

Abstract
We previously engineered the tryptophan synthase β-subunit (TrpB), which catalyzes the condensation of l-serine and indole to l-tryptophan, to synthesize a range of noncanonical amino acids from l-serine and indole derivatives or other nucleophiles. Here we employ directed evolution to engineer TrpB to accept 3-substituted oxindoles and form C–C bonds leading to new quaternary stereocenters. Initially, the variants that could use 3-substituted oxindoles preferentially formed N–C bonds by attacking N1 of the substrate. Protecting N1 encouraged evolution towards C-alkylation, which persisted when protection was removed. Six generations of directed evolution resulted in TrpB Pfquat with a 400-fold improvement in activity for alkylation of 3-substituted oxindoles and the ability to selectively form a new, all-carbon quaternary stereocenter at the γ-position of the amino acid products. The enzyme can also alkylate and form all-carbon quaternary stereocenters on structurally similar lactones and ketones, where it exhibits excellent regioselectivity for the tertiary carbon. The configurations of the γ-stereocenters of two of the products were determined via microcrystal electron diffraction (MicroED), and we report the MicroED structure of a small molecule obtained using the Falcon III direct electron detector. Highly thermostable and expressed at >500 mg/L E. coli culture, TrpB Pfquat offers an efficient, sustainable, and selective platform for the construction of diverse noncanonical amino acids bearing all-carbon quaternary stereocenters.
Keywords: TrpB, noncanonical amino acid, asymmetric catalysis, quaternary carbon, oxindole, C–C bond formation, directed evolution
Graphical Abstract

Introduction
In order to meet nature’s demand for chiral and selective biotransformations, enzymes have evolved to exert high levels of stereo-, regio-, and chemoselective control. Directed evolution can similarly tailor enzyme selectivity for target-oriented biocatalysis.1–2 We have used directed evolution to engineer variants of the β-subunit of tryptophan synthase (TrpB), a pyridoxal 5’-phosphate (PLP)-dependent enzyme, to synthesize a bevy of noncanonical amino acids (ncAAs). Natively, TrpB catalyzes the formation of a C–C bond between l-serine and indole to construct l-tryptophan (Figure 1a).3 We have evolved this enzyme to accept a variety of indole analogues and nitroalkanes with activated sp3-carbon atoms as nucleophiles (Figure 1b).4–9 However, these studies revealed limitations including chemo- and stereoselective control that need to be addressed in order to realize TrpB’s broad potential as a biocatalyst for C–C bond formation.
Figure 1.

a) Catalytic cycle of amino acid synthesis using TrpB. b) Previous biocatalytic reactions catalyzed by variants of TrpB6–7, 9 and proposed stereo- and chemo- selective reaction using engineered TrpB.
Our previous efforts first focused on engineering TrpB to accept nucleophiles bearing structural and electronical similarity to indole. We recently found we could branch out to nitroalkanes with some TrpB variants, taking advantage of the known nucleophilicity of these substrates that arises from the low pKa of the C–H bond adjacent to the electron-withdrawing nitro group.9,10 We observed, however, that TrpBs do not catalyze C–C bond formation if the substrate is only marginally nucleophilic. Nucleophilic atoms such as nitrogen and sulfur instead serve as competing donors for nucleophilic attack, leading to the formation of C–N and C–S bonds rather than the desired C–C bond in nonnatural substrates.3, 11
TrpB is highly stereoselective and retains the absolute configuration of the Cα of l-serine after product formation (Figure 1b).4–8 Substrates with nucleophilic sp3-hybridized carbons have the ability to form a new quaternary carbon at the γ-position, where TrpB exerts unknown stereoselectivity. This selectivity has not yet been explored because indole derivatives with an sp2-carbon lack chirality, and the γ carbon of nitroalkanes epimerizes (Figure 1b).9
All-carbon quaternary stereocenters are found in a raft of biologically active natural products,12 but are notoriously challenging to prepare. This is reflected by the fact that of the 200 top-grossing pharmaceuticals in 2012, none bear quaternary carbons introduced by chemical synthesis. The enzymes that synthesize quaternary carbon-bearing natural products include radical-SAM dependent enzymes, which use a Fe-S-cluster, prenyltransferases that prenylate the 3-position of tryptophan, and cyclases that synthesize natural products including valenic acid or cholesterol.13–16 Recent developments in synthetic chemistry have led to new strategies for enantioselective synthesis of quaternary carbon centers using chiral catalysts such as palladium complexes.17 The repertoire of C–C bond-forming enzymes used in biocatalysis is mostly limited to aldolases and ThDP-dependent enzymes.18 Because both of these enzyme classes use activated carbonyl compounds as nucleophiles, they are restricted in their substrate scope to aldehydes and ketones and cannot be used to form quaternary carbon centers.
