

Abstract
Objective: The objective of this study is to evaluate efficacy and safety of SPN-812 (extended-release viloxazine) for ADHD in children aged 6 to 12 years. Method: In an 8-week study, 222 participants were randomized to placebo or SPN-812 100, 200, 300, or 400 mg/day. Measurements included ADHD Rating Scale (RS)-IV total score and Clinical Global Impression-Severity (CGI-S) and Clinical Global Impression-Improvement (CGI-I) scores. Safety assessments included laboratory and electrocardiogram (ECG) measurements, suicidality monitoring (Columbia-Suicide Severity Rating Scale), and adverse event (AE) reporting. Results: Significant improvements in ADHD-RS-IV total score were observed for 200, 300, and 400 mg dose groups versus placebo (p < .05; effect size [ES] = 0.547, 0.596, and 0.623). CGI-I score for the 300 mg group and CGI-S score for all SPN-812 groups except for 100 mg improved significantly (p < .05) versus placebo. The most frequent AEs (≥15%) were somnolence, headache, and decreased appetite. Conclusion: SPN-812 significantly reduced the severity of ADHD symptoms and was well tolerated. The efficacy and safety of SPN-812 are being investigated in Phase III trials.
Keywords: attention deficit/hyperactivity disorder, ADHD, nonstimulant, viloxazine
Introduction
Attention deficit/hyperactivity disorder (ADHD) is one of the most common psychiatric disorders affecting children and adolescents, with an estimated 6.1 million U.S. children aged 2 to 17 years (9.4% of that population) having received an ADHD diagnosis in 2016 (Adesman, 2001; Danielson et al., 2018). Children experiencing symptoms of ADHD may have difficulties in the school setting that limit academic achievement; the symptoms may also affect the quality of life of these children and their families (Danckaerts et al., 2010; Harpin, 2005; Peasgood et al., 2016).
The American Academy of Pediatrics (AAP) practice guidelines for ADHD recommend pharmacotherapy with a Food and Drug Administration (FDA)-approved medication (Hervas et al., 2016; Subcommittee on Attention-Deficit/Hyperactivity Disorder et al., 2011). Approved stimulant medications have been shown to be more effective than currently available nonstimulants, making them the treatment of choice for most patients with ADHD (Briars & Todd, 2016; Catala-Lopez et al., 2017; Subcommittee on Attention-Deficit/Hyperactivity Disorder et al., 2011). However, an estimated 10% to 30% of patients do not respond optimally to stimulants, and some patients cannot tolerate side effects commonly associated with these medications (Briars & Todd, 2016). Furthermore, caution should be exercised when prescribing stimulants for patients with certain relative contraindications (e.g., Tourette syndrome, substance use disorders; Briars & Todd, 2016). Due to their potential for abuse, stimulants are classified as schedule II controlled substances by the United States Drug Enforcement Administration (Childress & Tran, 2016; U.S. Department of Justice, n.d.). FDA-approved nonstimulant medications, such as the norepinephrine reuptake inhibitor, atomoxetine, and the α2 agonists, extended-release guanfacine, and extended-release clonidine, do not have abuse liability and can be effective options for some patients precluded from stimulant treatment (Childress & Tran, 2016). However, they are generally considered to be less effective than stimulants based on their lower effect size, and they have a more gradual onset of action. In addition, nonstimulants may be associated with problematic side effects, such as rebound hypertension (α2 agonists), decreased appetite (atomoxetine), and sedation/somnolence (all) (Briars & Todd, 2016; Childress & Tran, 2016; Clemow & Bushe, 2015; Connor, Arnsten, Pearson, & Greco, 2014; Huss, Chen, & Ludolph, 2016; Shire US Inc., 2017). In short, although currently approved medications for ADHD are generally associated with moderate to robust symptom improvement, they may not be optimal for some patients (Briars & Todd, 2016). Consequently, there is a need for alternative nonstimulant options.
SPN-812 (extended-release viloxazine), a structurally distinct, bicyclic norepinephrine reuptake inhibitor with selective serotonergic activity, is a novel, once-daily, nonstimulant medication currently in Phase III development for the treatment of ADHD in children and adolescents (Johnson, Saylor, Brittain, Tulloch, & Liranso, 2015). Previously, the immediate-release formulation of viloxazine underwent a comprehensive clinical and safety review, revealing few safety concerns (Johnson, Saylor, Brittain, Tulloch, & Liranso, 2017). In addition, immediate-release viloxazine was evaluated and shown to be both efficacious and well tolerated in adults with ADHD in a Phase 2a, randomized, double-blind, placebo-controlled study (Johnson et al., 2015). Following these studies, SPN-812 was developed with the aim of reducing the therapeutic dose frequency to improve the plasma concentration profile over time and its associated tolerability in patients (Johnson et al., 2015). The Phase II study described here assessed the efficacy and safety of once-daily SPN-812 in children 6 to 12 years of age with ADHD, as measured by the change in ADHD Rating Scale-IV (ADHD-RS-IV) scores and Clinical Global Impression-Severity (CGI-S) and Clinical Global Impression-Improvement (CGI-I) scores versus placebo.
Method
This 8-week, randomized, double-blind, placebo-controlled, Phase II study (NCT02633527) was conducted between February 2016 and July 2016 in 32 centers in the United States. The study was conducted in accordance with the Helsinki Declaration and the International Council for Harmonisation (ICH) Note for Guidance on Good Clinical Practice. Study protocols, amendments, and informed consents were reviewed and approved by the appropriate Institutional Review Boards. Each participant’s parent/legal guardian provided written informed consent prior to screening assessments.
Study Participants
Children aged 6 to 12 years at the time of consent (inclusive) with a diagnosis of ADHD per the Diagnostic and Statistical Manual of Mental Disorders (4th ed.; DSM-IV; American Psychiatric Association, 1994) were enrolled in the study if they met defined inclusion and exclusion criteria. Inclusion criteria required participants to be deemed medically healthy through assessment of medical history, physical examination, clinical laboratory tests, and electrocardiogram (ECG). Participants, with a body weight of at least 20 kg, were required to have a minimum score of 26 on the ADHD-RS-IV and a minimum score of 4 on the CGI-S at Baseline. They also had to be free of ADHD medication for at least 1 week prior to Baseline. Participants could not take any ADHD medication or concomitant medication during the study period, with the exception of over-the-counter agents such as nutritional supplements, common transient treatments (e.g., ibuprofen or acetaminophen), or other select concomitant medications including selective β2-adrenoreceptor agonists (25 participants, 11.5% of the safety population). Key exclusion criteria included a history or presence of neuropsychiatric disease other than ADHD as the primary diagnosis (i.e., the condition most impacting the child), including major depressive disorder, bipolar disorder, personality disorder, Tourette disorder, pervasive developmental disorder, obsessive-compulsive disorder, post-traumatic stress disorder, or any other anxiety disorder or psychosis not otherwise specified. In addition, a history or presence of systemic disease including cardiovascular, pulmonary, hepatic, renal, hematologic, gastrointestinal, endocrine, immunologic, dermatologic, or other neurologic or psychiatric disease led to exclusion from the study. Additional exclusion criteria included evidence of suicidal attempt or ideation 6 months prior to screening or at screening.
