

Abstract
Here, we review recent insights into the neuronal presynaptic fusion machinery that releases neurotransmitter molecules into the synaptic cleft upon stimulation. The structure of the pre-fusion state of the SNARE/complexin-1/synaptotagmin-1 synaptic protein complex suggests a new model for the initiation of fast Ca2+-triggered membrane fusion. Functional studies have revealed roles of the essential factors Munc18 and Munc13, demonstrating that a part of their function involves the proper assembly of synaptic protein complexes. Near-atomic resolution structures of the NSF/αSNAP/SNARE complex provide first glimpses of the molecular machinery that disassembles the SNARE complex during the synaptic vesicle cycle. These structures show how this machinery captures the SNARE substrate and provide clues as to a possible processing mechanism.
Introduction
Synaptic transmission between pre-synaptic and post-synaptic neurons occurs when the pre-synaptic neuron terminal is temporarily depolarized upon arrival of an action potential, opening Ca2+ channels at the active zones of synapses. Because the extracellular Ca2+ concentration is much higher than the cytoplasmic concentration, Ca2+ will flow into the cytoplasm. In turn, Ca2+ will trigger fusion of neurotransmitter-filled synaptic vesicles with the presynaptic membrane in less than a millisecond [1,2]. Upon fusion, neurotransmitter molecules are released into the synaptic cleft and bound by receptors located in the postsynaptic membrane. Many, if not most, of the key factors of the core synaptic fusion machinery have been identified, including fusogenic SNARE (Soluble N-ethylmaleimide sensitive factor Attachment protein REceptor) proteins, the Ca2+-sensor synaptotagmin, the activator/regulator complexin, the assembly factors Munc18 and Munc13, and the disassembly factors NSF and SNAP. Yet, the molecular mechanisms of Ca2+-triggering, regulation, and membrane fusion remain unclear. Here, we review recent advances which have begun to reveal these molecular mechanisms.
Ca2+-triggered synaptic vesicle fusion
The SNARE complex assembly provides the energy for membrane fusion [3,4]. The SNARE proteins synaptobrevin-2/VAMP2, on the synaptic vesicle in conjunction with syntaxin-1A and SNAP-25A on the plasma membrane initiate vesicle fusion by forming a trans-SNARE complex before Ca2+-triggering. While SNAREs are essential for neurotransmitter release, a Ca2+-sensor to trigger fusion is also required because SNAREs themselves do not exhibit any Ca2+ sensitivity. In the case of fast synchronous neurotransmitter release, this task is accomplished by synaptotagmin-1 [5,6], a member of the synaptotagmin family of proteins. Synaptotagmins contain a single transmembrane-spanning domain and tandem C-terminal cytoplasmic C2 domains, termed C2A and C2B (or C2AB together) [7,8]. Additionally, the cytoplasmic protein complexin (we focus on complexin-1 in this review) also plays critical roles in regulating neurotransmitter release [9,10]. The characterization of all of these factors begs a number of mechanistic questions: How do SNAREs, synaptotagmins, and complexins achieve fusion in less than a millisecond upon Ca2+-triggering; how are they situated between the membranes, and how is the process regulated?
In the past, synaptotagmins have been primarily viewed as factors that activate fusion upon Ca2+-binding. However, recent structural and functional studies suggest that fast (submillisecond) Ca2+-triggered synaptic vesicle fusion begins with release of inhibition upon Ca2+ binding to synaptotagmin [11••,12]. Central to this new model of Ca2+-triggering are atomic resolution structures of the SNARE/synaptotagmin-1 [13••] and SNARE/complexin-1/synaptotagmin-1 complexes (Figure 1a) [11••], combined with functional studies in neuronal cultures and in reconstituted systems. The SNARE/complexin-1/synaptotagmin-1 structure was determined in the absence of Ca2+. We also note that membrane domains, the membrane proximal regions of the SNARE complex, and the Habc domain of syntaxin-1A were excluded in the constructs used for crystallization. In the trans-SNARE complex, the membrane proximal regions of syntaxin-1A and synaptobrevin-2 are probably flexible, sampling many conformations and allowing them to tether the trans-SNARE complex to their respective membrane domains. Thus, the crystal structure likely represents the well-folded core of the pre-fusion state before Ca2+-triggering.
Figure 1.
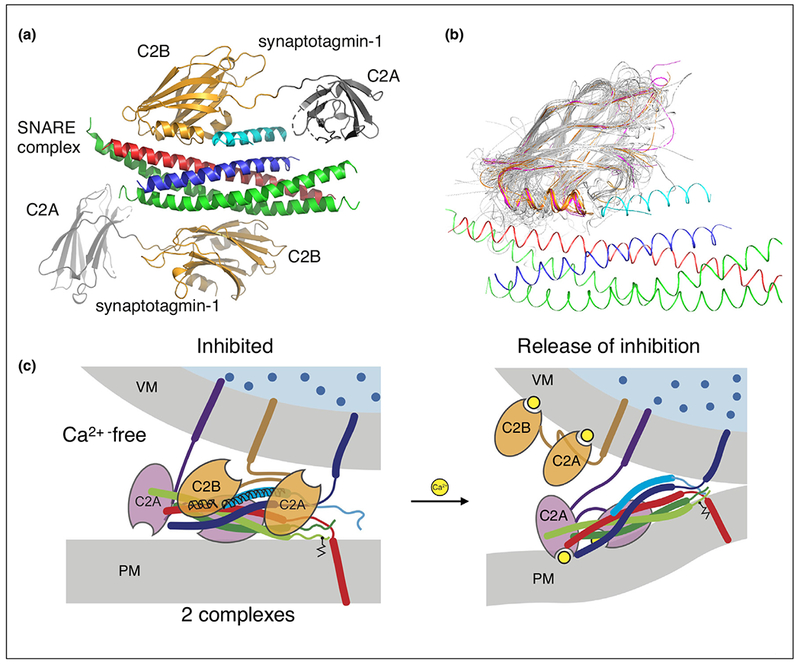
Synaptic fusion complex. (a) Crystal structure of the SNARE/complexin-1/synaptotagmin-1 complex [11••], PDB ID 5W5C (red: syntaxin-1A, green: SNAP-25A, blue: synaptobrevin-2, light blue: central helix of complexin-1, orange: C2B domains of synaptotagmin-1, gray: C2A domains of synaptotagmin-1. (b) Superposition of representative members of the C2 domain superfamily 2.60.40.150 generated by CATH [89] (http://www.cathdb.info) with the structure of the SNARE/complex-1/synaptotagmin-1 complex (red: syntaxin, green: SNAP-25, blue: synaptobrevin, gold: synaptotagmins, purple: Doc2 and Rabphilin, gray: 40 representative structures out of 346 C2 domains with known structure). (c) Release of inhibition model of Ca2+-triggering. Note that it is unknown as to which membrane the Ca2+-bound synaptotagmin-1 C2 domains might interact with. For illustration purposes, we placed it at the synaptic vesicle membrane, although the presence of PIP2 increases the binding affinity between Ca2+-bound synaptotagmin-1 and the membrane, so it also possible, or even likely, that all synaptotagmin-1 C2 domains localize to the plasma membrane upon Ca2+-binding.
