

SUMMARY
Non-recirculating tissue-resident memory T cells (TRMs) are the predominant T cell subset in diverse tissue sites, where they mediate protective immune responses in situ. Here, we reveal a role for TRM in maintaining immune homeostasis in the human pancreas through interactions with resident macrophages and the PD-1/PD-L1 inhibitory pathway. Using tissues obtained from organ donors, we identify that pancreas T cells comprise CD8+PD-1hi TRMs, which are phenotypically, functionally, and transcriptionally distinct compared to TRMs in neighboring jejunum and lymph node sites. Pancreas TRMs cluster with resident macrophages throughout the exocrine areas; TRM effector functions are enhanced bay macrophage-derived co-stimulation and attenuated by the PD-1/PD-L1 pathways. Conversely, in samples from chronic pancreatitis, TRMs exhibit reduced PD-1 expression and reduced interactions with macrophages. These findings suggest important roles for PD-1 and TRM-macrophage interactions in controlling tissue homeostasis and immune dysfunctions underlying inflammatory disease, with important implications for PD-1-based immunotherapies.
Graphical Abstract
In Brief
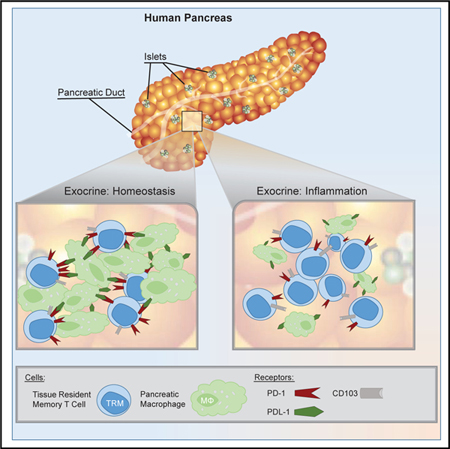
Non-recirculating tissue-resident memory T cells (TRMs) mediate immune responses in non-lymphoid tissues. Using a human organ donor tissue resource, Weisberg et al. reveal that PD-1hi pancreas TRMs are regulated by PD-L1+ macrophages during homeostasis. Comparison with chronic pancreatitis patient samples shows how pancreas TRM regulation is altered during inflammation.
INTRODUCTION
Tissue-resident memory T cells (TRMs) are generated in response to pathogens, allergens, vaccines, and autoantigens and persist as non-circulating populations in diverse sites, including barrier, mucosal, exocrine, lymphoid, and peripheral sites (for reviews, see Mackay and Kallies, 2017; Masopust and Soerens, 2019; Szabo et al., 2019). In mouse models, TRMs can mediate rapid protective responses to challenge with multiple types of pathogens and can also participate in immunopathology (Hondowicz et al., 2016; Schenkel et al., 2014; Teijaro et al., 2011). TRMs in mice and humans are distinguished from circulating memory T cells based on core phenotypic and transcriptional signatures, which include upregulated expression of tissue retention molecules (CD69), integrins (CD103 and CD49a), and downregulation of egress signals (Hombrink et al., 2016; Kumar et al., 2017; Mackay et al., 2015, 2016; Thome et al., 2014; Watanabe et al., 2015), establishing TRMs as a distinct subset.
In humans, populations of TRMs constitute the majority of T cells throughout the body and are present in constant frequencies over many decades of human life (Kumar et al., 2018a; Senda et al., 2019; Thome et al., 2014). Functionally, human TRMs can rapidly produce interleukin-2 (IL-2) and prion-flammatory cytokines but also express molecules that attenuate activation, including the inhibitory molecules PD-1 and CD101, and the regulatory cytokine IL-10 (Kumar et al., 2018b; Kumar et al., 2017; Miron et al., 2018). In addition, TRM can exhibit site-specific functional and transcriptional adaptations in certain tissues (Cheuk et al., 2017; Miron et al., 2018; Pallett et al., 2017). This evidence for site-specific long-term TRM maintenance and the dichotomous functional and regulatory properties suggest an important role for TRMs in mediating tissue homeostasis.
The pancreas is a critical organ for digestion and metabolism via its dual exocrine and endocrine components. Disruption of pancreas immune homeostasis has severe consequences for human health. In acute and chronic pancreatitis (CP), infiltration by adaptive and innate immune cells leads to inflammation, tissue destruction, and high rates of morbidity and mortality (Russo et al., 2004), while immune-mediated destruction of insulin-producing cells in the endocrine pancreas leads to type 1 diabetes (T1D). Chronic pancreatic inflammation is also associated with increased risk of pancreatic ductal adenocarcinoma (PDAC), an aggressive cancer refractory to immunotherapy (Kirkegård et al., 2017, 2018). Studies in humans and mouse models show that T cells play a central role in pancreas immune surveillance and immunopathology (Balachandran et al., 2017; Coppieters et al., 2012; Demols et al., 2000; Kuric et al., 2017; Rodriguez-Calvo et al., 2014). In T1D, CD8+ T cells are found in the exocrine pancreas and within islets and insulitic lesions (Damond et al., 2019; Kuric et al., 2017; Radenkovic et al., 2017; Rodriguez-Calvo et al., 2014; Wang et al., 2019). However, the functional capacity and role of T cells in the non-diseased pancreas is not clear. Studying immune cells within the human pancreas is further limited by tissue availability and the technical challenge of isolating viable cell populations from this enzyme-rich site.
We have established a collaboration with the local organ-procurement agency, LiveOnNY, to obtain mucosal, lymphoid, and exocrine tissues from human organ donors, enabling assessment of T cell compartmentalization and tissue-specific effects on T cell maintenance and function (Granot et al., 2017; Miron et al., 2018; Senda et al., 2019; Thome et al., 2014, 2016). Here, we use this resource to define how resident immune cells are maintained in the non-diseased pancreas compared to those in neighboring jejunum and their draining lymph nodes to establish a baseline of immune homeostasis from which to define disease-associated immune dysregulation. We identify CD8+ PD-1hi TRMs as the predominant T cell subset in the human pancreas, largely confined to exocrine areas and exhibiting tissue-specific phenotypes and transcriptional programs controlling T cell activation and metabolism. Pancreas TRMs cluster together with tissue macrophages, which represent the other major immune cell type in the exocrine pancreas. We further show that TRM effector functions are enhanced by macrophage-derived co-stimulation and attenuated by the PD-1/PD-L1 pathways. Importantly, these TRM regulatory mechanisms are altered during inflammation associated with CP; pancreas TRMs exhibit reduced PD-1 expression concomitant with a marked decrease in pancreas macrophages. Together, these findings demonstrate how TRMs, macrophages, and the PD-1 pathway contribute to in situ immune regulation and suggest a key role for TRMs in tissue homeostasis.
RESULTS
Localization and Subset Composition of T Cells in the Human Pancreas
We obtained non-diseased tissues from a human organ donor tissue resource (see STAR Methods), which we have previously described and extensively validated for immune cell studies over diverse anatomic sites and ages (Carpenter et al., 2018; Sathaliyawala et al., 2013; Thome et al., 2014, 2016). Whole pancreas along with samples of neighboring gastrointestinal (GI) and lymphoid sites, including jejunum, pancreas-draining lymph node (PLN), and mesenteric lymph node (MLN), were obtained from 32 donors of diverse race and ethnicity (Table S1). Donors ranged in age from 18 to 71 years (median age, 52 years), and none had a documented history of T1D or pancreatic disease. Donor BMI ranged from 16 to 47; 40% of donors (13/32) were obese (BMI >30 kg/m2), comparable to the US population (Hales et al., 2017).
Pancreatic tissue consists predominantly of exocrine components (85%) composed of acinar cells secreting digestive enzymes, while endocrine components (15%) consist of discrete islets of neuroendocrine cells producing insulin and glucagon. We used quantitative multiplex immunofluorescence (qmIF) to localize CD3+ T cells among CK19+ ductal epithelium (exocrine portion) and islets (chromogranin+, endocrine portion) (Figure 1A, left). High-density cellular areas between the ductal and endocrine components were classified as acinar. Computational analysis of images from multiple pancreas sections (see STAR Methods) shows that T cells are largely restricted to the periductal and acinar areas of the exocrine pancreas and are not within islets (Figure 1A, right). Therefore, the majority of T cells in the non-diseased pancreas are within the exocrine region.
Figure 1. Localization and Expression of Key Tissue-Residency Markers on T Cells in Human Pancreas.

(A) Representative qmIF composite image of a pancreas section stained with antibodies specific for CD3 (purple), the ductal marker CK19 (green), DAPI nuclear counterstain (gray), and the neuroendocrine marker chromogranin (white) are shown (left) adjacent to a representative single color CD3 image (middle). Acinar, ductal, and endocrine areas were defined based on CK19 and chromogranin staining. White bar, 100 µm for scale. Right: densities of CD3+ T cells were quantified in the three regions of pancreas using inForm software. Plots show mean ± SEM from 13 donors.
(B) T cells were analyzed in cell suspensions of pancreas (Panc), jejunum (Jej), pancreas-draining lymph node (PLN), and mesenteric lymph node (MLN). Shown are representative (left) and the compiled (right) CD4 and CD8 T cell frequencies (gated on DAPIlo CD45+CD3+ cells) from the four tissue sites. Bars indicate comparisons for CD8+ T cells.
(C) Expression of CD69 in conjunction with TRM signature markers CD103, CD49a, and PD-1 on CD8+ TEM cells (CD45RA−CCR7−) subsets isolated from indicated sites shown as representative flow cytometry plots (left) with the compiled frequencies ±SEM of the indicated subsets from three to eight donors (right). Bars indicate comparisons of the CD69+CD103+ (top), CD69+CD49a+ (middle), and CD69+PD-1hi (bottom) subsets.
(D) Expression of intracellular granzyme B (GZMB) in CD8+CD69+TEM cells isolated from pancreas, jejunum, and PLN shown as representative flow cytometry plots (left), and compiled frequencies ± SEM of GZMB+ cells from three to six donors for each tissue (right). Bars indicate comparisons of the GZMB+ frequencies within the indicated subsets.
**p < 0.001 as calculated by two-way ANOVA with Dunnett’s multiple comparisons test.
See also Figure S1.
Isolation of immune cells from pancreatic tissue is challenging due to the high enzyme content. We optimized a protocol for isolation of viable cells from the pancreas using a modified Ricordi chamber method (see STAR Methods) (Bugliani et al., 2004). Flow cytometry analysis showed that pancreas T cells are predominantly CD8+ (85% ± 1.5% CD3+ cells) compared to jejunum, which contains 54% ± 3.3% CD4+ T cells and associated lymph nodes (PLNs and MLNs) with prevalent CD4+ T cells (Figure 1B). Pancreas T cells, similar to jejunum, are largely effector memory (TEM) phenotype (CD45RA+CCR7−,92% ± 1.7%) whereas PLN and MLN T cells contain significant naive (CD45RA+CCR7+) and central memory (TCM; CD45RA− CCR7+) populations (Figure S1A). CD4+ regulatory T cells (Tregs) were not detected in the pancreas or jejunum (<0.5%) but were present in PLNs and MLNs (Figure S1B). These results show site-specific differences in T cell subset composition; notably, the pancreas contains predominant CD8+ TEM cells, distinct among neighboring GI and lymphoid tissues.