Study Design
The study included a Screening visit, a Baseline visit, a Titration period (3 weeks), and a Maintenance period (5 weeks; Figure 1). Two hundred and fifty-five participants were screened (Visit 1) within 28 days prior to the Baseline visit; screening included confirmation of the ADHD diagnosis, using the Mini International Neuropsychiatric Interview for Children and Adolescents (MINI-KID; Sheehan et al., 2010). The DSM-IV edition was used (as noted above) for consistency throughout the study because the MINI-KID employs DSM-IV ADHD criteria. Participants receiving ADHD drug therapy (including stimulants or nonstimulants such as atomoxetine) at Screening were given at least 7 days to wash out from any ADHD medication before the scheduled Baseline visit (Visit 2), when final assessments were made to ensure participants met all inclusion criteria and were eligible for the study.
Figure 1.

Schematic overview of the study design.
Note. V = visit; QD = once daily.
aParticipants receiving ADHD drug therapy at Screening were given at least 7 days to wash out.
bMaintenance period began at Weeks 2, 3, 4, or 5, depending on the dose group.
Participants who remained eligible were randomized (1:2:2:2:2) to placebo or 100, 200, 300, or 400 mg/day of SPN-812 (Figure 1), using an interactive web response system (WRS), and received their first dosing card. Treatment was double-blinded and dosing was dispensed in identical-appearing packages containing either placebo or 100 mg of SPN-812 sufficient for 7 days to minimize any potential placebo effects. During the Titration period, SPN-812 doses were increased by 100 mg/day/week until the target randomized dose was reached; the highest dose of 400 mg/day was attained by the beginning of Week 4 (Visit 5). The placebo group was titrated for the same period as the highest dose group, to minimize any potential placebo effects. During the Maintenance period, participants continued taking the randomized dose reached during Titration until the end of study ([EOS], Week 8). Participants who completed the study had the option to enroll in an open-label extension (OLE) study.
Efficacy Assessment Measures
The primary efficacy outcome measure was the change from Baseline to EOS in the ADHD-RS-IV total score. Each of the 18 items on the ADHD-RS-IV represents a symptom derived from diagnostic criteria for ADHD from the DSM-IV (Zhang, Faries, Vowles, & Michelson, 2005). The frequency/severity of each symptom is rated on a scale of 0 to 3, where 0 indicates “never or rarely,” 1 indicates “sometimes,” 2 indicates “often,” and 3 indicates “very often.” The ADHD-RS-IV scale contains two subscales, Hyperactivity/Impulsivity and Inattention, which were also evaluated as exploratory endpoints. Investigators administered the ADHD-RS-IV at Baseline (Visit 2) and at each visit during the 8-week, double-blind treatment period, with parents or caregivers as the respondents. Further analyses were performed on the intention-to-treat (ITT) population’s ADHD-RS-IV total scores to determine the number of responders (defined as a reduction from Baseline to EOS ≥25%), responders with symptomatic remission (total score ≤18), and responders with syndrome remission (total score ≤18 and CGI-S score ≤2). In addition, the ADHD-RS-IV total score was evaluated in patients who had missed at most one visit, had no major deviations from the study protocol, and had at least 80% medication adherence.
The secondary efficacy endpoints included change from Baseline in the CGI-S and CGI-I scores (Busner & Targum, 2007). The severity of illness (CGI-S) was evaluated by the investigator at Baseline (Visit 2) and at each visit during the 8-week, double-blind treatment period. CGI-S scores range from 0 to 7, with 0 meaning “not assessed” and disease severity increasing from 1 (“normal, not ill”) to 7 (“extremely ill”). Participants’ symptom improvement over the 8-week, double-blind treatment period was rated by the investigator at each visit post-Baseline using the CGI-I 7-point scale, with 1 representing “very much improved” and 7 representing “very much worse” (Guy, 1976).
Safety Assessments
Treatment-emergent adverse events (TEAEs) were those defined by the Medical Dictionary for Regulatory Activities (MedDRA), Version 18.1. TEAEs were monitored and recorded by the investigator at each visit after Screening and throughout the study. Safety assessments included incidence rates of overall TEAEs and TEAEs that were treatment-related, serious, and/or severe. Other safety assessments included clinical laboratory tests, measurements of vital signs, 12-lead ECGs, suicidality monitoring by the Columbia-Suicide Severity Rating Scale (C-SSRS), and physical examination findings.
Statistical Analyses
The sample size determination of the Phase II study was based on results of previous studies with SPN-812 in adults, which used investigator-rated Conners’ Adult ADHD Rating Scale (CAARS) as a primary efficacy measure (Johnson et al., 2015). From these results, it was assumed that approximately 200 participants randomized in a ratio of 1:2 to placebo or one of the four active dose groups would be needed to evaluate the efficacy and safety of the SPN-812 doses.
The safety population was defined as all randomized participants who received at least one dose of study medication. The ITT population consisted of all participants from the safety population who had a Baseline assessment and at least one post-randomization ADHD-RS-IV assessment. Baseline comparability among the study groups in demographic and clinical characteristics was assessed using chi-square tests and analysis of variance (ANOVA). Efficacy analyses were conducted using the ITT population, with missing data imputed using the last observation carried forward (LOCF) method. Because analysis of covariance (ANCOVA) did not meet the test assumptions of linearity and parallelism, the primary endpoint analysis was conducted using an ANOVA model, with treatment as a fixed effect. The same methods were also conducted on the ADHD-RS-IV Hyperactivity/Impulsivity and Inattention Subscale scores. The analyses of the secondary endpoints were based on the ITT population, with missing data imputed using the LOCF method. ANOVA was performed on the absolute CGI-I values and on change from Baseline to EOS in CGI-S.
Each of the active SPN-812 dose groups (100, 200, 300, and 400 mg) was compared with the placebo group. Pair-wise comparisons among the active treatment groups were also performed. The P values, least squares (LS) means of treatment groups, and differences between the LS means of SPN-812 dose groups and placebo are reported.
