repeat RNA and RAN proteins are present in most models, precluding a clean assessment of RNA toxicity. Unfortunately, even “RNA only constructs” are not optimal either (cf. Box 2). Different approaches to assess pure RNA toxicity, in a RAN‐devoid context, are highly needed. Possible paradigms include the use of specific RAN translation inhibitors or the induction of RAN peptide degradation. In this regard, further research into the exact mechanism underlying RAN translation will be crucial to develop these strategies. For the time being, an adequate assessment of RAN proteins in all models exploiting “non‐ATG repeat RNA constructs” is pivotal.
Another important issue is the need to assess all possible toxic repeat RNA species. Whereas research so far has been highly biased toward RNA foci, soluble repeat RNA has been given very little attention. Unfortunately, visualizing this soluble repeat RNA is very difficult. Approaches like MS2 tagging (Burguete et al, 2015) will need to be further refined and exploited.
Regarding rRBP dysfunction, for each non‐coding repeat expansion disorder the pool of rRBPs needs to be identified and validated, e.g., by performing additional pulldown approaches and bioinformatical modeling of the interactions. Moreover, the effect of its overexpression as well as knockdown needs to be determined for each identified rRBP in relevant disease models in order to assess its functional implication in disease pathogenesis. Similarly, rRBP subcellular localization as well as downstream disturbances needs to be investigated more extensively in disease models.
Altogether, disease models need to be exploited more efficiently. Ideally, each model should be completely and thoroughly characterized, and RAN peptide presence should be assessed systematically using an adequate methodology.
Post‐mortem research
So far, most research has been performed in disease models. As post‐mortem research has an obviously higher disease relevance, it should be given much more attention. Brain and spinal cord tissue should be obtained more systematically and used judiciously. Similar to research in disease models, more attention should be given to soluble repeat RNA species. Moreover, rRBP dysfunction assessment should comprise more than just assessment of colocalization with RNA foci. Downstream signatures (e.g., splicing defects) of rRBP dysfunction should also be investigated (e.g., by RNA sequencing).
Patient research
Research in living patients is of highest disease relevance. Imaging approaches aiming to visualize RAN proteins (e.g., through a PET (positron emission tomography) tracer) might provide valuable information and may be able to establish a temporal relationship with disease milestones. Also, fluid biomarkers (e.g., DPRs or repeat RNA in cerebrospinal fluid and/or blood) might be another approach to gauge the relation between these disease mechanisms and clinical aspects. These approaches should ideally be combined with prospective long‐term follow‐up studies of presymptomatic individuals carrying a C9ORF72 repeat expansion.
Conclusions
Non‐coding repeat expansion disorders in general
Three possible mechanisms might underlie pathogenesis in non‐coding repeat expansion disorders; RNA toxicity, RAN toxicity, and loss of function. With the currently available information, we conclude that RNA toxicity might contribute to the disease mechanism in all diseases. However, it has not been assessed in SCA12 so far. RAN toxicity seems possible in all diseases except SCA12 as presence of RAN protein inclusions in post‐mortem material was positively excluded. Contribution of RAN toxicity in SCA10 and SCA36 is still largely elusive as the presence of RAN proteins in post‐mortem tissue has not reliably been established yet. Loss of function could eventually only play a significant pathogenic role in SCA12, where an “alteration‐of‐function” instead of a loss‐of‐function is suspected.
Data so far have been inconclusive in determining the exact contribution of RNA toxicity, RAN toxicity, and loss of function in the pathogenesis of non‐coding repeat expansion disorders. As all non‐coding repeat expansion disorders probably share a similar underlying pathogenesis, a holistic research approach concerning these disorders needs to be implemented. Pathogenic differences and commonalities between these disorders are the key to unravel the exact contribution of RNA toxicity, RAN toxicity, and loss‐of‐function. Of interest, several rRBPs are shared between different repeat RNA interactomes, with MBNL1, CUGBP1, PURA, HNRNPA2, HNRNPK, SRSF1, and SRSF2 being shared by at least two disorders.
C9 ALS/FTD
While C9 ALS/FTD mainly seems to be a gain‐of‐function disease, it is currently unclear whether this is mediated by DPR and/or RNA toxicity. Given the toxicity in several in vitro and in vivo models, DPRs could be involved. While DPRs have been modeled extensively, research into RNA toxicity is still limited, mainly due to technical challenges in modeling RNA toxicity. Despite these methodological problems, we conclude that evidence favoring the existence of RNA toxicity is increasing and that this toxicity could induce alterations in splicing, nucleolar function, mRNA nuclear export, cytoplasmic RNA transport, and autophagy. Further research to establish the exact role of RNA toxicity in C9 ALS/FTD needs to be structured along the pathogenic cascade of repeat RNA toxicity. Such research will be crucial in guiding clinical research to develop new therapeutic approaches for C9 ALS/FTD.
Author contributions
BS performed the literature search and wrote the manuscript. LVDB and WR discussed the literature and co‐wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Acknowledgements
Research of the authors is supported by VIB, the University of Leuven (KU Leuven), the Research Foundation Flanders (FWO‐Vlaanderen), the Agency for Innovation by Science and Technology, the Muscular Dystrophy Association (MDA), the Thierry Latran Foundation, the ALS Association (ALSA), the ALS Liga (Belgium), and the Association Belge contre les Maladies Neuro‐Musculaires (ABMM). WR is supported through the E. von Behring Chair for Neuromuscular and Neurodegenerative Disorders and the “Hart voor ALS” Fund, KU Leuven. BS was a PhD Fellow of FWO‐Vlaanderen.
The EMBO Journal (2020) 39: e101112
See the Glossary for abbreviations used in this article.