In this study, we set out to address the limitations of TrpB by engineering the enzyme for unprecedented quaternary stereocenter formation with 3-substituted oxindoles. We challenged TrpB with 3-methyloxindole 1 (rac), which has a tertiary, sp3-hybridized carbon atom that could be used to form a quaternary carbon (Figure 1b). In addition, 3-methyloxindole 1 contains two nucleophilic atoms, C3 and N1, with which we could probe chemoselectivity for C–C bond formation. Engineering TrpB for activity on 3-methyloxindole 1 would provide biocatalytic access to 3,3-disubstituted oxindoles (Figure 2a), motifs present in a broad range of natural and synthetic biologically active compounds (Figure 2b).19–20 Various approaches to form 3,3-disubstituted oxindoles have been explored in synthetic chemistry, including C–H activation and chiral catalysis using phosphoramides or alkaloid-derived urea catalysts.21–23 These methods may require protection of the nitrogen and/or activation of C3 and almost always use high chiral catalyst loadings. In contrast, enzymes function under mild reaction conditions and can exert unparalleled stereocontrol. However, an enzyme with this activity has not been reported.
Figure 2.
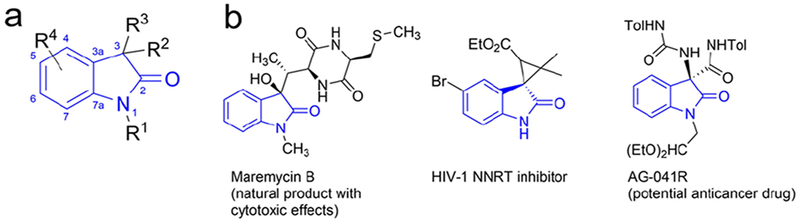
a) Core structure of substituted oxindoles. b) Examples of natural products/bioactive molecules derived from 3,3-disubstituted oxindoles.21, 24–25
Results
Screening for activity with 3-methyloxindole
In previous work where TrpB was engineered for the synthesis of tryptophan derivatives, enzymes from the thermophilic organisms Pyrococcus furiosus (PfTrpB) and Thermotoga maritima (TmTrpB) had sufficient activity with the indole derivatives to enable screening for beneficial mutations.3–4, 6–8 When we screened the wild-type enzymes and engineered variants from our collection via mass spectrometry for activity on 3-methyloxindole 1 as a nucleophile and l-serine 2 as an electrophile, several variants exhibited activity, with up to 28% HPLC yield for the expected mass (SI, Figure 1). However, these enzymes formed the N-alkylated oxindole 3a (Figure 3a) rather than the desired C-alkylated product 3b. Certain variants also produced traces of a second, more polar peak, which corresponds to the C-alkylation product 3b (SI, Figure 2). As both 3a and 3b have the same molecular weight and similar retention times on reverse-phase HPLC, rapid screening for the C-alkylation product 3b proved difficult.
Figure 3.
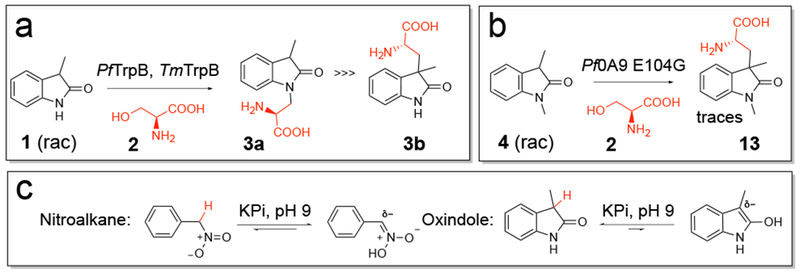
a) N-alkylation using PfTrpB and TmTrpB variants. b) Pf0A9 E104G shows trace activity for C-alkylation of 3-substituted oxindoles if the nitrogen of the nucleophile is protected. c) Tautomeric forms of nitromethylbenzene and 3-methyloxindole 1 in aqueous solution under standard reaction conditions. While both tautomers of the nitroalkane could be detected in NMR experiments, only the keto form of the oxindole was visible, indicating that the equilibrium is left-shifted.
Like nitroalkanes, oxindoles can theoretically tautomerize to activate C3 by forming an enolate species with an sp2-hybridized carbon center (Figure 3c). To investigate the equilibrium between the enolate and keto forms, NMR spectra were acquired for 3-methyloxindole 1 and nitromethylbenzene in aqueous solution. Only the nitroalkane displayed a shift in equilibrium towards sp2-hybridization at C3; no activated tautomer was observed for the oxindole (SI, Figure 3). This finding indicates that TrpB needs to overcome a high activation barrier to enable nucleophilic attack by the C3 of 3-methyloxindole 1.