We examined whether pancreas T cells express canonical TRM markers CD69 and CD103, along with additional core TRM signature markers defined previously (Kumar et al., 2017), including the collagen-binding integrin CD49a and inhibitory molecules PD-1 (Freeman et al., 2000) and CD101 (Schey et al., 2016). The vast majority (>85%) of CD8+ TEM cells from pancreas and jejunum co-expressed CD69 and CD103 and therefore exhibit a TRM phenotype. In PLNs, >70% of CD8+ TEM cells were CD69+ and largely CD103− (Figure 1C); these likely represent TRMs based on previous findings that human CD69+CD8+ T cells in mucosal tissues and peripheral lymph nodes (LNs) exhibit transcriptional features of TRMs (Buggert et al., 2018; Kumar et al., 2017; Miron et al., 2018) and results in mice showing that LN CD69+TEM cells are retained as TRMs (Beura et al., 2018). Pancreas TRMs also express CD101 and exhibited markedly higher expression of PD-1 and CD49a compared to TRMs in jejunum, PLN, and MLN (Figures 1C and S1C). Pancreas CD8+TRMs further exhibited constitutive expression of the cytotoxic effector molecule granzyme B (GZMB), while CD8+ TRMs from jejunum and PLN were mostly GZMB negative (Figure 1D). These results show that T cells within the non-diseased human pancreas exhibit high-level expression of multiple TRM markers and are distinct from TRMs in neighboring GI and lymphoid sites in elevated PD-1 and GZMB expression.
Pancreas TRMs Exhibit a Distinct Transcriptional Signature of Activation and Mitochondrial-Associated Genes
We further investigated the distinct state of pancreas TRMs by whole-transcriptome profiling of sorted CD8+ TRMs (defined as CD8+CD69+TEM cells) from donor-matched pancreata, jejunum, and PLNs compared to circulating blood CD8+ TEM cells from healthy living donors. Principal-component analysis (PCA) showed clustering of the pancreas, jejunum, and PLN CD8+ TRM expression signatures distinct from those of blood CD8+ TEM cells on the first principal component (PC1) and tissue-specific clustering of pancreas TRMs distinct from TRMs in jejunum and PLN based on PC2 (Figure 2A). Gene set enrichment analysis (GSEA) of the pancreas, jejunum, and PLN CD8+ TRMs compared to blood CD8+ TEM cells showed positive enrichment for genes shown to be upregulated in human TRMs (normalized enrichment score = 2, 2.3, and 2.2 with p = 0 for pancreas, PLN, and jejunum, respectively) and negative enrichment for genes downregulated in human TRMs (Kumar et al., 2017) (normalized enrichment score = −3, −3.5, and −3.7 with p = 0 for pancreas, PLN, and jejunum, respectively) (Figures 2B and S1A). We identified differentially expressed genes (DEGs) in tissues versus blood based on adjusted p value %0.05 and absolute value of log2 fold change (FC) R0.75. There was a strong correlation between the genes differentially expressed in pancreas TRMs versus blood TEM cells and in jejunum TRMs versus blood TEM cells (Figure 2C, top). A similar correlation was observed between genes differentially expressed in pancreas TRMs versus blood TEM cells and in PLN TRMs versus blood TEM cells (Figure S2B).
Figure 2. TRMs in the Pancreas Express a Tissue-Specific Transcriptional Profile Associated with Enhanced T Cell Function and Mitochondrial Mass.

CD8+ TRMs (CD8+CD69+ TEM) were sorted from pancreas, PLN, and jejunum along with blood (BL) CD8+CD69− TEM cells for whole-transcriptome profiling by RNA-seq.
(A) Principal-component analysis (PCA) of gene expression profiles of tissue CD8+ TRMs compared to blood CD8+CD69− TEM cells.
(B) Gene set enrichment analysis (GSEA) comparing expression of genes in pancreas TRMs versus blood CD8+ TEM cells to a core human TRM gene signature (Kumar et al., 2017).
(C) Biplots of all genes showing log FC of pancreas/blood versus log FC of jejunum/blood (top) and log FC of pancreas/PLN versus log FC of pancreas/jejunum (bottom). Significantly differentially expressed genes (DEGs) for each comparison were identified using DeSeq and functionally classified and are indicated as colored dots based on gene pathway.
(D) Heatmaps showing representative DEG from annotated gene sets T cell activation, proliferation, migration (left) and mitochondrion (right) which significantly overlap with DEGs in pancreas versus PLN and pancreas versus jejunum.
(E) Determination of mitochondrial mass using MitoTracker green staining in verapamil-treated CD8+ TRMs from matched donor sites pancreas, jejunum, and PLN. Representative histogram of MitoTracker green signal in matched sites from one donor is shown (top) with the compiled frequencies ±SEM from three donors for each tissue (bottom).
(F) Sorted CD8+CD69+ TEM subsets isolated from pancreas, jejunum, PLN, and sorted blood CD8+CD69− TEM cells and naive CD8+ T cells were stimulated with PMA and ionomycin, and intracellular staining was used to assess IFN-γ, IL-2, and TNF-α production. Profiles of IFN-γ and IL-2 are shown in representative flow cytometry plots for the indicated tissues (left). Graphs of compiled frequencies of IFN-γ -producing cells and the polyfunctionality index (see STAR Methods) ±SEM from three to six donors for each tissue (right).
**p < 0.01 as calculated by one-way ANOVA with Dunnett’s multiple comparisons test.
See also Figure S2.
Given the tissue-specific clustering observed on PC2 (Figure 2A), we further defined the gene expression differences between TRM from the different tissue sites. We identified 1,731 genes differentially expressed in pancreas TRMs with comparison to both PLN and jejunum TRMs (674 upregulated in both comparisons, 1,040 downregulated in both comparisons (adjusted p value % 0.05 and absolute value of log2 fold change R 0.75) (Table S2). A strong correlation was observed in the differentially upregulated and downregulated genes between pancreas versus jejunum TRMs and pancreas versus PLN TRMs, which together define a pancreas-associated gene signature (Figure 2C, bottom; Table S2). In contrast, the genes differentially expressed in pancreas TRMs versus blood TEM cells were not strongly correlated to those genes differentially expressed in pancreas compared to jejunum TRMs (Figure S2C), suggesting that the tissue residency gene signature and the pancreas-associated gene signature are unrelated. Using pathway and Gene Ontology (GO) analysis, we found significant overlap between the pancreas-associated gene signature and multiple annotated gene sets involved in T cell activation, T cell proliferation, T cell migration (Table S3), and mitochondrial function (GO Term: 0005739) (Figures 2D and S2D). Genes upregulated in pancreas TRMs that are involved in T cell function include those encoding cytotoxic effector molecules perforin (PRF1) and granulysin (GNLY) and genes involved in memory T cell maintenance, including IL-15 receptor components IL15RA and the shared IL2RB beta chain. Downregulated transcripts include those involved in T cell activation (e.g., FOS, JUN, NR4A1, EGR1, MYC, and MYB) and those associated with T cell migration (KLF2, CCR7, and CCR9) (Figure 2D, left; Table S3).
Notably, there was coordinate upregulation in pancreas TRMs of genes involved in a diverse array of mitochondrial functions, including mitochondrial ribosomes (e.g., MRPL17, MRPL28, MRPL34, MRPL2), enzymes of the respiratory chain (e.g., NDUFS8), synthesis of mitochondrial cardiolipin (PTPMT1), and enzymes mediating fatty acid beta-oxidation (ACADS) (Figure 2D, right; Table S2). In addition, genes encoding several transcription factors for mitochondrial gene expression and biogenesis (PPARA, PPARD, ESRRA, and RXRA) are significantly upregulated in pancreas TRMs compared to TRMs in jejunum and PLN (Figure S2E). This increase in mitochondria-associated genes in pancreas TRMs correlated with increased mitochondrial mass in pancreas TRMs compared to TRMs from jejunum and PLN based on staining with the mitochondrial dye MitoTracker green in the presence of verapamil to inhibit spurious dye efflux (de Almeida et al., 2017) (Figure 2E).
Because PD-1 expression is a hallmark of T cell exhaustion or hyporesponsiveness during chronic viral infections and cancer (Wherry et al., 2003; Wu et al., 2014), we investigated whether the gene expression profile of pancreas TRMs was enriched for exhaustion-associated transcripts previously defined from mouse models and human tumors (Schietinger et al., 2016). However, there was no significant enrichment of genes associated with exhausted T cells in pancreas TRMs, with the exception of the transcript encoding PD-1 (PDCD1) (Figure S2F). Pancreas TRMs are also highly functional. Upon activation with PMA/ionomycin, a high frequency of pancreas TRMs produced interferon-γ (IFN-γ), which was higher than the frequency of IFN-γ produced by CD8+ T cells in tissues and blood (Figure 2F). Pancreas CD8+ TRMs also produced high levels of IL-2 similar to CD8+ TRMs from jejunum and PLN and higher than blood CD8+ T cell subsets (Figure S2G). Moreover, pancreas CD8+ TRMs showed a high polyfunctionality index (Larsen et al., 2012) comparable to that of CD8+ TRMs from jejunum and PLN and greater than that of blood subsets (Figure 2F). These results show that pancreas TRMs exhibit a distinct transcriptional signature with enhanced mitochondrial mass and polyfunctional capacity relative to TRMs in neighboring tissues. Moreover, despite elevated PD-1 expression, pancreas TRMs exhibit no overt transcriptional or functional similarity to exhausted T cells.
Pancreas TRMs Exhibit Tissue-Specific Clonal Expansion and Markers of Replication History
To identify the clonal relationships between TRMs in pancreas and neighboring sites, we extracted T cell receptor (TCR) sequences from the RNA sequencing (RNA-seq) datasets (Bolotin et al., 2015) (see STAR Methods). While the majority (>50%) of TCR clones identified in pancreas and jejunum were also found in PLNs (Figure 3A, 3B, top), the TCR repertoires of the pancreas and jejunum TRMs had reduced clonal diversity compared to the TCR repertoires of PLNs (Figure 3B, bottom, and Figure S3A). Tracking individual clones among the three tissue sites revealed highly expanded clonotypes within TRMs in the pancreas that were either not detected or detected at low frequency in the other tissue sites (Figures 3A, 3C, and S3B). Jejunum TRMs also have expanded clonotypes, but these are distinct from those in the pancreas (Figure 3A, 3C, and S3B). Accordingly, the expanded clonotypes specific to the pancreas occupied >50% of the total TCR repertoire sampled (Figure 3C), suggesting pancreas-specific clonal expansion of TRMs.
Figure 3. Pancreas TRMs Exhibit Tissue-Specific Clonal Expansion with Phenotypic Features of Previous Replication.

