

Summary
Small molecules that directly target MYC and are also well tolerated in vivo will provide invaluable chemical probes and potential anti-cancer therapeutic agents. We developed a series of small molecule MYC inhibitors that engage MYC inside cells, disrupt MYC/MAX dimers, and impair MYC-driven gene expression. The compounds enhance MYC phosphorylation on threonine-58, consequently increasing proteasome-mediated MYC degradation. The initial lead, MYC inhibitor 361 (MYCi361), suppressed in vivo tumor growth in mice, increased tumor immune cell infiltration, upregulated PD-L1 on tumors, and sensitized tumors to anti-PD1 immunotherapy. However, 361 demonstrated a narrow therapeutic index. An improved analogue, MYCi975 showed better tolerability. These findings indicate the potential of small molecule MYC inhibitors as chemical probes and possible anti-cancer therapeutic agents.
Graphical abstract
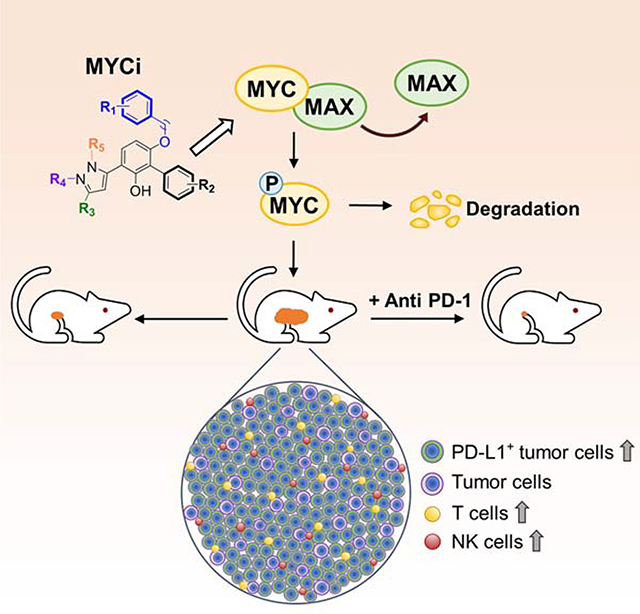
Introduction
MYC proteins, including MYC (also known as c-MYC), MYCL and MYCN, play critical roles in tumorigenesis and therapeutic resistance (Dang, 2012). MYC proteins are implicated in up to 70% of all human cancers via gene amplification, translocation, mRNA upregulation and protein stabilization (Dang, 2012; Dang et al., 2006). Notably, several oncogenic signaling pathways such as Wnt, Ras and PI3K/Akt may mediate their pro-tumorigenic functions through MYC (Karim et al., 2004; Kress et al., 2015). MYC heterodimerizes with MAX to bind to a consensus sequence DNA element, enhancer box (E-Box), and regulates downstream target genes primarily involved in proliferation, differentiation, cell cycle progression, metabolism, apoptosis and angiogenesis (Blackwell et al., 1990; Evan and Vousden, 2001; Meyer and Penn, 2008; Trumpp et al., 2001). Silencing MYC expression in multiple tumor models leads to tumor regression associated with remodeling of the tumor microenvironment (Dang, 2013; Jain et al., 2002; Shachaf and Felsher, 2005), and MYC is considered an attractive cancer therapeutic target (McKeown and Bradner, 2014). However, several conceptual and practical difficulties, including the lack of defined “pockets” in the MYC proteins and potential “on-target” toxicity to normal tissues have led to these proteins being regarded as “undruggable” (McKeown and Bradner, 2014). This latter concern has been alleviated by elegant in vivo genetic modeling studies using the dominant negative MYC peptide Omomyc, showing that a therapeutic window may exist for targeting MYC (Soucek et al., 2008). These observations are supported by more recent strategies of targeting MYC indirectly, such as with BRD4 or CDK7 inhibitors (Posternak and Cole, 2016). Nonetheless, the need for chemical probes that directly modulate MYC function and that can serve as possible therapeutic leads remains acute.
Despite the lack of clinical stage small molecule MYC inhibitors, pioneering studies from several groups have shown the feasibility of developing small molecules that can directly bind to and inhibit MYC activity (Fletcher and Prochownik, 2015). These molecules disrupt MYC/MAX dimerization and/or MYC/MAX/DNA complex formation but are limited by lack of potency and poor pharmacokinetic properties (Clausen et al., 2010; Fletcher and Prochownik, 2015; Guo et al., 2009). This deficiency has also hindered efforts to study the effects of small molecule MYC inhibitors on the tumor microenvironment. We reasoned that sampling a much larger chemical space coupled with the rapid screening of candidates in mice may facilitate the discovery of MYC inhibitors with in vivo efficacy.
Results
Identification of MYC inhibitors
To increase the probability of identifying MYC inhibitors with in vivo activity, we coupled the in silico screening of a large chemical library to a rapid in vivo screen in mice (Figure S1A). We built a 5-point pharmacophore model (Figure S1B) to screen a 16 million compound library. The library was generated by applying multiple filters including the Pan Assay Interference compounds (PAINS) filter (Baell and Holloway, 2010) to remove potentially toxic or metabolically unstable groups and non-drug like molecules from the ZINC database containing 35 million compounds (Sterling and Irwin, 2015). The screen identified 61 hits. The hits were then subjected to secondary screening assessing disruption of MYC/MAX/DNA complex formation (by Electrophoretic Mobility Shift Assay, EMSA); suppression of MYC transcriptional activity (E-box reporter assay); and inhibition of cell viability in a MYC/MAX-dependent manner. A previously reported small molecule MYC inhibitor, 10074-G5 (G5), was included for comparison (Yin et al., 2003). This approach yielded compound ZINC16293153, called Min9, that was active in all tested assays (Figure S1C-S1E) and fit well in the pharmacophore model (fitting score = 4.74, 95%; conformational energy = 3.4 kcal/mol). We then tested eight Min9 analogs and found that 5 out of the 8 analogs disrupted MYC/MAX/DNA complex formation (Figure S1F and S1G), validating the Min9 scaffold as an active MYC inhibitor series.
For subsequent lead optimization, we integrated rapid in vivo screening with the in vitro assays (Figure S1H). We engineered a MYC-dependent E-box luciferase reporter cell line, MycCaP E-box-Luc, that was used to establish allografts in mice (Figure S1I). This allowed monitoring of MYC transcriptional activity in the tumor grafts following compound treatment. The approach is illustrated by data for three compounds active in in vitro (342, 309 and 361) and a closely related inactive analog (360) (Figure S1J-S1L). Although compounds 342, 309 and 361 showed similar potencies in EMSA and cell viability assays (Figure S1K and S1L), they displayed vastly different effects on MYC activity and tumor growth in vivo (Figure S1M), presumably due to differences in pharmacokinetics. Compound 361 (MYCi361, NUCC-0196361) significantly reduced tumor size and E-box luciferase activity in vivo (Figure S1M), selectively inhibited E-box-luciferase but not CMV-luciferase activity in vitro (Figure S1N), and impaired MYC/MAX heterodimer but not the closely related MAX/MAX homodimer binding to E-box DNA (Figure S1K).
To examine MYC target engagement by 361 in cells using unlabeled protein and inhibitor, we performed the cellular thermal shift assay (CETSA). CETSA assesses drug-protein interaction in the protein’s native cellular environment, based on ligand-induced changes in protein thermal stability (Cimmperman et al., 2008; Martinez Molina et al., 2013). Treatment of PC3 cells with 361 (4–10 μM) or G5 (15–60 μM) for 30 min led to significant thermal destabilization of MYC protein while 360 (6 μM) had no effect (Figure 1A-1C, and Figure S2A-S2E). 361 and its inactive analog 360 are regioisomers, differing only in the position of the methyl group (Figure S1J). Notably, we have consistently observed the same activity pattern related to the position of this methyl group in active analogs and their regioisomers.
Figure 1.

Identification of MYC Inhibitors
(A) Chemical structures of compound 361, Biotin-361 and Phosphate-361.
(B) Melt curves of MYC protein in cellular thermal shift assay (CETSA) in PC3 cells treated with 361 or DMSO. The graph shows the quantification of MYC protein versus temperature points based on western blot analyses.
(C) 361 CETSA under isothermal condition. Graph shows the quantification of MYC protein at room temperature (RT) 25 °C or 42 °C from cells treated with indicated concentrations of 361.
(D) Western blots for recombinant MYC protein after Biotin-361 (5 μM) or control D-Biotin (5 μM) pulldown.
(E) Western blot analysis on endogenous MYC protein after Biotin-361 pulldown in PC3 cell lysates.
(F and G) Biotin-361 (5 μM) binding to MYC from PC3 cell lysate was analyzed after pre-treatment with Phosphate-361 (F) or compounds G5 or JKY-2–169 (JKY) (G).
(H) Illustration of MYC binding sites of reported MYC inhibitors including G5, JKY, 7594–0035, and F4, as well as 361 and 975 from this study.
(I) 361 binding affinity to MYC was assessed by fluorescence polarization (FP) competition assay. The graph shows 361 at varying concentrations (3.1–25 μM) against G5 (10 μM) binding to MYC353–439 in FP.
(J and K) Western blot showing (J) and quantification of (K) the levels of MAX co-immunoprecipitated with MYC in PC3 cells with or without 1 hr treatment of 361.
(L and M) Representative immunofluorescence (IF) images (L) and quantification (M) of proximity ligation assay (PLA) for MYC/MAX interaction in PC3 cells after 1 hr treatment of 361. Red signals indicate close proximity between MYC and MAX and green fluorescence shows MYC expression at same cell sections (scale bar, 5 μm).
Error bars represent mean ± SEM, n = 3 independent experiments for (B), (C), (I) and (K), n = ~ 200 cells counted/group for (M), and analyzed by two-way ANOVA for (B) and (C), “One site - Fit Ki” analysis and “Binding-competitive” suite for (I), unpaired t-test for (K) and (M) in Prism.***p < 0.001, ****p < 0.0001.