To confirm the robustness of the findings of the primary analysis, a sensitivity analysis was performed. This was based on the mixed model for repeated measures (MMRM) technique, using a restricted maximum likelihood (REML) estimation method. The model included the change from Baseline for each post-Baseline visit in ADHD-RS-IV total score as a dependent variable; Baseline ADHD-RS-IV total score as a covariate; treatment group, site, and visit as fixed-effect factors; a treatment-by-visit fixed-effect interaction; and an unstructured covariance matrix. Safety analyses were performed using the safety population and were summarized using descriptive statistics.
Results
Subject Disposition
The subject disposition is summarized in Figure 2. Of the 255 participants who were screened, 234 (8.2% screen failure) were randomized to placebo or the four SPN-812 treatment arms (100, 200, 300, or 400 mg/day). Twelve participants withdrew from the study prior to the Baseline visit, and therefore, the randomized population, defined as the subset of enrolled participants who were randomized and had a Baseline visit scheduled, consisted of 222 participants. Of these, 160 (72.1%) completed the study and 62 (27.9%) discontinued early. The primary reasons for early discontinuation were loss to follow-up (17 participants, 7.7%), withdrawal of consent (16 participants, 7.2%), and AEs (13 participants, 5.9%). Following the Baseline visit, 5 participants were excluded from the study (due to non-eligibility, non-compliance, or withdrawal of consent) and therefore not included in the safety population (N = 217) or the ITT population (N = 206). A breakdown of the underlying reasons for discontinuation from the study for each treatment group and placebo is provided in Figure 2.
Figure 2.

Subject disposition.
Note. ITT = intention-to-treat; PP = per-protocol.
aOne participant randomized to SPN-812 300 mg/day received 100 mg/day throughout the study; this participant is included in the 100 mg/day group in all safety analyses, but in the 300 mg/day group in all analyses based on the ITT population.
Participant Demographics and Clinical Characteristics at Baseline
As shown in Table 1, demographic and clinical characteristics were comparable across all treatment groups (ITT population) at Baseline. The median age was 9.0 years across all study groups except in the SPN-812 100 mg group, in which the median age was 8.0 years. The majority of participants were male (67.0%) and White (56.8%). Psychiatric disorders other than ADHD were reported in 21.7% of the participants, with oppositional defiant disorder and anxiety disorders being the most common. During the double-blind phase of the study, 53% of participants took at least one concomitant medication; the most frequently used (≥10% in safety population) included selective β2 adrenoceptor agonists (11.5%, indicated for asthma) and systemic antihistamines (10.1%, indicated for seasonal allergies/allergies).
Table 1.
Participant Demographics and Clinical Characteristics at Baseline (ITT Population).
| Parameter | Placebo (N = 24) | SPN-812 (mg/day) |
|||
|---|---|---|---|---|---|
| 100 (N = 45) | 200 (N = 46) | 300 (N = 47) | 400 (N = 44) | ||
| Age, years, median (range) | 9.0 (6-11) | 8.0 (6-12) | 9.0 (6-12) | 9.0 (6-12) | 9.0 (6-12) |
| Height, cm, median (range) | 134.2 (115-153) | 132.0 (116-169) | 138.8 (115-167) | 135.9 (117-172) | 136.8 (109-162) |
| Weight, kg, median (range) | 28.2 (21-49) | 29.1 (20-64) | 33.3 (21-66) | 33.1 (21-62) | 32.6 (20-59) |
| BMI, kg/m2, median (range) | 16.8 (14-22) | 17.0 (14-23) | 16.7 (13-26) | 17.7 (14-26) | 17.4 (13-24) |
| Sex, n (%) | |||||
| Male | 11 (45.8) | 27 (60.0) | 33 (71.7) | 36 (76.6) | 31 (70.5) |
| Female | 13 (54.2) | 18 (40.0) | 13 (28.3) | 11 (23.4) | 13 (29.5) |
| Race, n (%) | |||||
| White | 17 (70.8) | 22 (48.9) | 26 (56.5) | 24 (51.1) | 28 (63.6) |
| Black or African American | 7 (29.2) | 19 (42.2) | 16 (34.8) | 22 (46.8) | 15 (34.1) |
| American Indian or Alaska Native | 0 | 0 | 1 (2.2) | 1 (2.1) | 0 |
| Asian | 0 | 0 | 2 (4.3) | 0 | 0 |
| Multiple | 0 | 4 (8.9) | 1 (2.2) | 0 | 1 (2.3) |
| ADHD-RS-IV scores, M (SD) | |||||
| Total | 42.4 (7.8) | 42.4 (6.8) | 43.9 (7.5) | 41.3 (7.9) | 40.8 (7.9) |
| Hyperactivity/Impulsivity Subscale | 20.5 (4.4) | 20.3 (5.2) | 21.7 (5.1) | 19.4 (6.0) | 19.7 (4.4) |
| Inattention Subscale | 21.9 (4.7) | 22.1 (3.9) | 22.2 (3.6) | 21.8 (3.8) | 21.0 (4.7) |
| CGI-S Score, M (SD) | 4.7 (0.69) | 4.9 (0.59) | 4.9 (0.64) | 4.8 (0.70) | 4.8 (0.71) |
Note. ITT = intention-to-treat; BMI = body mass index; ADHD-RS-IV = ADHD Rating Scale-IV; M = mean; CGI-S = Clinical Global Impression-Severity Scale.
The Baseline median ADHD-RS-IV total scores were similar across treatment groups, and the overall mean score was 42.1 ± 7.6. Similarly, the Hyperactivity/Impulsivity and Inattention Subscale scores were comparable for placebo and SPN-812 treatment groups at Baseline; overall mean scores were 20.3 ± 5.1 and 21.8 ± 4.1, respectively. At Baseline, the mean CGI-S score ranged from 4.7 ± 0.7 to 4.9 ± 0.7 for the study groups.
Efficacy
ADHD-RS-IV total scores
The primary efficacy analysis showed that the mean ADHD-RS-IV total score decreased from Baseline to the end of the double-blind phase in all SPN-812 dose groups and in the placebo group (ITT population). The LS mean change from Baseline in ADHD-RS-IV total score to EOS was significantly greater for the SPN-812 200, 300, and 400 mg dose groups (p = .031, .027, and .021, respectively) compared with placebo (Table 2). The effect size compared with placebo increased with the dose of SPN-812 (100 mg, 0.453; 200 mg, 0.547; 300 mg, 0.596; 400 mg, 0.623; Table 2).