TCR sequences of CD8+ TRMs from pancreas, jejunum, and PLN were extracted from RNA-seq data using MIXR (see STAR Methods).
(A) Representative clone tracking analysis showing the distribution and degree of expansion of PLN clonotypes across tissues. Each line represents a tracked clone present in PLN (far left, arrow) and at least one of the other tissues. Line thickness indicates degree of clonal expansion at the indicated site and line color indicates the tissue site in which that clone exhibits the greatest degree of clonal expansion.
(B) The percentage of the total TCR sequences (±SEM from three to five donors for each tissue) detected in pancreas and jejunum TRM that are either shared or not shared with PLN clonotypes is shown (top). The diversity of the TCR repertoires in TRMs isolated from indicated tissues is depicted as the Chao1 index (bottom).
(C) The percent contribution of expanded clonotypes from TRMs isolated from pancreas (top) and jejunum (bottom) to the total TCR repertoire extracted from pancreas, jejunum, and PLN (±SEM from three to five donors for each tissue). **p < 0.01, *p < 0.05 as calculated by one-way ANOVA with Dunnett’s multiple comparisons test.
(D) Coordinate expression of CD127 and CD28 by CD8+CD69+ TEM cells isolated from pancreas, jejunum, PLN, and MLN shown as representative flow cytometry plots (left), with the compiled frequencies (±SEM) of indicated subsets from 9 to 20 donors for each tissue (right). **p < 0.01 as calculated by two-way ANOVA with Sidak’s multiple comparisons test. Bars indicate comparisons of the CD127+CD28− subset.
See also Figure S3.
To assess the extent of activation and replicative history of pancreas TRMs relative to other sites, we examined expression of co-stimulation molecules and markers of homeostasis and senescence. The co-stimulatory receptor CD28 is downregulated following activation and proliferation (Appay et al., 2002; Im et al., 2016; Powell et al., 2005), and accordingly, TRMs from both pancreas and jejunum were predominantly CD28− (Figure 3D) (consistent with the TCR results suggesting extensive clonal expansion). By contrast, TRMs in LN are CD28+ (Figure 3D). Expression of CD127, the IL-7 receptor, is associated with long-lived memory T cells and mediates their antigen-independent proliferation (Surh and Sprent, 2008). TRMs across all tissue sites were predominantly CD127+, including CD28− TRMs (Figure 3D). Furthermore, pancreas TRMs did not express CD57, a marker of senescence (Figure S3C). Together, these results show that pancreas TRMs exhibit clonal expansions in situ yet are not senescent and express IL-7R consistent with a resting memory T cell phenotype.
Distinct Properties of Pancreas Tissue Macrophages and Interactions with Pancreas TRMs
We hypothesized that the distinct features of pancreas TRMs could be due to specific interactions with immune and/or non-immune cells in the pancreas. The majority (65%) of CD45+ cells in unfractionated pancreas cell suspensions consist of CD14+CD64+ myeloid lineage cells (Figure 4A), consistent with previous findings that large numbers of macrophages are distributed throughout pancreas (Calderon et al., 2015; Wang et al., 2019). CD14+CD64+ cells in pancreas express high levels of the tissue macrophage markers CD163 and CD206 (Lau et al., 2004; A-Gonzalez et al., 2017) that are increased relative to counterparts in spleen, jejunum, and PLN (Figure 4B). Macrophages in the pancreas and jejunum also express increased levels of major histocompatibility complex class II (MHC class II) and the co-stimulatory molecule CD86 compared to macrophages in PLNs and spleen (Figure 4C), as well as CD13, CD11c, and CD141, but not CD1c (Figure S4A). For other myeloid subpopulations, CD14− conventional dendritic cell (cDC) subsets were present as CD11c+ MHC class II+ cells in jejunum and LN (Figure S4B), consistent with previous reports (Granot et al., 2017). In pancreas, cDC populations were not readily detected; >99% of CD11c+ MHC class II+ cells were CD14+ (Figure S4B), likely representing macrophages. Analysis of macrophage localization by qmIF showed a reticular pattern of CD163+ macrophage staining enriched in the exocrine areas (acinar and ductal) compared to the endocrine portions (Figure 4D). These results show that pancreas macrophages, like pancreas TRMs, localize to exocrine areas and exhibit distinct properties relative to neighboring GI and lymphoid sites.
Figure 4. Localization and Phenotype of Pancreas Macrophages.

(A) Light scatter—forward scatter (FSC) and side scatter (SSC)—profile of DAPIloCD45+ cells from a pancreas cell suspension after dissociation in a Ricordi chamber (left). The proportion of these cells expressing the myeloid lineage markers CD14 and CD64 is shown in a representative flow cytometry plot (center) with the compiled percentages of the indicated CD14+CD64+ subset (right) (± SEM from 12 to 16 donors for each tissue).
(B and C) Expression of tissue macrophage markers CD163 and CD206 (B), as well as MHC class II and CD86 (C), on the DAPIlo CD45+CD64+CD14+ myeloid cells isolated from pancreas, jejunum, PLN, and spleen. Shown are representative flow cytometry plots (left) with the compiled percentages of the indicated subsets (right) (±SEM from 12 to 16 donors for each tissue). **p < 0.01 as calculated by one-way ANOVA with Dunnett’s multiple comparisons test.
(D) Density of pancreas macrophages in endocrine and exocrine pancreas. Shown is a representative qmIF image with the single-marker CD163 (left) and the corresponding composite image (right) of CD163 (yellow), the ductal marker CK19 (green), and the neuroendocrine marker chromogranin (white). Acinar, ductal, and endocrine areas were defined using inForm software, and the densities of CD163+ macrophages were quantified in the indicated pancreatic tissue areas (right) (± SEM from 13 donors). **p < 0.01 as calculated by one-way ANOVA with Sidak’s multiple comparisons test. White bar, 100 µm for scale.
See also Figure S4.
To investigate potential co-localization of pancreas T cells and macrophages, we imaged sections of the pancreas with a qmIF panel to detect neuroendocrine (chromogranin+) cells, ductal cells (CK19+), macrophages (CD163+), T cells (CD3+), and TRMs (CD103+) from 13 individuals (Figure 5A, left). These images show macrophages as networks of yellow-pseudocolored reticular cells surrounding but only rarely within islets; CD103+ (purple) TRMs are dispersed throughout the exocrine areas and also found frequently found in clusters with macrophages (Figures 5A and S5A).
Figure 5. TRMs Cluster with Macrophages in the Exocrine Pancreas.

(A) Composite image of human pancreas acquired using the Vectra platform (left) for quantitative multiplex immunofluorescence (qmIF) showing the ductal marker CK19 (green), the neuroendocrine marker chromogranin (white), and the macrophage marker CD163 (yellow). CD3+CD103+ T cells are shown in purple, and CD3+CD103− T cells are shown in blue. A map of the ductal, neuroendocrine, and T cells identified using inForm software is shown (right).
(B) PCFs based on qmIF images were calculated between each pair of cell types. Each line represents the median PCF across samples, and shaded regions are 95% confidence intervals of the median (left). The area under the curve (AUC) for PCFs were compared among three cell pairs: (1) macrophages versus any cell (gray), (2) macrophages versus CD103− T cells (blue), and (3) macrophages versus CD103+ T cells (purple). AUC has units of microns, and a random Poisson-distributed sample would have an AUC of 50. ***p < 0.001, Mann-Whitney U test (right).
See also Figure S5.
To quantitate spatial proximity between immune cells and non-immune cells, we analyzed multiple pancreas images using the pair correlation function (PCF) (Zhao et al., 2019) (see STAR Methods). The PCFs of ductal and neuroendocrine cells showed substantial clustering among themselves, consistent with the formation of tubular ducts and neuroendocrine islets in the pancreas, while significant clustering was not observed between any immune cell type and the acinar, ductal, and neuroendocrine cells (Figure 5B, left). The immune cells in pancreas, however, did form clusters with each other—macrophages with T cells (both CD103+ and CD103−) compared to other cell types (p < 1e-38, Mann-Whitney U test on area under the curve [AUC]) (Figure 5B, right) and also with other macrophages and T cells with macrophages and also with other T cells (p < 1e-82, Mann-Whitney U test on AUC) (Figure 5B, left). Simulations with random permutations of cell labels demonstrated that this effect was not due to the intrinsic structure of the acinar compartment (p < 0.05) (Figure S5B). These imaging analyses show that TRMs in the pancreas interact in situ with tissue-resident macrophages.
Regulation of Pancreas TRM Effector Function by Macrophages and PD-1
We hypothesized that pancreas TRMs are functionally regulated by interactions with macrophages. Co-culture of purified pancreas TRMs with pancreas macrophages (CD14+CD64+ CD163+) in the presence of monomeric anti-CD3 antibody resulted in secretion of multiple cytokines (IL-2, IFN-γ, and tumor necrosis factor alpha [TNF-α]) comparable to levels produced following stimulation with anti-CD3/anti-CD28/anti-CD2-coated microbeads; TRMs cultured with macrophages alone did not trigger activation (Figure 6A). We investigated potential mediators of pancreas TRM-macrophage functional interactions by staining for co-inhibitory and potential costimulatory ligands on the macrophage surface. Indeed, CD14+CD163+ pancreas macrophages express high levels of PD-L1, the ligand for PD-1 (Figure 6B), as well as CD58 (Figure 6C), a ligand for the pan-T cell co-stimulatory receptor CD2, which is highly expressed by pancreas TRMs (Figure S6).
Figure 6. Functional Regulation of Pancreas TRMs by CD58 and PD-L1 Pathways.

Sorted pancreas TRMs (CD8+CD69+ TEM cells) were stimulated with monomeric TCR-activating anti-CD3 antibody (OKT3) in the presence or absence of sorted pancreas macrophages (MF; CD14+CD64+ CD163+) (1:1 ratio) or with microbeads coated with anti-CD3 and exogenous co-stimulation (activating anti-CD2 and anti-CD28 antibodies).
(A) Cytokine production (IFN-γ and IL-2) with the indicated stimulation conditions is shown in representative flow cytometry plots (left) with compiled data for each cytokine and the cytokine polyfunctionality index (right) (±SEM from three to five donors). **p < 0.01, as calculated by one-way ANOVA with Dunnett’s multiple comparisons test.
(B and C) Representative histograms of PD-L1 (B) and CD58 (C) expression on pancreatic macrophages (CD45+CD14+CD163+, red), non-myeloid immune cells (CD45+ CD14−, blue), and CD45− cells (gray) with graphs of compiled frequencies (±SEM from four to seven donors) expressing the indicated marker (right). **p < 0.01 macrophages versus both other cells types as calculated by one-way ANOVA with Dunnett’s multiple comparisons test.