This SNARE/complexin-1/synaptotagmin-1 structure reveals a tripartite interface between one synaptotagmin-1 C2B domain and both the SNARE complex and complexin-1 [11••] (Figure 1a). This tripartite interface only forms when complexin-1 is bound to the SNARE complex. Simultaneously, a second synaptotagmin-1 C2B domain interacts with the other side of the SNARE complex via a different, pairwise interface that is independent of complexin-1, as also described in structural studies with SNAREs and synaptotagmin-1 [13••]. Structure-guided mutagenesis coupled with isothermal titration calorimetry solution-binding studies and electrophysiological experiments in neuronal cultures showed that both Ca2+-triggered synchronous neurotransmitter release and suppression of spontaneous release depend on synaptotagmin-1 C2B residues involved in both the SNARE/complexin-1/synaptotagmin-1 tripartite and the SNARE/synaptotagmin-1 primary interfaces [11••,13••]. Moreover, in a reconstituted assay of synaptic vesicle fusion [14–16,17••] the effects of mutations, deletions, or truncations of these interfaces generally track with electrophysiological observations in neurons. Both interfaces map to distinct regions on the surface of the synaptotagmin-1 C2B domain, and they are separate from the polybasic region implicated in membrane interactions [18,19]. Interestingly, the Ca2+-binding loops of the synaptotagmin C2B domains are not involved in either interface.
Among the most striking structural features of the tripartite interface is the continuation of the complexin-1 central helix into the HA α-helix of synaptotagmin-1 (Figure 1a). The HA α-helix is structurally conserved in C2B domains of all synaptotagmins, as well as the homologous C2B domains of the synaptic proteins Doc2b and Rabphilin, both of which have tandem C2 domains similar to synaptotagmins. In contrast, there is no HA α-helix in all other C2 domains of known structure (Figure 1b). Note, that there is no HA α-helix in the so-called C2B domain of Munc13, but the three C2 domains of Munc13 are separated in sequence (i.e. there are no tandem C2 in Munc13) [20].
The primary SNARE/synaptotagmin-1 interface occurs in very different crystal forms [11••,13••], and regardless of the presence of complexin bound to the other side of the SNARE complex. The primary interface is identical for apo-bound, Ca2+-bound, or Mg2+-bound synaptotagmin-1. Additionally, the importance of some of the residues involved in the primary interface was subsequently confirmed in neurons by two other groups [21,22]. In addition to the primary SNARE/synaptotagmin-1 interface [13••], other bipartite interactions between the SNARE complex and the synaptotagmin-1 C2B domain may exist, as suggested by single molecule FRET experiments in both the absence or presence of Ca2+ [23] and by solution NMR studies in the presence of Ca2+ [24••]. However, the data from both studies were insufficient to determine unique high-resolution structures of these alternative interfaces. Moreover, the placement of the lanthanide labels on SNAP-25 (D166C and D41C) and mutations of synaptotagmin-1 (R398Q and R399Q) in the NMR study [24••] likely disrupt the primary interface [13••]. Nevertheless, both solution studies [24••,25] suggest possible alternative conformations of the SNARE/synaptotagmin complex, especially in the presence of Ca2+.
At least two SNARE complexes are probably involved in Ca2+-triggered fusion [26,27], but it has not yet been possible to visualize these complexes. For example, the resolution of cryo-electron tomography images (cryo-ET) of synapses [28,29] was insufficient to localize synaptic protein complexes. However, cryo-ET studies of proteoliposomes with reconstituted synaptic proteins revealed proteinaceous contacts with a variety of morphologies between the vesicle membranes [30]. The observed contacts span inter-membrane distances of 40–60 Å. At that separation, lipids alone would be unable to bridge the membranes, as the critical distance at which lipid stalks form is <9 Å [31]. Thus, one can conclude from this study that the pre-fusion SNARE/complex-1/synaptotagmin-1 complex keeps the membranes in close juxtaposition, but short of actually promoting fusion. Two or more synaptic complexes involved in Ca2+-triggered fusion could potentially interact with each other via Syt1 C2B domains [11••,12], but the precise quarternary arrangement remains to be elucidated.
On the basis of the available structures and functional data, a new model of action-potential evoked, Ca2+-triggered synaptic vesicle fusion has emerged [12] (Figure 1c). In the pre-fusion state, the SNARE/complexin-1/synaptotagmin-1 complex is formed. Upon Ca2+-binding, the synaptotagmin-1 molecule involved in the tripartite interface is dislodged, for example by binding and inserting into a nearby membrane or by binding to a different region on the SNARE complex. We note that it is unknown as to which membrane the Ca2+-bound C2 domains of synaptotagmin-1 would bind to, although the presence of PIP2 in the plasma membrane increases the binding affinity [18,19]. In turn, the primary SNARE/synaptotagmin-1 interface may assist in inducing membrane curvature upon Ca2+-binding [13••,32,33]. With the disruption of the tripartite interface, the trans-SNARE complex then fully zippers, and the membranes move close enough to fuse. This release-of-inhibition model of Ca2+-triggered fusion may explain the rapid (submillisecond) speed of the process because all the factors to promote fusion are already in place once the fusion block is released. We note that this release-of-inhibition model is independent of the actual number of SNARE complexes involved at the docking site of a primed synaptic vesicle, assuming that the presence of one or more inhibited complexes would prevent fusion.
This model of the inhibited pre-fusion SNARE/complexin-1/synaptotagmin-1 complex provides a mechanistic explanation as to why certain mutations of the Ca2+-binding region of the C2B domain of synaptotagmin-1 have dominant negative effects on both evoked and spontaneous neurotransmitter release [11••,34,35]. In other words, expression of mutant synaptotagmin-1 reduces evoked release and upregulates spontaneous release in the presence of endogenous wildtype synaptotagmin-1. Also, complexin knockdown abrogates these dominant negative effects [11••]. When some of the tripartite complexes with mutant synaptotagmin-1 molecules participate in a docked synaptic vesicle, these mutant complexes would prevent the membranes from moving close enough for triggered fusion because Ca2+ cannot bind to these mutant synaptotagmin-1 molecules.
We note that the molecular mechanisms of spontaneous release are probably, or even likely, different from those of evoked release and involve a Ca2+ sensor other than synaptotagmin-1 [36]. The release-of-inhibition model is meant to explain action-potential evoked synchronous release. However, we note that disruption of either the tripartite or the primary interface in the SNARE/complexin-1/synaptotagmin-1 complexes increases spontaneous release rate [11••], so the presence of such complexes may also modulate spontaneous release.
Regulated assembly of the synaptic pre-fusion complex
As discussed, the pre-fusion SNARE/complexin-1/synaptotagmin-1 complex (Figure 1a) is probably important for establishing the primed state of synaptic vesicles (i.e. for the association of synaptic vesicles with presynaptic proteins that enables them to undergo fast Ca2+-triggered fusion). Regulation of the formation of the primed state is critical as well. Several factors are involved in priming, including Munc18 and Munc13, but the molecular basis of the priming function of these molecules had been unclear until recently. Munc18 and its homologues (also referred to as SM proteins) are required components for all membrane trafficking pathways, as exemplified by the complete block of synaptic vesicle fusion upon Munc18-1 knockout in mice [37]. At the molecular level, Munc18 captures free syntaxin-1A, locking it into a heterodimeric complex that kinetically prevents formation of the ternary SNARE complex [38,39] (Figure 2a).