See also Figures S1 and S2
To further study 361 binding to MYC protein, we synthesized a biotinylated derivative of 361 (Biotin-361) and a soluble compound 361 (Phosphate-361) (Figure 1A). Biotin-361 pulled down recombinant MYC and endogenous MYC, but not MAX or another bHLH protein HIF-1α from cell lysates (Figure 1D, 1E and S2F). MYC binding to Biotin-361 was competed by excess phosphate-361 , G5 or another reported MYC binding compound JKY-2–169 (Figure 1F and 1G), but not by another MYC inhibitor, 10058-F4 or “F4” (Figure S2G). G5 and JKY-2–169 have been shown to bind to amino acids 366–378 of the MYC protein while F4 binds to amino acids 402–409 (Follis et al., 2008; Jung et al., 2015). Our results, therefore, localize 361 binding to the same region as G5 and JKY-2–169 (Figure 1H). This region of MYC has been shown to bind multiple structurally diverse MYC inhibitors including 10075-G5, JKY-2–169 and 7594–0035 (Carabet et al., 2018).
To determine the binding affinity of 361 to MYC protein, we used a fluorescence polarization competition assay against 10074-G5, which displays intrinsic fluorescence (Hammoudeh et al., 2009). The results indicate a KD value for 361 binding to MYC of 3.2 μM (Figure1I). To examine the effect of 361 on MYC/MAX interaction inside cells, we performed co-immunoprecipitation (co-IP) and proximity ligation assays (PLA). Treatment of PC3 cells with 361 at 6 μM for 1 hr led to disruption of the MYC/MAX interaction when assayed by co-IP (Figure 1J and 1K) and the PLA assay (Figure 1L and 1M). These studies establish that 361 binds to MYC and disrupts MYC/MAX complex formation.
361 decreases MYC protein stability by modulating MYC-threonine 58 phosphorylation
We noticed that treatment of multiple cell lines expressing MYC and MYCN with 361 led to a reduction in MYC and MYCN, but not MAX, protein levels (Figure S3A-S3E). However, MYC mRNA levels were not altered (Figure S3F). The reduction in MYC protein caused by 361 treatment can be rescued by proteasome inhibitor MG132, indicating that 361 affects MYC protein stability (Figure 2A). Using a cycloheximide (CHX) chase assay, we found that 361 reduced MYC protein half-life from 66 min to 28 min in PC3 cells (Figure 2B and 2C). We hypothesize that interaction of compound with MYC and/or disruption of MYC/MAX heterodimerization may promote MYC degradation. MYC protein stability is regulated by several mechanisms, prominent among which is an ordered phosphorylation cascade where phosphorylation of MYC on serine 62 (pS62) by kinases such as ERK, CDK and JNK primes MYC for subsequent phosphorylation on threonine 58 (pT58) by GSK3β (Zhou et al., 2015). MYC pT58 is recognized by E3 ubiquitin ligases and degraded by the 26S proteasome (Farrell and Sears, 2014). We examined whether 361 affects MYC protein stability through this mechanism. We found that 361 treatment (6 μM) selectively increased T58 but not S62 phosphorylation (Figure 2D-2F). This increase in pT58 preceded the reduction in MYC protein levels (Figure 2D). We next considered whether the increased MYC phosphorylation is due to off-target effect of MYCi on GSK3β activity. This is unlikely due to the following reasons: First, 361 did not affect levels of regulatory GSK3β S9 phosphorylation (Figure 2D). Second, 361 did not affect phosphorylation of another GSK3β substrate, β-Catenin, at S33/37/T41 (Wu and Pan, 2010) (Figure S4A). Third, a kinome screen against 468 kinases, including GSK3β, after treatment with 361 (6 μM), was negative (Figure S4B and Table S1). Additionally, 361 was negative in a phosphatase inhibition screening panel (10-dose 3-fold serial dilutions starting at 100 μM) (Table S1). MYC T58 phosphorylation is critical for 361-induced MYC degradation, as MYCT58A (threonine-to-alanine) mutant that cannot be phosphorylated at this site is not readily degraded by 361 (6 μM) (Figure 2G and 2H). The resistance of the MYCT58A mutant to 361-induced degradation is not due to lack of interaction of the small molecule with the mutant protein, as confirmed by CETSA with 361 treatment (6 μM) (Figure 2I, S4C and S4D). Furthermore, a MYCS62A mutant that could not be phosphorylated on S62 and consequently could not be recognized and phosphorylated on T58 by GSK3β is also resistant to degradation (Figure S4E). In sum, these results indicate that 361 promotes MYC degradation by enhancing MYC phosphorylation on T58.
Figure 2.

361 Decreases MYC Protein Stability by Modulating MYC-threonine 58 Phosphorylation
(A) MYC protein levels in PC3 cells treated with 361 in the absence or presence of proteasome inhibitor MG132 determined by western blot.
(B) PC3 cells were pretreated with 361 or DMSO for 3 hr, followed by cycloheximide (CHX) treatment. Cells were harvested at indicated time points and MYC levels determined by western blot.
(C) MYC protein degradation kinetic curves based on the quantification of MYC levels in (B).
(D) Western blots for MYC, phosphorylated MYC T58 and S62, GSK3β and phosphorylated GSK3β S9 in 361 treated PC3 cells at indicated time points.
(E and F) Ratios of pT58 to pS62 (E) and pT58 or pS62 to total MYC protein levels (F) from experiment in (D).
(G and H) Western blot analysis (G) and quantification (H) of Flag-tagged MYC T58 alanine mutant (Flag-MYCT58A) or Flag-tagged wild-type MYC (Flag-MYC) levels in PC3 cells stably expressing the indicated constructs after 361 (6 μM) treatment at the indicated time points.
(I) Melt curve of MYCT58A in MYCT58A-expressing PC3 cells treated with DMSO or 361 at indicated temperature points in CETSA.
(J) Phosphorylated MYC T58 levels by GSK3β were assessed by western blot in in vitro kinase assay where recombinant MYC was first phosphorylated on S62 by activated recombinant ERK2, then incubated with GSK3β kinase and 6 μM of 361 or inactive analog 360.
Error bars represent mean ± SEM, n = 3 independent experiments for (C, E, H and I), n = 2 independent experiments for (F), Half-life of MYC protein calculated by “one phase decay” analysis in Prism for (C), and analyzed by two-way ANOVA in Prism for (I). Data are representative of two independent experiments with similar results for (J).
Next, we sought to determine whether 361 could directly promote MYC T58 phosphorylation in an in vitro reconstituted system. We established an in vitro kinase assay where recombinant MYC was first phosphorylated on S62 by activated recombinant ERK2, then incubated with GSK3β kinase and 6 μM of 361 or inactive analog 360. 361, but not 360, significantly increased pT58 levels, indicating that interaction with 361 enhances MYC phosphorylation at this site (Figure 2J).
The availability of the MYCT58A mutant that is resistant to 361-induced degradation allowed us to examine the effect of 361 on MYC/MAX interaction in cells without the confounding effects of MYC protein degradation. As shown earlier, MYCT58A can interact with 361 (6 μM) in cells by CETSA (Figure 2I, S4C and S4D). In PLA assays, 361 (6 μM) disrupted MYCT58A/MAX interactions (Figure S4F and S4G). Similar results were obtained by co-IP studies at same concentration (Figure S4H). 361 treatment (6 μM, 24 hr) also potently suppressed the expression of MYC target genes CDC25A and MYB (Dzikiewicz-Krawczyk et al., 2017; Galaktionov et al., 1996) in MYCT58A-expressing cells (Figure S4I). These results indicate that 361 impairs MYC/MAX complex formation and MYC-dependent gene expression independent of its effects on MYC protein stability.
361 inhibits MYC-dependent cancer cell viability and tumorigenicity
We assessed the selectivity of 361 by using a panel of MYC-dependent and -independent cell lines. 361 inhibited the viability of MYC-dependent cancer cells including prostate cancer (MycCaP, LNCaP, PC3), leukemia (MV4–11), lymphoma (HL-60, P493–6) and neuroblastoma (SK-N-B2) with low micromolar IC50s, but had little effect on pheochromocytoma PC12 cells, which does not require MYC/MAX dimer for proliferation (Figure 3A and 3B). We also tested G5 and androgen receptor inhibitor enzalutamide in certain cell lines for comparison. Myc knockout Rati fibroblasts (HO15.19) were more resistant to 361 compared to wild-type Rat1 fibroblasts (TGR.1) (Figure S5A). To further examine 361 selectivity, we generated prostate organoids from MycCaP cells or their parental normal prostate epithelial cells of FVB mice (Watson et al., 2005). MycCaP organoids were more sensitive to 361 than the FVB mouse prostate organoids (Figure 3C). Additionally, in the P493–6 lymphoma model in which MYC protein levels could be titrated with tetracycline, sensitivity to 361 was inversely correlated with MYC levels (Figure 3D and 3E). Finally, we examined 361 and several of its analogs in the NCI60 cell line panel cell growth screen. We found that compounds with high inhibition of MYC/MAX/DNA complex formation in EMSA showed more potent inhibition of cell growth in the NCI60 panel (Figure S5B and S5C). Activity of 361 in the NCI60 panel cells also showed a trend for inverse correlation with MYC expression levels (Figure S5D).
Figure 3.

361 Inhibits MYC-dependent Cancer Cell Proliferation and Tumorigenicity
(A) Anti-proliferative effects of 361 on prostate cancer cell lines and MYC/MAX complex independent cell line PC12 following 5 days of treatment.
(B) IC50s of 361, G5 and enzalutamide in cell lines with 5 days treatment.
(C) Representative images of established organoids formed from normal FVB mouse prostate epithelial cells or MycCaP cells treated with 361 for 4 days (scale bar, 10 μm).
(D) Western blots show MYC levels in P493–6 cells maintained in 0–10 ng/ml of tetracycline.