Table 2.
Results of Analysis of Change From Baseline to End of Study in ADHD-RS-IV Total Score (ITT Population).
| ADHD-RS-IV scores | Placebo (N = 24) | SPN-812 (mg/day) |
|||
|---|---|---|---|---|---|
| 100 (N = 45) | 200 (N = 46) | 300 (N = 47) | 400 (N = 44) | ||
| Primary analysis | |||||
| LS meana | −10.5 | −16.7 | −18.4 | −18.6 | −19.0 |
| Placebo-adjusted treatment effectb | — | −6.2 | −7.9 | −8.1 | −8.5 |
| Effect size | — | 0.453 | 0.547 | 0.596 | 0.623 |
| p valueb,c | — | .0899 | .0310d | .0268d | .0209d |
| Hyperactivity/Impulsivity Subscale | |||||
| LS meana | −4.7 | −7.9 | −9.0 | −9.5 | −9.8 |
| Placebo-adjusted treatment effectb | — | −3.1 | −4.3 | −4.8 | −5.1 |
| p valueb,c | — | .1007 | .0254d | .0121d | .0086d |
| Inattention Subscale | |||||
| LS meana | −5.7 | −8.9 | −9.0 | −9.4 | −9.5 |
| Placebo-adjusted treatment effectb | — | −3.2 | −3.2 | −3.7 | −3.7 |
| p valueb,c | — | .1014 | .0909 | .0552 | .0533 |
| Sensitivity analysis using MMRM | |||||
| LS meane | −10.7 | −16.2 | −17.3 | −18.6 | −19.5 |
| Placebo-adjusted treatment effectb | — | −5.5 | −6.6 | −7.9 | −8.8 |
| p valueb,e | — | .0961 | .0473 | .0167 | .0092 |
Note. ADHD-RS-IV = ADHD Rating Scale-IV; ITT = intention-to-treat; LS = least squares; MMRM = mixed model for repeated measures; ANOVA = analysis of variance.
Estimated from ANOVA including fixed effect for treatment.
LS mean of SPN-812 dose group minus LS mean of placebo group.
Raw (unadjusted for multiplicity) p values.
p < .05 versus placebo, estimated by ANOVAd and MMRMe.
The mean change from Baseline over time in ADHD-RS-IV total score is shown in Figure 3. Based on MMRM as a sensitivity analysis, change from Baseline to each post-Baseline visit in ADHD-RS-IV total score demonstrated a statistically significant difference (p < .05) from placebo that was based on dose. The difference was significant at Week 4 only in the SPN-812 100 mg dose group; from Week 4 to Week 6 in the SPN-812 200 mg dose group; and from Week 4 to EOS in the SPN-812 300 and 400 mg dose groups (Figure 3). Further analyses revealed that based on the ADHD-RS-IV total score, responder rates (reduction from Baseline of at least 25%) ranged from 60.0% to 68.2% across the 100 to 400 mg dose groups, compared with 45.8% in the placebo group, whereas symptomatic remission rates (scores ≤18) ranged from 37.8% to 47.7% across the 100 to 400 mg dose groups, compared with 16.7% in the placebo group (Supplemental Table 1). Symptomatic remission rates in the 300 and 400 mg dose groups were significantly higher than in the placebo group (Supplemental Table 1).
Figure 3.
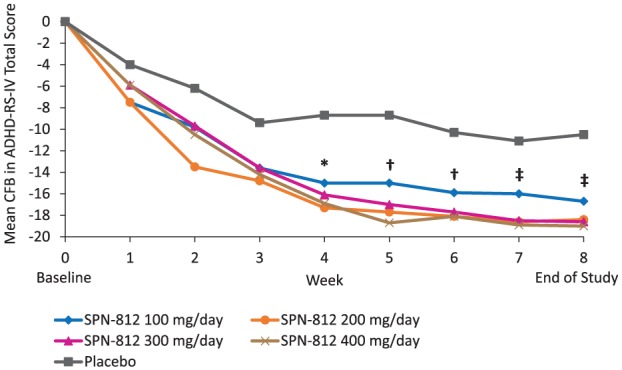
Mean change from Baseline over time in ADHD-RS-IV total score by treatment group (ITT population).
Note. ADHD-RS-IV = ADHD Rating Scale-IV; ITT = intention-to-treat; CFB = change from Baseline; ANOVA = analysis of variance.
Statistics based on sensitivity analysis using a mixed model for repeated measures analysis:
*p < .05 for the 100, 200, 300, and 400 mg/day dose groups versus placebo (ANOVA).
†p < .05 only for the 200, 300, and 400 mg/day dose groups versus placebo (ANOVA).
‡p < .05 only for the 300 and 400 mg/day dose groups versus placebo (ANOVA).
ADHD-RS-IV total score was also evaluated, as a second sensitivity analysis, in patients who missed at most one visit, had at least 80% study medication adherence overall, and had no major protocol deviation (per-protocol [PP] population, N = 149). The analysis of change from Baseline to EOS in ADHD-RS-IV total score demonstrated a statistically significant improvement in all SPN-812 doses compared with placebo (p < .05; ANCOVA model, treatment and Baseline as fixed effect; Supplemental Table 2). The LS mean change was −18.3, −20.7, −19.2, and −23.3 for the SPN-812 100, 200, 300, and 400 mg dose groups, respectively, compared with −9.4 for the placebo group.
ADHD-RS-IV subscales
As shown in Table 2, the mean change from Baseline to EOS in ADHD-RS-IV Hyperactivity/Impulsivity Subscale scores improved (decreased) in all SPN-812 dose groups and in the placebo group. Differences in the LS mean change from Baseline to EOS were statistically significant for the SPN-812 200, 300, and 400 mg dose groups compared with placebo (p = .025, .012, and .009, respectively; Table 2). The placebo-adjusted treatment effect (i.e., LS mean of SPN-812 dose minus LS mean of placebo) increased with the dose of SPN-812 (100 mg, −3.1; 200 mg, −4.3; 300 mg, −4.8; 400 mg, −5.1).
Mean ADHD-RS-IV Inattention Subscale scores also improved (decreased) throughout treatment in all study groups (Table 2). The LS mean change from Baseline to EOS did not reach statistical significance between placebo and SPN-812 dose group (100 mg, p = .101; 200 mg, p = .091; 300 mg, p = .055; 400 mg, p = .053); however, there was a trend for the scores to be lower in the 300 and 400 mg SPN-812 dose groups.