Figure 2.
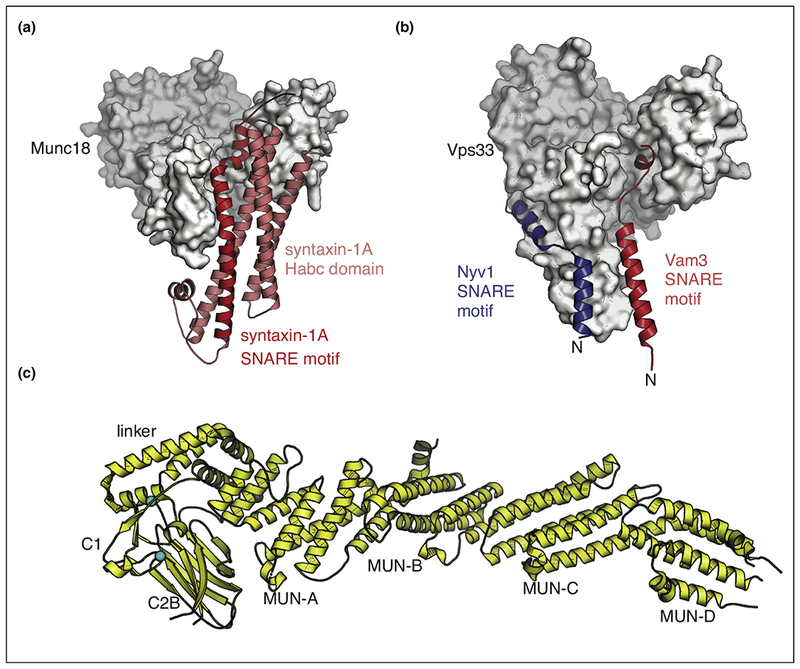
Munc18 and Munc13 are chaperones of SNARE complex assembly. (a) Crystal structure of the Munc18/syntaxin-1A complex [38,39] (PDB ID: 3C98) (red: syntaxin-1, gray: Munc18). (b) Interactions between SNAREs and Munc18. Composite model based on the crystal structures of the Vps33/Nyv1 and Vps33/Vam3 complexes [56••] (PDB IDs 5BUZ and 5BV0) (gray: Vps33, red: Vam3, blue: Nyv1). (c) Crystal structure of the C1C2BMUN fragment of Munc13 [60••] (PDB ID 5UE8).
To enable ternary neuronal SNARE complex formation, another factor is required. One such factor is Munc13, a primarily brain-specific, cytoplasmic protein in the presynaptic terminal implicated in synaptic vesicle priming and short-term synaptic plasticity [1,40]. At the molecular level, Munc13 accomplishes two tasks: (1) catalyzing the transit of syntaxin from the syntaxin/Munc18 complex into the ternary trans-SNARE complex [41–43] and (2) promoting proper assembly of the SNARE complex in conjunction with Munc18 (i.e. ensuring the parallel configuration of all components of the SNARE complex) [17••]. In vivo, both Munc13 and Munc18 are required to promote proper SNARE complex assembly because genetic deletion of Munc13 cannot be fully rescued with a mutant of syntaxin that bypasses the Munc13 requirement in vitro [17••,44].
Why is it necessary to assist the proper assembly of the SNARE complex? When soluble fragments of SNARE proteins are simply mixed in solution, improper (e.g. anti-parallel) configurations may occur [45,46••]. It turns out that Munc13-1 promotes the proper parallel subconfiguration of syntaxin-1A and synaptobrevin-2 when assembling the ternary SNARE complex [17••]. Additionally, Munc13 also cooperates with Munc18 to promote the proper syntaxin-1A/SNAP-25A subconfiguration within the assembled ternary SNARE complex [17••]. Together, Munc13 and Munc18 can be viewed as assembly factors for establishing the functional subconfiguration of the ternary SNARE complex assembly.
The cooperation of Munc18 and Munc13 in promoting the proper SNARE complex assembly explains the severe effect of deletion of Munc18 and Munc13 in neurons [37,47]. Moreover, the localization of Munc13 to the plasma membrane and to synaptic vesicle docking sites may be regulated by Ca2+ binding to the C2 domains of Munc13 [48], as well as by interactions with other factors, including calmodulin [49] and Doc2 [50]. Such a membrane localization may in turn modulate the functions of Munc13 in priming, pre-synaptic plasticity, and augmentation. Finally, and consistent with their molecular functions, Munc18 and Munc13 substantially increased the efficiency of Ca2+-triggered vesicle fusion in a reconstituted fusion assay [17••].
The molecular mechanism by which Munc13 and Munc18 promote proper SNARE complex assembly is still unclear, although there is some information about their molecular interactions [17••,43,51–54,55•]. To begin, syntaxin-1A forms a tight complex with Munc18 [38,39] (Figure 2a), although this complex appears specific to neuronal exocytosis. For example, the situation is quite different for a yeast homologue of Munc18, the yeast homotypic fusion and vacuole protein-sorting (HOPS) protein Vps33. Crystal structures of the yeast SNARE proteins Vam3 and Nyv1 bound to Vps33 show fairly different interactions and suggest that Vps33 alone may promote proper assembly of ternary yeast SNARE complexes [56••] (Figure 2b). Similar interactions may also exist for the neuronal SNAREs and Munc18 as suggested by NMR [52] and single molecule pulling experiments [57••]. However, Munc18 alone appears to be insufficient to promote the proper configurations of all components of the neuronal SNARE complex because a constitutively ‘open’ mutant of syntaxin-1A does not rescue the essential role of Munc13 in neurons [17••]. Although there is no homolog for the MUN of Munc13 in yeast, the molecular architecture of entire HOPS complex [58] includes a relatively long, flat central region that bears some superficial resemblance to the shape of the MUN domain. Interestingly, the fold of the MUN domain is similar to that of several other proteins, including the Sec3, Sec15, Exo70 components of the exocyst tethering complex [59].
How does Munc13 then cooperate with Munc18 to promote proper assembly of the neuronal SNARE complex? The most complete structure of Munc13 to date is the structure of its C1C2BMUN fragment [60••] (Figure 2c). In this structure, the C2B domain interacts with the MUN domain of Munc13. Both syntaxin-1A and synaptobrevin-2 weakly interact with the MUN domain [17••,51,53,54,55•]. The membrane proximal region of synaptobrevin-2 binds to the MUN domain; this interaction is essential for Munc13 function [55•].
Deletion of either the C1 or C2B domain of Munc13, or Ca2+ binding to the C2B domain, increases evoked release in Caenorhabditis elegans, suggesting that the molecule exists in two states—an autoinhibited state and an activated state [61••]. The crystal structure of the C1C2BMUN domain of Munc13 was determined in the presence of Ca2+, so it presumably corresponds to the active state. Of course, it possible that the activation relates to the interactions between Munc13 and the membrane as opposed to a conformational change in Munc13 [62].