References
- Allen EG, He W, Yadav‐Shah M, Sherman SL (2004) A study of the distributional characteristics of FMR1 transcript levels in 238 individuals. Hum Genet 114: 439–447 [DOI] [PubMed] [Google Scholar]
- Almeida S, Gascon E, Tran H, Chou HJ, Gendron TF, Degroot S, Tapper AR, Sellier C, Charlet‐Berguerand N, Karydas A et al (2013) Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC‐derived human neurons. Acta Neuropathol 126: 385–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amick J, Ferguson SM (2017) C9orf72: at the intersection of lysosome cell biology and neurodegenerative disease. Traffic 18: 267–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki Y, Manzano R, Lee Y, Dafinca R, Aoki M, Douglas AGL, Varela MA, Sathyaprakash C, Scaber J, Barbagallo P et al (2017) C9orf72 and RAB7L1 regulate vesicle trafficking in amyotrophic lateral sclerosis and frontotemporal dementia. Brain 140: 887–897 [DOI] [PubMed] [Google Scholar]
- Ash PE, Bieniek KF, Gendron TF, Caulfield T, Lin WL, Dejesus‐Hernandez M, van Blitterswijk MM, Jansen‐West K, Paul JW, Rademakers R et al (2013) Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron 77: 639–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atanasio A, Decman V, White D, Ramos M, Ikiz B, Lee HC, Siao CJ, Brydges S, LaRosa E, Bai Y et al (2016) C9orf72 ablation causes immune dysregulation characterized by leukocyte expansion, autoantibody production, and glomerulonephropathy in mice. Sci Rep 6: 23204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayhan F, Perez BA, Shorrock HK, Zu T, Banez‐Coronel M, Reid T, Furuya H, Clark HB, Troncoso JC, Ross CA et al (2018) SCA8 RAN polySer protein preferentially accumulates in white matter regions and is regulated by eIF3F. EMBO J 37: e99023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balendra R, Isaacs AM (2018) C9orf72 ‐mediated ALS and FTD: multiple pathways to disease. Nat Rev Neurol 2018: 544–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batra R, Charizanis K, Manchanda M, Mohan A, Li M, Finn DJ, Goodwin M, Zhang C, Sobczak K, Thornton CA et al (2014) Loss of MBNL leads to disruption of developmentally regulated alternative polyadenylation in RNA‐mediated disease. Mol Cell 56: 311–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeynaems S, Bogaert E, Michiels E, Gijselinck I, Sieben A, Jovičić A, De Baets G, Scheveneels W, Steyaert J, Cuijt I et al (2016) Drosophila screen connects nuclear transport genes to DPR pathology in c9ALS/FTD. Sci Rep 6: 20877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boivin M, Willemsen R, Hukema RK, Sellier C (2018) Potential pathogenic mechanisms underlying Fragile X Tremor Ataxia Syndrome: RAN translation and/or RNA gain‐of‐function? Eur J Med Genet 61: 674–679 [DOI] [PubMed] [Google Scholar]
- Botta A, Vallo L, Rinaldi F, Bonifazi E, Amati F, Biancolella M, Gambardella S, Mancinelli E, Angelini C, Meola G et al (2007) Gene expression analysis in myotonic dystrophy: indications for a common molecular pathogenic pathway in DM1 and DM2. Gene Expr 13: 339–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buijsen RA, Sellier C, Severijnen LA, Oulad‐Abdelghani M, Verhagen RF, Berman RF, Charlet‐Berguerand N, Willemsen R, Hukema RK (2014) FMRpolyG‐positive inclusions in CNS and non‐CNS organs of a fragile X premutation carrier with fragile X‐associated tremor/ataxia syndrome. Acta Neuropathol Commun 2: 162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burberry A, Suzuki N, Wang JY, Moccia R, Mordes DA, Stewart MH, Suzuki‐Uematsu S, Ghosh S, Singh A, Merkle FT et al (2016) Loss‐of‐function mutations in the C9ORF72 mouse ortholog cause fatal autoimmune disease. Sci Transl Med 8: 347ra93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burguete AS, Almeida S, Gao FB, Kalb R, Akins MR, Bonini NM (2015) GGGGCC microsatellite RNA is neuritically localized, induces branching defects, and perturbs transport granule function. Elife 4: e08881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celona B, von Dollen J, Vatsavayai SC, Kashima R, Johnson JR, Tang A, Hata A, Miller BL, Huang EJ, Krogan NJ et al (2017) Suppression of C9orf72 RNA repeat‐induced neurotoxicity by the ALS‐associated RNA‐binding protein Zfp106. Elife 10: e19032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlet‐B N, Savkur RS, Singh G, Philips AV, Grice EA, Cooper TA (2002) Loss of the muscle‐specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol Cell 10: 45–53 [DOI] [PubMed] [Google Scholar]
- Chen W, Wang Y, Abe Y, Cheney L, Udd B, Li YP (2007) Haploinsuffciency for Znf9 in Znf9+/− mice is associated with multiorgan abnormalities resembling myotonic dystrophy. J Mol Biol 368: 8–17 [DOI] [PubMed] [Google Scholar]
- Chew J, Gendron TF, Prudencio M, Sasaguri H, Zhang Y, Castanedes‐Casey M, Lee CW, Jansen‐West K, Kurti A, Murray ME et al (2015) C9ORF72 repeat expansions in mice cause TDP‐43 pathology, neuronal loss and behavioral deficits. Science 5: 1151–1154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon EG, Lu L, Sharma A, Yamazaki T, Tang T, Shneider NA, Manley JL (2016) The C9ORF72 GGGGCC expansion forms RNA G‐quadruplex inclusions and sequesters hnRNP H to disrupt splicing in ALS patient brains. Elife 5: e17820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper‐Knock J, Higginbottom A, Connor‐Robson N, Bayatti N, Bury JJ, Kirby J, Ninkina N, Buchman VL, Shaw PJ (2013) C9ORF72 transcription in a frontotemporal dementia case with two expanded alleles. Neurology 81: 1719–1721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper‐Knock J, Walsh MJ, Higginbottom A, Robin Highley J, Dickman MJ, Edbauer D, Ince PG, Wharton SB, Wilson SA, Kirby J et al (2014) Sequestration of multiple RNA recognition motif‐containing proteins by C9orf72 repeat expansions. Brain 137: 2040–2051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper‐Knock J, Bury JJ, Heath PR, Wyles M, Higginbottom A, Gelsthorpe C, Highley JR, Hautbergue G, Rattray M, Kirby J et al (2015a) C9ORF72 GGGGCC expanded repeats produce splicing dysregulation which correlates with disease severity in amyotrophic lateral sclerosis. PLoS One 10: e0127376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper‐Knock J, Higginbottom A, Stopford MJ, Highley JR, Ince PG, Wharton SB, Pickering‐Brown S, Kirby J, Hautbergue GM, Shaw PJ (2015b) Antisense RNA foci in the motor neurons of C9ORF72‐ALS patients are associated with TDP‐43 proteinopathy. Acta Neuropathol 130: 63–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daughters RS, Tuttle