Reasoning that protecting the nitrogen could coerce the enzyme to favor C-alkylation, we investigated whether the enzyme could accept 1,3-dimethyloxindole 4 (rac) (Figure 3b). This reaction would construct 3,3-disubstituted oxindoles with a methylated nitrogen, a common motif in natural products (Figure 2b). A panel of engineered and wild-type TrpB enzymes was evaluated and the highest activity for formation of the 3,3-dialkylated oxindole 13 was found with Pf0A9 E104G, a variant previously evolved for nitrotryptophan synthesis.6 However, even this top variant showed only trace activity (total turnover number (TTN) <1).
Engineering TrpB Pf0A9 E104G for improved C-alkylation activity
To improve the activity of Pf0A9 E104G on the N-protected 1,3-dimethyloxindole 4, we introduced random mutations via error-prone PCR and assayed variants for activity with an LC-MS based screen (SI, Figure 4). One variant was identified with two mutations (W172R and S185T) that gave a 6-fold improvement in product formation compared to parent. Interestingly, W172R and S185T were not beneficial as single mutations, indicating epistasis (SI, Figure 5). To evaluate whether our strategy for modifying chemoselectivity was effective, we tested this variant for activity on 3-methyloxindole 1, which lacks protection on the nitrogen. Gratifyingly, the main product was the C-alkylated oxindole (Figure 4a). Thus, a single generation of directed evolution was sufficient to switch the chemoselectivity from N- to C-alkylation.
Figure 4.

a) Ratio of N- to C-alkylation of 3-methyloxindole 1 using the parent enzyme and the best variants after the first two generations of directed evolution. b) Proximity of R172 and T185 to the active site of TrpB as shown on the structure of an evolved variant of TrpB with the PLP-bound amino-acrylate (PDB ID: 5vm5). c) Locations of all beneficial mutations collected after six generations of directed evolution (See SI Section 9.4 for screening effort). d) Total turnover number (TTN) for the best variant after each generation of directed evolution, determined using 100 mM 3-methyloxindole 1 and l-serine 2 as substrates and 20 μM of purified enzyme. epPCR = error-prone PCR.
Notably, only the S185T mutation lies proximal to the active site and can interact directly with the substrate (Figure 4b). By contrast, W172R is 15 Å distal from the catalytic site and a direct interaction with T185 seems unlikely. One of the benefits of directed evolution is that it explores mutations that cannot be predicted easily.26–27 It is difficult to explain the effects of these mutations on the activity and chemoselectivity of TrpB, and they would not have been found with targeted mutagenesis. A second generation of random mutagenesis uncovered two additional mutations (R170C and V221A) that further doubled activity for oxindoles with and without nitrogen protection (Figure 4a+d).
Since only one of the four mutations in the best variant at this point is close to the active site, we postulated that the substrate binding pocket might still be optimized for indole, the natural nucleophile of TrpB, rather than for oxindole. We therefore constructed site-saturation mutagenesis libraries at seven residues that cover the nucleophile-binding site (SI, Figure 6a) and were previously shown to affect the activity of TrpB with nitroalkanes.9 Screening these libraries on the unprotected 3-methyloxindole 1 identified three beneficial mutations: G104D, I183E and V187W (SI, Figure 6b). A recombination library was constructed; the best variant contained only one of these mutations (V187W), which gave a 3-fold improvement in product formation.
Three further generations of directed evolution resulted in final variant Pfquat (Figure 4c+d) that exhibits >99% chemoselectivity toward C3. TrpB Pfquat expresses at high levels (>500 mg/L of Escherichia coli culture) and can be purified easily by heat treatment at 75 °C. As a result, we were able to synthesize 122 mg (52% isolated yield) of product 3b from a reaction using 1 mmol substrate 1 and Pfquat in cell lysate from 100 mL of E. coli culture.
Substrate scope
We next evaluated the substrate scope of TrpB Pfquat, which was evolved for activity on 1. We chose oxindoles with halogen substituents on the aromatic ring, a variety of substituents at the 3-position, and structurally related lactones and ketones (Figure 5a). Substrates with alkyl chains at the 3-position ranging from methyl to butyl (products 3b, 9-11) were accepted. Although longer alkyl groups led to reduced product formation, even bulky oxindoles are accepted by the enzyme. 3-hydroxyl oxindole is a structural motif in maremycin A, B, C and D and represents the core structure of the protease inhibitor TMC-95A.28 We tested and found activity for 3-hydroxyl oxindole, leading to product 12. The activity uncovered for these substrates can serve as a starting point for further directed evolution.