CGI-I analyses
Improvements (decreases) in CGI-I scores were observed during the first 5 weeks of study medication and remained consistent thereafter in all study groups and in the placebo group (ITT population, Figure 4). Differences in the LS means of CGI-I scores at the end of the double-blind phase showed a significant improvement for the SPN-812 300 mg dose group compared with placebo (p = .009; Figure 4, Table 3). Analysis of the LS means of CGI-I scores at EOS in the PP population demonstrated a statistically significant difference between all doses of SPN-812 and placebo (p = .034, p = .020, p = .012, p = .008, for doses 100, 200, 300 and 400 mg, respectively; Supplemental Table 2).
Figure 4.
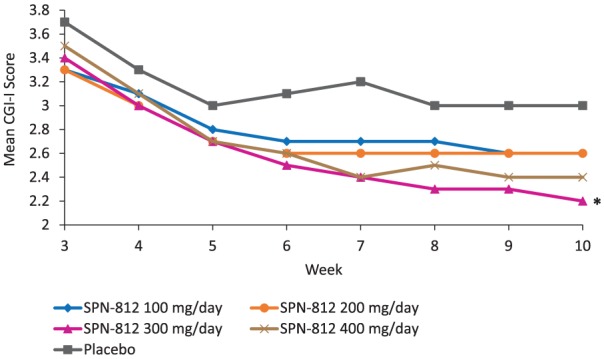
Mean CGI-I scores by treatment group (ITT population).
Note. CGI-I = Clinical Global Impressions Scale-Improvement; ITT = intention-to-treat; EOS = end of study.
Statistics based on analysis at EOS: *p < .05 for the 300 mg/day dose group versus placebo (ANOVA, unadjusted p value).
Table 3.
Results of Analysis of CGI Score: Absolute Values for CGI-I at End of Study and Change From Baseline to End of Study for CGI-S (ITT Population).
| CGI | Placebo (N = 24) | SPN-812 (mg/day) |
|||
|---|---|---|---|---|---|
| 100 (N = 45) | 200 (N = 46) | 300 (N = 47) | 400 (N = 44) | ||
| CGI-I | |||||
| LS meana | 3.0 | 2.6 | 2.6 | 2.2 | 2.4 |
| Placebo-adjusted treatment effectb | — | −0.4 | −0.4 | −0.8 | −0.6 |
| p valueb,c | — | .1305 | .1376 | .0090d | .0546 |
| CGI-S | |||||
| LS meana | −0.8 | −1.4 | −1.5 | −1.6 | −1.7 |
| Placebo-adjusted treatment effectb | — | −0.6 | −0.8 | −0.8 | −0.9 |
| p valueb,c | — | .0708 | .0309d | .0148d | .0136d |
Note. CGI = Clinical Global Impression; CGI-I = Clinical Global Impression-Improvement Scale; CGI-S = Clinical Global Impression-Severity Scale; ITT = intention-to-treat; LS = least squares; ANOVA = analysis of variance.
ANOVA including fixed effect for treatment.
Comparison between each SPN-812 dose group and placebo.
Raw (unadjusted for multiplicity) p values.
p < .05 versus placebo, estimated by ANOVA.
CGI-S analyses
The mean CGI-S score decreased in all groups, suggesting that by the end of the double-blind phase, ADHD symptoms had improved. The improvement in LS mean change from Baseline to EOS in CGI-S score was statistically significant compared with placebo for all SPN-812 dose groups except 100 mg (p < .05; Table 3, ITT population). When considering the PP population, a statistically significant improvement in LS mean change from Baseline to EOS in CGI-S score was observed for all SPN-812 dose groups compared with placebo (p = .030, p = .016, p = .019, p = .004, for doses 100, 200, 300, and 400 mg, respectively; Supplemental Table 2). Responder analyses indicated that syndrome remission rates (ADHD-RS-IV score ≤18 and CGI-S score ≤2) were 26.7% to 34.1% across dose groups compared with 12.5% in the placebo group (Supplemental Table 1).
Safety
The most frequently reported AEs (≥15.0% of participants) were somnolence, headache, and decreased appetite, and the incidence of somnolence and headache appeared to be dose related for the SPN-812 treatment groups (Table 4). The most common AE for the placebo group was decreased appetite. Discontinuation from the study due to TEAEs occurred in four (8.3%), three (6.3%), one (2.1%), and five (10.2%) participants in the SPN-812 100, 200, 300, and 400 mg dose groups, respectively, for a total of 13 participants (6.7%). These TEAEs occurred mostly in one participant each, except for headache (three participants) and irritability (two participants), and all events resolved with discontinuation of the study medication. There were no discontinuations due to TEAEs in the placebo group (0%).
Table 4.
AEs Occurring in ≥5% of Participants in Any Treatment Group (Safety Population).
| System organ class Preferred term, n (%) |
Placebo (N = 24) | SPN-812 (mg/day) |
|||
|---|---|---|---|---|---|
| 100 (N = 48) | 200 (N = 48) | 300 (N = 48) | 400 (N = 49) | ||
| Any AE | 11 (45.8) | 34 (70.8) | 30 (62.5) | 33 (68.8) | 35 (71.4) |
| Nervous system disorders | 1 (4.2) | 12 (25.0) | 16 (33.3) | 17 (35.4) | 22 (44.9) |
| Somnolence | 1 (4.2) | 7 (14.6) | 10 (20.8) | 11 (22.9) | 12 (24.5) |
| Headache | 1 (4.2) | 4 (8.3) | 6 (12.5) | 6 (12.5) | 9 (18.4) |
| Gastrointestinal disorders | 1 (4.2) | 8 (16.7) | 7 (14.6) | 10 (20.8) | 8 (16.3) |
| Vomiting | 0 | 2 (4.2) | 4 (8.3) | 3 (6.3) | 5 (10.2) |
| Nausea | 0 | 2 (4.2) | 1 (2.1) | 5 (10.4) | 2 (4.1) |
| Upper abdominal pain | 0 | 3 (6.3) | 3 (6.3) | 0 | 0 |
| Diarrhea | 0 | 0 | 0 | 3 (6.3) | 1 (2.0) |
| Psychiatric disorders | 0 | 9 (18.8) | 10 (20.8) | 9 (18.8) | 9 (18.4) |
| Irritability | 0 | 1 (2.1) | 4 (8.3) | 2 (4.2) | 1 (2.0) |
| Infections and infestations | 4 (16.7) | 5 (10.4) | 6 (12.5) | 5 (10.4) | 10 (20.4) |
| Gastroenteritis viral | 1 (4.2) | 0 | 1 (2.1) | 0 | 3 (6.1) |
| Metabolism and nutrition disorders | 2 (8.3) | 5 (10.4) | 7 (14.6) | 4 (8.3) | 9 (18.4) |
| Decreased appetite | 2 (8.3) | 5 (10.4) | 7 (14.6) | 4 (8.3) | 8 (16.3) |
| General disorders and administration site conditions | 2 (8.3) | 3 (6.3) | 3 (6.3) | 3 (6.3) | 5 (10.2) |
| Fatigue | 0 | 2 (4.2) | 2 (4.2) | 1 (2.1) | 5 (10.2) |
| Investigations | 2 (8.3) | 1 (2.1) | 2 (4.2) | 2 (4.2) | 6 (12.2) |
| Weight decreased | 0 | 0 | 1 (2.1) | 0 | 4 (8.2) |
Note. AE = adverse event.