DL, Gao W, Ikeda Y, Moseley ML, Ebner TJ, Swanson MS, Ranum LP (2009) RNA gain‐of‐function in spinocerebellar ataxia type 8. PLoS Genet 5: e1000600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson YS, Barker H, Robinson AC, Thompson JC, Harris J, Troakes C, Smith B, Al‐Saraj S, Shaw C, Rollinson S et al (2014) Brain distribution of dipeptide repeat proteins in frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9ORF72. Acta Neuropathol Commun 2: 70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson Y, Robinson AC, Liu X, Wu D, Troakes C, Rollinson S, Masuda‐Suzukake M, Suzuki G, Nonaka T, Shi J et al (2016) Neurodegeneration in frontotemporal lobar degeneration and motor neurone disease associated with expansions in C9orf72 is linked to TDP‐43 pathology and not associated with aggregated forms of dipeptide repeat proteins. Neuropathol Appl Neurobiol 42: 242–254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson YS, Flood L, Robinson AC, Nihei Y, Mori K, Rollinson S, Richardson A, Benson BC, Jones M, Snowden JS et al (2017) Heterogeneous ribonuclear protein A3 (hnRNP A3) is present in dipeptide repeat protein containing inclusions in Frontotemporal Lobar Degeneration and Motor Neurone disease associated with expansions in C9orf72 gene. Acta Neuropathol Commun 5: 31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BM, McCurrach ME, Taneja KL, Singer RH, Housman DE (1997) Expansion of a CUG trinucleotide repeat in the 3’ untranslated region of myotonic dystrophy protein kinase transcripts results in nuclear retention of transcripts. Proc Natl Acad Sci USA 94: 7388–7393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus‐Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, Nicholson AM, Finch NA, Flynn H, Adamson J et al (2011) Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p‐linked FTD and ALS. Neuron 72: 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeJesus‐Hernandez M, Finch NA, Wang X, Gendron TF, Bieniek KF, Heckman MG, Vasilevich A, Murray ME, Rousseau L, Weesner R et al (2017) In‐depth clinico‐pathological examination of RNA foci in a large cohort of C9ORF72 expansion carriers. Acta Neuropathol 134: 255–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Wu JJ, Wu ZY (2015) Identification of 46 CAG repeats within PPP2R2B as probably the shortest pathogenic allele for SCA12. Parkinsonism Relat Disord 21: 398–401 [DOI] [PubMed] [Google Scholar]
- Donnelly CJ, Zhang P‐W, Pham JT, Haeusler AR, Mistry NA, Vidensky S, Daley EL, Poth EM, Hoover B, Fines DM et al (2013) RNA toxicity from the ALS/FTD C9ORF72 expansion is mitigated by antisense intervention. Neuron 80: 415–428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fifita JA, Zhang KY, Galper J, Williams KL, McCann EP, Hogan AL, Saunders N, Bauer D, Tarr IS, Pamphlett R et al (2017) Genetic and pathological assessment of hnRNPA1, hnRNPA2/B1, and hnRNPA3 in familial and sporadic amyotrophic lateral sclerosis. Neurodegener Dis 17: 304–312 [DOI] [PubMed] [Google Scholar]
- Fratta P, Poulter M, Lashley T, Rohrer JD, Polke JM, Beck J, Ryan N, Hensman D, Mizielinska S, Waite AJ et al (2013) Homozygosity for the C9orf72 GGGGCC repeat expansion in frontotemporal dementia. Acta Neuropathol 126: 401–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freibaum BD, Lu Y, Lopez‐Gonzalez R, Kim NC, Almeida S, Lee KH, Badders N, Valentine M, Miller BL, Wong PC et al (2015) GGGGCC repeat expansion in C9orf72 compromises nucleocytoplasmic transport. Nature 525: 129–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freibaum BD, Taylor JP (2017) The role of dipeptide repeats in C9ORF72‐related ALS‐FTD. Front Mol Neurosci 10: 35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freyermuth F, Rau F, Kokunai Y, Linke T, Sellier C, Nakamori M, Kino Y, Arandel L, Jollet A, Thibault C et al (2016) Splicing misregulation of SCN5A contributes to cardiac‐conduction delay and heart arrhythmia in myotonic dystrophy. Nat Commun 7: 11067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fugier C, Klein AF, Hammer C, Vassilopoulos S, Ivarsson Y, Toussaint A, Tosch V, Vignaud A, Ferry A, Messaddeq N et al (2011) Misregulated alternative splicing of BIN1 is associated with T tubule alterations and muscle weakness in myotonic dystrophy. Nat Med 17: 720–725 [DOI] [PubMed] [Google Scholar]
- Gendron TF, Bieniek KF, Zhang YJ, Jansen‐West K, Ash PE, Caulfield T, Daughrity L, Dunmore JH, Castanedes‐Casey M, Chew J et al (2013) Antisense transcripts of the expanded C9ORF72 hexanucleotide repeat form nuclear RNA foci and undergo repeat‐associated non‐ATG translation in c9FTD/ALS. Acta Neuropathol 126: 829–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendron TF, van Blitterswijk M, Bieniek KF, Daughrity LM, Jiang J, Rush BK, Pedraza O, Lucas JA, Murray ME, Desaro P et al (2015) Cerebellar c9RAN proteins associate with clinical and neuropathological characteristics of C9ORF72 repeat expansion carriers. Acta Neuropathol 130: 559–573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gijselinck I, Van Langenhove T, van der Zee J, Sleegers K, Philtjens S, Kleinberger G, Janssens J, Bettens K, Van Cauwenberghe C, Pereson S et al (2012) A C9orf72 promoter repeat expansion in a Flanders‐Belgian cohort with disorders of the frontotemporal lobar degeneration‐amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol 11: 54–65 [DOI] [PubMed] [Google Scholar]
- Gomez‐Deza J, Lee YB, Troakes C, Nolan M, Al‐Sarraj S, Gallo JM, Shaw CE (2015) Dipeptide repeat protein inclusions are rare in the spinal cord and almost absent from motor neurons in C9ORF72 mutant amyotrophic lateral sclerosis and are unlikely to cause their degeneration. Acta Neuropathol Commun 3: 38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenstein PE, Vonsattel JP, Margolis RL, Joseph JT (2007) Huntington's disease like‐2 neuropathology. Mov Disord 22: 1416–1423 [DOI] [PubMed] [Google Scholar]
- Guo W, Naujock M, Fumagalli L, Vandoorne T, Baatsen P, Boon R, Ordovas L, Patel A, Welters M, Vanwelden T et al (2017) HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS‐ALS patients. Nat Commun 8: 861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeusler AR, Donnelly CJ, Periz G, Simko EA, Shaw PG, Kim MS, Maragakis NJ, Troncoso JC, Pandey A, Sattler R et al (2014) C9orf72 nucleotide repeat structures initiate molecular cascades of disease. Nature 507: 195–200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Handa V, Yeh HJ, McPhie P, Usdin K (2005) The AUUCU repeats responsible for spinocerebellar ataxia type 10 form unusual RNA hairpins. J Biol Chem 280: 29340–29345 [DOI] [PubMed] [Google Scholar]