Post-fusion SNARE complex disassembly
Following vesicle fusion, the ternary SNARE complex must be disassembled so that the individual SNARE proteins can be recycled. In concert with the adaptor protein SNAP, the ATPase NSF disassembles the ternary SNARE complex upon ATP hydrolysis [63–65]. NSF is a member of the AAA+ family which consists of two ATPase rings (D1 and D2 domains, known as Type II AAA+) and an N-terminal domain. Most eukaryotic organisms have only one NSF gene. By contrast, there are three homologues of SNAP, with αSNAP being the most widely studied. NSF, SNAPs, and SNAREs form the so-called 20S complex, the starting state for the disassembly process. The molecular mechanism of NSF-mediated SNARE complex disassembly is still unknown.
Years ago, crystal structures of the N and D2 domains of NSF were determined [66–69]. Subsequently, crystal structures of the distantly related ATPase p97 [70–72] were determined. Early EM reconstructions of NSF and the NSF/SNAP/SNARE complex followed [73,74]. However, these earlier EM studies suffered from relatively low resolution and the imposition of symmetry on the reconstructions that obscured the SNARE complex and its interactions with SNAPs and NSF (see Extended Data Figure 5 and Supplemental Discussion in Ref. [75••]), and despite many attempts, crystallization of full-length NSF and the NSF/αSNAP/SNARE complex was unsuccessful. In 2015, a major breakthrough was achieved by determining cryo-EM structures of NSF at 4.2 Å and of the NSF/αSNAP/SNARE complex at 7.6 Å resolution without imposing symmetry [75••]. Two key factors led to these higher resolution EM reconstructions: (1) much improved sample homogeneity and (2) advances in cryo-EM technology, especially the availability of direct electron detectors. The higher sample homogeneity was achieved by ensuring a specific nucleotide-bound state [75••] and by building on previous biochemical and biophysical studies of the NSF-mediated SNARE complex disassembly [76,77].
Recently, an even higher-resolution reconstruction of the NSF/αSNAP/SNARE complex was obtained at 4.5 Å resolution [78••]. This reconstruction showed the interaction between the SNAREs with both NSF and αSNAPs in unprecedented detail (Figure 3a). Two αSNAP molecules bind to the ternary SNARE complex primarily along a large, negatively charged region at the center of the SNARE complex. Although the αSNAP stoichiometry is different for the two reconstructions of the NSF/αSNAP/SNARE complex (related to the absence of the SNAP-25A linker in the previous reconstruction [75••]), the interactions of the common two αSNAP molecules are nearly identical for all structures determined to date. The interacting regions on the SNARE complex and the two αSNAP molecules have complementary electrostatic features that may be a key contributor to this preferred binding mode. Taken together, these structures suggest that the 2:1 αSNAP: SNARE configuration is likely conserved in all NSF/αSNAP/SNARE complexes.
Figure 3.
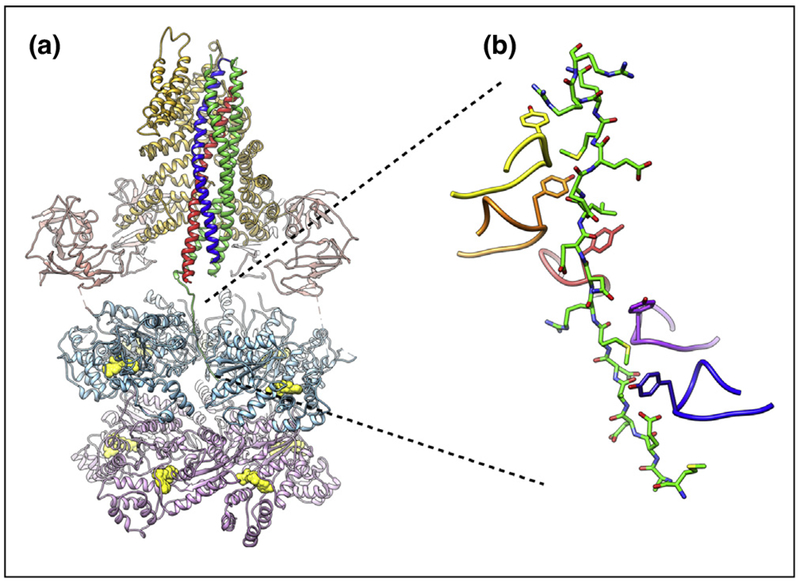
SNARE complex disassembly machinery. (a) EM structure of NSF/αSNAP/SNARE complex at ~3.9 Å resolution [78••] (PDB ID 6MDM). The N, D1, and D2 domains of NSF are colored salmon, light blue, and light purple, respectively. Nucleotides in the D1 and D2 rings are colored yellow. The two αSNAP molecules are shown in gold. The SNARE proteins syntaxin-1a, synaptobrevin-2, and SNAP-25A are colored blue, red, and green, respectively. (b) Close-up view of the interaction between the N-terminal residues of SNAP-25A and the pore of the D1 ATPase ring of NSF. An amino acid essential for SNARE complex disassembly (Y294) is found at the apex of the pore loop of each ATPase subunit; five of six of these tyrosines intercalate between the side chains of the SNAP25 N-terminus, seemingly locking it in place.
Perhaps most importantly, a series of direct interactions between the SNARE complex and NSF were observed. A large portion of the SNAP-25A N-terminus was found within the pore of the D1 ATPase ring of NSF, consistent with a ratchet-like threading mechanism before disassembly (Figure 3b). This configuration is reminiscent of other recent structures of Type II AAA+ proteins in complex with peptides, suggesting a common, conserved mechanism for substrate unfolding. Some recent examples of AAA proteins with model substrates include: casein for Hsp104 [79] and Vps4 binding a peptide from the substrate Vps2 [80]); inhibited-substrate complexes with the 26S proteasome [81,82], and a substrate complex with inhibited PAN-proteasome [83]. More broadly, this suggests that different parts of AAA proteins and their adapters are responsible for substrate recognition and initiation of the unfolding or disassembly process.
A single molecule FRET disassembly assay enabled characterization of the disassembly process under a variety of conditions [84]. These studies revealed that NSF/αSNAP is capable of also disassembling anti-parallel SNARE complexes, suggesting a role in quality control of SNARE complex formation in conjunction with Munc18 and Munc13. Moreover, complexin-1 reduces the rate of NSF/αSNAP -mediated SNARE complex disassembly [84,85], probably by interfering with binding of one of the two conserved SNAP molecules found in structures of the NSF/αSNAP/SNARE complex. The interference by complexin on disassembly is more pronounced for a trans-SNARE complex situated between a synaptic vesicle and the plasma membranes [86•].
Summary and outlook
In summary, recent structures of the pre-fusion SNARE/complexin-1/synaptotagmin-1 complex, the C1C2BMUN fragment of Munc13, and the NSF/αSNAP/SNARE complex are important new pieces in the puzzle of neuronal exocytosis (Figure 4). Yet, the molecular mechanisms are still unclear.
Figure 4.