Figure 5.
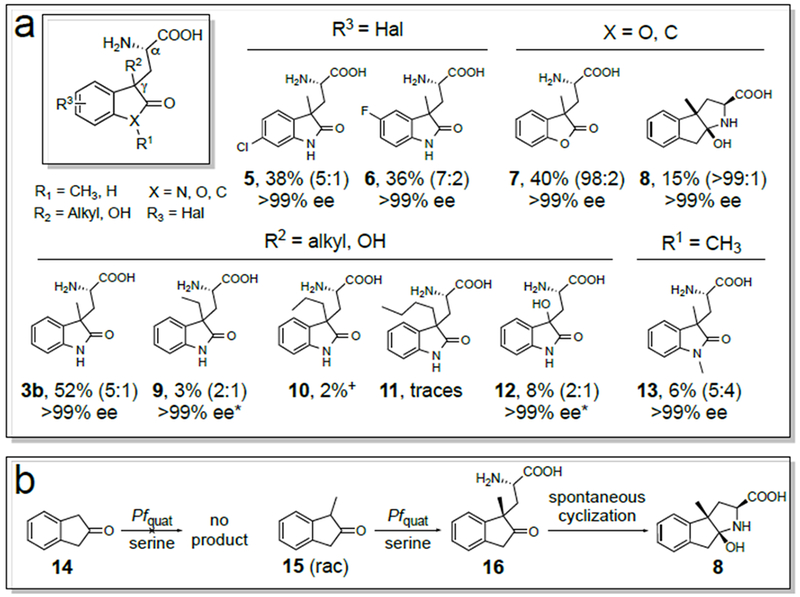
a) Scope of amino acid products using 50 mM of oxindole, lactone, or ketone nucleophile, 2 eq. of l-serine 2 and 0.2 mol% of purified TrpB Pfquat. The percentages reflect isolated yields. For compound 3b the reaction and purification conditions are further optimized (vide supra). +HPLC yield (See SI Section 9.5 for details). The diastereomeric ratio (in parentheses) refers to the configuration at the γ-position (S:R). The ee values represent the enantiomeric purity for both diastereomers. *The ee of only one diastereomer could be determined. b) Activity of TrpB Pfquat on 2-indanones.
In order to explore the ability of the enzyme to accept nucleophiles beyond oxindoles, we tested a tertiary lactone and a ketone. The versatility of this substrate manifold is demonstrated by the high conversion of a lactone to product 7, despite the purification conditions being optimized for product 3b. We challenged TrpB Pfquat with two different ketones, 2-indanone 14 and 1-methyl-2-indanone 15 (rac) (Figure 5b). Interestingly, while no activity was observed for substrate 14, which lacks a tertiary C1, Pfquat converted the 1-methylated ketone 15 to product 16 with 15% isolated yield and >95% regioselectivity for the tertiary carbon. Spontaneous nucleophilic attack of the free amino group on the carbonyl carbon leads to the final cyclic product 8.
A list of substrates not accepted by Pfquat for C–C-bond formation is shown in SI, Figure 7.
Determination of relative configuration
Since the products shown in Figure 5a have two stereocenters at the α- and γ-positions, four stereoisomers are theoretically possible. HPLC and NMR analysis of the products confirmed the formation of two diastereomers. Both diastereomers exhibit >99% ee, indicating that two of four possible stereoisomers predominate. From previous studies of TrpB in nature and biocatalysis, it is known that the enzyme does not catalyze the formation of d-amino acids at a measurable level. Thus, we inferred by analogy that the α-position remains S-configured while the γ-carbon is responsible for the two diastereomers (Figure 5a).
We employed MicroED to determine the configuration of the new quaternary stereocenter at the γ-position.29–30 Lyophilized powder of the isolated, major diastereomer of product 3b with >99% ee was applied directly onto an EM grid. Needle-shaped nanocrystals were identified, and continuous rotation MicroED30–31 data were collected. The data from two nanocrystals were merged to increase completeness, and the structure was solved by ab initio direct methods to 0.9 Å resolution (Figure 6a). We were surprised to discover that the structure represented two enantiomers in a centric space group, where both stereocenters are either S- or R-configured. We reasoned that this compound only crystallizes as a racemic mixture under these conditions. Because MicroED is a very sensitive method which allows structural determination using nanocrystals, traces of the R-configured amino acid were sufficient for the formation of the racemic crystals.
Figure 6.