- Hao M, Akrami K, Wei K, De Diego C, Che N, Ku JH, Tidball J, Graves MC, Shieh PB, Chen F (2008) Muscleblind‐like 2 (Mbnl2) ‐deficient mice as a model for myotonic dystrophy. Dev Dyn 237: 403–410 [DOI] [PubMed] [Google Scholar]
- Harms MB, Cady J, Zaidman C, Cooper P, Bali T, Allred P, Cruchaga C, Baughn M, Libby RT, Pestronk A et al (2013) Lack of C9ORF72 coding mutations supports a gain of function for repeat expansions in amyotrophic lateral sclerosis. Neurobiol Aging 34: 2234.e13–2234.e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hautbergue GM, Castelli LM, Ferraiuolo L, Sanchez‐Martinez A, Cooper‐Knock J, Higginbottom A, Lin YH, Bauer CS, Dodd JE, Myszczynska MA et al (2017) SRSF1‐dependent nuclear export inhibition of C9ORF72 repeat transcripts prevents neurodegeneration and associated motor deficits. Nat Commun 8: 16063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho TH, Bundman D, Armstrong DL, Cooper TA (2005) Transgenic mice expressing CUG‐BP1 reproduce splicing mis‐regulation observed in myotonic dystrophy. Hum Mol Genet 14: 1539–1547 [DOI] [PubMed] [Google Scholar]
- Huichalaf C, Sakai K, Jin B, Jones K, Wang GL, Schoser B, Schneider‐Gold C, Sarkar P, Pereira‐Smith OM, Timchenko N et al (2010) Expansion of CUG RNA repeats causes stress and inhibition of translation in myotonic dystrophy 1 (DM1) cells. FASEB J 24: 3706–3719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hukema RK, Buijsen RA, Raske C, Severijnen LA, Nieuwenhuizen‐Bakker I, Minneboo M, Maas A, de Crom R, Kros JM, Hagerman PJ et al (2014) Induced expression of expanded CGG RNA causes mitochondrial dysfunction in vivo . Cell Cycle 13: 2600–2608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hukema RK, Buijsen RA, Schonewille M, Raske C, Severijnen LA, Nieuwenhuizen‐Bakker I, Verhagen RF, van Dessel L, Maas A, Charlet‐Berguerand N et al (2015) Reversibility of neuropathology and motor deficits in an inducible mouse model for FXTAS. Hum Mol Genet 24: 4948–4957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda Y, Ohta Y, Kobayashi H, Okamoto M, Takamatsu K, Ota T, Manabe Y, Okamoto K, Koizumi A, Abe K (2012) Clinical features of SCA36: a novel spinocerebellar ataxia with motor neuron involvement (Asidan). Neurology 79: 333–341 [DOI] [PubMed] [Google Scholar]
- Ishiguro T, Sato N, Ueyama M, Fujikake N, Sellier C, Kanegami A, Tokuda E, Zamiri B, Gall‐Duncan T, Mirceta M et al (2017) Regulatory role of RNA chaperone TDP‐43 for RNA misfolding and repeat‐associated translation in SCA31. Neuron 94: 108–124.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen G, Groenen PJ, Bächner D, Jap PHK, Coerwinkel M, Oerlemans F, van den Broek W, Gohlsch B, Pette D, Plomp JJ et al (1996) Abnormal myotonic dystrophy protein kinase levels produce only mild myopathy in mice. Nat Genet 13: 316–324 [DOI] [PubMed] [Google Scholar]
- Jiang J, Zhu Q, Gendron TF, Saberi S, McAlonis‐Downes M, Seelman A, Stauffer JE, Jafar‐Nejad P, Drenner K, Schulte D et al (2016) Gain of toxicity from ALS/FTD‐linked repeat expansions in C9ORF72 is alleviated by antisense oligonucleotides targeting GGGGCC‐containing RNAs. Neuron 90: 535–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin P, Zarnescu DC, Zhang F, Pearson CE, Lucchesi JC, Moses K, Warren ST (2003) RNA‐mediated neurodegeneration caused by the fragile X premutation rCGG repeats in Drosophila . Neuron 39: 739–747 [DOI] [PubMed] [Google Scholar]
- Jin P, Duan R, Qurashi A, Qin Y, Tian D, Rosser TC, Liu H, Feng Y, Warren ST (2007) Pur α binds to rCGG repeats and modulates repeat‐mediated neurodegeneration in a Drosophila model of Fragile X Tremor/Ataxia Syndrome. Neuron 55: 556–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones K, Jin B, Iakova P, Huichalaf C, Sarkar P, Schneider‐Gold C, Schoser B, Meola G, Shyu AB, Timchenko N et al (2011) RNA Foci, CUGBP1, and ZNF9 are the primary targets of the mutant CUG and CCUG repeats expanded in myotonic dystrophies type 1 and type 2. Am J Pathol 179: 2475–2489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanadia RN, Johnstone KA, Mankodi A, Lungu C, Thornton CA, Esson D, Timmers AM, Hauswirth WW, Swanson MS (2003) A muscleblind knockout model for myotonic dystrophy. Science 302: 1978–1980 [DOI] [PubMed] [Google Scholar]
- Kenneson A, Zhang F, Hagedorn CH, Warren ST (2001) Reduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediate‐length and premutation carriers. Hum Mol Genet 10: 1449–1454 [DOI] [PubMed] [Google Scholar]
- Keren B, Jacquette A, Depienne C, Leite P, Durr A, Carpentier W, Benyahia B, Ponsot G, Soubrier F, Brice A et al (2010) Evidence against haploinsuffiency of human ataxin 10 as a cause of spinocerebellar ataxia type 10. Neurogenetics 11: 273–274 [DOI] [PubMed] [Google Scholar]
- Kobayashi H, Abe K, Matsuura T, Ikeda Y, Hitomi T, Akechi Y, Habu T, Liu W, Okuda H, Koizumi A (2011) Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am J Hum Genet 89: 121–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppers M, Blokhuis AM, Westeneng HJ, Terpstra ML, Zundel CA, Vieira de Sá R, Schellevis RD, Waite AJ, Blake DJ, Veldink JH et al (2015) C9orf72 ablation in mice does not cause motor neuron degeneration or motor deficits. Ann Neurol 78: 426–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krans A, Kearse MG, Todd PK (2016) Repeat‐associated non‐AUG translation from antisense CCG repeats in fragile X tremor/ataxia syndrome. Ann Neurol 80: 871–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, Hasan GM, Hassan I (2017) Unraveling the role of RNA mediated toxicity of C9orf72 repeats in C9‐FTD/ALS. Front Neurosci 11: 711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuyumcu‐Martinez NM, Wang GS, Cooper TA (2007) Increased steady‐state levels of CUGBP1 in myotonic dystrophy 1 are due to PKC‐mediated hyperphosphorylation. Mol Cell 28: 68–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon I, Xiang S, Kato M, Wu L, Theodoropoulos P, Wang T, Kim J, Yun J, Xie Y, McKnight SL (2014) Poly‐dipeptides encoded by the C9ORF72 repeats bind nucleoli, impede RNA biogenesis, and kill cells. Science 345: 1139–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagier‐Tourenne C, Baughn M, Rigo F, Sun S, Liu P, Li HR, Jiang J, Watt AT, Chun S, Katz M et al (2013) Targeted degradation of sense and antisense C9orf72 RNA foci as therapy for ALS and frontotemporal degeneration. PNAS 110: E4530–E4539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CT, Chiu