The incidence of TEAEs was similar for all SPN-812 treatment groups. For the SPN-812 treatment groups and the placebo group, the incidence of TEAEs reported during the Titration period (52.8% and 37.5%, respectively) was higher than that reported during the Maintenance period (41.2% and 16.7%, respectively). The majority of TEAEs reported were mild or moderate in severity; three severe TEAEs (tearfulness, irritability, and decreased appetite) were reported in two (1.0%) SPN-812–treated participants. Treatment-related TEAEs were reported in 105 (54.4%) SPN-812–treated participants and in three (12.5%) placebo participants. No deaths or serious TEAEs (SAEs) were reported at any point during the Phase II study.
There were no clinically significant trends in clinical laboratory tests, vital signs, or ECG results over time in participants from any of the treatment groups. There was a low incidence of individual clinically relevant changes to ECG, wherein seven participants (3.6%) receiving SPN-812 and one participant receiving placebo experienced increases in heart rate or tachycardia that were reported as TEAEs. Specifically, seven of the participants had a high QRS duration (as did the one placebo participant, defined as a duration ≥90 ms) and one had a high PR interval (≥180 ms). All cases were mild in severity, with the exception of one moderate case. Of these participants, three were in the 400 mg group, two were in the 300 mg group, and there was one each in the 100 and 200 mg groups. One participant with increased heart rate, considered mild in severity, discontinued treatment, and was withdrawn from the study due to the AE, which resolved after 5 days. One participant experienced a mild increase in blood pressure, which resolved on subsequent visits. Four participants (1.8%) in the SPN-812 100 or 200 mg group (two participants each) had positive responses to the C-SSRS inquiry on suicidal ideation. Suicidal ideation was reported as mild and was not considered related to treatment in three of the participants. In one participant (SPN-812 100 mg dose group), the TEAE of suicidal ideation was considered possibly related to the treatment by the investigator and led to the participant’s discontinuation from the study. The TEAE resolved following withdrawal of treatment.
Discussion
The results of this randomized, placebo-controlled, Phase II study (N = 222) provide preliminary evidence that once-daily dosing of SPN-812 may produce ADHD symptom improvement in children aged 6 to 12 years, as measured by the ADHD-RS-IV total score. Specifically, in children who received SPN-812 200, 300, or 400 mg/day for 8 weeks, a statistically significant reduction in the ADHD-RS-IV total score was observed compared with those given placebo. The level of improvement demonstrated a dose response, as children who received the 400 mg/day dose had the greatest reduction in ADHD-RS-IV total score, followed by those in the 300 and 200 mg/day dose groups. The placebo-adjusted effect size ranged from 0.45 to 0.62 across the four SPN-812 dose groups. Similarly, treatment with SPN-812 200, 300, or 400 mg/day yielded statistically significant improvement in the ADHD-RS-IV Hyperactivity/Impulsivity Subscale and CGI-S scores. Although the ITT population did not achieve statistical significance for each dose of SPN-812 on the ADHD-RS-IV Inattention Subscale and the CGI-I scale, trends for improvement were observed. Taken together, these results suggest that SPN-812 is a nonstimulant medication that may have therapeutic value for children diagnosed with the symptoms of ADHD.
Improvements in ADHD-RS-IV total scores from Baseline to EOS seen in this Phase II study after 8 weeks of treatment are similar to those observed in two recent Phase III studies examining 10 weeks of treatment with extended-release guanfacine in children (Hervas et al., 2014; Huss, Sikirica, et al., 2016). The three highest doses of SPN-812 yielded significant improvement from Baseline to EOS in the ITT population. Indeed, responder rates for symptomatic remission in the 300 and 400 mg dose groups were significant compared with placebo. Findings on both the Hyperactivity/Impulsivity and Inattention subscales of the ADHD-RS-IV favorably parallel those for the total score, with the caveat that treatment differences in the ADHD-RS-IV Inattention Subscale score did not achieve statistical significance with SPN-812. The ADHD-RS-IV Hyperactivity/Impulsivity Subscale score was found to be significantly different for SPN-812 200, 300, and 400 mg/day. It is possible that the smaller effect size and the failure to achieve statistical significance for the Inattention Subscale score were due to the ages of the children, the duration of the treatment, or, alternatively, because the number of participants per group may not have provided sufficient power to see changes in inattention. The robust sample size in the ongoing Phase III studies in children and adolescents with ADHD should provide further insight into the effect of SPN-812.
Secondary analyses using the CGI ratings buttress the findings for ADHD symptom improvement seen in the primary analyses. Significance was observed for the SPN-812 300 mg dose, and the mean absolute CGI-I scores decreased for all SPN-812 dose groups at the end of the 8-week study. The mean end-of-treatment CGI-I scores were 2.6 (100 and 200 mg), 2.2 (300 mg), and 2.4 (400 mg), indicating that a large number of children were rated as at least “much improved.” Furthermore, illness severity, as evaluated by CGI-S, decreased from Baseline to the end of the 8-week study by approximately two points (range = 1.5-1.7) in the 200, 300, and 400 mg SPN-812 treatment groups compared with the placebo group, who had a reduction of approximately one point (M = 0.8). These results suggest clinically meaningful global changes (two-point change; Kelly, 2010) in ADHD symptoms with SPN-812.