(E) Cell viability of P493–6 cells with different MYC levels from (B) upon treatment with 4 μM 361 for 72 hr.
Error bars represent mean ± SEM, n = 4 replicates for (A-C) and (E), and analyzed by unpaired t test in Prism for (E). ****p < 0.0001.
See also Figure S5
We next examined the impact of 361 on MYC transcriptional activity by RNA-seq analysis. Gene Set Enrichment Analysis (GSEA) on six MYC target gene sets showed MYC target gene expression was significantly down regulated in all gene sets after 361 treatment (6 μM, 24 hr) (Figure S5E). In gene ontology (GO) biological process analysis, cell cycle is the most strongly negatively enriched category (Figure S5F). Most MYC target genes involved in cell cycle regulation (Bretones et al., 2015) were significantly differentially expressed in 361-treated cells including downregulation of CDC25, Cyclins, E2F, CDKs, and Skp2 and upregulation of p21, p15 and p16 (Figure S5G and S5H). To show that 361 effect on cell cycle progression is not due to intercalation of DNA and activation of a DNA damage response, we assessed γ-H2AX status after 24-hour treatment with 10 μM 361. We did not observe induction of γ-H2AX in contrast to doxorubicin-treated controls (Figure S5I).
361 shows favorable pharmacokinetics and inhibits MYC-driven tumor growth in vivo
The initial in vivo rapid screen results demonstrated that 361 inhibited MycCaP tumor growth, indicating that it has suitable pharmacokinetic properties in vivo to show efficacy. In agreement with this, pharmacokinetic analyses after intraperitoneal (i.p.) or oral (p.o.) dosing of 361 in mice indicate plasma half-lives of 44 hr and 20 hr, respectively (Figure 4A), with maximum plasma concentrations (Cmax) of 27200 ng/ml (46 μM) i.p. and 13867 ng/ml (23 μM) p.o. (Table S2). At 24 hr post-exposure, the plasma concentration was 12733 ng/ml (21 μM) for i.p. and 5283 ng/ml (9 μM) for p.o. (Table S2). 361 treatment of FVB mice bearing established MycCaP tumor allografts at 100 mg/kg/day induced tumor regression (Figure 4B). With treatment however, mice lost an average 10% of their body weight (Figure S6A). When treatment was stopped, mice regained weight. Treatment was re-started at a dose of 70 mg/kg/day after tumors had attained the original size. 361 was again effective in controlling tumor growth without additional loss in mouse body weight. Further studies confirmed 361 anti-tumor efficacy, including against a prostate PDX model with modest MYC expression as shown in the gene expression prolife (Jackson laboratory Model ID: TM00298) (Figure 4C). Ki67 proliferation marker was decreased and MYC pT58 level was increased in tumor tissues after 361 treatment (Figure 4D). Importantly, enhanced pT58 levels in tumor tissue after 361 treatment is consistent with in vitro observations, and provides pharmacodynamic evidence of 361 engaging MYC in tumor tissue. Next, we compared 361 anti-tumor efficacy in immune-competent FVB mice versus immunocompromised NSG mice. FVB and NSG mice bearing MycCaP tumor grafts were treated with 361 at 50 mg/kg/day for 4 days. 361 exhibited a stronger tumor inhibitory effect in immunocompetent FVB mice than in the immunodeficient NSG mice (Figure 4E), suggesting that full anti-tumor efficacy of 361 is dependent on an intact immune system.
Figure 4.

361 Shows Favorable Pharmacokinetics and Inhibits MYC-dependent Tumor Growth in Vivo
(A) Pharmacokinetic (PK) analysis in C57BL/6 mice treated p.o. or i.p. with 50 mg/kg of 361. Plasma concentration of 361 was determined at the indicated time points up to 24 hr after a single dose administration.
(B) Average tumor volumes of MycCaP allografts in FVB mice after treatment with 361 initially at 50 mg/kg twice daily for 2 days, then 70 mg/kg/day for 9 days as indicated.
(C) Average of tumor growth percentage of human prostate cancer patient derived xenografts (PDX) after 361 treatment (55 mg/kg/day, 3 consecutive days a week for 2 weeks).
(D) Representative images of H&E and IF staining for Ki67 and pT58 in MycCaP tumor tissue after 361 treatment from the study in (B) (scale bar, 50 μm).
(E) Tumor volume fold change of MycCaP allografts in FVB mice and xenografts in NSG mice after 4 days treatment with 361 at 50 mg/kg/day. Dotted line indicates threshold of 10% of fold change and numbers in parentheses indicate how many tumors were under the 10% threshold out of total number of tumors.
Error bars represent mean ± SEM, n = 3 mice at each time point in (A), n = 6–8 grafts/group (from 3–4 mice) in (B and D), n = 9–10 grafts/group (from 5 mice) in (C) from two independent experiments, n = 8–12 grafts/group (from 4–6 mice) in (E), and analyzed by two-way ANOVA in Prism for (C). *p < 0.05.
361 modulates the tumor immune microenvironment and enhances anti-PD1 immunotherapy
MYC inhibition may affect the host anti-tumor response via various mechanisms, such as by modulating tumor cell expression of CD274, encoding PD-L1, CD47, or cytokines or by inducing immunogenic cell death (Casey et al., 2018; Casey et al., 2016; Kortlever et al., 2017; Zou et al., 2018). To assess the effects of MYC inhibition on the tumor microenvironment, we examined MycCaP tumors post-361 treatment. We observed enhanced tumor infiltration of CD3+ T cells and upregulation of PD-L1 expression on tumor cells in 361 treated mice (Figure 5A and 5B). Immunophenotyping of 361-treated tumors by flow cytometry showed, in addition to an increase in overall percentage of CD3+ cells, an increase in CD3+CD4+ and CD3+CD8+ T cells, IFNγ-expressing CD4+ and CD8+ T cells, TNFα-expressing CD8+ cells, dendritic cells and NK cells (Figure 5C and 5D). In addition, there was a trend for a decrease in regulatory T (Treg) cells while MDSCs were increased (Figure 5D). By contrast, no changes were seen in lymph nodes in any of the examined cellular parameters (Figure S6B). Gating strategy of flow cytometry analysis is shown in Figure S6C and S6D.
Figure 5.

361 Modulates the Tumor Immune Microenvironment and Potentiates Anti-PD1 Immunotherapy
(A) Representative IHC staining of CD3 in the MycCaP tumor tissue after 361 treatment from the study in Figure 4B (scale bar, 50 μm), and the quantification of CD3 positive cells per field of version (FOV).
(B) Representative IF images of PDL-L1 staining in the MycCaP tumor tissue after 361 treatment from the study in Figure 4B (scale bar, 50 μm).
(C) Scheme for tumor immunophenotyping from FVB mice bearing established MycCaP allografts treated with 361 (50 mg/kg/day, 2 days on/2 days off for 2 rounds). TIL, tumor infiltrating lymphocytes.
(D) Flow cytometry analysis of immune cells in MycCaP allografts treated with 361 or vehicle as described in (C), shown by percent of parent gates.
(E) Western blot analysis shows cleaved Caspase-3 in 361 treated MycCaP cells for 48 hr.
(F) Immunogenic cell death (ICD) was assessed in vitro in MycCaP cells treated with 4 μM 361 for 72 hr via HMGB1 release (ELISA), ATP release (luminescence assay), and cell surface calreticulin expression (flow cytometry). Data are representative of two independent experiments with similar results.
(G) Scheme for combination treatment of 361 with anti-PD1 antibody in MycCaP allografts. FVB mice bearing established MycCaP tumors were treated with alternating doses of 361 at 50 mg/kg/day for 2 days, then anti-PD1 or IgG2a isotype control at 100 μg/day for 2 days, for a total of 4 cycles.
(H) Average of tumor growth percentage of the grafts under the combination treatment described in (G).
(I) Individual tumor growth trajectories of study in (H).
Error bars represent mean ± SEM, n = 4–6 grafts/group (from 3 mice) and 6 FOVs/group were analyzed in in (A) and (B), n= 3–4 mice/group in (D), n = 3 replicates in (F), and n = 4–6 mice/group in (H), and analyzed by unpaired t test for (A), (D) and (F), and two-way ANOVA for (H) in Prism. *p < 0.05, **p < 0.01, ***P < 0.001, ****p < 0.0001.
See also Figure S6
Next we examined whether 361 treatment, which at 6 μM induces cancer cell death with activated caspase-3 expression (Figure 5E), may induce immunogenic cell death (ICD). Induction of ICD could activate the immune response in tumors (Kepp et al., 2014), leading to immune cell infiltration. We found that treatment of MycCaP cells with 361 (4 μM) led to significant upregulation of cell surface calreticulin expression, and release of HMGB1 and ATP (Figure 5F), which are all markers of ICD. Overall, these data are consistent with induction of immunogenic cell death of tumor cells by 361 provoking an immune response that subsequently results in upregulation of tumor PD-L1 possibly due to hyperexpression of cytokines such as IFNγ (Mimura et al., 2018; Pardoll, 2012).
These findings prompted us to examine the effect of combining MYC inhibition with anti-PD-1 immune checkpoint blockade. We have previously shown that MycCaP tumors are resistant to anti-PD1 therapy (Anker et al., 2018). We treated FVB mice bearing established MycCaP tumors with alternating doses of 361 at 50 mg/kg/day for 2 days, followed by anti-PD1 at 100 μg/d for 2 days, for a total of 4 cycles (Figure 5G). The sub-optimal dosing for 361 in this study was chosen to avoid toxicity and to allow assessment of possible synergy with anti-PD1 treatment. Mice that received vehicle or single agent displayed no significant differences in tumor growth while the combination treatment resulted in synergistic suppression of tumor growth as shown by average of tumor volume growth percentage (Figure 5H) or individual tumor trajectories (Figure 5I). Treatment at this dose was well-tolerated by the mice (Figure S6E). Overall, these results indicate the potential for combining MYC inhibitors with immune checkpoint blockade.