YW, Wang KC, Hwang CS, Lin KH, Lee IT, Tsai CP (2013a) Riluzole and prognostic factors in amyotrophic lateral sclerosis long‐term and short‐term survival: a population‐based study of 1149 cases in Taiwan. J Epidemiol 23: 35–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YB, Chen HJ, Peres JN, Gomez‐Deza J, Attig J, Stalekar M, Troakes C, Nishimura AL, Scotter EL, Vance C et al (2013b) Hexanucleotide repeats in ALS/FTD form length‐dependent RNA foci, sequester RNA binding proteins, and are neurotoxic. Cell Rep 5: 1178–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YB, Baskaran P, Gomez‐Deza J, Chen HJ, Nishimura AL, Smith BN, Troakes C, Adachi Y, Stepto A, Petrucelli L et al (2017) C9orf72 poly GA RAN‐translated protein plays a key role in amyotrophic lateral sclerosis via aggregation and toxicity. Hum Mol Genet 26: 4765–4777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefave CV, Squatrito M, Vorlova S, Rocco GL, Brennan CW, Holland EC, Pan YX, Cartegni L (2011) Splicing factor hnRNPH drives an oncogenic splicing switch in gliomas. EMBO J 30: 4084–4097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Miller JW, Mankodi A, Kanadia RN, Yuan Y, Moxley RT, Swanson MS, Thornton CA (2006) Failure of MBNL1‐dependent post‐natal splicing transitions in myotonic dystrophy. Hum Mol Genet 15: 2087–2097 [DOI] [PubMed] [Google Scholar]
- Liu W, Ikeda Y, Hishikawa N, Yamashita T, Deguchi K, Abe K (2014) Characteristic RNA foci of the abnormal hexanucleotide GGCCUG repeat expansion in spinocerebellar ataxia type 36 (Asidan). Eur J Neurol 21: 1377–1386 [DOI] [PubMed] [Google Scholar]
- Liu F, Liu Q, Lu CX, Cui B, Guo XN, Wang RR, Liu MS, Li XG, Cui LY, Zhang X (2016) Identification of a novel loss‐of‐function C9orf72 splice site mutation in a patient with amyotrophic lateral sclerosis. Neurobiol Aging 47: 219.e1–219.e5 [DOI] [PubMed] [Google Scholar]
- Lopez‐Morato M, Brook JD, Wojciechowska M (2018) Small molecules which improve pathogenesis of myotonic dystrophy type 1. Front Neurol 9: 349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackenzie IR, Arzberger T, Kremmer E, Troost D, Lorenzi S, Mori K, Weng SM, Haass C, Kretzschmar HA, Edbauer D et al (2013) Dipeptide repeat protein pathology in C9ORF72 mutation cases: clinico‐pathological correlations. Acta Neuropathol 126: 859–879 [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Frick P, Neumann M (2014) The neuropathology associated with repeat expansions in the C9ORF72 gene. Acta Neuropathol 127: 347–357 [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Frick P, Grässer FA, Gendron TF, Petrucelli L, Cashman NR, Edbauer D, Kremmer E, Prudlo J, Troost D et al (2015) Quantitative analysis and clinico‐pathological correlations of different dipeptide repeat protein pathologies in C9ORF72 mutation carriers. Acta Neuropathol 130: 845–861 [DOI] [PubMed] [Google Scholar]
- Mahadevan MS, Yadava RS, Yu Q, Balijepalli S, Frenzel‐McCardell CD, Bourne TD, Phillips LH (2006) Reversible model of RNA toxicity and cardiac conduction defects in myotonic dystrophy. Nat Genet 38: 1066–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malgowska M, Gudanis D, Kierzek R, Wyszko E, Gabelica V, Gdaniec Z (2014) Distinctive structural motifs of RNA G‐quadruplexes composed of AGG, CGG and UGG trinucleotide repeats. Nucleic Acids Res 42: 10196–10207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandrile G, Di Gregorio E, Goel H, Giachino D, De Mercanti S, Iudicello M, Rolando M, Losa S, De Marchi M, Brusco A (2016) Heterozygous deletion of KLHL1/ATX8OS at the SCA8 locus is unlikely associated with cerebellar impairment in humans. Cerebellum 15: 208–212 [DOI] [PubMed] [Google Scholar]
- Mankodi A, Urbinati CR, Yuan QP, Moxley RT, Sansone V, Krym M, Henderson D, Schalling M, Swanson MS, Thornton CA (2001) Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum Mol Genet 10: 2165–2170 [DOI] [PubMed] [Google Scholar]
- Mankodi A, Takahashi MP, Jiang H, Beck CL, Bowers WJ, Moxley RT, Cannon SC, Thornton CA (2002) Expanded CUG repeats trigger aberrant splicing of ClC‐1 chloride channel pre‐mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol Cell 10: 35–44 [DOI] [PubMed] [Google Scholar]
- Margolis RL, Holmes SE, Rosenblatt A, Gourley L, O'Hearn E, Ross CA, Seltzer WK, Walker RH, Ashizawa T, Rasmussen A et al (2004) Huntington's disease‐like 2 (HDL2) in North America and Japan. Ann Neurol 56: 670–674 [DOI] [PubMed] [Google Scholar]
- Matsuzono K, Imamura K, Murakami N, Tsukita K, Yamamoto T, Izumi Y, Kaji R, Ohta Y, Yamashita T, Abe K et al (2017) Antisense oligonucleotides reduce RNA foci in spinocerebellar ataxia 36 patient iPSCs. Mol Ther Nucleic Acids 8: 211–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMillan CT, Russ J, Wood EM, Irwin DJ, Grossman M, McCluskey L, Elman L, Van Deerlin V, Lee EB (2015) C9orf72 promoter hypermethylation is neuroprotective. Neurology 84: 1622–1630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JW, Urbinati CR, Teng‐Umnuay P, Stenberg MG, Byrne BJ, Thornton CA, Swanson MS (2000) Recruitment of human muscleblind proteins to (CUG)n expansions associated with myotonic dystrophy. EMBO J 19: 4439–4448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizielinska S, Lashley T, Norona FE, Clayton EL, Ridler CE, Fratta P, Isaacs AM (2013) C9orf72 frontotemporal lobar degeneration is characterised by frequent neuronal sense and antisense RNA foci. Acta Neuropathol 126: 845–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizielinska S, Grönke S, Niccoli T, Ridler CE, Clayton EL, Devoy A, Moens T, Norona FE, Woollacott IOC, Pietrzyk J et al (2014) C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine‐rich proteins. Science 345: 1192–1194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moens TG, Mizielinska S, Niccoli T, Mitchell JS, Thoeng A, Ridler CE, Grönke S, Esser J, Heslegrave A, Zetterberg H et al (2018) Sense and antisense RNA are not toxic in Drosophila models of C9orf72‐associated ALS/FTD. Acta Neuropathol 135: 445–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori K, Arzberger T, Grässer FA, Gijselinck I, May S, Rentzsch K, Weng SM, Schludi MH, van der Zee J, Cruts M et al (2013a) Bidirectional transcripts of the expanded C9orf72 hexanucleotide repeat are translated into aggregating dipeptide repeat proteins. Acta Neuropathol 126: 881–893 [DOI] [PubMed] [Google Scholar]