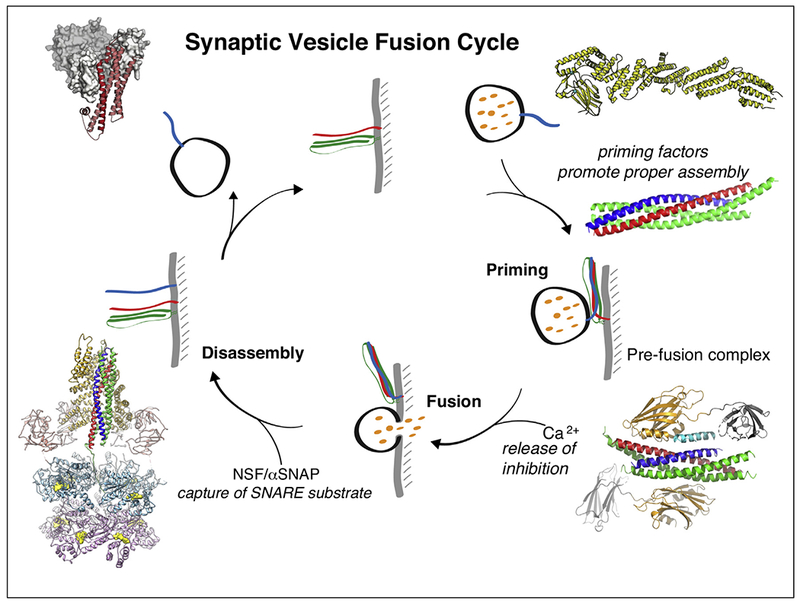
Synaptic vesicle cycle. Superimposed on the cycle are structures of the neuronal SNARE complex [3] (PDB ID 1SFC), the SNARE/complexin-1/synaptotagmin-1 complex [11••] (PDB ID 5W5C), the NSF/αSNAP/SNARE complex [78••] (PDB ID 6MDM), the Munc18/syntaxin-1A complex [38,39] (PDB ID: 3C98), and the C1C2BMUN fragment of Munc13 [60••] (PDB ID 5UE8).
The key roles of Munc13 in priming and short-term plasticity may be related to the role of the MUN domain in proper assembly of the SNARE complex [17••]. This function of the MUN domain may in turn be subject to regulation by the other domains of Munc13. For example, Ca2+-binding to the C2B domain [87], the synergy between C1, C2B, and C2C domains [48], and the Ca2+-dependent interaction between calmodulin and Munc13 [49] may localize Munc13 to sites of docked synaptic vesicles and increase its ability to assist in proper SNARE complex assembly. Clearly, the underlying molecular mechanisms of Munc13 and Munc18 remain to be uncovered.
The conformation of the trans-SNARE/complexin-1/synaptotagmin-1 complex within its native environment (that is, between the synaptic and plasma membranes) is unknown. In accordance with the abovementioned cryo-ET studies [30], the SNARE/complexin-1/synaptotagmin-1 complexes form a protein stalk that juxtaposes the membranes but keeps them far enough away to reduce the chances of membrane fusion. Yet, the resolution of the cryo-ET study was insufficient to draw any conclusions about the quaternary arrangements. Finally, the molecular steps after Ca2+-binding to the synaptotagmin C2 domains should be visualized. In other words, what happens to the synaptotagmin C2 domains, the SNARE complex, complexin, and the membranes around the emerging fusion pore?
The recent structures of the NSF/αSNAP/SNARE complex provide evidence for loading of the full-length neuronal SNARE complex via the N-terminal residues of one of the SNARE proteins (SNAP-25A). How does complete disassembly then proceed? Is the N-terminus of SNAP-25A further translocated or threaded through the pore of the D1 ATPase ring? Such a translocation would exert a pulling force on the remaining membrane-anchored SNAP-25A, likely destabilizing interactions with syntaxin-1A and synaptobrevin-2—a model appealing in its simplicity. In any case, regardless of the mechanism, single molecule disassembly studies suggest that the maximum number of ATPs required for complete disassembly cannot exceed 12 [88], ruling out processive mechanisms requiring many rounds of ATP hydrolysis. Future studies of conformational intermediates will be essential in testing these models and for uncovering the molecular mechanism of NSF/αSNAP-mediated disassembly of the SNARE complex.
Acknowledgements
We acknowledge support by the National Institutes of Health (R37MH63105 to A.T.B., K99MH113764 to Q.Z.), a grant by the Stanford ADRC Zaffaroni Alzheimer’s Disease Translational Research Program to A.T.B. (ADRC grant #5P50AG047366-02), and a postdoctoral fellowship from the Helen Hay Whitney Foundation supported by the Howard Hughes Medical Institute awarded to K.I.W.
Footnotes
Conflict of interest statement
Nothing declared.
References
- 1.Südhof TC: Neurotransmitter release: the last millisecond in the life of a synaptic vesicle. Neuron 2013, 80:675–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rothman JE: The principle of membrane fusion in the cell (Nobel Lecture). Angew Chem Int Ed 2014, 53:12676–12694. [DOI] [PubMed] [Google Scholar]
- 3.Sutton RBB, Fasshauer D, Jahn R, Brunger AT: Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 Å resolution. Nature 1998, 395:347–353. [DOI] [PubMed] [Google Scholar]
- 4.Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Söllner TH, Rothman JE: SNAREpins: minimal machinery for membrane fusion. Cell 1998, 92:759–772. [DOI] [PubMed] [Google Scholar]
- 5.Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, Südhof TC: Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell 1994, 79:717–727. [DOI] [PubMed] [Google Scholar]
- 6.Fernáandez-Chacón R, Königstorfer A, Gerber SHH, García J, Matos MFF, Stevens CFF, Brose N, Rizo J, Rosenmund C, Südhof TCC: Synaptotagmin I functions as a calcium regulator of release probability. Nature 2001, 410:41–49. [DOI] [PubMed] [Google Scholar]
- 7.Perin MSS, Fried VAA, Mignery GAA, Jahn R, Südhof TCC: Phospholipid binding by a synaptic vesicle protein homologous to the regulatory region of protein kinase C. Nature 1990, 345:260–263. [DOI] [PubMed] [Google Scholar]
- 8.Perin MS, Johnston PA, Ozcelik T, Jahn R, Francke U, Südhof TC: Structural and functional conservation of synaptotagmin (p65) in Drosophila and humans. J Biol Chem 1991, 266:615–622. [PubMed] [Google Scholar]
- 9.Mohrmann R, Dhara M, Bruns D: Complexins: small but capable. Cell Mol Life Sci 2015. 10.1007/s00018-015-1998-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trimbuch T, Rosenmund C: Should I stop or should I go? The role of complexin in neurotransmitter release. Nat Rev Neurosci 2016, 17:118–125. [DOI] [PubMed] [Google Scholar]
- 11.••.Zhou Q, Zhou P, Wang AL, Wu D, Zhao M, Südhof TC, Brunger AT: The primed SNARE-complexin-synaptotagmin complex for neuronal exocytosis. Nature 2017, 548:420–425. [DOI] [PMC free article] [PubMed] [Google Scholar]; The crystal structure of the tripartite complex between SNAREs, synaptotagmin-1, and complexin-1 reveals two interfaces. In addition to the primary interface between synaptotagmin-1 and the SNARE complex, a tripartite interface between SNAREs, complexin-1 and synaptotagmin-1 was discovered, explaining the cooperation between all three components in evoked synaptic vesicle fusion.