MYCi975 is a close analog of 361 with improved therapeutic index
Our animal studies with 361 suggested that it may not be well tolerated for prolonged periods at doses necessary for single-agent efficacy. Acute toxicity studies indicated a maximum tolerated dose (MTD) of 240 mg/kg/day p.o. (Table S2). Histopathological analysis of major organs of 361-treated mice showed suppression of the splenic white pulp and hepatocyte hypertrophy (Table S2). Thus while 361 shows efficacy in vivo, it is hampered by a narrow therapeutic index. This prompted us to undertake an additional medicinal chemistry campaign with the goal of developing better tolerated analogs of 361. Various analogs were synthesized by modifying substituents of different regions such as the central phenol ring, the p-chlorobenzyl group, the bis-trifluoromethylphenyl group and the trifluoromethyl substituent on the pyrazole moiety. The analogs were systematically explored for efficacy using in vitro and in vivo assays described above. This iterative medicinal chemistry optimization resulted in compound NUCC-0200975 (MYCi975 or 975) as a lead compound (Figure 6A) which showed similar activity as 361 with increased tolerability at significantly higher doses as discussed below.
Figure 6.

975, a Close Analog of 361 with Improved Therapeutic Index
(A) Structures of 975 and its soluble analog used to facilitate NMR studies.
(B) Melt curves of MYC in PC3 cells after 975 (8 μM) or DMSO treatment by CETSA. Error bars represent mean ± SEM, n = 3 independent experiments, and analyzed by two-way ANOVA in Prism.
(C) Saturation-Transfer Difference (STD) NMR analysis of 975 soluble analog (100 μM) with MYC (5 μM) or MAX (5 μM) protein.
(D) 975 at varying concentrations (3.1–25 μM) against G5 (10 μM) binding to MYC353–439 in FP assay. Error bars represent mean ± SEM, n = 3 independent experiments, and analyzed by “One site - Fit Ki” analysis and “Binding-competitive” suite in Prism.
(E) MYC levels after 48 hr treatment of 975 in PC3 cells, assessed by western blot.
(F) Western blot analysis for MYC T58 and S62 phosphorylation status in 975 treated PC3 cells at indicated time points.
(G) Phosphorylated MYC T58 levels by GSK3β were assessed by western blot in in vitro kinase assay with the treatment of 6 μM 975.
(H) Mass spectrometry analysis of common proteins bound to Biotin-361 (10 μM) and Biotin-975 (10 μM) in PC3 and P493–6 cells with MYC in the “on” or “off” condition.
We confirmed MYC target engagement by 975 (8 μM) in cells by CETSA (Figure 6B and S7A) and a biotinylated derivative of 975 (Biotin-975, 10 μM) pulled down MYC protein in PC3 cells (Figure S7B and S7C). We further validated 975 binding to recombinant MYC protein by Saturation Transfer Difference (STD) Nuclear Magnetic Resonance (NMR) spectroscopy. A 975 phosphate analog generated to increase aqueous solubility for NMR studies (and which retained MYC inhibitory activity) bound to MYC but not MAX at 100 μM (Figure 6A and 6C). We also confirmed interaction of G5 (200 μM) with MYC but not MAX (Figure S7D). Importantly, 975 competed with G5 binding to MYC353–439 with a KD value of 2.5 μM in the fluorescence polarization assay (Figure 6D), indicating that 975 also localizes to the same region of the MYC protein as G5, 361 and several other reported MYC binders (Figure 1I). Moreover, treatment of cells with 975 (8 μM) enhanced MYC degradation and phosphorylation on T58 (Figure 6E and 6F). Finally, like 361, 975 also directly increased GSK3β-mediated MYC pT58 in the in vitro kinase assay at 6 μM (Figure 6G).
361 and 975 may interact with multiple proteins inside the cell in addition to MYC to mediate the observed anti-tumor effects. To investigate this, we performed unbiased mass spectrometric analysis of compound-bound proteins. We examined proteins bound to biotinylated-361 and −975 from PC3 cells and P493–6 cells in the MYC-off and MYC-on conditions. Although the sensitivity of the assay was insufficient for MYC detection as demonstrated by recombinant protein spike-in controls (Figure S7E), we detected a total of 135 common proteins bound by Biotin-361 and Biotin-975 (Figure 6H, Table S6). Of these, 38% have been reported to be part of the MYC interactome (Agrawal et al., 2010; Ewing et al., 2007; Kalkat et al., 2018; Koch et al., 2007; Mathivanan et al., 2006). The binding of 61 out of the 135 proteins was lost (n = 46) or reduced (n = 15) when MYC is suppressed by tetracycline treatment in P493–6 cells. The interaction of the remaining proteins was unchanged or increased in the MYC-off condition, indicating these as possible MYC-independent targets of the MYCi compounds. However, these binding studies should be interpreted in light of functional pathway analyses from RNA-seq data described next, which indicate limited modulation of non-MYC target genes by MYCi.
975 inhibits MYC-dependent cancer cell viability and suppresses MYC transcriptional activity
975 inhibited cell viability in a MYC-dependent manner (Figure 7A, S8A and S8B) and selectively suppressed E-box-luciferase activity (Figure 7B). To assess the molecular pathways modulated by MYCi treatment in an unbiased manner, we performed RNA-seq experiments using P493–6 and PC3 cells. The ability to repress MYC with tetracycline treatment in the P493–6 model allowed us to directly compare empirical MYC targets in these cells after turning MYC “off” to the genes regulated by 975 treatment. We also included the dataset (Dang_2018) for MYC target genes in P493–6 cells identified in a previous study (Lu et al., 2018). The results, shown in Figure 7C, indicate that 975 affected the expression of 3647 genes, the majority (69%) of which are MYC responsive. Among the 975-regulated genes that did not respond to MYC (and may therefore represent off-target effects), the top altered pathways were related to small molecule compound metabolism process, consistent with a general cellular response to exposure to organic small molecule (Figure 7D). Next, we compared the effects of 975 to those of 361 by RNA-seq in PC3 cells. 975 affected the expression of a smaller number of genes (n = 3095) compared to 361 (n = 5033), of which 66.4% were common between the two compounds (Figure 7E). GO biological process analysis of the common genes showed that cell cycle and DNA replication were among the top down regulated pathways, while pathways related to cell death, response to organic compound and ER stress were upregulated (Table S3). GSEA analysis of genes uniquely regulated by 361 (n = 2978) showed suppression of several sets that all share common leading edge genes encoding HIST1H proteins and the TCA cycle/respiratory electron transport (Table S4). However, no gene sets were significantly enriched in GSEA analysis of genes uniquely regulated by 975 in PC3 cells. These findings may partly explain the improved tolerability of 975 compared to 361 as will be shown below.
Figure 7.

975 Selectively Inhibits MYC-dependent Cancer Cell Viability and the MYC Transcriptional Program.
(A) Anti-proliferative effects of 975 on prostate cancer cells and PC12 following 5 days of treatment.
(B) Dose response effect of 975 on MYC transcriptional activity in E-box luciferase reporter assay compared to CMV-luciferase reporter.
(C) Venn diagram showing overlap of genes regulated in P493–6 cells by: 1) silencing MYC by Tetra 0.1 μg/ml for 48 hr, log fold change > 0.5 from Dang_2018; 2) silencing MYC by Tetra, 0.1 μg/ml for 24 hr, adj-p<0.05, this study; and 3) 975 treatment at 6 μM for 24 hr, adj-p<0.05, from this study.
(D) GO biological process analysis on 975 uniquely regulated genes (1128) in P493–6 cells.
(E) Venn diagram showing overlap of genes regulated by 361 (6 μM, 24 hr) and 975 (8 μM, 24 hr) treatment of PC3 cells from RNA-seq. Genes with adj-p < 0.05, and log fold change > 0.5 were included.
Error bars represent mean ± SEM, n = 4 replicates in (A) and (B), data are representative of two to three independent experiments with similar results. RNA-seq data was assessed in triplicates (C-E).
975 pharmacokinetics, anti-tumor efficacy and tolerability
975 exhibited excellent pharmacokinetic profiles following p.o., i.p. or i.v. administration (Figure S8C and S8D). The half-lives observed were 7 hr and 12 hr when dosed at 100 mg/kg and 250 mg/kg p.o. respectively. The Cmax values attained were 41533 ng/ml (74 μM) and 54000 ng/ml (96 μM) respectively. 975 significantly inhibited tumor growth (Figures 8A) and increased survival (Figures 8B) in the MycCaP allograft model with animals tolerating a 100 mg/kg/day i.p. dosing for 14 days. Analysis of tumor tissue showed increased pT58 and PD-L1 levels (Figure 8C) and enhanced tumor infiltration of CD3+ T cells (Figure 8D), B220+ B Cells (Figure 8E), and NKp46+ NK cells (Figure 8F) after 975 treatment. Therefore, we examined the effect of combining 975 with anti-PD1 treatment. 975 alone dosed at 100 mg/kg/day, 2 days on/2 days off slowed tumor growth, while the combination treatment with anti-PD1 (100 μg/day, on alternating 2 days on/2 days off) resulted in a synergistic suppression of tumor growth (Figure 8G and S8E). Similar to 361, 975 treatment inhibited MycCaP tumors grown in immunocompetent FVB mice more strongly than in immunodeficient NSG mice (Figure 8H), indicating that full anti-tumor efficacy of 975 is also dependent on an intact immune system. Treatment of Lewis Lung Carcinoma (LLC1)-bearing mice with 975 (100 mg/kg/day) inhibited tumor growth with no changes in body weight (Figure 8I and S8F). NSG mice bearing MV-411 AML xenografts were treated with 975 (50 mg/kg/day) or Ara-C (20 mg/kg/day) 5 days a week. In this model, the lower dose of 975 and the immunodeficient host background may explain reduced efficacy as a single agent. 975 synergized with Ara-C with no obvious impact on mouse body weight (Figure 8J and S8G).
Figure 8.