- Mori K, Lammich S, Mackenzie IR, Forné I, Zilow S, Kretzschmar H, Edbauer D, Janssens J, Kleinberger G, Cruts M et al (2013b) hnRNP A3 binds to GGGGCC repeats and is a constituent of p62‐positive/TDP43‐negative inclusions in the hippocampus of patients with C9orf72 mutations. Acta Neuropathol 125: 413–423 [DOI] [PubMed] [Google Scholar]
- Mori K, Nihei Y, Arzberger T, Zhou Q, Mackenzie IR, Hermann A, Hanisch F, German Consortium for Frontotemporal Lobar Degeneration, Bavarian Brain Bank Alliance , Kamp F et al (2016) Reduced hnRNPA3 increases C9orf72 repeat RNA levels and dipeptide‐repeat protein deposition. EMBO Rep 17: 1314–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moseley ML, Zu T, Ikeda Y, Gao W, Mosemiller AK, Daughters RS, Chen G, Weatherspoon MR, Clark HB, Ebner TJ et al (2006) Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat Genet 38: 758–769 [DOI] [PubMed] [Google Scholar]
- Nadel J, Athanasiadou R, Lemetre C, Wijetunga NA, Ó Broin P, Sato H, Zhang Z, Jeddeloh J, Montagna C, Golden A et al (2015) RNA:DNA hybrids in the human genome have distinctive nucleotide characteristics, chromatin composition, and transcriptional relationships. Epigenetics Chromatin 8: 46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niblock M, Smith BN, Lee YB, Sardone V, Topp S, Troakes C, Al‐Sarraj S, Leblond CS, Dion PA, Rouleau GA et al (2016) Retention of hexanucleotide repeat‐containing intron in C9orf72 mRNA: implications for the pathogenesis of ALS/FTD. Acta Neuropathol Commun 4: 18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niimi Y, Takahashi M, Sugawara E, Umeda S, Obayashi M, Sato N, Ishiguro T, Higashi M, Eishi Y, Mizusawa H et al (2013) Abnormal RNA structures (RNA foci) containing a penta‐nucleotide repeat (UGGAA)n in the Purkinje cell nucleus is associated with spinocerebellar ataxia type 31 pathogenesis. Neuropathology 33: 600–611 [DOI] [PubMed] [Google Scholar]
- O'Donnell WT, Warren ST (2002) A decade of molecular studies of fragile X syndrome. Annu Rev Neurosci 25: 315–338 [DOI] [PubMed] [Google Scholar]
- O'Hearn EE, Hwang HS, Holmes SE, Rudnicki DD, Chung DW, Seixas AI, Cohen RL, Ross CA, Trojanowski JQ, Pletnikova O et al (2015) Neuropathology and cellular pathogenesis of spinocerebellar ataxia type 12. Mov Disord 30: 1813–1824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Rourke JG, Bogdanik L, Muhammad AKMG, Gendron TF, Kim KJ, Austin A, Cady J, Liu EY, Zarrow J, Grant S et al (2015) C9orf72 BAC transgenic mice display typical pathologic features of ALS/FTD. Neuron 88: 892–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Rourke JG, Bogdanik L, Yanez A, Lall D, Wolf AJ, Muhammad AK, Ho R, Carmona S, Vit JP, Zarrow J et al (2016) C9orf72 is required for proper macrophage and microglial function in mice. Science 351: 1324–1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obayashi M, Stevanin G, Synofzik M, Monin ML, Duyckaerts C, Sato N, Streichenberger N, Vighetto A, Desestret V, Tesson C et al (2015) Spinocerebellar ataxia type 36 exists in diverse populations and can be caused by a short hexanucleotide GGCCTG repeat expansion. J Neurol Neurosurg Psychiatry 86: 986–995 [DOI] [PubMed] [Google Scholar]
- Oh SY, He F, Krans A, Frazer M, Taylor JP, Paulson HL, Todd PK (2015) RAN translation at CGG repeats induces ubiquitin proteasome system impairment in models of fragile X‐associated tremor ataxia syndrome. Hum Mol Genet 24: 4317–4326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olney NT, Spina S, Miller BL (2017) Frontotemporal dementia. Neurol Clin 35: 339–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onishi H, Kino Y, Morita T, Futai E, Sasagawa N, Ishiura S (2008) MBNL1 associates with YB‐1 in cytoplasmic stress granules. J Neurosci Res 86: 1994–2002 [DOI] [PubMed] [Google Scholar]
- Park H, González ÀL, Yildirim I, Tran T, Lohman JR, Fang P, Guo M, Disney MD (2015) Crystallographic and computational analyses of AUUCU repeating RNA that causes spinocerebellar ataxia type 10 (SCA10). Biochemistry 54: 3851–3859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perbellini R, Greco S, Sarra‐Ferraris G, Cardani R, Capogrossi MC, Meola G, Martelli F (2011) Dysregulation and cellular mislocalization of specific miRNAs in myotonic dystrophy type 1. Neuromuscul Disord 21: 81–88 [DOI] [PubMed] [Google Scholar]
- Pettersson OJ, Aagaard L, Jensen TG, Damgaard CK (2015) Molecular mechanisms in DM1 – A focus on foci. Nucleic Acids Res 43: 2433–2441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philips AV, Timchenko LT, Cooper TA (1998) Disruption of splicing regulated by a CUG‐binding protein in myotonic dystrophy. Science 280: 737–741 [DOI] [PubMed] [Google Scholar]
- Prudencio M, Belzil VV, Batra R, Ross CA, Gendron TF, Pregent LJ, Murray ME, Overstreet KK, Piazza‐Johnston AE, Desaro P et al (2015) Distinct brain transcriptome profiles in C9orf72‐associated and sporadic ALS. Nat Neurosci 18: 1175–1182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy S, Smith DB, Rich MM, Leferovich JM, Reilly P, Davis BM, Tran K, Rayburn H, Bronson R, Cros D et al (1996) Mice lacking the myotonic dystrophy protein kinase develop a late onset progressive myopathy. Nat Genet 13: 325–335 [DOI] [PubMed] [Google Scholar]
- Renton AE, Majounie E, Waite A, Simón‐Sánchez J, Rollinson S, Gibbs JR, Schymick JC, Laaksovirta H, van Swieten JC, Myllykangas L et al (2011) A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21‐linked ALS‐FTD. Neuron 72: 257–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renton AE, Chiò A, Traynor BJ (2014) State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 17: 17–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi S, Serrano A, Gerbino V, Giorgi A, Di Francesco L, Nencini M, Bozzo F, Schininà ME, Bagni C, Cestra G et al (2015) Nuclear accumulation of mRNAs underlies G4C2 repeat‐induced translational repression in a cellular model of C9orf72 ALS. J Cell Sci 128: 1787–1899 [DOI] [PubMed] [Google Scholar]
- Rudnicki DD, Holmes SE, Lin MW, Thornton CA, Ross CA, Margolis RL (2007) Huntington's disease‐like 2 is associated with CUG repeat‐containing RNA foci. Ann Neurol 61: 272–282 [DOI] [PubMed] [Google Scholar]
- Rudnicki DD, Pletnikova O, Vonsattel JP, Ross CA, Margolis RL (2008) A comparison of huntington disease and huntington disease‐like 2 neuropathology. J Neuropathol Exp Neurol 67: 366–374 [DOI] [PubMed] [Google Scholar]