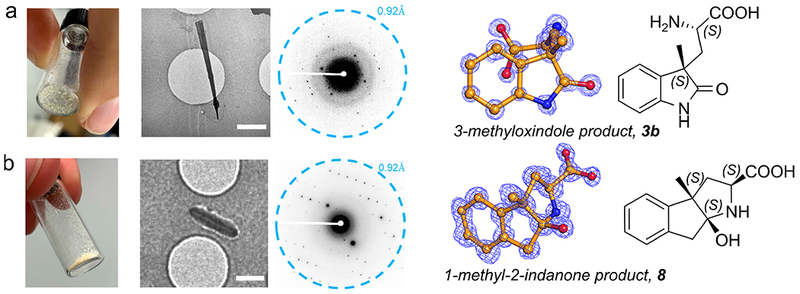
Structures of a) the ncAA product derived from 3-methyloxindole 1, which was obtained as a resin-like material and b) the cyclized ncAA product derived from 1-methyl-2-indanone 15, which was obtained as a white/yellow powder. Both products 3b and 8 formed nanocrystals on an EM grid. These nanocrystals diffracted to 0.9 Å resolution. Scale bars are 1 μm. 2Fo-Fc density is contoured at the 2σ level.
Based on the overwhelming preference of TrpB to form amino acids with an S-configured α-carbon, we deduced that the S, S stereo centered mirror was the predominant configuration. In order to validate this observation with a second, independent structural determination, we also collected MicroED data from compound 8 (Figure 6b). This structure was solved using ab initio direct methods in an acentric space group using data from two nanocrystals, with a single molecule in the asymmetric unit. The structure was solved with the second quaternary center in the S-configuration. We used the Falcon III direct electron detector, which is more commonly used in imaging applications, to determine this structure. The first MicroED structures of a protein and peptide using this detector have recently been obtained32–33 and the aforementioned structure is the first of a small molecule solved using the Falcon III detector. By analogy to these two structures using chemical shifts from 1H NMR data, we assigned the relative configuration of the other products shown in Figure 5a. In all cases, the S-configuration at the γ-position predominates.
Discussion
Unlocking new reactivity of TrpB
TrpB has been engineered to synthesize a diverse array of l-tryptophan analogues and other noncanonical amino acids. The enormous variety of ncAAs found in natural products and useful for biotechnological applications extends far beyond the current scope of TrpB,34–35 which has mostly been limited to specific sp2–sp3 carbon bond formation between l-serine and indole analogues.
In this study, we extended TrpB’s scope to the reaction between l-serine 2 and a variety of oxindoles, a lactone, and a ketone. While TrpB variants capable of using 3-methyloxindole 1 form an N–C bond between l-serine and N1, directed evolution generated a Pyrococcus furiosus TrpB variant that exhibits excellent chemoselectivity for C–C bond formation and stereoselective control at both the α- and γ-position of the product. The stereoselectivity of Pfquat is substrate-dependent and decreases when bulkier substituents are present at the α-position or at N1. Gratifyingly, both the lactone- and the ketone-based products 7 and 8 were produced with excellent diastereoselectivity of more than 95:5.
The desired oxindole-based ncAAs were obtained from an enzyme that attains >400-fold improved total turnover numbers while retaining high thermostability and expression level (>500 mg protein/L E. coli culture). The range of biotechnologically relevant ncAAs encompasses those with sp3 or sp2 C–C bonds as well as C–N, C–S and C–O bonds.35 This engineering approach demonstrates that the chemoselectivity of TrpB can be switched between different, competing functional groups. Our strategy provides proof of principle that the chemoselectivity and stereoselectivity of TrpB may be tailored to expand its portfolio of noncanonical amino acid products well beyond analogues of tryptophan.
Biocatalytic synthesis of 3,3-disubstituted oxindoles
We reasoned that TrpB should be able to use activated tertiary carbons as nucleophiles to form quaternary carbon centers. Oxindoles were chosen because they possess an activated tertiary carbon and 3,3-disubstituted oxindoles represent a structural motif found in many natural products.21, 24–25 Two previous studies describe enzymatic approaches to the formation of molecules containing the oxindole substructure. Pietruszka et al. used laccase-driven oxidation of catechols which undergo a non-enzymatic Michael addition with N-protected oxindoles to form 3,3-disubstituted oxindoles;36 the enzyme did not directly catalyze arylation of the oxindole. Recently, Black et al. reported that ‘ene’-reductases can catalyze an abiological, radical cyclization reaction to form 3,3-disubstituted oxindoles from N-protected α-haloamides.37 The engineered TrpB described here directly functionalizes unprotected oxindoles at C3.