(D) Pancreas TRMs in macrophage co-cultures were stimulated with anti-CD3 in the presence or absence of PD1 blocking antibody (nivolumab) or CD58 blocking antibody followed by intracellular cytokine staining. Shown are flow cytometry profiles of TRM IFN-γ and IL-2 production (top) with compiled data showing the frequencies of TRMs producing each cytokine and the cytokine polyfunctionality index (bottom) (±SEM from four donors). **p < 0.01, *p < 0.05 as calculated by one-way ANOVA with Dunnett’s multiple comparisons test.
See also Figure S6.
To examine the role of macrophage PD-L1 and CD58 in regulating TRM functional responses in the presence of macrophages as accessory cells, we used blocking antibodies in the co-culture system. Inhibiting the PD-1 pathway with the blocking antibody nivolumab significantly enhanced anti-CD3-induced IFN-γ, TNF-α, and IL-2 production and the polyfunctionality index of pancreas TRMs (Figure 6D). By contrast, inhibiting the CD2-CD58 pathway reduced TRM-mediated cytokine production to anti-CD3 and macrophage co-culture (Figure 6D). The increased response of pancreas TRMs to PD-1 inhibition indicates that their high constitutive expression of PD-1 is functionally important.
Chronic Pancreatitis Is Associated with Reduced PD-1 Expression and Macrophage Density
We asked whether the specific features of pancreas TRMs were altered in the context of immune dysregulation and inflammation in samples of unfractionated pancreatic cell suspensions and tissue sections from individuals with CP undergoing pancreatectomy (Table S4). Analysis of the pancreatic immune cell composition in CP showed an increased T cell abundance with substantially decreased frequency of macrophages compared to organ donor controls without pancreatic disease (Figure 7A), resulting in an increased T cell to macrophage ratio in CP compared to control pancreas (Figure 7B, top). Macrophages in CP also showed qualitative changes including decreased expression of CD163, a molecule associated with tissue repair (Akahori et al., 2015), and increased expression of MHC class II (Figure S7A).
Figure 7. Alterations in Pancreatic Immune Cell Composition and T Cell Regulation in Patients with Chronic Pancreatitis.

Samples of unfractionated pancreatic cell suspensions were obtained from research subjects undergoing total pancreatectomy with auto-islet transplantation.
(A) Pancreas immune cells from patients with CP and age-matched organ donor controls without evidence of pancreatic disease. Shown are representative (left) and compiled frequencies (right, ±SEM) of total T cells (first column, gated on DAPIloCD45+CD3+ cells) and macrophages (second column, gated on DAPIlo CD45+Lin(CD19/20/56/15/16/66b)−CD14+CD64+ cells).
(B) Graphs of compiled data show the T cell to macrophage ratio (top) and frequencies of CD8+ TRMs (bottom, gated on DAPIloCD45+CD3+CD8+CD45RA− CCR7−CD69+) in control and CP pancreata. Data shown are from 19 controls and 8 CP patients.
(C) Representative micrographs (203 magnification) of single-color immunohistochemistry showing CD8 (top) and CD163 (bottom) staining in control (left) and CP pancreas (right). White bar, 100 µm for scale.
(D) Expression of PD-1 on CD8+ TRMs (left) and PD-L1 on macrophages (right) from pancreas of control (blue) and CP patients (red) shown in representative histograms (top), with graphs of compiled mean fluorescence intensities (MFIs).
(E) Expression of T-bet in CD8+ TRMs is shown from pancreas of controls (blue) and CP patients (red) in representative histograms (left) and graphs of compiled MFIs (±SEM from five controls and five CP patients). **p < 0.01, *p < 0.05 as calculated by unpaired t test.
(F) Linear regression showing the relationship between PD-1 and T-bet MFI on pancreas CD8+ TRMs of CP patients and controls with 95% confidence bands of the best-fit line.
See also Figure S7.
Within the T cell compartment, the frequency of CD8+ TRMs was similar in CP and controls (Figure 7B, bottom); however, in some CP samples, there was an increase in CD4+ T cell and CD4+ TRM frequency (Figure S7B). Consistent with the cellular data, immunohistochemistry showed decreased density of CD163+ tissue macrophages relative to CD8+ T cells in pancreatic parenchyma of CP patients relative to controls (Figure 7C). Moreover, surface expression of PD-1 was decreased on CD8+ TRMs of CP compared to controls (Figure 7D, left), whereas surface expression of macrophage PD-L1 was unchanged in CP (Figure 7D, right). To investigate the mechanism for PD-1 downregulation on TRMs, we examined expression of T-bet, an inflammation-associated transcription factor known to repress PD-1 expression in CD8+ T cells (Kao et al., 2011). Indeed, T-bet was increased in pancreatic CD8+ TRMs from CP patients compared to controls, and the degree of T-bet upregulation was significantly correlated with PD-1 downregulation (Figures 7E and 7F). Together, these changes in pancreatic TRMs and tissue macrophages are associated with altered immune regulation in CP.
DISCUSSION
TRMs are maintained throughout life as the predominant T cell subset in diverse human tissue sites, suggesting roles in immune homeostasis. Here, we elucidated how TRMs in the human pancreas exhibit tissue-specific adaptations and mediate functional regulation in situ that is disrupted during chronic inflammation. Our findings reveal that human pancreas maintains a population of CD8+ TRMs with a distinct organ-specific transcriptional, phenotypic, and functional profile compared to those in jejunum and PLN. Pancreas TRMs localize almost exclusively in exocrine areas, where they cluster with tissue macrophages, which can regulate TRM activation via PD-L1/PD-1 signaling. Moreover, we show that pancreatic inflammation in patients with CP is associated with alterations in these TRM regulatory mechanisms, including downregulation of PD-1 expression and reduced interactions with macrophages. Together, our results reveal mechanisms of pancreatic immune regulation with implications for understanding the immune dysfunctions underlying pancreatic disease.
Our tissue resource enables cross-tissue comparison of immune cells in the pancreas relative to neighboring GI and lymphoid tissue sites within the same individual. We show predominantly CD8+ T cells in the human pancreas, consistent with previous studies (Damond et al., 2019; Rodriguez-Calvo et al., 2014; Wang et al., 2019), and that pancreatic CD8+ T cells exhibit uniform and high level expression of key TRM surface markers such as CD103, CD49a, and PD-1 compared to memory T cells in other sites. While pancreas TRMs retain the core molecular profile previously defined for human TRMs (Kumar et al., 2017), they also exhibit a pancreas-specific gene expression signature associated with T cell activation and mitochondrial biogenesis and corresponding polyfunctional cytokine profiles and increased mitochondrial mass relative to TRMs in other sites. Increased mitochondrial mass has been associated with enhanced T cell effector function (Fischer et al., 2018; Scharping et al., 2016), and mitochondrial metabolism is essential for memory T cell maintenance; skin TRMs are particularly dependent on mitochondrial fatty acid oxidation (Pan et al., 2017; van der Windt et al., 2012). Our results suggest a distinct metabolic profile in pancreas TRMs that may reflect their residency in an organ that both senses and regulates systemic nutrient metabolism and influences their functional capacity.
The CD8+ T cell bias and the known susceptibility of the human pancreas to multiple common viruses, including cytomegalovirus, enteroviruses, Epstein-Barr virus, and hepatitis (Alidjinou et al., 2017; Chan et al., 2014; Rawla et al., 2017; Sarmiento et al., 2017), suggests that pancreas TRMs may originate from antiviral immune responses. In this way, TRMs could be important for rapid in situ clearance of viruses with pancreatic tropism. TRMs can also play important roles in tumor immunity (Brooks et al., 2018; Ganesan et al., 2017; Nizard et al., 2017). Rare cases of long-term survival in patients with PDAC are associated with development of high-quality anti-tumor T cell responses (Balachandran et al., 2017). The functional potency of pancreatic TRMs suggests their potential for targeting in anti-tumor immunity.
The immune checkpoint molecule PD-1 is expressed by human TRMs in multiple sites, including lung, liver, spleen, and brain (Hombrink et al., 2016; Kumar et al., 2017; Pallett et al., 2017; Shwetank et al., 2017). Pancreas TRMs display uniformly high surface PD-1 expression yet do not exhibit transcriptional or functional features of T cell ‘‘exhaustion’’ associated with PD-1 expression in chronic infection or cancer (Wherry, 2011). We further show that pancreas TRMs are localized near pancreatic resident macrophages, which themselves express PD-L1, and that inhibiting PD-1/PD-L1 in pancreas TRM-macrophage co-cultures results in enhanced TRM-mediated functional responses. We propose that TRM interaction with macrophages in situ leads to their functional regulation through PD-1 during homeostasis. By contrast, in pancreata of patients with CP, there was decreased expression of surface PD-1 on CD8+ TRMs concomitant with a significant decline in macrophage density, implicating loss of TRM-macrophage interactions and decreased PD-1 signaling. Decreased PD-1 expression on TRMs was significantly correlated with increased expression of the inflammation-associated transcription factor T-bet, previously shown to directly repress PD-1 expression in CD8+ T cells (Kao et al., 2011). Our results suggest a potential role for PD-1 in TRM regulation in situ and in the development or pathogenesis of CP, consistent with results in mouse models demonstrating that a deficiency of PD-1 expression resulted in pancreatitis (Zhang et al., 2016).
Our results have direct implications for PD-1 targeted immunotherapies, particularly for PD-1/PD-L1 checkpoint blockade therapies, which have shown remarkable success in treating certain cancers (Sharma and Allison, 2015). Adverse events due to checkpoint inhibition include autoimmune and inflammatory syndromes in different tissue sites (Pauken et al., 2019). Interestingly, T1D and pancreatic inflammation of varying severities have been reported in a small number of cancer patients treated with these therapies (Araújo et al., 2017; Miyoshi et al., 2016; Stamatouli et al., 2018). Our results suggest that blocking the PD-1/PD-L1 interaction in vivo could disrupt in situ tissue homeostasis in the pancreas and perhaps other sites by releasing PD-1-mediated control of TRMs. Conversely, the elevated PD-1 expression by pancreas TRMs suggests that checkpoint blockade therapies could have potential in harnessing TRMs for treating PDAC.
Together, our findings reveal how tissue-specific TRM adaptations shape distinct modes of TRM regulation that can be governed by interactions with tissue macrophages, with implications for understanding the immune dysfunctions underlying pancreas inflammatory disease and cancer and the overall regulation of tissue homeostasis.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for reagents should be directed to and will be fulfilled by lead author Donna L. Farber (df2396@cumc.columbia.edu).
Materials Availability Statement
This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human samples
Tissues were obtained from deceased organ donors as part of organ acquisition for clinical transplantation through an approved protocol and material transfer agreement with LiveOnNY as described previously (Carpenter et al., 2018; Gordon et al., 2017; Granot et al., 2017; Kumar et al., 2017; Miron et al., 2018; Senda et al., 2019). Donors were free of cancer, chronic diseases, seronegative for hepatitis B, C, and HIV, and represented diverse ages (Table S1). Use of organ donor tissues does not qualify as ‘‘human subjects’’ research, as confirmed by the Columbia University IRB as tissue samples were obtained from brain-dead (deceased) individuals.