- Saberi S, Stauffer JE, Schulte DJ, Ravits J (2015) Neuropathology of amyotrophic lateral sclerosis and its variants. Neurol Clin 33: 855–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saberi S, Stauffer JE, Jiang J, Garcia SD, Taylor AE, Schulte D, Ohkubo T, Schloffman CL, Maldonado M, Baughn M et al (2018) Sense‐encoded poly‐GR dipeptide repeat proteins correlate to neurodegeneration and uniquely co‐localize with TDP‐43 in dendrites of repeat‐expanded C9orf72 amyotrophic lateral sclerosis. Acta Neuropathol 135: 459–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakae N, Bieniek KF, Zhang YJ, Ross K, Gendron TF, Murray ME, Rademakers R, Petrucelli L, Dickson DW (2018) Poly‐GR dipeptide repeat polymers correlate with neurodegeneration and Clinicopathological subtypes in C9ORF72‐related brain disease. Acta Neuropathol Commun 6: 63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salisbury E, Schoser B, Schneider‐Gold C, Wang GL, Huichalaf C, Jin B, Sirito M, Sarkar P, Krahe R, Timchenko NA et al (2009) Expression of RNA CCUG repeats dysregulates translation and degradation of proteins in myotonic dystrophy 2 patients. Am J Pathol 175: 748–762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sareen D, O'Rourke JG, Meera P, Muhammad AK, Grant S, Simpkinson M, Bell S, Carmona S, Ornelas L, Sahabian A et al (2013) Targeting RNA foci in iPSC‐derived motor neurons from ALS patients with C9ORF72 repeat expansion. Sci Transl Med 5: 208ra149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato N, Amino T, Kobayashi K, Asakawa S, Ishiguro T, Tsunemi T, Takahashi M, Matsuura T, Flanigan KM, Iwasaki S et al (2009) Spinocerebellar ataxia type 31 is associated with ‘inserted’ penta‐nucleotide repeats containing (TGGAA)n. Am J Hum Genet 85: 544–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savkur RS, Philips AV, Cooper TA (2001) Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet 29: 40–47 [DOI] [PubMed] [Google Scholar]
- Seixas AI, Holmes SE, Takeshima H, Pavlovich A, Sachs N, Pruitt JL, Silveira I, Ross CA, Margolis RL, Rudnicki DD (2012) Loss of junctophilin‐3 contributes to huntington disease‐like 2 pathogenesis. Ann Neurol 71: 245–257 [DOI] [PubMed] [Google Scholar]
- Sellier C, Rau F, Liu Y, Tassone F, Hukema RK, Gattoni R, Schneider A, Richard S, Willemsen R, Elliott DJ et al (2010) Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J 29: 1248–1261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellier C, Freyermuth F, Tabet R, Tran T, He F, Ruffenach F, Alunni V, Moine H, Thibault C, Page A et al (2013) Sequestration of DROSHA and DGCR132 by expanded CGG RNA repeats alters microRNA processing in fragile X‐associated tremor/ataxia syndrome. Cell Rep 3: 869–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellier C, Campanari ML, Julie Corbier C, Gaucherot A, Kolb‐Cheynel I, Oulad‐Abdelghani M, Ruffenach F, Page A, Ciura S, Kabashi E et al (2016) Loss of C9ORF72 impairs autophagy and synergizes with polyQ Ataxin‐2 to induce motor neuron dysfunction and cell death. EMBO J 35: 1276–1297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellier C, Buijsen RAM, He F, Natla S, Jung L, Tropel P, Gaucherot A, Jacobs H, Meziane H, Vincent A et al (2017) Translation of expanded CGG repeats into FMRpolyG is pathogenic and may contribute to fragile X tremor ataxia syndrome. Neuron 93: 331–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaraj BT, Livesey MR, Zhao C, Gregory JM, James OT, Cleary EM, Chouhan AK, Gane AB, Perkins EM, Dando O et al (2018) C9ORF72 repeat expansion causes vulnerability of motor neurons to Ca2+‐permeable AMPA receptor‐mediated excitotoxicity. Nat Commun 9: 347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Lin S, Staats KA, Li Y, Chang WH, Hung ST, Hendricks E, Linares GR, Wang Y, Son EY et al (2018) Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat Med 24: 313–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiina N, Nakayama K (2014) RNA granule assembly and disassembly modulated by nuclear factor associated with double‐stranded RNA 2 and nuclear factor 45. J Biol Chem 289: 21163–21180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofola OA, Jin P, Qin Y, Duan R, Liu H, de Haro M, Nelson DL, Botas J (2007) RNA‐binding proteins hnRNP A2/B1 and CUGBP1 suppress fragile X CGG premutation repeat‐induced neurodegeneration in a Drosophila model of FXTAS. Neuron 55: 565–571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stopford MJ, Higginbottom A, Hautbergue GM, Cooper‐knock J, Mulcahy PJ, De Vos KJ, Renton AE, Calvo A, Chio A, Traynor BJ et al (2017) C9ORF72 hexanucleotide repeat exerts toxicity in a stable, inducible motor neuronal cell model, which is rescued by partial depletion of Pten. Hum Mol Genet 26: 1133–1145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudria‐Lopez E, Koppers M, de Wit M, van der Meer C, Westeneng HJ, Zundel CA, Youssef SA, Harkema L, de Bruin A, Veldink JH et al (2016) Full ablation of C9orf72 in mice causes immune system‐related pathology and neoplastic events but no motor neuron defects. Acta Neuropathol 132: 145–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinnen B, Robberecht W (2014) The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol 10: 661–670 [DOI] [PubMed] [Google Scholar]
- Swinnen B, Bento‐Abreu A, Gendron TF, Boeynaems S, Bogaert E, Nuyts R, Timmers M, Scheveneels W, Hersmus N, Wang J et al (2018) A zebrafish model for C9orf72 ALS reveals RNA toxicity as a pathogenic mechanism. Acta Neuropathol 135: 427–443 [DOI] [PubMed] [Google Scholar]
- Taneja KL, McCurrach M, Schalling M, Housman D, Singer RH (1995) Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J Cell Biol 128: 995–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B, White RJ, Xia T, Welle S, Turner DH, Mathews MB, Thornton CA (2000) Expanded CUG repeat RNAs form hairpins that activate the double‐stranded RNA‐dependent protein kinase PKR. RNA 6: 79–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd PK, Oh SY, Krans A, Pandey UB, Di Prospero NA, Min KT, Taylor JP, Paulson HL (2010) Histone deacetylases suppress cgg repeat‐induced neurodegeneration via transcriptional silencing in models of Fragile X Tremor Ataxia Syndrome. PLoS Genet 6: e1001240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd PK, Oh SY, Krans A, He F, Sellier C, Frazer M, Renoux AJ, Chen KC, Scaglione KM, Basrur V et al (2013) CGG repeat associated translation mediates neurodegeneration in Fragile X Tremor Ataxia Syndrome. Neuron 78: 440–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi BK, Surabhi S, Bhaskar PK, Mukherjee A, Mutsuddi M (2016) The RNA binding KH domain of Spoonbill depletes pathogenic non‐coding spinocerebellar ataxia 8 transcripts and suppresses neurodegeneration in Drosophila . Biochim Biophys Acta – Mol Basis Dis 1862: 1732–1741 [DOI] [PubMed] [Google Scholar]