Treatment SPN-812 was generally well tolerated at all doses throughout the study. Somnolence, headache, and decreased appetite were the most frequently reported TEAEs and were similar to events described by Hervas et al. (2014) with extended-release guanfacine (somnolence and headache) and atomoxetine (decreased appetite), highlighting that there were no unexpected TEAEs with SPN-812 in children aged 6 to 12 years with ADHD. Further evidence to support the tolerability of SPN-812 includes the absence of SAEs and deaths throughout the study and the incidence of TEAEs leading to discontinuation of SPN-812 (6.7%) not pertaining to any particular AE. In addition, treatment with SPN-812 was not associated with any clinically significant trends in laboratory, vital sign, or ECG abnormalities. As noted, individual incidences of changes to ECG were low and related to tachycardia, where only 3.6% of SPN-812–treated participants experienced TEAEs of increased QRS duration (seven participants, as well as one placebo participant) or PR interval (one SPN-812 participant). One potential limitation regarding the identification of TEAEs occurring at greater rates than in the placebo group, however, is the relatively small size of the placebo group compared with the collective size of the treatment group. Nonetheless, this study’s findings are consistent with somnolence, headache, and decreased appetite being the most common AEs associated with SPN-812.
As with many studies of children and adolescents with ADHD, a potential limitation of the current study is that the exclusion criteria screen out participants with certain neuropsychiatric conditions that can be comorbid with ADHD (such as bipolar disorder, major depressive disorder, or post-traumatic stress disorder). As noted, 21.7% of patients in this study had a concomitant psychiatric disorder, most commonly oppositional defiant disorder or an anxiety disorder. Therefore, while this sample population did include patients with concomitant disorders, requiring exclusion of participants with other more severe neuropsychiatric disorders may affect the comparability of our representative population with the general population with ADHD. Although these exclusion criteria are necessary controls, it is important to take into consideration the differences between the ADHD population in a clinical trial setting and the general population.
This 8-week Phase II study indicates that for children diagnosed with ADHD, SPN-812 has the potential to be a much-needed addition to the current armamentarium of ADHD therapies. The findings of this study show that SPN-812 is tolerable and improved ADHD symptoms as indicated by the ADHD-RS-IV. Ongoing Phase III studies will provide further insight into SPN-812 treatment in children and adolescents with ADHD.
Supplemental Material
Supplemental material, Supplemental_Table_1 for A Phase II Double-Blind, Placebo-Controlled, Efficacy and Safety Study of SPN-812 (Extended-Release Viloxazine) in Children With ADHD by Janet K. Johnson, Tesfaye Liranso, Keith Saylor, Gabriela Tulloch, Toyin Adewole, Stefan Schwabe, Azmi Nasser, Robert L. Findling and Jeffrey H. Newcorn in Journal of Attention Disorders
Author Biographies
Janet K. Johnson, PhD, was formerly a Senior Director, Clinical Research, at Supernus Pharmaceuticals, Inc.
Tesfaye Liranso, PhD, is a Senior Director, Biostatistics, at Supernus Pharmaceuticals, Inc.
Keith Saylor, PhD, ScM, is President, Chief Executive Officer, NeuroScience, Inc., a clinical psychologist, and principal investigator for investigational medication research studies for adults, adolescents, and children.
Gabriela Tulloch, RN, was formerly a Senior Clinical Program Manager at Supernus Pharmaceuticals, Inc.
Toyin Adewole, MD, MPH, is an Assistant Director, Drug Safety, at Supernus Pharmaceuticals, Inc.
Stefan Schwabe, MD, PhD, is Chief Medical Officer and Vice President for Research and Development at Supernus Pharmaceuticals, Inc.
Azmi Nasser, PhD, is a Senior Director, Clinical Research, at Supernus Pharmaceuticals, Inc.
Robert L. Findling, MD, MBA, is the Leonard and Helen R. Stulman Professor in Child & Adolescent Psychiatry, professor and vice chair in the Department of Psychiatry and Behavioral Sciences, professor of pediatrics at Johns Hopkins University; and Vice President of Psychiatric Services and Research at the Kennedy Krieger Institute.
Jeffrey H. Newcorn, MD, is professor of Psychiatry and Pediatrics and Director, Division of ADHD and Learning Disorders, Icahn School of Medicine at Mount Sinai; and Director of Pediatric Psychopharmacology, Mount Sinai Health System.
Footnotes
Declaration of Conflicting Interests: The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: J.K.J. is a former employee of Supernus Pharmaceuticals, Inc.; T.L. is an employee of Supernus Pharmaceuticals, Inc.; K.S. received clinical trial research support from Otsuka and Supernus Pharmaceuticals, Inc. and serves as a consultant and advisory board member for Supernus Pharmaceuticals, Inc.; G.T. is a former employee of Supernus Pharmaceuticals, Inc.; T.A. is an employee of Supernus Pharmaceuticals, Inc.; S.S. is an employee of Supernus Pharmaceuticals, Inc.; A.N. is an employee of Supernus Pharmaceuticals, Inc.; R.L.F. discloses past or present relationships with the following organizations in the past 24 months: Research support, and/or consultant, and/or speaker’s bureau for Aevi, Akili, Alcobra, Amerex, American Academy of Child & Adolescent Psychiatry, Bracket, Daiichi Sankyo, ePharmaSolutions, Forest, Genentech, Ironshore, KemPharm, Lundbeck, U.S. National Institutes of Health, Neurim, Nuvelution, Otsuka, PCORI, Pfizer, Physicians Postgraduate Press, Roche, Shire, Sunovion, Supernus Pharmaceuticals, SyneuRx, Teva, Tris, TouchPoint, Validus; and received royalties from American Psychiatric Press and Sage. J.H.N. discloses the following relationships: Akili Interactive (consultant), Alcobra (consultant), Arbor (consultant), Cingulate Therapeutics (consultant), Enzymotec (consultant, research support), KemPharm (consultant), Lundbeck (consultant, research support), Medice (consultant), NLS Pharma (consultant), Pfizer (consultant), Rhodes (consultant), Shire (consultant, research support), Sunovion (consultant), and Supernus Pharmaceuticals Inc. (consultant). He received research support from Enzymotec, Lundbeck, and Shire.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Editorial support was provided by IMPRINT Science, New York, NY, USA, with funding from Supernus Pharmaceuticals, Inc.
Supplemental Material: Supplemental material for this article is available online.