Human subjects undergoing total pancreatectomy with auto-islet transplantation provided informed consent for collection of specimens and data for research under a protocol approved by the Institutional Review Board of CUIMC. Table S4 shows the list of patients and their characteristics.
METHOD DETAILS
Isolation and preparation of single cell suspensions from tissue samples
Tissue samples were maintained in cold saline or CoStorSol® (University of Wisconsin (UW) solution (Preservation Solutions, Elkhorn, WI, Cat# PS004), and transported to the laboratory within 2–4 hours of organ procurement. Lymphocytes were isolated from blood and BM samples by density centrifugation using lymphocyte separation medium (Corning cat# 25–072-CI) for recovery of mononuclear cells. All non-lymphoid tissues were carefully inspected for the presence of lymph nodes and these were removed and processed separately. Spleen, PLN, MLN and jejunum samples were processed using enzymatic and mechanical digestion, resulting in high yields of live leukocytes, as previously described (Carpenter et al., 2018; Gordon et al., 2017; Granot et al., 2017; Kumar et al., 2017; Miron et al., 2018; Senda et al., 2019).
Human pancreatic tissue was dissociated by enzymatic digestion based on a modified human islet isolation method (Bugliani et al., 2004). Briefly, the body and tail portion of the pancreas was perfused intraductally and digested with enzyme solution containing Premium Grade Collagenase (SERVA Electrophoresis GmbH, Heidelberg, Germany, Cat#17455.1) and Neutral Protease (SERVA Electrophoresis GmbH, Cat#30301.5) until the majority of the islets were separated from exocrine tissue as examined microscopically by Dithizone-staining (Sigma, Cat#D5130). The pancreas digest, in a 650ml Ricordi chamber (Biorep Technologies, Inc., Miami Lakes, FL, Cat# RC3–600-MUL) with a 450 µm stainless steel mesh filter, was diluted, washed, and collected using at least 8 l of circulating RPMI (Mediatech, Cat# 99–595-CM). Single cell suspensions of the pancreas digest were prepared by mechanical disruption of cell clusters by vigorous pipetting in the presence of DNase I (Sigma, Cat# DN25–5G) (50–100 µg/ml) followed by filtration through 70 µm mesh.
Flow cytometry analysis and cell sorting
For flow cytometric analysis, cells were stained in 5mL polystyrene tubes in the dark using antibody panels (see key resources table). Surface staining was done for 20min at room temperature (RT). For intracellular staining, surface stained cells were fixed for 60min at 4°C in fixing buffer (BD Biosciences Cat# 51–2090KZ), followed by staining in permeabilization buffer (BD Biosciences Cat# 51–2091KZ) at RT for 30 min. Controls were unstained and single-fluorochrome stained samples. Flow cytometry data were acquired on a BD Fortessa and analyzed using FlowJo (Treestar, Ashland, OR).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies, Conventional Flow Cytometry and Sorting | ||
| Anti-Human CCR7 AF488 | Biolegend | G043H7; RRID: AB_10918624 |
| Anti-Human CD101 APC | Biolegend | BB27; RRID:AB_2121761 |
| Anti-Human CD103 PE | Biolegend | Ber-ACT8; RRID:AB_10641843 |
| Anti-Human CD103 BV605 | Biolegend | Ber-ACT8; RRID:AB_2564283 |
| Anti-Human CD11c PerCP-Cy5.5 | Biolegend | Bu15; RRID:AB_1279069 |
| Anti-Human CD11c BV785 | Biolegend | Bu15 |
| Anti-Human CD127 BV510 | Biolegend | A019D5; RRID:AB_2562304 |
| Anti-Human CD14 APC-Cy7 | Biolegend | HCD14; RRID:AB_830692 |
| Anti-Human CD14 BV650 | Biolegend | HCD14 |
| Anti-Human CD14 PerCP-Cy5.5 | Biolegend | HCD14; RRID:AB_893252 |
| Anti-Human CD141 APC | Biolegend | M80; RRID:AB_10899578 |
| Anti-Human CD15 BV510 | Biolegend | W6D3; RRID:AB_2561526 |
| Anti-Human CD163 BV605 | Biolegend | GHI/61; RRID:AB_2562712 |
| Anti-Human CD163 APC | Biolegend | GHI/61; RRID:AB_2074533 |
| Anti-Human CD19 APC | Biolegend | HIB19; RRID:AB_314241 |
| Anti-Human CD19 BV510 | Biolegend | HIB19; RRID:AB_2561381 |
| Anti-Human CD1c AF488 | Biolegend | L161; RRID:AB_10719095 |
| Anti-Human CD20 BV510 | Biolegend | 2H7; RRID:AB_2561941 |
| Anti-Human CD206 PE | Biolegend | 15-2; RRID:AB_571911 |
| Anti-Human CD25 BV650 | Biolegend | BC96; RRID:AB_11218989 |
| Anti-Human CD28 PE-Dazzle 594 | Biolegend | CD28.2; RRID:AB_2564235 |
| Anti-Human CD3 PE-Cy7 | Biolegend | UCHT1; RRID:AB_439781 |
| Anti-Human CD3 BV510 | Biolegend | UCHT1; RRID:AB_2563467 |
| Anti-Human CD39 PE-Dazzle 594 | Biolegend | A1; RRID:AB_2564319 |
| Anti-Human CD4 APC-Cy7 | Biolegend | RPA-T4; RRID:AB_314086 |
| Anti-Human CD4 BV510 | Biolegend | RPA-T4; RRID:AB_2563313 |
| Anti-Human CD45 AF700 | Biolegend | HI30; RRID:AB_493760 |
| Anti-Human CD45 BV711 | Biolegend | HI30; RRID:AB_2563466 |
| Anti-Human CD45RA BV605 | Biolegend | HI100; RRID:AB_2563814 |
| Anti-Human CD45RA BV785 | Biolegend | HI100; RRID:AB_2563816 |
| Anti-Human CD49A PE | BD Bioscience | SR84; RRID:AB_397288 |
| Anti-Human CD56 BV510 | Biolegend | HCD56; RRID:AB_2561944 |
| Anti-Human CD57 PE-Dazzle 594 | Biolegend | HNK-1; RRID:AB_2564062 |
| Anti-Human CD58 PerCP-Cy5.5 | Biolegend | TS2/9; RRID:AB_2650811 |
| Anti-Human CD64 PE-Cy7 | Biolegend | 10.1; RRID:AB_2561585 |
| Anti-Human CD69 BV711 | BD Bioscience | FN50 |
| Anti-Human CD69 BV711 | Biolegend | FN50; RRID:AB_2566466 |
| Anti-Human CD8 PerCP-Cy5.5 | BD Bioscience | RPA-T8; RRID:AB_1727513 |
| Anti-Human CD8 BV650 | Biolegend | RPA-T8; RRID:AB_11125174 |
| Anti-Human CD86 BV650 | Biolegend | IT2.2; RRID:AB_11126752 |
| Anti-Human HLA-DR AF700 | Biolegend | LN3; RRID:AB_893565 |
| Anti-Human PD-1 BB700 | BD Bioscience | EH12.1 |
| Anti-Human PD-L1 BV711 | Biolegend | 29E.2A3; RRID:AB_2565763 |
| Anti-Human PD-L1 PE-Dazzle 594 | Biolegend | 29E.2A3; RRID:AB_2616888 |
| Anti-Human Granzyme B APC | Biolegend | QA16A02; RRID:AB_2687027 |
| Anti-Human IL-2 PE | Biolegend | MQ1-17H12; RRID:AB_315093 |
| Anti-Human IL-2 APC-R700 | BD Bioscience | MQ1-17H12 |
| Anti-Human IFNγ BV421 | BD Bioscience | B27 |
| Anti-Human TNFα PE-Cy7 | Biolegend | MAb11; RRID:AB_1727578 |
| Antibodies, Multispectral Staining | ||
| Anti-Human CK19 | Sigma Aldrich | A53-B/A2.26; RRID:AB_1158236 |
| Anti-Human CD163 | Biocare Medical | 10D6 |
| Anti-Human CD3 | Leica | LN10; RRID:AB_10554454 |
| Anti-Human Chromogranin A | Abcam | Cat#Ab45179; RRID:AB_726879 |
| Anti-Human CD103 | Abcam | EPR4166(2); RRID:AB_2128596 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Human TrueStain FcX | BioLegend | 422302 |
| Zombie NIR Fixable Viability Kit | BioLegend | 423105 |
| PMA | Sigma | P1585 |
| Ionomycin | Sigma | I9657 |
| RPMI 1640 | Corning | 10-040-CM |
| Deposited Data | ||
| Raw and analyzed data | This study | GEO: GSE135582 |
| Software and Algorithms | ||
| spatstat | Baddeley et al., 2016 | https://spatstat.org/ |
| DEseq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| VDJtools | Shugay et al., 2015 | https://vdjtools-doc.readthedocs.io/en/master/ |
| MiXCR | Bolotin et al., 2015 | https://mixcr.readthedocs.io/en/master/ |
| tcR | Nazarov et al., 2015 | http://imminfo.github.io/tcr/ |
| Enrichr | Chen et al., 2013 | https://amp.pharm.mssm.edu/Enrichr/ |
| Rstudio version 1.2.1335 | R Studio, Inc. (2019) | https://rstudio.com |
| inform Version 2.3 | PerkinElmer | https://www.perkinelmer.com/CMSResources/Images/44-140143BRO_010576_01_PRD_inForm.pdf |
| Functional Grade Antibodies | ||
| Anti-CD58 | BioLegend | TS2/9; RRID:AB_2291391 |
| Anti-PD-1 | Biovision | Nivolumab |
| Purified Mouse IgG1, k Isotype Ctrl Antibody | BioLegend | MG1-45; RRID:AB_2801451 |
| Ultra-LEAF Purified Human IgG4 Isotype Control Recombinant Antibody | BioLegend | QA16A15 |
For sorting, cells were stained with the antibodies described in the key resources table. The stained cells were sorted using a BD Influx Cell Sorter. Sorted cells were used either for functional assays or RNA isolation.
In vitro T cell stimulation
For PMA/ionomycin stimulation, 50,000 – 100,000 sorted T cells from tissue sites were plated cultured 96-well U-bottom plates in 200 µL of complete RPMI-1640 medium (containing fetal bovine serum, 10%) containing PMA (Sigma, Cat#P1585) (50 ng/ml) plus ionomycin (Sigma, Cat#I9657) (1 µg/ml) in 96-well round-bottom plates. For macrophage co-culture experiments, 50,000 – 100,000 sorted TRM (DAPI-CD45+Lin−CD4/8+CD45RA−CCR7−CD69+) were plated at 1:1 ratio with sorted autologous pancreatic macrophages (DAPI−CD45+Lin−CD14+CD64+CD163+) in 200 µL of complete RPMI and rested overnight prior to stimulation with 1 µg/ml monomeric anti-CD3 (OKT3, Biolegend) in the presence or absence of 10 µg/ml functional grade blocking anti-CD58 (TS2/9, Biolegend) or 10 µg/ml anti-PD-1 (Nivolumab, Biovision) antibody. Positive stimulation controls were sorted TRM cultured with microparticles coated with anti-CD2, -CD3, -CD28 antibodies (Miltenyi Biotec, Cat#130–091-441). Stimulated cells were incubated at 37°C for 4–6hrs in the presence of GolgiStop (BD Biosciences, cat# 554724) and GolgiPlug (BD Bioscience cat# 555029). Cells were washed, stained with the fixable viability marker (Zombie-NIR, Biolegend, Cat# 423105), prior to surface and intracellular staining.