975 Inhibits Tumor Progression, Increases Immune Cell Infiltration and Potentiates Anti-PD1 Immunotherapy
(A) Average tumor volumes of MycCaP allografts after treatment with 975 at 100 mg/kg/day for 14 days.
(B) Survival curves of animals from the study shown in (A).
(C) Representative images of MYC pT58 and PD-L1 levels assessed by IF in the tumor tissues from the study in (A) (scale bar, 50 μm).
(D) Representative images of CD3 by IHC and quantification of the positive cells/FOV in the tumor tissues from the study in (A) (scale bar, 100 μm).
(E) Representative images of B220 by IHC and quantification of the positive cells/FOV in the tumor tissues from the study in (A) (scale bar, 100 μm).
(F) Representative images of NK cells (NKp46+) by IF and quantification of the positive cells/FOV in the tumor tissues from the study in (A) (scale bar, 50 μm).
(G) Average tumor volumes of MycCaP allografts after treatment with alternating doses of 975 at 50 mg/kg, twice daily for 2 days, then anti-PD1 for 2 days for a total of 5 cycles.
(H) Fold change of tumor size of MycCaP allografts in FVB mice and xenografts in NSG mice after 3 days treatment with 975 at 50 mg/kg, twice daily. Tumor numbers under the 10% threshold out of total number of tumors in each group indicated.
(I) Average tumor volumes of LLC1 allografts in C57BL/6 mice after treatment with 975 at 50 mg/kg, twice daily for 12 days.
(J) Average tumor volumes of MV411 xenografts after treatment with lower dose 975 (50 mg/kg/day) alone or combined with Ara-C (20 mg/kg/day) for 3 three weeks (5 days a week).
Error bars represent mean ± SEM n = 6–7 grafts (from 4 mice) /group in (A-F), most affected 1–3 FOVs/graft were analyzed in (D-F), n = 5–7 mice/group in (G), n = 7–11 grafts (from 4 to 6 mice)/group in (H), n = 7 mice/group in (I), n = 4 mice/group in (J). Data were analyzed by Two-way ANOVA for (A, G, I and J), by survival curve comparison for (B), by unpaired t test for (D-F) in Prism, *p < 0.05, ****p < 0.0001.
To further evaluate 975 tolerability, we performed acute toxicity studies in mice where the inhibitor was dosed p.o. a single dose at 50, 100, 250, 500 or 1000 mg/kg. 975 was well tolerated up to 1000 mg/kg (Figure S8H). Additional toxicology analysis was then performed under the same experimental conditions employed in the MycCaP anti-tumor efficacy studies (100 mg/kg daily for 14 days) and mice analyzed one week later. The results indicate normal complete blood count and differential, normal blood chemistry, and normal kidney and liver function (Table S5). There were also no obvious pathologic abnormalities from gross and histological analysis of various organs including brain, heart, lung, liver, spleen, kidney, intestine and skin (Table S5). Collectively, these data indicate that 975 shows significant in vivo efficacy at higher exposure compared to 361, and could be a promising starting point for the development of MYC inhibitor therapeutics.
Discussion
We have taken advantage of the expanding structural diversity of MYC-MAX inhibitors to conduct a pharmacophore-based in silico screen of a large compound library linked to a rapid in vivo screen. The in vivo screen was key in excluding compounds with poor pharmacokinetics, poor pharmacodynamics or both at early stages of the inhibitor development. We identified a promising chemical scaffold, which, resulted in the discovery of closely related MYC inhibitors 361 and 975 with significant in vivo anti-tumor efficacy. These inhibitors disrupt MYC/MAX interaction while also decreasing MYC protein stability. This dual mechanism of action leads to significant inhibition of MYC-dependent cancer-cell proliferation in vitro with suppression of global MYC target gene expression and inhibition of tumor-growth in vivo. More importantly, both inhibitors showed excellent PK profiles, with long terminal half-lives, high peak plasma concentration and tumor penetration as evidenced by pharmacodynamic markers such as MYC T58 phosphorylation.
Checkpoint blockade therapy has revolutionized the field of cancer immunotherapy as a therapeutic strategy to overcome mechanisms of tumor immune escape (Pardoll, 2012). However, response to checkpoint inhibition is limited to immunogenic tumors that express checkpoint proteins such as PD-1 ligand (PD-L1) and/or contain tumor infiltrating T cells within the local tumor microenvironment (Pfirschke et al., 2016). In the case of nonimmunogenic tumors, it has been shown that induction of immunogenic conditions is possible and that the antitumor immune response can be primed by immunogenic cell death (ICD) and stimulation of type I interferon responses (Dosset et al., 2018; Pfirschke et al., 2016). Our studies show that MYCi induces ICD in tumor cells and allows increased T cell infiltration and subsequent upregulation of PD-L1 in the tumor microenvironment. Accordingly, MYCi treatment sensitized otherwise refractory tumors to immune checkpoint blockade. One may envision a future treatment regimen in which a MYC inhibitor is given to patients for a limited period of time followed by immune checkpoint blockade, thus avoiding potential toxicities to normal tissues that may arise from prolonged MYC inhibition. In summary, these studies illustrate a pathway for the development of viable MYC inhibitors for future mechanistic studies and therapeutic interventions.
STAR★Methods
Lead Contact and Materials Availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Sarki A. Abdulkadir (Sarki.abdulkadir@northwestern.edu ).
Experimental Model and Subject Details
Mice
All animal experiments and procedures were performed in compliance with ethical regulations and the approval of the Northwestern University Institutional Animal Care and Use Committee (IACUC). FVB mice, prostate PDX model (TM00298), NSG mice, C57BL/6 were obtained from the Jackson Laboratory. CB17/Icr-Prkdcscid/IcrIcoCrl mice and CD-1 were from Charles River. All the mice were housed in a pathogen-free animal barrier facility. All the in vivo experiments were initiated with mice of age 6 to 8 weeks.
Cell lines
MycCaP, PC3, LNCaP, PC12, MV411, SK-N-BE (2) and 293T cells lines were purchased from ATCC and P493–6 B cells were kindly provided by Professor Chi Van Dang from the University of Pennsylvania. LLC1 cells were from Professor Bin Zhang (Northwestern University). TGR-1 and HO15.19 Rat-1 cells were gift from Professor John Sedivy in Brown University. Cells were verified to be mycoplasma-free (Lonza) at multiple times throughout the study. MycCaP, PC3, LNCaP and P493–6 B cells were cultured in RPMI1640; LLC1 and 293T cells in DMEM (Gibco); SK-N-BE(2) cells in F12 (ATCC); MV411 cells in IMDM (ATCC), all supplemented with 10% heat inactivated fetal bovine serum (FBS, Gibco), 1% Penicillin-Streptomycin (10,000U/ml, Life Technologies). PC12 cells were grown in F-12K Medium (ATCC) with 2% heat inactivated FBS, 12.5% of horse serum (Thermo), and Rat-1 cells were cultured in DMEM with 10% calf serum. All cell culture was performed in a 37°C 5% CO2 incubator.
Method Details
Pharmacophore model used to identify MYC inhibitors
We selected 32 reported compounds (Table S7) to build a pharmacophore query (called a hypothesis) for screening a drug-like compound database. Since the reported MYC-MAX inhibitors are from different sources, we did not consider building an activity-based pharmacophore; rather, we preferred to construct a pharmacophoric features based hypothesis. In addition to using the compounds reported as direct MYC-MAX inhibitors to build our model, we also included 3 BET bromodomain inhibitors as decoys. The decoys are compounds having similar physiochemical features but dissimilar 2D-topology in the testing set. These three compounds were chosen as decoys because they are known to indirectly inhibit MYC and do not directly bind MYC or MAX. They also have similar chemotypes to several known inhibitors of MYC-MAX (Kiessling et al., 2006) and occupy similar conformational energy space. We considered these three compounds as challenging decoys in the test set and they served as negative controls to validate our model. The 3D chemical structures of all 32 compounds were generated using Discovery Studio 4.1 (http://www.3dsbiovia.com/resource-center) along with 250 conformations for each compound. The “best conformer” generation algorithm was used to generate the conformers within the energy range 0–10 kcal/mol from the global minimum. The generated conformers were used to align the common molecular features to construct the pharmacophore hypothesis. This method generates the hypothesis considering the most common chemical features present in the set of active compounds without considering the activity. The features associated with the hypothesis/query have geometrical constraints. A molecule matches the hypothesis if and only if, it possesses conformations and structural features that can be superimposed within certain tolerance from the corresponding ideal locations. This method also has the capability to partially match to the compounds having more diverse structures (Krovat et al., 2005; Yildiz et al., 2008). We divided the 32 compounds into training and test sets of compounds. The training set contained 20 compounds and the test set contained 12 compounds including the 3 decoys to challenge the model. Using the training set of compounds and specifying a principal value of 2 and maximum omitting feature to 0, we constructed 5 hypotheses. All the hypotheses were tested with the test set of compounds. We found that one of the hypotheses consisting of 5 point pharmacophoric features was able to map all 12 test set of compounds, which has one aromatic hydrophobic (ArHy), 2 hydrogen bond donors (HBD), one hydrogen bond acceptor (HBA) and one hydrophobic feature. The maximum fitting value was set at 5 giving equal weight to each pharmacophoric feature. The fitting values for all 12 test set compounds were in the range of 3.8–4.9 (76–98%) with the 3 decoys having fitting values less than 80% and more than 12 kcal/mol conformational energy values.
Conformational Database Creation
The drug-like database searching using a pharmacophore query (hypothesis) requires the conformational flexibility of each and every compound present in the database. Hence, a pre-computed conformer database is required for screening of potential hits using the query. We considered the ZINC database (Sterling and Irwin, 2015) containing 35 million drug-like compounds and then applied different filters including PAINS (Baell and Holloway, 2010) and generated a set of 16 million compounds. To generate a conformer database of 16 million compounds requires huge storage space and hence we carried out a diversity analysis keeping the diversity index to 80%. The diverse set contained ~ 1.2 million compounds. Then we applied the ‘fast search” algorithm implemented in Discovery Studio and generated 100 conformers for each compound using an energy cutoff 0–7 kcal/mol from global minimum energy.