TrpB for selective quaternary carbon bond formation
The evolved TrpB variant Pfquat is not limited in its nucleophile scope to oxindoles. Reasoning that TrpB might also utilize heterocycles with similar properties to oxindoles, we tested the enzyme on non-oxindole substrates. Replacing N1 of 3-methyloxindole 1 with carbon or oxygen leads to lactone and ketone structures (Figure 5) that were also converted by TrpB Pfquat to the corresponding products 7 and 8. These results reveal a broader ability of Pfquat to utilize a diverse suite of carbonyl-containing nucleophiles bearing a tertiary C1. They also demonstrate the exquisite chemo- and regioselectivity of Pfquat for quaternary carbon bond formation.
Highlighting the regioselectivity of Pfquat is its ability to exclusively functionalize the tertiary carbon rather than the secondary carbon of ketone 15. Ketones and lactones bearing quaternary carbon centers at the α-position are structural motifs found in many natural products with medicinal properties.17, 38–39 Hence, much effort in organic synthesis has been invested to enable regioselective alkylation of α-substituted ketones to form quaternary carbon centers. Synthesizing such compounds, especially with high enantiomeric purity, can be very challenging, as the substituted product is more sterically hindered and thus kinetically disfavored.17, 40–41 By showing that substrate 1 is not enantioenriched over the course of an enzymatic reaction (SI, Figure 8a+b), we have ruled out classical kinetic resolution as the mechanism behind the exquisite enantio-control exerted by Pfquat. Thus, the enzyme is controlling the configuration of both stereocenters.
Pfquat represents a powerful biocatalytic platform for functionalizing α-carbons of ketones and lactones. Coupled with its ability to synthesize an array of 3,3-disubstituted oxindoles bearing all-carbon quaternary stereocenters, this genetically encoded catalyst is capable of providing access to valuable products with diverse properties. These products highlight the ability of TrpB to address chemo- and regioselectivity challenges while expanding the synthetic space available to biocatalysis.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the Deutsche Forschungsgemeinschaft (DFG) and by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number R01GM125887. The Gonen lab is supported by funds from the Howard Hughes Medical Institute. The authors thank David C. Miller and Nathaniel W. Goldberg for assistance with experiments. The authors thank David K. Romney, Patrick J. Almhjell, Susanne Bahr, Nicholas J. Porter, Professor Brian M. Stoltz (Caltech) and Sanel Suljic for helpful discussions. [Placeholder: Accession codes for MicroED structures.]
Footnotes
Publisher's Disclaimer: Just Accepted
Publisher's Disclaimer: “Just Accepted” manuscripts have been peer-reviewed and accepted for publication. They are posted online prior to technical editing, formatting for publication and author proofing. The American Chemical Society provides “Just Accepted” as a service to the research community to expedite the dissemination of scientific material as soon as possible after acceptance. “Just Accepted” manuscripts appear in full in PDF format accompanied by an HTML abstract. “Just Accepted” manuscripts have been fully peer reviewed, but should not be considered the official version of record. They are citable by the Digital Object Identifier (DOI®). “Just Accepted” is an optional service offered to authors. Therefore, the “Just Accepted” Web site may not include all articles that will be published in the journal. After a manuscript is technically edited and formatted, it will be removed from the “Just Accepted” Web site and published as an ASAP article. Note that technical editing may introduce minor changes to the manuscript text and/or graphics which could affect content, and all legal disclaimers and ethical guidelines that apply to the journal pertain. ACS cannot be held responsible for errors or consequences arising from the use of information contained in these “Just Accepted” manuscripts.
The authors declare the following competing financial interest: The contents of this paper are the subject of a patent application submitted by Caltech (M.D. inventor).
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Additional figures (Figures S1–S8), experimental procedures, crystallographic information (PDF) and NMR spectra (PDF).