References
- Adesman A. R. (2001). The diagnosis and management of attention-deficit/hyperactivity disorder in pediatric patients. Primary Care Companion to the Journal of Clinical Psychiatry, 3(2), 66-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychiatric Association. (1994). Diagnostic and Statistical Manual of Mental Disorders (4th ed.). Washington, DC: Author. [Google Scholar]
- American Psychiatric Association. (2013). Diagnostic and Statistical Manual of Mental Disorders (5th ed.). Washington, DC: American Psychiatric Publishing. [Google Scholar]
- Briars L., Todd T. (2016). A review of pharmacological management of attention-deficit/hyperactivity disorder. Journal of Pediatric Pharmacology and Therapeutics, 21, 192-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busner J., Targum S. D. (2007). The clinical global impressions scale: Applying a research tool in clinical practice. Psychiatry, 4(7), 28-37. [PMC free article] [PubMed] [Google Scholar]
- Catala-Lopez F., Hutton B., Nunez-Beltran A., Page M. J., Ridao M., Macías Saint-Gerons D., . . . Moher D. (2017). The pharmacological and non-pharmacological treatment of attention deficit hyperactivity disorder in children and adolescents: A systematic review with network meta-analyses of randomised trials. PLoS ONE, 12(7), e0180355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childress A., Tran C. (2016). Current investigational drugs for the treatment of attention-deficit/hyperactivity disorder. Expert Opinion on Investigational Drugs, 25, 463-474. [DOI] [PubMed] [Google Scholar]
- Clemow D. B., Bushe C. J. (2015). Atomoxetine in patients with ADHD: A clinical and pharmacological review of the onset, trajectory, duration of response and implications for patients. Journal of Psychopharmacology, 29, 1221-1230. [DOI] [PubMed] [Google Scholar]
- Connor D. F., Arnsten A. F., Pearson G. S., Greco G. F. (2014). Guanfacine extended release for the treatment of attention-deficit/hyperactivity disorder in children and adolescents. Expert Opinion on Pharmacotherapy, 15, 1601-1610. [DOI] [PubMed] [Google Scholar]
- Danckaerts M., Sonuga-Barke E. J., Banaschewski T., Buitelaar J., Döpfner M., Hollis C., . . . Coghill D. (2010). The quality of life of children with attention deficit/hyperactivity disorder: A systematic review. European Child & Adolescent Psychiatry, 19, 83-105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielson M. L., Bitsko R. H., Ghandour R. M., Holbrook J. R., Kogan M. D., Blumberg S. J. (2018). Prevalence of parent-reported ADHD diagnosis and associated treatment among U.S. children and adolescents, 2016. Journal of Clinical Child & Adolescent Psychology, 47, 199-212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy W. (1976). ECDEU assessment manual for psychopharmacology. Rockville, MD: National Institute of Mental Health, U.S. Department of Health, Education, and Welfare. [Google Scholar]
- Harpin V. A. (2005). The effect of ADHD on the life of an individual, their family, and community from preschool to adult life. Archives of Disease in Childhood, 90(Suppl. 1), i2-i7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hervas A., de Santos T., Quintero J., Ruíz-Lázaro P. M., Alda J. A., Fernández-Jaén A., Ramos-Quiroga J. A. (2016). Delphi consensus on attention deficit hyperactivity disorder (ADHD): Evaluation by a panel of experts. Actas Españolas de Psiquiatría, 44(6), 231-243. [PubMed] [Google Scholar]
- Hervas A., Huss M., Johnson M., McNicholas F., van Stralen J., Sreckovic S., . . . Robertson B. (2014). Efficacy and safety of extended-release guanfacine hydrochloride in children and adolescents with attention-deficit/hyperactivity disorder: A randomized, controlled, phase III trial. European Neuropsychopharmacology, 24, 1861-1872. [DOI] [PubMed] [Google Scholar]
- Huss M., Chen W., Ludolph A. G. (2016). Guanfacine extended release: A new pharmacological treatment option in Europe. Clinical Drug Investigation, 36(1), 1-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huss M., Sikirica V., Hervas A., Newcorn J. H., Harpin V., Robertson B. (2016). Guanfacine extended release for children and adolescents with attention-deficit/hyperactivity disorder: Efficacy following prior methylphenidate treatment. Neuropsychiatric Disease and Treatment, 112, 1085-1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson J. K., Saylor K., Brittain S. T., Tulloch G., Liranso T. (2015, October 26-31). Double-blind, randomized, placebo-controlled study of immediate-release viloxazine (SPN-812 IR) as a novel non-stimulant therapy in adults with attention-deficit/hyperactivity disorder (ADHD). Poster presented at American Academy of Child and Adolescent Psychiatry, San Antonio, TX. [Google Scholar]
- Johnson J. K., Saylor K., Brittain S. T., Tulloch G., Liranso T. (2017, October 23-28). A double-blind, placebo-controlled, dose-ranging study of extended-release viloxazine (SPN-812 ER) in children with ADHD. Abstract presented at American Academy of Child and Adolescent Psychiatry, Washington, DC. [Google Scholar]
- Kelly R. (2010). Calculating clinically significant change: Applications of the Clinical Global Impressions (CGI) Scale to evaluate client outcomes in private practice. Clinical Psychologist, 14, 107-111. [Google Scholar]
- Peasgood T., Bhardwaj A., Biggs K., Brazier J. E., Coghill D., Cooper C. L., . . . Sonuga-Barke E. J. (2016). The impact of ADHD on the health and well-being of ADHD children and their siblings. European Child & Adolescent Psychiatry, 25, 1217-1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shire US Inc. (2017). INTUNIV® (guanfacine) extended-release tablets, for oral use, prescribing information. Lexington, MA: Author. [Google Scholar]
- Sheehan D. V., Sheehan K. H., Shytle R. D., Janavs J., Bannon Y., Rogers J. E., . . . Wilkinson B. (2010). Reliability and validity of the Mini International Neuropsychiatric Interview for Children and Adolescents (MINI-KID). Journal of Clinical Psychiatry, 71, 313-326. [DOI] [PubMed] [Google Scholar]
- Subcommittee on Attention-Deficit/Hyperactivity Disorder, Steering Committee on Quality Improvement Management, Wolraich M., Brown L., Brown R. T., DuPaul G., . . . Visser S. (2011). ADHD: Clinical practice guideline for the diagnosis, evaluation, and treatment of attention-deficit/hyperactivity disorder in children and adolescents. Pediatrics, 128, 1007-1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. Department of Justice. (n.d.). List of controlled substances. Retrieved from https://www.deadiversion.usdoj.gov/schedules
- Zhang S., Faries D. E., Vowles M., Michelson D. (2005). ADHD Rating Scale IV: Psychometric properties from a multinational study as a clinician-administered instrument. International Journal of Methods in Psychiatric Research, 14, 186-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental material, Supplemental_Table_1 for A Phase II Double-Blind, Placebo-Controlled, Efficacy and Safety Study of SPN-812 (Extended-Release Viloxazine) in Children With ADHD by Janet K. Johnson, Tesfaye Liranso, Keith Saylor, Gabriela Tulloch, Toyin Adewole, Stefan Schwabe, Azmi Nasser, Robert L. Findling and Jeffrey H. Newcorn in Journal of Attention Disorders