- Udd B, Krahe R (2012) The myotonic dystrophies: molecular, clinical, and therapeutic challenges. Lancet Neurol 11: 891–905 [DOI] [PubMed] [Google Scholar]
- van Blitterswijk M, Gendron TF, Baker MC, DeJesus‐Hernandez M, Finch NA, Brown PH, Daughrity LM, Murray ME, Heckman MG, Jiang J et al (2015) Novel clinical associations with specific C9ORF72 transcripts in patients with repeat expansions in C9ORF72. Acta Neuropathol 130: 863–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verkerk A, Pieretti M, Sutcliffe J, Fu YH, Kuhl DP, Pizzuti A, Reiner O, Richards S, Victoria MF, Zhang FP et al (1991) Identification of a gene (FMR‐1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 65: 905–914 [DOI] [PubMed] [Google Scholar]
- Waite AJ, Bäumer D, East S, Neal J, Morris HR, Ansorge O, Blake DJ (2014) Reduced C9orf72 protein levels in frontal cortex of amyotrophic lateral sclerosis and frontotemporal degeneration brain with the C9ORF72 hexanucleotide repeat expansion. Neurobiol Aging 35: 1779.e5–1779.e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakamiya M, Matsuura T, Liu Y, Schuster GC, Gao R, Xu W, Sarkar PS, Lin X, Ashizawa T (2006) The role of ataxin 10 in the pathogenesis of spinocerebellar ataxia type 10. Neurology 67: 607–613 [DOI] [PubMed] [Google Scholar]
- Wandrey F, Montellese C, Koos K, Badertscher L, Bammert L, Cook AG, Zemp I, Horvath P, Kutay U (2015) The NF45/NF90 heterodimer contributes to the biogenesis of 60S ribosomal subunits and influences nucleolar morphology. Mol Cell Biol 35: 3491–3503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang YC, Lee CM, Lee LC, Tung LC, Hsieh‐Li HM, Lee‐Chen GJ, Su MT (2011) Mitochondrial dysfunction and oxidative stress contribute to the pathogenesis of spinocerebellar ataxia type 12 (SCA12). J Biol Chem 286: 21742–21754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward AJ, Rimer M, Killian JM, Dowling JJ, Cooper TA (2010) CUGBP1 overexpression in mouse skeletal muscle reproduces features of myotonic dystrophy type 1. Hum Mol Genet 19: 3614–3622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster CP, Smith EF, Bauer CS, Moller A, Hautbergue GM, Ferraiuolo L, Myszczynska MA, Higginbottom A, Walsh MJ, Whitworth J et al (2016) The C9orf72 protein interacts with Rab1a and the ULK1 complex to regulate initiation of autophagy. EMBO J 35: 1656–1676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster CP, Smith EF, Grierson AJ, De Vos KJ (2018) C9orf72 plays a central role in Rab GTPase‐dependent regulation of autophagy. Small GTPases 9: 399–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen X, Tan W, Westergard T, Krishnamurthy K, Markandaiah SS, Shi Y, Lin S, Shneider NA, Monaghan J, Pandey UB et al (2014) Antisense proline‐arginine RAN dipeptides linked to C9ORF72‐ALS/FTD form toxic nuclear aggregates that initiate in vitro and in vivo neuronal death. Neuron 84: 1213–1225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westergard T, Jensen BK, Wen X, Cai J, Kropf E, Iacovitti L, Pasinelli P, Trotti D (2016) Cell‐to‐cell transmission of dipeptide repeat proteins linked to C9orf72‐ALS/FTD. Cell Rep 17: 645–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White MC, Gao R, Xu W, Mandal SM, Lim JG, Hazra TK, Wakamiya M, Edwards SF, Raskin S, Teive HA et al (2010) Inactivation of hnRNP K by expanded intronic AUUCU repeat induces apoptosis via translocation of PKCδ to mitochondria in spinocerebellar ataxia 10. PLoS Genet 6: e1000984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White M, Xia G, Gao R, Wakamiya M, Sarkar PS, McFarland K, Ashizawa T (2012) Transgenic mice with SCA10 pentanucleotide repeats show motor phenotype and susceptibility to seizure — A toxic RNA gain‐of‐function model. J Neurosci Res 90: 706–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilburn B, Rudnicki DD, Zhao J, Weitz TM, Cheng Y, Gu X, Greiner E, Park CS, Wang N, Sopher BL et al (2011) An antisense CAG repeat transcript at JPH3 locus mediates expanded polyglutamine protein toxicity in Huntington's disease‐like 2 Mice. Neuron 70: 427–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willemsen R, Hoogeveen‐Westerveld M, Reis S, Holstege J, Severijnen LA, Nieuwenhuizen IM, Schrier M, van Unen L, Tassone F, Hoogeveen AT et al (2003) The FMR1 CGG repeat mouse displays ubiquitin‐positive intranuclear neuronal inclusions; implications for the cerebellar tremor/ataxia syndrome. Hum Mol Genet 12: 949–959 [DOI] [PubMed] [Google Scholar]
- Xiao S, MacNair L, McGoldrick P, McKeever PM, McLean JR, Zhang M, Keith J, Zinman L, Rogaeva E, Robertson J (2015) Isoform specific antibodies reveal distinct subcellular localizations of C9orf72 in amyotrophic lateral sclerosis. Ann Neurol 78: 568–583 [DOI] [PubMed] [Google Scholar]
- Xu Z, Poidevin M, Li X, Li Y, Shu L, Nelson DL, Li H, Hales CM, Gearing M, Wingo TS et al (2013) Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. PNAS 110: 7778–7783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Liang C, Swaminathan K, Herrlinger S, Lai F, Shiekhattar R, Chen JF (2016) A C9ORF72/SMCR164‐containing complex regulates ULK1 and plays a dual role in autophagy. Sci Adv 2: e1601167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin S, Lopez‐Gonzalez R, Kunz RC, Gangopadhyay J, Borufka C, Gygi SP, Gao FB, Reed R (2017) Evidence that C9ORF72 dipeptide repeat proteins associate with U2 snRNP to cause mis‐splicing in ALS/FTD patients. Cell Rep 19: 2244–2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, Donnelly CJ, Haeusler AR, Grima JC, Machamer JB, Steinwald P, Daley EL, Miller SJ, Cunningham KM, Vidensky S et al (2015) The C9orf72 repeat expansion disrupts nucleocytoplasmic transport. Nature 525: 56–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu T, Gibbens B, Doty NS, Gomes‐Pereira M, Huguet A, Stone MD, Margolis J, Peterson M, Markowski TW, Ingram MA et al (2011) Non‐ATG‐initiated translation directed by microsatellite expansions. PNAS 108: 260–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu T, Liu Y, Bañez‐coronel M, Reid T, Pletnikova O, Lewis J, Miller TM, Harms MB, Falchook AE, Subramonya SH et al (2013) RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. PNAS 110: E4968–E4977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zu T, Cleary JD, Liu Y, Bañez‐Coronel M, Bubenik JL, Ayhan F, Ashizawa T, Xia G, Clark HB, Yachnis AT et al (2017) RAN translation regulated by muscleblind proteins in myotonic dystrophy type 2. Neuron 95: 1292–1305.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]