REFERENCES
- 1.Devine PN; Howard RM; Kumar R; Thompson MP; Truppo MD; Turner NJ, Extending the application of biocatalysis to meet the challenges of drug development. Nature Reviews Chemistry 2018, 2 (12), 409–421. [Google Scholar]
- 2.Zeymer C; Hilvert D, Directed Evolution of Protein Catalysts. Annu Rev Biochem 2018, 87, 131–157. [DOI] [PubMed] [Google Scholar]
- 3.Buller AR; Brinkmann-Chen S; Romney DK; Herger M; Murciano-Calles J; Arnold FH, Directed evolution of the tryptophan synthase beta-subunit for stand-alone function recapitulates allosteric activation. Proc Natl Acad Sci U S A 2015, 112 (47), 14599–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Herger M; van Roye P; Romney DK; Brinkmann-Chen S; Buller AR; Arnold FH, Synthesis of beta-Branched Tryptophan Analogues Using an Engineered Subunit of Tryptophan Synthase. J Am Chem Soc 2016, 138 (27), 8388–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murciano-Calles J; Romney DK; Brinkmann-Chen S; Buller AR; Arnold FH, A Panel of TrpB Biocatalysts Derived from Tryptophan Synthase through the Transfer of Mutations that Mimic Allosteric Activation. Angew Chem Int Ed Engl 2016, 55 (38), 11577–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Romney DK; Murciano-Calles J; Wehrmuller JE; Arnold FH, Unlocking Reactivity of TrpB: A General Biocatalytic Platform for Synthesis of Tryptophan Analogues. J Am Chem Soc 2017, 139 (31), 10769–10776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boville CE; Romney DK; Almhjell PJ; Sieben M; Arnold FH, Improved Synthesis of 4-Cyanotryptophan and Other Tryptophan Analogues in Aqueous Solvent Using Variants of TrpB from Thermotoga maritima. J Org Chem 2018, 83 (14), 7447–7452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Boville CE; Scheele RA; Koch P; Brinkmann-Chen S; Buller AR; Arnold FH, Engineered Biosynthesis of beta-Alkyl Tryptophan Analogues. Angew Chem Int Ed Engl 2018, 57 (45), 14764–14768. [DOI] [PubMed] [Google Scholar]
- 9.Romney DK; Sarai NS; Arnold FH, Nitroalkanes as Versatile Nucleophiles for Enzymatic Synthesis of Noncanonical Amino Acids. ACS Catal 2019, 9 (9), 8726–8730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matthews WS; Bares JE; Bartmess JE; Bordwell FG; Cornforth FJ; Drucker GE; Margolin Z; McCallum RJ; McCollum GJ; Vanier NR, Equilibrium acidities of carbon acids. VI. Establishment of an absolute scale of acidities in dimethyl sulfoxide solution. J Am Chem Soc 1975, 97 (24), 7006–7014. [Google Scholar]
- 11.Phillips RS, Synthetic applications of tryptophan synthase. Tetrahedron-Asymmetr 2004, 15 (18), 2787–2792. [Google Scholar]
- 12.Quasdorf KW; Overman LE, Catalytic enantioselective synthesis of quaternary carbon stereocentres. Nature 2014, 516 (7530), 181–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fan AL; Winkelblech J; Li SM, Impacts and perspectives of prenyltransferases of the DMATS superfamily for use in biotechnology. Appl Microbiol Biot 2015, 99 (18), 7399–7415. [DOI] [PubMed] [Google Scholar]
- 14.Yeo YS; Nybo SE; Chittiboyina AG; Weerasooriya AD; Wang YH; Gongora-Castillo E; Vaillancourt B; Buell CR; DellaPenna D; Celiz MD; Jones AD; Wurtele ES; Ransom N; Dudareva N; Shaaban KA; Tibrewal N; Chandra S; Smillie T; Khan IA; Coates RM; Watt DS; Chappell J, Functional identification of valerena-1,10-diene synthase, a terpene synthase catalyzing a unique chemical cascade in the biosynthesis of biologically active sesquiterpenes in Valeriana officinalis. J Biol Chem 2013, 288 (5), 3163–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yokoyama K; Lilla EA, C-C bond forming radical SAM enzymes involved in the construction of carbon skeletons of cofactors and natural products. Nat Prod Rep 2018, 35 (7), 660–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nes WD, Biosynthesis of cholesterol and other sterols. Chem Rev 2011, 111 (10), 6423–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Y; Han SJ; Liu WB; Stoltz BM, Catalytic enantioselective construction of quaternary stereocenters: assembly of key building blocks for the synthesis of biologically active molecules. Acc Chem Res 2015, 48 (3), 740–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schmidt NG; Eger E; Kroutil W, Building Bridges: Biocatalytic C-C-Bond Formation toward Multifunctional Products. ACS Catal 2016, 6 (7), 4286–4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y; Zou Y; Brock NL; Huang T; Lan Y; Wang X; Deng Z; Tang Y; Lin S, Characterization of 2-Oxindole Forming Heme Enzyme MarE, Expanding the Functional Diversity of the Tryptophan Dioxygenase Superfamily. J Am Chem Soc 2017, 139 (34), 11887–11894. [DOI] [PubMed] [Google Scholar]