- 12.Brunger AT, Leitz J, Zhou Q, Choi UB, Lai Y: Ca2+-triggered synaptic vesicle fusion initiated by release of inhibition. Trends Cell Biol 2018, 28:631–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.••.Zhou Q, Lai Y, Bacaj T, Zhao M, Lyubimov AY, Uervirojnangkoorn M, Zeldin OB, Brewster AS, Sauter NK, Cohen AE et al. : Architecture of the synaptotagmin–SNARE machinery for neuronal exocytosis. Nature 2015, 525:62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]; High-resolution crystal structure of the complex between synaptotagmin-1 and the neuronal SNARE complex. The main interface (so-called primary interface) was found to be essential for evoked neurotransmitter release.
- 14.Kyoung M, Srivastava A, Zhang Y, Diao J, Vrljic M, Grob P, Nogales E, Chu S, Brunger AT: In vitro system capable of differentiating fast Ca2+-triggered content mixing from lipid exchange for mechanistic studies of neurotransmitter release. Proc Natl Acad Sci U S A 2011, 108:E304–E313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Diao J, Grob P, Cipriano DJ, Kyoung M, Zhang Y, Shah S, Nguyen A, Padolina M, Srivastava A, Vrljic M et al. : Synaptic proteins promote calcium-triggered fast transition from point contact to full fusion. eLife 2012, 1:e00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lai Y, Diao J, Cipriano DJ, Zhang Y, Pfuetzner RA, Padolina MS, Brunger AT: Complexin inhibits spontaneous release and synchronizes Ca2+-triggered synaptic vesicle fusion by distinct mechanisms. eLife 2014, 3:e03756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.••.Lai Y, Choi UB, Leitz J, Rhee HJ, Lee C, Altas B, Zhao M, Pfuetzner RA, Wang AL, Brose N et al. : Molecular mechanisms of synaptic vesicle priming by Munc13 and Munc18. Neuron 2017, 95:591–607.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]; Single molecule FRET studies and fusion experiments with reconstituted SNAREs, synaptotagmin-1, and complexin-1 show that Munc18 and Munc13 cooperate to properly assembly the ternary SNARE complex. Either Munc18 or Munc13 alone is insufficient for proper assembly.
- 18.Pérez-Lara Á, Thapa A, Nyenhuis SBB, Nyenhuis DAA, Halder P, Tietzel M, Tittmann K, Cafiso DSSD, Jahn R, Ames B et al. : PtdInsP2 and PtdSer cooperate to trap synaptotagmin-1 to the plasma membrane in the presence of calcium. eLife 2016, 5:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bai J, Tucker WC, Chapman ER: PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nat Struct Mol Biol 2004, 11:36–44. [DOI] [PubMed] [Google Scholar]
- 20.Koch H, Hofmann K, Brose N: Definition of Munc13-homology-domains and characterization of a novel ubiquitously expressed Munc13 isoform. Biochem J 2000, 349:247–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schupp M, Malsam J, Ruiter M, Scheutzow A, Wierda KDB, Sollner TH, Sorensen JB: Interactions between SNAP-25 and synaptotagmin-1 are involved in vesicle priming, clamping spontaneous and stimulating evoked neurotransmission. J Neurosci 2016, 36:11865–11880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guan Z, Bykhovskaia M, Jorquera RA, Sutton RB, Akbergenova Y, Littleton JT: A synaptotagmin suppressor screen indicates SNARE binding controls the timing and Ca2+ cooperativity of vesicle fusion. eLife 2017, 6:1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choi UB, Strop P, Vrljic M, Chu S, Brunger AT, Weninger KR: Single-molecule FRET-derived model of the synaptotagmin 1-SNARE fusion complex. Nat Struct Mol Biol 2010, 17:318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.••.Brewer KD, Bacaj T, Cavalli A, Camilloni C, Swarbrick JD, Liu J, Zhou A, Zhou P, Barlow N, Xu J et al. : Dynamic binding mode of a Synaptotagmin-1-SNARE complex in solution. Nat Struct Mol Biol 2015, 22:555–564. [DOI] [PMC free article] [PubMed] [Google Scholar]; NMR studies of complexes between Ca2+-bound synaptotagmin-1 and the SNARE complex suggest that there are multiple binding modes between these molecules in solution.
- 25.Choi UB, Strop P, Vrljic M, Chu S, Brunger AT, Weninger KR: Single-molecule FRET-derived model of the synaptotagmin 1-SNARE fusion complex. Nat Struct Mol Biol 2010, 17:318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shi L, Shen Q-T, Kiel A, Wang J, Wang H-W, Melia TJ, Rothman JE, Pincet F : SNARE proteins: one to fuse and three to keep the nascent fusion pore open. Science 2012, 335:1355–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sinha R, Ahmed S, Jahn R, Klingauf J: Two synaptobrevin molecules are sufficient for vesicle fusion in central nervous system synapses. Proc Natl Acad Sci U S A 2011, 108:14318–14323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fernández-Busnadiego R, Asano S, Oprisoreanu AM, Sakata E, Doengi M, Kochovski Z, Zürner M, Stein V, Schoch S, Baumeister W et al. : Cryo-electron tomography reveals a critical role of RIM1 α in synaptic vesicle tethering. J Cell Biol 2013, 201:725–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cole AA, Chen X, Reese TS: A network of three types of filaments organizes synaptic vesicles for storage, mobilization, and docking. J Neurosci 2016, 36:3222–3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gipson P, Fukuda Y, Danev R, Lai Y, Chen D-H, Baumeister W, Brunger AT: Morphologies of synaptic protein membrane fusion interfaces. Proc Natl Acad Sci USA 2017, 114:9110–9115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aeffner S, Reusch T, Weinhausen B, SaldittT: Energetics of stalk intermediates in membrane fusion are controlled by lipid composition. Proc Natl Acad Sci USA 2012, 109:E1609–E1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martens S, Kozlov MM, McMahon HT: How synaptotagmin promotes membrane fusion. Science (80-) 2007, 316:1205–1208. [DOI] [PubMed] [Google Scholar]
- 33.Hui E, Johnson CP, Yao J, Dunning FM, Chapman ER: Synaptotagmin-mediated bending of the target membrane is a critical step in Ca2+-regulated fusion. Cell 2009, 138:709–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee J, Guan Z, Akbergenova Y, Littleton JT: Genetic analysis of synaptotagmin C2 domain specificity in regulating spontaneous and evoked neurotransmitter release. J Neurosci 2013, 33:187–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wu D, Bacaj T, Morishita W, Goswami D, Arendt KL, Xu W, Chen L, Malenka RC, Sudhof TC: Postsynaptic synaptotagmins mediate AMPA receptor exocytosis during LTP. Nature 2017, 544:316–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kavalali ET: The mechanisms and functions of spontaneous neurotransmitter release. Nat Rev Neurosci 2015, 16:5–16. [DOI] [PubMed] [Google Scholar]
- 37.Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M et al. : Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science (New York, NY) 2000, 287:864–869. [DOI] [PubMed] [Google Scholar]
- 38.Misura KM, Scheller RH, Weis WI: Three-dimensional structure of the neuronal-Sec1-syntaxin 1a complex. Nature 2000, 404:355–362. [DOI] [PubMed] [Google Scholar]
- 39.Burkhardt P, Hattendorf DA, Weis WI, Fasshauer D: Munc18a controls SNARE assembly through its interaction with the syntaxin N-peptide. EMBO J 2008, 27:923–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sudhof TC: The presynaptic active zone. Neuron 2012, 75:11–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Basu J, Shen N, Dulubova I, Lu J, Guan R, Guryev O, Grishin NV, Rosenmund C, Rizo J: A minimal domain responsible for Munc13 activity. Nat Struct Mol Biol 2005, 12:1017–1018. [DOI] [PubMed] [Google Scholar]
- 42.Ma C, Su L, Seven AB, Xu Y, Rizo J: Reconstitution of the vital functions of Munc18 and Munc13 in neurotransmitter release. Science 2013, 339:421–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang X, Wang S, Sheng Y, Zhang M, Zou W, Wu L, Kang L, Rizo J, Zhang R, Xu T et al. : Syntaxin opening by the MUN domain underlies the function of Munc13 in synaptic-vesicle priming. Nat Struct Mol Biol 2015, 22:547–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hammarlund M, Palfreyman MT, Watanabe S, Olsen S, Jorgensen EM: Open syntaxin docks synaptic vesicles. PLoS Biol 2007, 5:e198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weninger K, Bowen ME, Chu S, Brunger AT: Single-molecule studies of SNARE complex assembly reveal parallel and antiparallel configurations. Proc Natl Acad Sci USA 2003, 100:14800–14805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.••.Choi UB, Zhao M, Zhang Y, Lai Y, Brunger AT: Complexin induces a conformational change at the membrane-proximal C-terminal end of the SNARE complex. eLife 2016, 5:e16886. [DOI] [PMC free article] [PubMed] [Google Scholar]; Single molecule FRET studies show that both properly and improperly assembled ternary SNARE complexes are disassembled by NSF/αSNAP.