Searching the conformer database
The database searching is a two-step filtering technique where the first step is being the elimination of the compounds based on the feature-types, feature-counts and a quick geometrical distance and angle checking. The second step is the matching of the 3D features with the conformers of the compounds. This computational alignment step is a very time consuming and slow process hence the pre-filtering in the first step is essential. We considered the query/hypothesis generated before to screen this curated database. The screening resulted in 61 potential hits having a fitting score ranging from 4–5 (80–100%) with low energy score ranging from 2–7 kcal/mol.
Expression and purification of recombinant MYC353–439 and MAX
Human MYC bHLHZip domain (residues 353–439), human MAX isoforms, MAX(L) (160 amino acids) and MAX(S) (151 amino acids), were introduced into backbone vector pET151D/-TOPO with an N-terminal hexa-histidine (His 6) tag separated by a TEV (Tobacco Etch Virus) protease digestion site, and expressed in bacteria BL21-CodonPlus strain. The expressing bacterial stocks for all three constructs were kindly provided by Dr. Prochownik from University of Pittsburgh. Bacterial culture and protein purification were performed by following published protocols: B21-CodonPlus competent cells instruction manual, QIAexpressionist (2003) and previous study (Wang et al., 2007). Briefly, 20 μl of bacterial stocks were grown in 10 ml of LB medium with 100 μg/ml of ampicillin and 50 μg/ml of chloramphenicol at 37C°, 225 rpm, overnight. Next day, culture volume was scaled up by 20x with fresh LB for a further 3 to 4 hr culture to reach an A600-0.6 to 0.8, and then expression induced by adding 0.5 mM isopropyl-L-thio-B-D-galactopyranoside (IPTG, Sigma) for 5 hr. Cultures were harvested and lysed in a buffer containing 8 M urea, 100 mM NaH2PO4, and 10 mM Tris (pH 8.0). Proteins were purified on NTA-Ni-agarose (Qiagen) column (Qiagen) with a pH gradient elution as instructed in QIAexpressionist. MAX protein was further dialyzed in the buffer (Hepes 50 mM, NaCI 500 mM, P-mercapethanol 10 mM, Glyserol 5%) using dialysis cassette (Thermo) overnight, replaced with fresh buffer twice and followed by addition of TEV protease (Sigma) at 1:100 (w:w) for another overnight incubation to cleave HisX6 tag. HisX6 tag removed MAX protein was purified on NTA-Ni-agarose by eluting with gradient imidazole 10 to 80 mM containing dialysis buffer, and combined elutes containing 20 mM to 40 mM of imidazole with enriched Max protein. Purified MYC bHLHZip and MAX proteins were quantified using Nanodrop and utilized in EMSA assay.
EMSA assay
E-box containing dsDNA oligonucleotide with one strand labeled with hexachlorofluorescein for fluorescence visualization was synthesized by IDT, Inc. The sequence of the oligonucleotide is 5-CACCCGGTCACGTGGCCTACAC-3 as previously reported (Wang et al., 2007). The binding reaction buffer consists of 0.005% IGEPAL CA-630 (sigma), 5% glycerol, 1 mM EDTA in 1xPBS. The concentration of MYC (residues 353–439), referred to as MYC in EMSA assay, MAX(S) and MAX(L) (as negative control) was 60 nM, and the Oligo was 20nM in the assay. All compounds were dissolved in DMSO at 20 or 40 mM and stored in −20 C°. To prepare the reaction mixture, compounds were further diluted into reaction buffer/DMSO 6:4 to make 10 times the final concentration; 2 μl of prepared compound was added into 18 μl of reaction buffer containing either MYC or MAX and incubated for 1 hr at room temperature. The oligo was added to MAX(S) reaction mix before incubation with compounds since MAX(S) does not bind to oligo by itself. The MYC/compound solution was finally mixed with MAX(S)/oligo/compound solution and the binding reaction allowed to proceed for 15 min before loading 20ul of sample to native gel prepared with 8% of acrylamide/bis-acrylamide (80:1), 10% glycerol in 0.5xTris-borate EDTA (TBE) buffer. The gel was run for 45 min at constant voltage (80 V), and scanned with Alexa Fluor 546 on a Bio-Rad FX molecular imager (Bio-Rad). Data were analyzed with Image J software.
Proliferation assay and NCI 60 panel screen
Cell viability was estimated using the MTS kit, CellTiter 96 AQueous One Solution (Promega) or by counting viable cells. According to cell type and experimental setting, 1000 to 5000 cells/well were seeded in 96 well plates. For rat fibroblast cells, HO15.19 (1000/well) and TGR.1 (3000/well) were seeded in 48 well plates. After 2 to 7 days following the treatment, viable cells were counted or MTS reagent was added and absorption at 490 was measured using plate reader (Perkin Elmer Victor 3V). NCI-60 human tumor cell lines screen for the compounds were performed by the Developmental Therapeutics Program of the National Cancer Institute. MYC expression levels of NCI-60 cell lines were analyzed using gene transcript level Z score analysis tool (https://discover.nci.nih.gov/cellminer/).
Organoid culture and treatment
To isolate mouse prostate epithelial cells, all lobes of prostates were isolated from 8–10 weeks old FVB male mice (Jackson Laboratory), minced and digested with collagenase (Gibco) in RPMI1640 media with 10% FBS for 2 hr at 37 °C. Subsequently, digested tissues were incubated with Trypsin and DNase I (Sigma), and then passed through 40 μm cell strainer to obtain single cells. Dissociated single cells were stained with anti-CD326 (EpCAM)-APC (BioLegend, 118214), anti-CD31-FITC (eBioscience, 11–0311-85), anti-CD45-FITC (eBioscience, 11–0451-85), and anti-Ter119-FITC (eBioscience, 11–5921-85) on ice for 30 min with occasional shaking. EpCAM+Lin (CD45/CD31/Ter119)− cells were sorted to obtain mouse prostate epithelial cells. Both sorted normal epithelial cells and prostate cancer MycCaP cells were resuspended in Hepatocyte Defined Medium (Corning) supplemented with 10 ng/ml epidermal growth factor (Corning), 5% FBS, 1x Glutamax (Gibco), 5% matrigel (Corning), 10 μM ROCK inhibitor (Y-27632, STEMCELL Technologies), 100 nM DHT (Sigma), and 1x Gentamicin/Amphotericin (Lonza), as described in the previous study (Unno et al., 2017). Cells were plated in Ultra-Low Attachment Surface plates (Corning) at 5,000 cells for normal epithelial cells and 1000 cells for MycCaP cells per 100 μl media. Additional 100 μl media was added at day 4. After organoids formed at day 7 for normal epithelial cells, and day 4 for MycCaP cells, organoids were centrifuged at 300 rcf for 5 min. After supernatant was carefully removed, organoids were gently resuspended in 200 μl organoid culture media with 361 or DMSO. At day 4 after treatment, representative bright field images were taken on a ZEISS Axiovert 200 microscope.
Immunogenic cell death assays
MycCaP cells were treated with 4 μM 361 for 72 hr, and supernatants were collected. Cell counts were performed for quantifying secreted ATP (Bioluminescent Assay Kit, Sigma) and high mobility group protein B1 (HMGB1; Elisa, Tecan Trading). For detection of surface Calreticulin, cells were incubated with rabbit anti-Calreticulin (1:1000, Abcam, ab2907) for 60 min and then incubated with Alexa Flour 488 anti-rabbit secondary antibody (Invitrogen, A11008, 1 μg/ml), and analyzed by flow cytometry.
Western blot analysis
Western blot analysis was carried out as previously described (Anker et al., 2018). For MG132 experiment, cells were treated with 10 μM MG132 (VWR) for 3 hr before 361 was added for another 2 hr treatment, and cells were collected for western blot analysis. For cycloheximide (CHX) chase studies, cells were treated with 361 for 3 hr, then 50 μg/ml of CHX was added, and cells were collected at indicated time points for western blot analysis. Primary antibodies used (see Key Resources Table): MYC (Y69) (Abcam, ab32072), MYCN (C-19) (Santa Cruz, sc-764), HIF-1α (Novus Biologicals, NB100–134SS), MAX (H-2) (Santa Cruz, sc-8011), Max (S20) (Cell Signaling, 4739S), MYC (phospho T58) (Abcam, ab185655), MYC (phospho S62) (Abcam, ab185656), ANTI-Flag (Sigma-Aldrich, F1804), β-Catenin (BD Bioscience, 610153), Phospho β-Catenin (Ser33/37/Thr41) (Cell Signaling, 9561T), active-β-Catenin (nonphosphorylated) (EMD Millipore, 05–665), Cleaved Caspase-3 (Asp175) (Cell Signaling, 9661S), β-actin (Cell Signaling, 5125S).