- 20.Kaur M; Singh M; Chadha N; Silakari O, Oxindole: A chemical prism carrying plethora of therapeutic benefits. Eur J Med Chem 2016, 123, 858–894. [DOI] [PubMed] [Google Scholar]
- 21.Cao ZY; Zhou F; Zhou J, Development of Synthetic Methodologies via Catalytic Enantioselective Synthesis of 3,3-Disubstituted Oxindoles. Acc Chem Res 2018, 51 (6), 1443–1454. [DOI] [PubMed] [Google Scholar]
- 22.Saraswat P; Jeyabalan G; Hassan MZ; Rahman MU; Nyola NK, Review of synthesis and various biological activities of spiro heterocyclic compounds comprising oxindole and pyrrolidine moities. Synth Commun 2016, 46 (20), 1643–1664. [Google Scholar]
- 23.Liu YL; Zhou J, Organocatalytic asymmetric synthesis of 3-difluoroalkyl 3-hydroxyoxindoles. Chem Commun (Camb) 2012, 48 (13), 1919–21. [DOI] [PubMed] [Google Scholar]
- 24.Jiang T; Kuhen KL; Wolff K; Yin H; Bieza K; Caldwell J; Bursulaya B; Wu TY; He Y, Design, synthesis and biological evaluations of novel oxindoles as HIV-1 non-nucleoside reverse transcriptase inhibitors. Part I. Bioorg Med Chem Lett 2006, 16 (8), 2105–8. [DOI] [PubMed] [Google Scholar]
- 25.Liu Y; Zhang L; Jia Y, Total synthesis of maremycins A, B, C1/C2, D1, and D2. Tetrahedron Lett 2012, 53 (6), 684–687. [Google Scholar]
- 26.Tracewell CA; Arnold FH, Directed enzyme evolution: climbing fitness peaks one amino acid at a time. Curr Opin Chem Biol 2009, 13 (1), 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilding M; Hong N; Spence M; Buckle AM; Jackson CJ, Protein engineering: the potential of remote mutations. Biochem Soc Trans 2019, 47 (2), 701–711. [DOI] [PubMed] [Google Scholar]
- 28.Albrecht BK; Williams RM, A concise, total synthesis of the TMC-95A/B proteasome inhibitors. Proc Natl Acad Sci U S A 2004, 101 (33), 11949–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi D; Nannenga BL; Iadanza MG; Gonen T, Three-dimensional electron crystallography of protein microcrystals. Elife 2013, 2, e01345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nannenga BL; Shi D; Leslie AGW; Gonen T, High-resolution structure determination by continuous-rotation data collection in MicroED. Nat Methods 2014, 11 (9), 927–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones CG; Martynowycz MW; Hattne J; Fulton TJ; Stoltz BM; Rodriguez JA; Nelson HM; Gonen T, The CryoEM Method MicroED as a Powerful Tool for Small Molecule Structure Determination. ACS Cent Sci 2018, 4 (11), 1587–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hattne J; Martynowycz MW; Penczek PA; Gonen T, MicroED with the Falcon III direct electron detector. IUCrJ 2019, 6 (Pt 5), 921–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ting CP; Funk MA; Halaby SL; Zhang Z; Gonen T; van der Donk WA, Use of a scaffold peptide in the biosynthesis of amino acid-derived natural products. Science 2019, 365 (6450), 280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Almhjell PJ; Boville CE; Arnold FH, Engineering enzymes for noncanonical amino acid synthesis. Chem Soc Rev 2018, 47 (24), 8980–8997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu CC; Schultz PG, Adding new chemistries to the genetic code. Annu Rev Biochem 2010, 79, 413–44. [DOI] [PubMed] [Google Scholar]
- 36.Pietruszka J; Wang C, Laccase-catalyzed 3-Arylation of 3-Substituted Oxindoles. ChemCatChem 2012, 4 (6), 782–785. [Google Scholar]
- 37.Black MJ; Biegasiewicz KF; Meichan AJ; Oblinsky DG; Kudisch B; Scholes GD; Hyster TK, Asymmetric Redox-Neutral Radical Cyclization Catalyzed by Flavin-Dependent ‘Ene’-Reductases. 10.26434/chemrxiv.8307629.v1 2019. [DOI] [PMC free article] [PubMed]
- 38.Liu W; Ali SZ; Ammann SE; White MC, Asymmetric Allylic C-H Alkylation via Palladium(II)/ cis-ArSOX Catalysis. J Am Chem Soc 2018, 140 (34), 10658–10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li Y; Li X; Cheng J-P, Catalytic Asymmetric Synthesis of Chiral Benzofuranones. Adv Synth Catal 2014, 356 (6), 1172–1198. [Google Scholar]
- 40.Doyle AG; Jacobsen EN, Enantioselective alkylations of tributyltin enolates catalyzed by Cr(salen)Cl: access to enantiomerically enriched all-carbon quaternary centers. J Am Chem Soc 2005, 127 (1), 62–3. [DOI] [PubMed] [Google Scholar]
- 41.Saito S; Ito M; Yamamoto H, Highly Regioselective Alkylation at the More-Hindered α-Site of Unsymmetrical Ketones by the Combined Use of Aluminum Tris(2,6-diphenylphenoxide) and Lithium Diisopropylamide. J Am Chem Soc 1997, 119 (3), 611–612. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.