- 47.Imig C, Min S-W, Krinner S, Arancillo M, Rosenmund C, Sudhof TC, Rhee J, Brose N, Cooper BH: The morphological and molecular nature of synaptic vesicle priming at presynaptic active zones. Neuron 2014, 84:416–431. [DOI] [PubMed] [Google Scholar]
- 48.Liu X, Seven AB, Camacho M, Esser V, Xu J, Trimbuch T, Quade B, Su L, Ma C Rosenmund C et al. : Functional synergy between the Munc13 C-terminal C1 and C2 domains. eLife 2016, 5:1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Junge HJ, Rhee J, Jahn O, Varoqueaux F, Spiess J, Waxham MN, Rosenmund C, Brose N: Calmodulin and Munc13 Form a Ca2+ sensor/effector complex that controls short-term synaptic plasticity. Cell 2004, 118:389–401. [DOI] [PubMed] [Google Scholar]
- 50.Xue R, Briguglio JS, Figueroa AG, Pearce RA, Ruhl DA, Chapman ER: Doc2-mediated superpriming supports synaptic augmentation. Proc Natl Acad Sci uSa 2018, 115:E5605–E5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ma C, Li W, Xu Y, Rizo J: Munc13 mediates the transition from the closed syntaxin-Munc18 complex to the SNARE complex. Nat Struct Mol Biol 2011, 18:542–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sitarska E, Xu J, Park S, Liu X, Quade B, Stepien K, Sugita K, Brautigam CA, Sugita S, Rizo J: Autoinhibition of Munc18–1 modulates synaptobrevin binding and helps to enable Munc13-dependent regulation of membrane fusion. eLife 2017, 6:618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weninger K, Bowen ME, Choi UB, Chu S, Brunger AT: Accessory proteins stabilize the acceptor complex for synaptobrevin, the 1:1 Syntaxin/SNAP-25 complex. Structure 2008, 16:308–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guan R, Dai H, Rizo J: Binding of the Munc13–1 MUN domain to membrane-anchored SNARE complexes. Biochemistry 2008, 47:1474–1481. [DOI] [PubMed] [Google Scholar]
- 55.•.Wang S, Li Y, Gong J, Ye S, Yang X, Zhang R, Ma C: Munc18 and Munc13 serve as a functional template to orchestrate neuronal SNARE complex assembly. Nat Commun 2019, 10:69. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study reveals an interaction between the membrane proximal region of synaptobrevin and Munc13, and that this interaction is important for Munc13 function.
- 56.••.Baker RW, Jeffrey PD, Zick M, Phillips BP, Wickner WT, Hughson FM: A direct role for the Sec1/Munc18-family protein Vps33 as a template for SNARE assembly. Science (80-) 2015, 349:1111–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]; Structures of complexes between the Munc18 homolog Vps33 and the vacuolar yeast SNARE Vam3 and between Vps33 and Nyv1 suggest that Vps33 provides a template for SNARE complex assembly.
- 57.••.Jiao J, He M, Port SA, Baker RW, Xu Y, Qu H, Xiong Y, Wang Y, Jin H, Eisemann TJ et al. : Munc18-1 catalyzes neuronal SNARE assembly by templating SNARE association. eLife 2018, 7:1–32. [DOI] [PMC free article] [PubMed] [Google Scholar]; Single molecule force measurements suggest that Munc18 helps to promote proper assembly of the SNARE complex.
- 58.Brocker C, Kuhlee A, Gatsogiannis C, Balderhaar HJ, Honscher C, Engelbrecht-Vandre S, Ungermann C, Raunser S: Molecular architecture of the multisubunit homotypic fusion and vacuole protein sorting (HOPS) tethering complex. Proc Natl Acad Sci U S A 2012, 109:1991–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mei K, Li Y, Wang S, Shao G, Wang J, Ding Y, Luo G, Yue P, Liu J-J, Wang X et al. : Cryo-EM structure of the exocyst complex. Nat Struct Mol Biol 2018, 25:139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.••.Xu J, Camacho M, Xu Y, Esser V, Liu X, Trimbuch T, Pan Y-Z, Ma C, Tomchick DR, Rosenmund C et al. : Mechanistic insights into neurotransmitter release and presynaptic plasticity from the crystal structure of Munc13–1 C 1 C 2 BMUN. eLife 2017, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]; In this work, the first structure of the C1C2BMUN fragment of Munc13 is presented.
- 61.••.Michelassi F, Liu H, Hu Z, Dittman JS, Michelassi F, Liu H, Hu Z, Dittman JS: Article article A C1-C2 module in Munc13 inhibits calcium-dependent neurotransmitter release. Neuron 2017, 95:577–590. [DOI] [PMC free article] [PubMed] [Google Scholar]; Deletion of the C1 or C2B domains of Munc13 increases evoked release.