Key Resources Table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| MYC (Y69) | Abcam | ab32072 |
| MYC (N-262) | Santa Cruz | sc-764 |
| MYCN (C-19) | Santa Cruz | sc-791 |
| Max (H-2) | Santa Cruz | sc-8011 |
| Max (S20) | Cell Signaling | 4739S |
| Cleaved Caspase-3 (Asp175) | Cell Signaling | 9661S |
| Phospho-Histone H2A.X (Ser139) (20E3) | Cell Signaling | 9718S |
| Monoclonal ANTI-FLAG® M2 | Sigma-Aldrich | F1804 |
| MYC (phospho T58) (EPR17923) | Abcam | ab185655 |
| MYC (phospho S62) (EPR17924) | Abcam | ab185656 |
| Ki-67 (SolA15) | eBioscience | 14–5698–80 |
| PD-L1 | Cell Signaling | 13684 |
| PD-1 (CD279) | BioXcell | BE0146 |
| IgG2a isotype control, anti-trinitrophenol | BioXcell | BE0089 |
| CD3 (2GV6) | Ventana | 790–4341 |
| B220/CD45R | BD | 550286 |
| CD335/NKp46 (29A1.4) | Biolegend | 137601 |
| Goat Anti-Mouse IgG (H + L)-HRP Conjugate | Bio-Rad | 1706516 |
| Goat Anti-Rabbit IgG (H + L)-HRP Conjugate | Bio-Rad | 1706515 |
| Alexa Fluor 488 goat anti-rabbit | Life Technologies | A11008 |
| β-actin (13E5) Rabbit mAb (HRP Conjugate) | Cell Signaling Technology | 5125S |
| Phospho-GSK-3-beta (Ser9) (D3A4) | Cell Signaling Technology | 9322S |
| GSK-3β (3D10) | Cell Signaling Technology | 9832S |
| β-Catenin | BD Bioscience | 610153 |
| Phospho β-Catenin (Ser33/37/Thr41) | Cell Signaling Technology | 9561T |
| Active-β-Catenin (nonphosphorylated) | EMD Millipore | 05–665 |
| HIF-1α | Novus Biologicals | NB100–134SS |
| CD326 (EpCAM)-APC* | BioLegend | 118214 |
| CD31-FITC (390) * | eBioscience | 11–0311–85 |
| CD45-FITC (30-F11) * | eBioscience | 11–0451–85 |
| Ter119-FITC * | eBioscience | 11–5921–85 |
| Calreticulin* | Abcam | ab2907 |
| Propidium Iodide* | eBioscience | 00–6690 |
| CD45-PE (30-F11)* | BD | 553081 |
| CD3e-V500 (500A2)* | BD | 560771 |
| CD4-BV786 (RM4–5)* | BD | 563727 |
| CD8a-BUV395 (53–6.7)* | BD | 563786 |
| B220-BV786* | BD | 563894 |
| CD25-BV421 (PC61)* | BD | 562606 |
| FoxP3-eFluor 660 (FJK-16s)* | eBioscience | 50–5773–80 |
| NKp46-Alexa Fluor 700 (29A1.4)* | BD | 561169 |
| F4/80-BV421 (T45–2342)* | BD | 565411 |
| CD11c-BV786 (HL3)* | BD | 563735 |
| CD11b-Alexa Fluor 700 (M1/70)* | BD | 557960 |
| Gr-1-BUV395 (RB6–8C5)* | BD | 563849 |
| IFNγ-Alexa Fluor 488 (XMG1.2)* | BioLegend | 505813 |
| TNFα-Alexa Fluor 700 (MP6-XT22)* | BD | 558000 |
| Bacterial and Virus Strains | ||
| Cignal Lenti Myc Reporter (luc) | Qiagen | CLS-012L-1 |
| Human MYC bHLHZip domain (residues 353–439) BL21-CodonPlus stain | (Wang et al., 2007) | N/A |
| Human Max isoform, Max(L) (160 amino acids) BL21-CodonPlus strain | (Wang et al., 2007) | N/A |
| Max(S) (151 amino acids) BL21-CodonPlus strain | (Wang et al., 2007) | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant human MYC protein | Abcam | ab169901 |
| Recombinant activated human ERK2 protein | Sigma Aldrich | E1283 |
| Recombinant human GSK3β protein | Abcam | ab60863 |
| 3X FLAG Peptide | Sigma Aldrich | F4799 |
| ATP disodium salt hydrate | Sigma Aldrich | A26209 |
| iTaq™ Universal SYBR® Green Supermix | Bio-Rad | 172–5122 |
| Corning Matrigel | Thomas Scientific | 354234 |
| Y-27632 (Dihydrochloride) | STEMCELL Technologies | 72302 |
| Pierce™ Streptavidin Magnetic Beads | Thermo Fisher Scientific | 88817 |
| TWEEN80 | MP Biomedicals | 02194725.1 – 100 ml |
| Lipofectamine® 2000 Transfection Reagent | Invitrogen | 11668–019 |
| TEV Protease | Sigma-Aldrich | T4455–1MG |
| Anti-FLAG® M2 Magnetic Beads | Sigma-Aldrich | M8823–1ML |
| MG132 | VWR | 80053–196 |
| Phosphatase Inhibitor Cocktail Tablets | Roche | 04906845001 |
| Protease Inhibitor Cocktail Tablets | Roche | 04693124001 |
| MDV 3100 enzalutamide | Axon Medchem | 1613 |
| 10058-F4 | Sigma-Aldrich | F3680–5MG |
| 10074-G5 | Sigma-Aldrich | G3798–5MG |
| JKY-2–169 | Dr. Edward V. Prochownik | N/A |
| IGEPAL® CA-630 | Sigma-Aldrich | I8896–50ML |
| Corn oil | Sigma-Aldrich | C8267–500ML |
| Isopropyl beta-D-thio galactopyranoside (IPTG) solution | Sigma-Aldrich | I1284–5ML |
| Polypropylene Columns | Qiagen | 34964 |
| Ni-NTA Agarose | Qiagen | 30210 |
| Collagenase | Gibco | 17018–029 |
| DNase I | Sigma | 10104159001 |
| Critical Commercial Assays | ||
| Steady-Glo® Luciferase Assay | Promega | E2510 |
| CellTiter 96 AQueous One Solution Cell Proliferation Assay | Promega | G3580 |
| ATP Determination Kit | Thermo Fisher Scientific | A22066 |
| Dynabeads® Co-Immunoprecipitation Kit | Thermo Fisher Scientific | 14321D |
| Nuclear Complex Co-IP Kit | Active Motif | 54001 |
| HMGB1 ELISA | Tecan Trading | ST51011 |
| Nuclear Complex Co-IP Kit | Active Motif | 54001 |
| RNeasy Plus Mini Kit | Qiagen | 74134 |
| Duolink® In Situ Red Starter Kit | Sigma-Aldrich : Aldrich | DUO92101–1KT |
| Deposited Data | ||
| RNAseq | GEO | GSE135877 |
| Proteomic | Peptideatlas | PASS01427 |
| Experimental Models: Mouse strain/Cell Lines | ||
| FVB mice | Jackson Laboratory | Stock No: 001800 |
| Prostate patient derived xenograft (PDX) model | Jackson Laboratory | Model ID: TM00298 |
| NSG mice | Jackson Laboratory | Stock No: 005557 |
| CB17/Icr-Prkdcscid/IcrIcoCrl mice | Charles River | Strain Code: 251 |
| C57BL/6 | Jackson Laboratory | Stock No: 000664 |
| CD-1 | Charles River | Strain Code 022 |
| MycCaP | ATCC | CRL-3255™ |
| HL-60 | ATCC | CCL-240™ |
| SK-N-BE(2) | ATCC | CRL-2271™ |
| PC12 adh cell line | ATCC | CRL-1721.1™ |
| PC3 | ATCC | CRL-1435™ |
| MV411 | ATCC | CRL-9591 |
| 293T | ATCC | CRL-3216™ |
| LLC1 | Professor Bin Zhang | N/A |
| P493–6 B | Professor Chi Van Dang | N/A |
| TGR-1 and HO15.19 Rat-1 cells | Professor John Sedivy | N/A |
| Oligonucleotides | ||
| HEX_Ebox_One strand: 5′-hexachlorofluorescenine-CACCCGGTCACGTGGCCTACAC Complementary strand: GTGTAGGCCACGTGACCGGGTG |
(Wang et al., 2007) | N/A |
| Human 18SrRNA primer (5′ to 3′) F: GTAACCCGTTGAACCCCATT R: CCATCCAATCGGTAGTAGCG |
This study | N/A |
| Human MYC primer F: GTCAAGAGGCGAACACACAAC R: TTGGACGGACAGGATGTATGC |
This study | N/A |
| Human CDC25A primer F: GTGAAGGCGCTATTTGGCG R: TGGTTGCTCATAATCACTGCC |
This study | N/A |
| Human MYB primer F: CCAACTGTTCACGCAGACCT R: CTTCTGATGCTGGTGCCATT |
This study | N/A |
| Recombinant DNA | ||
| Lentiviral constructs expressing Flag-tagged MYC, Flag-tagged MYCT58A and Flag-tagged MYC-S62A | (Fang et al., 2017) | N/A |
| pCS2-MYC | (Welcker et al., 2004) | N/A |
| Myc-responsive pGL-M4 luciferase reporter | (Kapeli and Hurlin, 2011) | N/A |
| Software and Algorithms | ||
| GraphPad Prism 7 | GraphPad Software | https://www.graphpad.com |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| FlowJo Software | FlowJo LLC | https://www.flowjo.com |
| Other | ||
| Ultra Low Attachment Microplate | Corning | 3474 |
| Slide-A-Lyzer™ Dialysis Cassettes | Thermo Fisher Scientific | 66810 |
| Zeba Spin Desalting Columns | Thermo Fisher Scientific | 89882 |
Primary antibodies used for flow cytometry
Cellular thermal shift assay (CETSA)
To determine target engagement of MYC by compound within cells, PC3 cells with 70 to 80% confluence in 15cm culture dish were treated with compounds or vehicle (DMSO) for 30 min. Cells were harvested and washed once with PBS, then suspended in 1 ml of PBS supplemented with proteinase and phosphatase inhibitors (Roche) and also maintained with same dose of compounds or DMSO as initial treatment. The cell suspension was distributed into seven to ten 0.2-ml PCR tubes with 100 μl volume (about 1 million cells) and each tube was designated a temperature point. Samples were heated at their designated temperatures for 2 min in AB 96-well thermal cycler. Immediately after heating, tubes were removed and incubated at room temperature for 3 min. After this 3 min incubation, tubes were immediately snap-frozen in liquid nitrogen, and stored at −80 C°. In order to lyse the cells, three freeze and thaw cycles in liquid nitrogen was performed. The tubes were vortexed briefly after each thawing. Cell lysate was collected and cell debris together with precipitated and aggregated proteins were removed by centrifuging samples at 20,000 g for 20 min at 4 °C. Cell lysate samples were boiled for 5 min at 90 °C after addition of loading buffer, and subjected to Western Blot analysis. The MYC antibody was from Abcam (Y69, Ab32072) and protein intensity was quantified through Image J software.