Multispectral staining and imaging of pancreas tissue
Representative sections 0.5–1.0 cm in thickness from the pancreas body and proximal jejunum were removed within 12 hours of organ procurement and placed in zinc-buffered formalin (Anatech Ltd.) for 48 hours prior to dehydration and embedding in paraffin. These pancreas samples were sectioned at 5-µm thickness and stained using 7-color multispectral Opal reagents (Akoya Biosciences, Cat# NEL811001KT) as previously described (Gartrell et al., 2018). The multiplex panel included DAPI for nuclear counterstaining, CD3 (clone LN10; Leica; 1:150 dilution; Opal 540 fluorophore), CK19 (clone A53-B/A2.26; Sigma Aldrich; 1:600 dilution; Opal 690 fluorophore), CD163 (clone 10D6; Biocare Medical; 1:200 diluation; Opal 570 fluorophore); Chromogranin A (polyclonal; Abcam Cat#Ab45179; 1:300; Opal 520 fluorophore), and CD103 [clone EPR4166(2); Abcam; 1:900 dilution; Opal 650 fluorophore]. Single controls and an unstained slide were stained with each group of slides. After staining, the sections were mounted in Vectashield Hard Set mounting media (Vector Labs, Cat#H1600) and stored at 4°C for up to 48 hours prior to image acquisition.
Multispectral imaging and acquisition at 20x magnification (numerical aperture 0.75) was performed using the integrated Vectra 3 automated quantitative pathology imaging system (PerkinElmer) as previously described (Gartrell et al., 2018). Images were analyzed using inForm software (PerkinElmer). Images depicting single color immunohistochemistry were created using the ‘pathology view’ function within the inForm software which pseudocolors the indicated marker brown and the DAPI nuclear counter stain blue. Representative areas (5–10) from each donor including both exocrine and endocrine regions were chosen for analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
Whole-Transcriptome Profiling by RNA-Seq
CD3+CD8+CD69+TEM (CD45RA−CCR7−) cells were sorted from the pancreas, jejunum and PLN of five individual donors (D304, D318, D325, D332, and D333; see Table S1), and CD8+TEM cells (CD45RA−CCR7−CD69−) were sorted from peripheral blood. RNA was isolated from cell pellets using the RNeasy Mini Kit (QIAGEN) and quantitated using an Agilent 2100 Bioanalyzer (Agilent Technologies), and library preparation and RNA-seq was performed by the Columbia Genome Center as previously described (Kumar et al., 2017). RNA-Seq reads were mapped using TopHat (Trapnell et al., 2009) with default parameters to the human reference genome build hg19, performed data quality control using RNA-SeQC (Wang et al., 2012) computed read counts using HTSeq (Anders et al., 2015). For a quality control (QC) summary of RNA-seq samples, see Table S5.
Tissue specific transcripts from non T cell components were identified as those transcripts most highly enriched in whole pancreas and jejunum compared to sorted T cells. Gene Transcripts per Million (TPM) values from pancreas and jejunum tissue were obtained from RNaseq data collected by the Genotype-Tissue Expression (GTEx) Project (Consortium and GTEx Consortium, 2013) and compared to TPM values from RNaseq datasets of sorted human CD8+TRM (Kumar et al., 2017). Genes found to be more than 4 fold enriched in pancreas or jejunum tissue compared to sorted CD8+TRM were excluded from downstream PCA and pathway analysis as potential non-T cell tissue specific contaminants. Differential gene expression analysis and principal component analysis was performed with DESeq2 (Love et al., 2014), and pathway analysis was performed with Ingenuity Pathway Analysis software (IPA; QIAGEN), Gene Ontology Analysis and Enrichr (Chen et al., 2013; Kuleshov et al., 2016; Mi et al., 2019). For GSEA analysis, the GSEAPreranked tool was used with the absolute value of log2 fold change between pancreas TRM and blood TEM cells as the ranking metric to assess enrichment of the indicated gene sets (Subramanian et al., 2005).
Extraction and analysis of TCR repertoire from T cell RNA-seq data
The MiXCR analysis pipeline was used to extract TCR repertoire data from the RNA-seq datasets as described above (Bolotin et al., 2015). Using the default parameters, sequencing reads within the fastq files were aligned against reference V, D, J and C genes, followed by contig assembly and export of all TCR clonotypes with associated clone counts. The extracted TCR clonotypes were then used for analysis of TCR repertoire diversity, and clonal overlap between tissue sites. Plots showing the percent of clonal space occupied by clones of different sizes were generated using the ‘clonal.space.homeostasis’ function in the R package tcR (Nazarov et al., 2015). Estimation of TCR repertoire diversity was performed using the ‘CalcDiversityStats’ function of VDJtools (Shugay et al., 2015). Clone tracking across TRM in PLN, pancreas and Jejunumwas performed using the TrackClonotypes function of VDJtools with the ‘intersect-type’ parameter set to ‘strict’ indicating that clonal overlap is defined as matching CDR3 nucleotide sequence, V and J genes (Shugay et al., 2015). To calculate the contribution of PLN clonotypes to the clonal space of pancreas and jejunum, and to generate the clone tracking map, the TrackClonotypes function was used with the PLN set as the index sample followed by plotting via running R scripts as described (Shugay et al., 2015). To calculate the contribution of expanded pancreas and jejunum clones to the clonal space of the other tissues the TrackClonotypes function was applied, with pancreas and jejunum, respectively, set as the index samples.
Gene Set Enrichment Analysis
For GSEA analysis, the GSEAPreranked tool was used with the absolute value of log2 fold change between pancreas TRM and blood TEM cells as the ranking metric to assess enrichment of the indicated gene sets (Subramanian et al., 2005). Genes ranked by absolute value of log2-fold change between comparator groups were used as input to run pre-ranked GSEA using the Broad Institute GSEA Java web application. The TRM signature gene sets were created from lung and spleen CD8+TRM transcription profiles, as previously determined (Kumar et al., 2017).
Polyfunctionality index
For assessment of polyfunctionality, boolean combination gates were generated using FlowJo for T cells positive for all possible cytokine combinations. Gates for cytokine positivity were drawn based on unstimulated cells stained in parallel. Calculation of the cytokine polyfunctionality index was then performed as described (Larsen et al., 2012).
Image analysis
Tissue segmentation was performed using inForm software (Version 2.3, PerkinElmer), by highlighting examples of CK19+ ductal structures, and chromogranin+ islets. The highly cellular CK19-/chromogranin- areas between the ducts and islets were classified as ‘acinar’. Manual tissue segmentation was performed on 10–20 representative fields allowing the algorithm to ‘‘learn’’ each tissue type and segment the image into the three main pancreas tissue components. Immune cell constituents within each tissue segment were defined by the DAPI nuclear counterstain to define the nucleus of each cell, with each associated membrane detected via presence of a specific stain (CD3, CD103, and/or CD163). Cell segmentation was adjusted as previously described to accurately locate all cells and minimize nuclear hypersegmentation and hyposegmentation (Gartrell et al., 2018). Cells were then phenotyped by training the phenotyping algorithm of inForm software, identifying: macrophage (CD163+, yellow dots), T cells (CD3+CD103−, blue dots; CD3+CD103+ purple dots), ductal cells (CK19+, green dots), and neuroendocrine cells (chromogranin+, white dots). The cell segmentation data summary provided densities of each cell type in the 3 pancreas tissue segments and the full cell segmentation data file provided the X and Y coordinates of each phenotyped cell.
Based on spatial maps generated from the images, PCFs were calculated between each pair of cell types. Positive deviations of the PCF above the reference value of 1 provide evidence of clustering at a particular radius, and the area under the curve (AUC) summarizes the overall degree of clustering. The cell segmentation data file containing positions and phenotypes of cells was processed into pair correlation functions (PCFs) using the spatstat R package (Baddeley et al., 2016). Inhomogeneous PCFs were calculated up to a radius of 50 microns between each pair of cell types, as long as there were at least 20 cells of that type in the sample. Isotropic edge correction and a normalization power of 2 were used. The area under the curve (AUC) for each PCF was used as a summary statistic for quantifying clustering, and plots represent the point-wise median PCF across samples with 95% confidence intervals obtained via bootstrapping. In addition, simulations were performed to determine the null distribution of macrophages and T cells, assuming that they were confined to the acinar areas of the pancreas. In each of 40 iterations, macrophages, T cells, and acinar cells had their labels randomly permuted for each sample, and PCFs were recalculated. The median PCFs of these simulated samples were then compared to those that were actually observed.
Statistical analysis and data visualization
Descriptive statistics of compiled flow cytometry data, graphs and statistical testingwere performed using GraphPad Prism (Graphpad software, San Diego, CA). To compare expression of surface and intracellular markers across different tissue sites, a one-way ANOVA test was run followed by Dunnett’s multiple comparison testing. For comparing expression of markers between 2 groups (CP patients versus controls) we performed an unpaired t test with unequal variance. P values below 0.05 were considered as statistically significant. For all figures *** = p value < 0.001, ** = p value < 0.01, and * = p value < 0.05.
DATA AND CODE AVAILABILITY
The accession number for the data reported in this paper is GEO: GSE135582.
Supplementary Material
Highlights.
The human pancreas contains CD8+ TRMs exhibiting tissue-specific molecular signatures
Pancreas TRMs express high levels of PD-1 yet maintain strong effector function
During homeostasis, pancreas TRMs are regulated by PD-L1+ tissue macrophages
In chronic pancreatitis, TRM PD-1 levels and PD-L1+ macrophage density are reduced
ACKNOWLEDGMENTS
This work was supported by a research grant from Bristol-Myers Squibb and by NIH grant AI106697 awarded to D.L.F. S.P.W. was supported by NIH grant K08 DK122130. M.E.S. was supported by NIH grant T32 HL105323, the American Society of Transplantation TIRN award, and a Parker B. Francis Foundation fellowship. P.D. was supported by a CRI Irvington Postdoctoral Fellowship. A.X.C. was supported by NIH grant T32GM007367. R.D.G.-C. was supported by NIH grant KL2TR001874 and Swim Across America. These studies were performed in the CCTI Flow Cytometry Core funded in part through NIH S10 shared instrumentation grants 1S10RR027050 (LSRII), S10OD020056 (Influx), and 5P30DK063608. The Human Studies Core at the Columbia Center for Translational Immunology assisted with patient-informed consent and patient sample procurement. We thank Leonora Peryero and the staff of the immunohistochemistry lab in the CUMC Department of Pathology for technical assistance with performing immunostains on human pancreas samples. We thank Yvette Tanhehco, Shanlong Jiang, and the staff of the CUMC cell therapy lab for technical assistance with pancreas cell processing and clinical sample procurement. We wish to gratefully acknowledge the generosity of the human research subjects who agreed to participate in this study, the organ donor families, and the outstanding efforts of LiveOnNY transplant coordinators and staff for making this study possible.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.11.056.