- 62.Rizo J: Mechanism of neurotransmitter release coming into focus. Protein Sci 2018, 27:1364–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.SollnerTH Bennett MK, Whiteheart SW Scheller RH, Rothman JE: A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell 1993, 75:409–418. [DOI] [PubMed] [Google Scholar]
- 64.Mayer A, Wickner W, Haas A: Sec18p (NSF)-driven release of Sec17p (alpha-SNAP) can precede docking and fusion of yeast vacuoles. Cell 1996, 85:83–94. [DOI] [PubMed] [Google Scholar]
- 65.Hanson PI, Roth R, Morisaki H, Jahn R, Heuser JE: Structure and conformational changes in NSF and its membrane receptor complexes visualized by quick-freeze/deep-etch electron microscopy. Cell 1997, 90:523–535. [DOI] [PubMed] [Google Scholar]
- 66.Lenzen C, Steinmann D, Whiteheart S, Weis W: Crystal structure of the hexamerization domain of N-ethylmaleimide-sensitive fusion protein. Cell 1998, 94:525–536. [DOI] [PubMed] [Google Scholar]
- 67.Yu RC, Hanson PI, Jahn R, Brünger AT: Structure of the ATP-dependent oligomerization domain of N-ethylmaleimide sensitive factor complexed with ATP. Nat Struct Biol 1998, 5:803–811. [DOI] [PubMed] [Google Scholar]
- 68.May AP, Misura KM, Whiteheart SW, Weis WI: Crystal structure of the amino-terminal domain of N-ethylmaleimide-sensitive fusion protein. Nat Cell Biol 1999, 1:175–182. [DOI] [PubMed] [Google Scholar]
- 69.Yu RC, Jahn R, Brunger AT: NSF N-terminal domain crystal structure. Mol Cell 1999, 4:97–107. [DOI] [PubMed] [Google Scholar]
- 70.DeLaBarre B, Brunger ATAT: Complete structure of p97/valosin-containing protein reveals communication between nucleotide domains. Nat Struct Mol Biol 2003, 10:856–863. [DOI] [PubMed] [Google Scholar]
- 71.Davies JM, Brunger AT, Weis WI: Improved structures of full-length p97, an AAA ATPase: implications for mechanisms of nucleotide-dependent conformational change. Structure 2008, 16:715–726. [DOI] [PubMed] [Google Scholar]
- 72.Huyton T, Pye VE, Briggs LC, Flynn TC, Beuron F, Kondo H, Ma J, Zhang X, Freemont PS: The crystal structure of murine p97/VCP at 3.6 A. J Struct Biol 2003, 144:337–348. [DOI] [PubMed] [Google Scholar]
- 73.Furst J: Electron cryomicroscopy structure of N-ethylmaleimide sensitive factor at 11A resolution. EMBO J 2003, 22:4365–4374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chang L-F, Chen S, Liu C-C, Pan X, Jiang J, Bai X-C, Xie X, Wang H-W, Sui S-F: Structural characterization of full-length NSF and 20S particles. Nat Struct Mol Biol 2012, 19:268–275. [DOI] [PubMed] [Google Scholar]
- 75.••.Zhao M, Wu S, Zhou Q, Vivona S, Cipriano DJ, Cheng Y, Brunger AT: Mechanistic insights into the recycling machine of the SNARE complex. Nature 2015, 518:61–67. [DOI] [PMC free article] [PubMed] [Google Scholar]; This work describes the first single particle structure of the NSF/αSNAP/SNARE revealing the interactions between αSNAP and NSF, and αSNAP and the neuronal SNARE complex. These interactions had been elusive in all previous studies.
- 76.Vivona S, Cipriano DJ, O’Leary S, Li YH, Fenn TD, Brunger AT: Disassembly of All SNARE complexes by N-ethylmaleimide-sensitive factor (NSF) is initiated by a conserved 1:1 interaction between a-soluble NSF attachment protein (SNAP) and SNARE complex. J Biol Chem 2013, 288:24984–24991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Cipriano DJ, Jung J, Vivona S, Fenn TD, Brunger AT, Bryant Z: Processive ATP-driven substrate disassembly by the N-ethylmaleimide-sensitive factor (NSF) molecular machine. J Biol Chem 2013, 288:23436–23445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.••.White KI, Zhao M, Choi UB, Pfuetzner RA, Brunger AT: Structural principles of SNARE complex recognition by the AAA+ protein NSF. eLife 2018, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]; The EM structure of the NSF/αSNAP/SNARE complex at ~3.9 Å resolution reveals a direct interaction between SNAP-25A and the D1 ring pore of NSF.
- 79.Gates SN, Yokom AL, Lin J, Jackrel ME, Rizo AN, Kendsersky NM, Buell CE, Sweeny EA, Mack KL, Chuang E et al. : Ratchet-like polypeptide translocation mechanism of the AAA+ disaggregase Hsp104. Science 2017, 1052:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Han H, Monroe N, Sundquist WI, Shen PS, Hill CP: The AAA ATPase Vps4 binds ESCRT-III substrates through a repeating array of dipeptide-binding pockets. eLife 2017, 6:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.de la Pena AH, Goodall EA, Gates SN, Lander GC, Martin A: Substrate-engaged 26 S proteasome structures reveal mechanisms for ATP-hydrolysis-driven translocation. Science (80-) 2018, 362:eaav0725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dong Y, Zhang S, Wu Z, Li X, Wang WL, Zhu Y, Stoilova-McPhie S, Lu Y, Finley D, Mao Y: Cryo-EM structures and dynamics of substrate-engaged human 26S proteasome. Nature 2019, 565:49–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Majumder P, Rudack T, Beck F, Danev R, Pfeifer G, Nagy I, Baumeister W: Cryo-EM structures of the archaeal PAN-proteasome reveal an around-the-ring ATPase cycle. Proc Natl AcadSci U S A 2018. 10.1073/pnas.1817752116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Choi UB, Zhao M, White KI, Pfuetzner RA, Esquivies L, Zhou Q, Brunger AT: NSF-mediated disassembly of on- and off-pathway SNARE complexes and inhibition by complexin. eLife 2018, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Winter U, Chen X, Fasshauer D: A conserved membrane attachment site in alpha-SNAP facilitates N-ethylmaleimide-sensitive factor (NSF)-driven SNARE complex disassembly. J Biol Chem 2009, 284:31817–31826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.•.Prinslow EA, Stepien KP, Pan Y-Z, Xu J, Rizo J: Multiple factors • maintain assembled trans-SNARE complexes in the presence of NSF and αSNAP. eLife 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study shows that the properly assembled trans-SNARE complex is protected against NSF/αSNAP disassembly when Munc18 and Munc13, complexin-1, and synaptotagmin-1 are present.
- 87.Shin O-H, Lu J, Rhee J-S, Tomchick DR, Pang ZP, Wojcik SM, Camacho-Perez M, Brose N, Machius M, Rizo J et al. : Munc13 C2B domain is an activity-dependent Ca2+ regulator of synaptic exocytosis. Nat Struct Mol Biol 2010, 17:280–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ryu J-K, Min D, Rah S-H, Kim SJ, Park Y, Kim H, Hyeon C, Kim HM, Jahn R, Yoon T-Y: Spring-loaded unraveling of a single SNARE complex by NSF in one round of ATP turnover. Science (80-) 2015, 347:1485–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Sillitoe I, Dawson N, Lewis TE, Das S, Lees JG, Ashford P, Tolulope A, Scholes HM, Senatorov I, Bujan A et al. : CATH: expanding the horizons of structure-based functional annotations for genome sequences. Nucleic Acids Res 2018, 47:280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]