DECLARATION OF INTERESTS
The work described in this study was supported, in part, by a grant from Bristol-Myers Squibb to Donna L. Farber.
REFERENCES
- A-Gonzalez N, Quintana JA, Garcia-Silva S, Mazariegos M, Gonzalez de la Aleja A, Nicolas-Avila JA, Walter W, Adrover JM, Crainiciuc G, Kuchroo VK, et al. (2017). Phagocytosis imprints heterogeneity in tissue-resident macrophages. J. Exp. Med 214, 1281–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akahori H, Karmali V, Polavarapu R, Lyle AN, Weiss D, Shin E, Husain A, Naqvi N, Van Dam R, Habib A, et al. (2015). CD163 interacts with TWEAK to regulate tissue regeneration after ischaemic injury. Nat. Commun 6, 7792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alidjinou EK, Engelmann I, Bossu J, Villenet C, Figeac M, Romond MB, Sané F, and Hober D (2017). Persistence of Coxsackievirus B4 in pancreatic ductal-like cells results in cellular and viral changes. Virulence 8, 1229–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appay V, Dunbar PR, Callan M, Klenerman P, Gillespie GM, Papagno L, Ogg GS, King A, Lechner F, Spina CA, et al. (2002). Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat. Med 8, 379–385. [DOI] [PubMed] [Google Scholar]
- Araújo M, Ligeiro D, Costa L, Marques F, Trindade H, Correia JM, and Fonseca C (2017). A case of fulminant Type 1 diabetes following anti-PD1 immunotherapy in a genetically susceptible patient. Immunotherapy 9, 531–535. [DOI] [PubMed] [Google Scholar]
- Baddeley A, Rubak E, and Turner R (2016). Spatial Point Patterns: Methodology and Applications with R (CRC Press, Taylor & Francis Group; ). [Google Scholar]
- Balachandran VP, Łuksza M, Zhao JN, Makarov V, Moral JA, Remark R, Herbst B, Askan G, Bhanot U, Senbabaoglu Y, et al. ; Australian Pancreatic Cancer Genome Initiative; Garvan Institute of Medical Research; Prince of Wales Hospital; Royal North Shore Hospital; University of Glasgow; St Vincent’s Hospital; QIMR Berghofer Medical Research Institute; University of Melbourne, Centre for Cancer Research; University of Queensland, Institute for Molecular Bioscience; Bankstown Hospital; Liverpool Hospital; Royal Prince Alfred Hospital, Chris O’Brien Lifehouse; Westmead Hospital; Fremantle Hospital; St John of God Healthcare; Royal Adelaide Hospital; Flinders Medical Centre; Envoi Pathology; Princess Alexandria Hospital; Austin Hospital; Johns Hopkins Medical Institutes; ARC-Net Centre for Applied Research on Cancer (2017). Identification of unique neoantigen qualities in long-term survivors of pancreatic cancer. Nature 551, 512–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beura LK, Wijeyesinghe S, Thompson EA, Macchietto MG, Rosato PC, Pierson MJ, Schenkel JM, Mitchell JS, Vezys V, Fife BT, et al. (2018). T cells in nonlymphoid tissues give rise to lymph-node-resident memory T cells. Immunity 48, 327–338.e325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolotin DA, Poslavsky S, Mitrophanov I, Shugay M, Mamedov IZ, Putintseva EV, and Chudakov DM (2015). MiXCR: software for comprehensive adaptive immunity profiling. Nat. Methods 12, 380–381. [DOI] [PubMed] [Google Scholar]
- Brooks J, Fleischmann-Mundt B, Woller N, Niemann J, Ribback S, Peters K, Demir IE, Armbrecht N, Ceyhan GO, Manns MP, et al. (2018). Perioperative, spatiotemporally coordinated activation of T and NK cells prevents recurrence of pancreatic cancer. Cancer Res 78, 475–488. [DOI] [PubMed] [Google Scholar]
- Buggert M, Nguyen S, Salgado-Montes de Oca G, Bengsch B, Darko S, Ransier A, Roberts ER, Del Alcazar D, Brody IB, Vella LA, et al. (2018). Identification and characterization of HIV-specific resident memory CD8+ T cells in human lymphoid tissue. Sci. Immunol 3, eaar4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bugliani M, Lupi R, Del Guerra S, Boggi U, Marselli L, Sbrana S, Vistoli F, Torri S, Del Chiaro M, Signori S, et al. (2004). An alternative and simple method to consistently prepare viable isolated human islets for clinical transplantation. Transplant. Proc 36, 605–606. [DOI] [PubMed] [Google Scholar]
- Calderon B, Carrero JA, Ferris ST, Sojka DK, Moore L, Epelman S, Murphy KM, Yokoyama WM, Randolph GJ, and Unanue ER (2015). The pancreas anatomy conditions the origin and properties of resident macrophages. J. Exp. Med 212, 1497–1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter DJ, Granot T, Matsuoka N, Senda T, Kumar BV, Thome JJC, Gordon CL, Miron M, Weiner J, Connors T, et al. (2018). Human immunology studies using organ donors: impact of clinical variations on immune parameters in tissues and circulation. Am. J. Transplant 18, 74–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan A, Bazerbachi F, Hanson B, Alraies MC, and Duran-Nelson A (2014). Cytomegalovirus hepatitis and pancreatitis in the immunocompetent. Ochsner J 14, 295–299. [PMC free article] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheuk S, Schlums H, Gallais Sérézal I, Martini E, Chiang SC, Marquardt N, Gibbs A, Detlofsson E, Introini A, Forkel M, et al. (2017). CD49a expression defines tissue-resident CD8+ T cells poised for cytotoxic function in human skin. Immunity 46, 287–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium GT, and GTEx Consortium. (2013). The Genotype-Tissue Expression (GTEx) project. Nat. Genet 45, 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppieters KT, Dotta F, Amirian N, Campbell PD, Kay TW, Atkinson MA, Roep BO, and von Herrath MG (2012). Demonstration of islet-autoreactive CD8 T cells in insulitic lesions from recent onset and long-term type 1 diabetes patients. J. Exp. Med 209, 51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damond N, Engler S, Zanotelli VRT, Schapiro D, Wasserfall CH, Kusmartseva I, Nick HS, Thorel F, Herrera PL, Atkinson MA, et al. (2019). A map of human type 1 diabetes progression by imaging mass cytometry. Cell Metab 29, 755–768.e755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida MJ, Luchsinger LL, Corrigan DJ, Williams LJ, and Snoeck HW (2017). Dye-independent methods reveal elevated mitochondrial mass in hematopoietic stem cells. Cell Stem Cell 21, 725–729.e724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demols A, Le Moine O, Desalle F, Quertinmont E, Van Laethem JL, and Devière J (2000). CD4(+)T cells play an important role in acute experimental pancreatitis in mice. Gastroenterology 118, 582–590. [DOI] [PubMed] [Google Scholar]
- Fischer M, Bantug GR, Dimeloe S, Gubser PM, Burgener AV, Grählert J, Balmer ML, Develioglu L, Steiner R, Unterstab G, et al. (2018). Early effector maturation of naïve human CD8+ T cells requires mitochondrial biogenesis. Eur. J. Immunol 48, 1632–1643. [DOI] [PubMed] [Google Scholar]
- Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, et al. (2000). Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J. Exp. Med 192, 1027–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesan AP, Clarke J, Wood O, Garrido-Martin EM, Chee SJ, Mellows T, Samaniego-Castruita D, Singh D, Seumois G, Alzetani A, et al. (2017). Tissue-resident memory features are linked to the magnitude of cytotoxic T cell responses in human lung cancer. Nat. Immunol 18, 940–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartrell RD, Marks DK, Hart TD, Li G, Davari DR, Wu A, Blake Z, Lu Y, Askin KN, Monod A, et al. (2018). Quantitative analysis of immune infiltrates in primary melanoma. Cancer Immunol. Res 6, 481–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon CL, Miron M, Thome JJ, Matsuoka N, Weiner J, Rak MA, Igarashi S, Granot T, Lerner H, Goodrum F, and Farber DL (2017). Tissue reservoirs of antiviral T cell immunity in persistent human CMV infection. J. Exp. Med 214, 651–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granot T, Senda T, Carpenter DJ, Matsuoka N, Weiner J, Gordon CL, Miron M, Kumar BV, Griesemer A, Ho SH, et al. (2017). Dendritic cells display subset and tissue-specific maturation dynamics over human life. Immunity 46, 504–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hales CM, Carroll MD, Fryar CD, and Ogden CL (2017). Prevalence of obesity among adults and youth: united states, 2015–2016. NCHS Data Brief 288, 1–8. [PubMed] [Google Scholar]
- Hombrink P, Helbig C, Backer RA, Piet B, Oja AE, Stark R, Brasser G, Jongejan A, Jonkers RE, Nota B, et al. (2016). Programs for the persistence, vigilance and control of human CD8+ lung-resident memory T cells. Nat. Immunol 17, 1467–1478. [DOI] [PubMed] [Google Scholar]
- Hondowicz BD, An D, Schenkel JM, Kim KS, Steach HR, Krishnamurty AT, Keitany GJ, Garza EN, Fraser KA, Moon JJ, et al. (2016). Interleukin-2-dependent allergen-specific tissue-resident memory cells drive asthma. Immunity 44, 155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, et al. (2016). Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao C, Oestreich KJ, Paley MA, Crawford A, Angelosanto JM, Ali MA, Intlekofer AM, Boss JM, Reiner SL, Weinmann AS, and Wherry EJ (2011). Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8+ T cell responses during chronic infection. Nat. Immunol 12, 663–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkegård J, Mortensen FV, and Cronin-Fenton D (2017). Chronic pancreatitis and pancreatic cancer risk: a systematic review and meta-analysis. Am. J. Gastroenterol 112, 1366–1372. [DOI] [PubMed] [Google Scholar]
- Kirkegård J, Cronin-Fenton D, Heide-Jørgensen U, and Mortensen FV (2018). Acute pancreatitis and pancreatic cancer risk: a nationwide matched-cohort study in denmark. Gastroenterology 154, 1729–1736. [DOI] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44 (W1), W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar BV, Ma W, Miron M, Granot T, Guyer RS, Carpenter DJ, Senda T, Sun X, Ho SH, Lerner H, et al. (2017). Human tissue-resident memory t cells are defined by core transcriptional and functional signatures in lymphoid and mucosal sites. Cell Rep 20, 2921–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar BV, Connors TJ, and Farber DL (2018a). Human T cell development, localization, and function throughout life. Immunity 48, 202–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar BV, Kratchmarov R, Miron M, Carpenter DJ, Senda T, Lerner H, Friedman A, Reiner SL, and Farber DL (2018b). Functional heterogeneity of human tissue-resident memory T cells based on dye efflux capacities. JCI Insight 3, 123568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuric E, Seiron P, Krogvold L, Edwin B, Buanes T, Hanssen KF, Skog O, Dahl-Jørgensen K, and Korsgren O (2017). Demonstration of tissue resident memory CD8 T cells in insulitic lesions in adult patients with recent-onset type 1 diabetes. Am. J. Pathol 187, 581–588. [DOI] [PubMed] [Google Scholar]
- Larsen M, Sauce D, Arnaud L, Fastenackels S, Appay V, and Gorochov G (2012). Evaluating cellular polyfunctionality with a novel polyfunctionality index. PLoS ONE 7, e42403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau SK, Chu PG, and Weiss LM (2004). CD163: a specific marker of macrophages in paraffin-embedded tissue samples. Am. J. Clin. Pathol 122, 794–801. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay LK, and Kallies A (2017). Transcriptional regulation of tissue-resident lymphocytes. Trends Immunol 38, 94–103. [DOI] [PubMed] [Google Scholar]
- Mackay LK, Braun A, Macleod BL, Collins N, Tebartz C, Bedoui S, Carbone FR, and Gebhardt T (2015). Cutting edge: CD69 interference with sphingosine-1-phosphate receptor function regulates peripheral T cell retention. J. Immunol 194, 2059–2063. [DOI] [PubMed] [Google Scholar]
- Mackay LK, Minnich M, Kragten NA, Liao Y, Nota B, Seillet C, Zaid A, Man K, Preston S, Freestone D, et al. (2016). Hobit and Blimp1 instruct a universal transcriptional program of tissue residency in lymphocytes. Science 352, 459–463. [DOI] [PubMed] [Google Scholar]
- Masopust D, and Soerens AG (2019). Tissue-resident T cells and other resident leukocytes. Annu. Rev. Immunol 37, 521–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Muruganujan A, Ebert D, Huang X, and Thomas PD (2019). PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res 47 (D1), D419–D426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miron M, Kumar BV, Meng W, Granot T, Carpenter DJ, Senda T, Chen D, Rosenfeld AM, Zhang B, Lerner H, et al. (2018). Human lymph nodes maintain TCF-1hi memory T cells with high functional potential and clonal diversity throughout life. J. Immunol 201, 2132–2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi Y, Ogawa O, and Oyama Y (2016). Nivolumab, an anti-programmed cell death-1 antibody, induces fulminant type 1 diabetes. Tohoku J. Exp. Med 239, 155–158. [DOI] [PubMed] [Google Scholar]
- Nazarov VI, Pogorelyy MV, Komech EA, Zvyagin IV, Bolotin DA, Shugay M, Chudakov DM, Lebedev YB, and Mamedov IZ (2015). tcR: an R package for T cell receptor repertoire advanced data analysis. BMC Bioinformatics 16, 175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nizard M, Roussel H, Diniz MO, Karaki S, Tran T, Voron T, Dransart E, Sandoval F, Riquet M, Rance B, et al. (2017). Induction of resident memory T cells enhances the efficacy of cancer vaccine. Nat. Commun 8, 15221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallett LJ, Davies J, Colbeck EJ, Robertson F, Hansi N, Easom NJW, Burton AR, Stegmann KA, Schurich A, Swadling L, et al. (2017). IL-2high tissue-resident T cells in the human liver: Sentinels for hepatotropic infection. J. Exp. Med 214, 1567–1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Y, Tian T, Park CO, Lofftus SY, Mei S, Liu X, Luo C, O’Malley JT, Gehad A, Teague JE, et al. (2017). Survival of tissue-resident memory T cells requires exogenous lipid uptake and metabolism. Nature 543, 252–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauken KE, Dougan M, Rose NR, Lichtman AH, and Sharpe AH (2019). Adverse events following cancer immunotherapy: obstacles and opportunities. Trends Immunol 40, 511–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell DJ Jr., Dudley ME, Robbins PF, and Rosenberg SA (2005). Transition of late-stage effector T cells to CD27+ CD28+ tumor-reactive effector memory T cells in humans after adoptive cell transfer therapy. Blood 105, 241–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radenkovic M, Uvebrant K, Skog O, Sarmiento L, Avartsson J, Storm P, Vickman P, Bertilsson PA, Fex M, Korgsgren O, and Cilio CM (2017). Characterization of resident lymphocytes in human pancreatic islets. Clin. Exp. Immunol 187, 418–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawla P, Bandaru SS, and Vellipuram AR (2017). Review of infectious etiology of acute pancreatitis. Gastroenterol. Res 10, 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Calvo T, Ekwall O, Amirian N, Zapardiel-Gonzalo J, and von Herrath MG (2014). Increased immune cell infiltration of the exocrine pancreas: a possible contribution to the pathogenesis of type 1 diabetes. Diabetes 63, 3880–3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo MW, Wei JT, Thiny MT, Gangarosa LM, Brown A, Ringel Y, Shaheen NJ, and Sandler RS (2004). Digestive and liver diseases statistics, 2004. Gastroenterology 126, 1448–1453. [DOI] [PubMed] [Google Scholar]
- Sarmiento L, Frisk G, Anagandula M, Hodik M, Barchetta I, Netanyah E, Cabrera-Rode E, and Cilio CM (2017). Echovirus 6 infects human exocrine and endocrine pancreatic cells and induces pro-inflammatory innate immune response. Viruses 9, E25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sathaliyawala T, Kubota M, Yudanin N, Turner D, Camp P, Thome JJ, Bickham KL, Lerner H, Goldstein M, Sykes M, et al. (2013). Distribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity 38, 187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, Ferris RL, and Delgoffe GM (2016). The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity 45, 374–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenkel JM, Fraser KA, Beura LK, Pauken KE, Vezys V, and Masopust D (2014). T cell memory. Resident memory CD8 T cells trigger protective innate and adaptive immune responses. Science 346, 98–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schey R, Dornhoff H, Baier JL, Purtak M, Opoka R, Koller AK, Atreya R, Rau TT, Daniel C, Amann K, et al. (2016). CD101 inhibits the expansion of colitogenic T cells. Mucosal Immunol 9, 1205–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schietinger A, Philip M, Krisnawan VE, Chiu EY, Delrow JJ, Basom RS, Lauer P, Brockstedt DG, Knoblaugh SE, Hämmerling GJ, et al. (2016). Tumor-specific T cell dysfunction is a dynamic antigen-driven differentiation program initiated early during tumorigenesis. Immunity 45, 389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Senda T, Dogra P, Granot T, Furuhashi K, Snyder ME, Carpenter DJ, Szabo PA, Thapa P, Miron M, and Farber DL (2019). Microanatomical dissection of human intestinal T-cell immunity reveals site-specific changes in gut-associated lymphoid tissues over life. Mucosal Immunol 12, 378–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, and Allison JP (2015). The future of immune checkpoint therapy. Science 348, 56–61. [DOI] [PubMed] [Google Scholar]
- Shugay M, Bagaev DV, Turchaninova MA, Bolotin DA, Britanova OV, Putintseva EV, Pogorelyy MV, Nazarov VI, Zvyagin IV, Kirgizova VI, et al. (2015). VDJtools: unifying post-analysis of T cell receptor repertoires. PLoS Comput. Biol 11, e1004503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shwetank A, Abdelsamed HA, Frost EL, Schmitz HM, Mockus TE, Youngblood BA, and Lukacher AE (2017). Maintenance of PD-1 on brain-resident memory CD8 T cells is antigen independent. Immunol. Cell Biol 95, 953–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatouli AM, Quandt Z, Perdigoto AL, Clark PL, Kluger H, Weiss SA, Gettinger S, Sznol M, Young A, Rushakoff R, et al. (2018). Collateral damage: insulin-dependent diabetes induced with checkpoint inhibitors. Diabetes 67, 1471–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surh CD, and Sprent J (2008). Homeostasis of naive and memory T cells. Immunity 29, 848–862. [DOI] [PubMed] [Google Scholar]
- Szabo PA, Miron M, and Farber DL (2019). Location, location, location: tissue resident memory T cells in mice and humans. Sci. Immunol 4, eaas9673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teijaro JR, Turner D, Pham Q, Wherry EJ, Lefrançois L, and Farber DL (2011). Cutting edge: Tissue-retentive lung memory CD4 T cells mediate optimal protection to respiratory virus infection. J. Immunol 187, 5510–5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thome JJ, Yudanin N, Ohmura Y, Kubota M, Grinshpun B, Sathaliyawala T, Kato T, Lerner H, Shen Y, and Farber DL (2014). Spatial map of human T cell compartmentalization and maintenance over decades of life. Cell 159, 814–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thome JJ, Grinshpun B, Kumar BV, Kubota M, Ohmura Y, Lerner H, Sempowski GD, Shen Y, and Farber DL (2016). Longterm maintenance of human naive T cells through in situ homeostasis in lymphoid tissue sites. Sci. Immunol 1, eaah6506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, and Salzberg SL (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Windt GJ, Everts B, Chang CH, Curtis JD, Freitas TC, Amiel E, Pearce EJ, and Pearce EL (2012). Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 36, 68–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Wang S, and Li W (2012). RSeQC: quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185. [DOI] [PubMed] [Google Scholar]
- Wang YJ, Traum D, Schug J, Gao L, Liu C, Consortium H, Atkinson MA, Powers AC, Feldman MD, Naji A, et al. (2019). Multiplexed in situ imaging mass cytometry analysis of the human endocrine pancreas and immune system in type 1 diabetes. Cell Metab 29, 769–783.e764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe R, Gehad A, Yang C, Scott LL, Teague JE, Schlapbach C, Elco CP, Huang V, Matos TR, Kupper TS, and Clark RA (2015). Human skin is protected by four functionally and phenotypically discrete populations of resident and recirculating memory T cells. Sci. Transl. Med 7, 279ra39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ (2011). T cell exhaustion. Nat. Immunol 12, 492–499. [DOI] [PubMed] [Google Scholar]
- Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, and Ahmed R (2003). Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol 77, 4911–4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Zhang H, Xing Q, Cui J, Li J, Li Y, Tan Y, and Wang S (2014). PD-1(+) CD8(+) T cells are exhausted in tumours and functional in draining lymph nodes of colorectal cancer patients. Br. J. Cancer 111, 1391–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Chikuma S, Hori S, Fagarasan S, and Honjo T (2016). Nonoverlapping roles of PD-1 and FoxP3 in maintaining immune tolerance in a novel autoimmune pancreatitis mouse model. Proc. Natl. Acad. Sci. USA 113, 8490–8495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, Bordbar D, Shan D, Samanamud J, Mahajan A, et al. (2019). Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med 25, 462–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the data reported in this paper is GEO: GSE135582.