Fluorescence polarization competition and fluorescence measurements
Human MYC bHLHZip domain (residues 353–439) was expressed and purified as described above, followed by an additional buffer exchange step using 7K MWCO Zeba Spin Desalting Columns (Thermo) and the following buffer: pH 8.0 50 mM NaH2PO4, 10 mM Tris base, 10 mM NaCl, 5 mM EDTA, 2 mM DTT, which is used as reaction buffer in this experiment. Samples were analyzed on a PC1 ISS spectrofluorimeter (ISS Inc., Champaign, IL) equipped with UV grade Glan-Thompson polarizers in the L format, under temperature control. All measurements were performed at 25 °C. Polarization measurements were conducted at an excitation wavelength of 470 nm and an emission wavelength of 560 nM, with the spectral width of excitation and emission slits set at 1 nm. Sample volume was 60 μl, in 100 μl quartz glass cuvettes with 10×2 mm optical path length. Competition affinity experiments were performed over a range of concentrations (3 – 25 μM) of the nonfluorescent inhibitor 361 or 975 being titrated against 10 μM 10074-G5 in the presence of 10 μM MYC353–439. Data was analyzed using the “One site - Fit Ki” analysis, part of the “Binding-competitive” suite in Prism 7.
In vitro pull down assay
To confirm 361/975 direct binding to endogenous MYC protein in cell lysate complex or binding to purified recombinant MYC protein, biotin conjugated 361/975 (Biotin361/975) was synthesized. Nuclear complex from exponentially growing PC3 and P493–6 cells was extracted using Nuclear Complex Co-IP Kit (Active Motif) and the Low co-IP buffer with addition of salt (final concentration of 150mM NaCl) was used for the pull down experiment. Nuclear extracts were pre-cleared with streptavidin beads (Thermo, 88817) for 1 hr at 4C°. Note that for MYC recombinant protein binding assay, the pull down buffer was supplemented with 1% BSA and the pre-cleared step was skipped. About 100 μg nuclear extract or 0.5μg MYC protein was applied to each sample and incubated with 1 to 10 μM of Biotin361, 10 μM of D-Biotin or DMSO on a rotator over night at 4C°. For the competition binding assay, the nuclear extract was pretreated with 80X more Phosphate361 or other MYC inhibitors (10074-G5, 10058-F4 or JKY-2–169) for 30 min, then Biotin361 was added. Next day, 60 μl of streptavidin beads was added to each sample and further rotated for 1 hr at 4C°. Beads were washed with wash buffer containing 0.1%BSA for 3 times and another 3 times with wash buffer without BSA, then eluted with 2x sample buffer and boiled at 95C° for 5 min. The supernatant was subjected to Western Blot and proteomic analysis.
Proteomic analysis
LC-MS/MS analysis was performed by Northwestern Proteomics Core Facility. In the pull down experiment, 10 μM of Biotin-361 or −975 was used. For MYC off condition, P493–6 cells were treated with 0.1 μg/ml tetracycline for 3 days before the nuclear extraction. Serial dilution of recombinant MYC protein in p493–6 MYC off extracts was used to generate a standard curve to quantify MYC levels in the pull down elution. Same amount of recombinant MYC was added to the Biotin-975 duplicate pull down sample in MYC off condition before LC-MS/MS analysis. The pull down samples were loaded onto stacking gel for 5min, and gel lane holding the total loaded proteins was excised and submitted to the facility. The proteins were digested with trypsin and analyzed by LC-MS/MS using a Dionex UltiMate 3000 Rapid Separation nanoLC and an Orbitrap Elite Mass Spectrometer (Thermo Fisher Scientific Inc, San Jose, CA) following the standard protocol in the Proteomics Core Facility. All MS/MS samples were analyzed using Mascot (Matrix Science, London, UK; version 2.5.1). Mascot was set up to search the uniprot-SP-human_20180326_20190417 database (selected for Homo sapiens, 20303 entries) assuming the use of the digestion enzyme trypsin. Mascot was searched with a fragment ion mass tolerance of 0.60 Da and a parent ion tolerance of 10.0 PPM. Scaffold (version Scaffold_4.8.9, Proteome Software Inc., Portland, OR) was used to validate MS/MS based peptide and protein identifications. Peptide identifications were established at greater than 90.0% probability by the Peptide Prophet algorithm with Scaffold delta-mass correction. Protein identifications were established at greater than 99.0% probability to achieve an FDR less than 1.0% and contained at least 2 identified peptides. Protein probabilities were assigned by the Protein Prophet. Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony (version Scaffold_4.8.9, publish). All samples were analyzed on the basis of protein spectral counts. For each pull down sample, spectral counts of proteins identified in the control sample (DMSO) were subtracted from proteins identified from the corresponding MYCi pull down samples, and the proteins with ≥5 spectral counts were considered as real bindings. Moreover, all identified proteins were filtered by Contaminant Repository for Affinity Purification (CRAPome) database following the workflow 1 instructions (www.crapome.org). The proteins with over 20% frequency and maximum counts less than in CRAPome database were considered as nonspecific bindings and removed from the list.
Co-immunoprecipitation Assay (Co-IP)
Cells were treated with the compounds at indicated time points and lysed with lysis buffer (20 mM TrisHCl [pH 8.0], 100 mM NaCl, 1 mM EDTA, and 0.5% Nonidet P-40). The insoluble pellets from the crude lysis step was treated with enzymatic shearing cocktail from Nuclear Complex Co-IP Kit (Active Motif) for 90 min at 4 C° to release nuclear proteins. Both cell lysate fractions were combined and 1mg of protein from the lysate was incubated with either MYC antibody (Santa Cruz, N-262) pre-coated magnetic beads following Dynabeads® Co-Immunoprecipitation Kit protocol (Thermo) or with Anti-FLAG® M2 Magnetic Beads (Sigma) over night at 4 C°. Samples then were washed 3 times with lysis buffer, one time with Last wash buffer and eluted with elute buffer for MYC pre-coated beads. For Anti-FLAG® M2 Magnetic Beads binding proteins, 3X FLAG Peptide (Sigma) was used to release the binding proteins. The eluted fractions were analyzed by Western blotting.
Proximity ligation assay (PLA)
PC3 or PC3 T58A cells were grown in chamber slides and treated with 361 for 1 to 2 hr, fixed in 4% paraformaldehyde (PFA) and permeabilized with 0.5% Triton-X. After wash with PBS, the cells were incubated with primary antibody against MYC (Y69, Abcam) 1:500 and Max (H-2, Santa cruz) 1:500 for 1 hr in a humidity chamber at 37C° and necessary subsequent procedures were performed according to the instructions of Duolink kit (DUO92101, Sigma). During the amplification step, an additional fluorescently secondary antibody (Alexa Fluor 488 goat anti-rabbit, Life Technologies A11006) was added 1:2000 to the amplification solution to counterstain for MYC protein. Image J software was used to quantify the number of PLA signals per cell.
In vitro kinase assay
The phosphorylation assays were performed in 1x Kinase Buffer (Cell Signaling Technology, 9802) containing 25 mM Tris-HCl (pH 7.5), 5 mM beta-glycerophosphate, 2 mM DTT, 0.1 mM sodium vanadate, 10 mM MgCl2, in a total volume of 20 μl at room temperature, for a total duration of 2 hr. For each individual reaction, first recombinant ERK (0.1 mg/ml) (Sigma, E1283) and MYC protein (0.5 mg/ml) (Abcam, ab169901) stocks are diluted 4x with 1x kinase buffer and 1 μl of each is added to 10 μl deionized water, 2 μl of 10x kinase buffer and 4 μl of 1 mM ATP (Sigma Aldrich, A26209). After 1 h of incubation at room temperature, 1 μl of GSK3β (0.1 mg/ml) (Abcam, ab60863) stock diluted 4x with 1x kinase buffer and 1 μl of either DMSO or compound at 20x desired concentration are added and reaction allowed to run for another hour at room temperature. Final concentrations of proteins are as follows: 118 nM MYC, 18 nM ERK2 and 17 nM GSK30. Reactions were terminated by addition of 20 μl 2x Laemmli sample buffer and boiling for 5 min, followed by Western Blot analysis.
DNA constructs, Lentivirus production and lentiviral transduction of cell lines
MYC E-box-luciferase reporter Lentivirus was acquired from Qiagen (Cat. 336851). Lentiviral constructs expressing Flag-tagged MYC (Flag-MYC), Flag-tagged T58A-MYC (Flag-MYCT58A) and Flag-tagged S62A-MYC (Flag-MYCS62A) were kindly provided by Dr. Bao from Lerner Research Institute (Fang et al., 2017). Viral particles were produced in 293T cells transfected with the expressing vector, Δ8.9 packaging vector and VSVG envelope vector (2:1:1) using Lipofectamine 2000 (Invitrogen) in Opti-MEM media (Gibco) as described (Anker et al., 2018). PC3 cells were transduced with the virus carrying Flag-MYC, Flag-MYCT58A or Flag-MYCS62A, and 1.5 μg/ml of puromycin was added to select stably expressing cells. MycCaP cells were transduced with E-box-luciferase reporter Lentivirus, and the stable expressing clone was established by maintaining cells in 12 μg/ml of puromycin.
E-Box luciferase assay
With 293T system, 12000 cells were seeded into 96-well white-wall plate. Next day, 20ul of transfection reagents mixture (pCS2-MYC plasmid; 1μg, Myc-responsive pGL-M4 luciferase reporter plasmid; 1μg, Lipofactamine2000; 5 μl, Opti-mem medium; 500 μl) was added and compounds were treated following the day. After 24h treatment, luminescence signal was determined using Steady-Glo® Luciferase Assay System (E2510, Promega), and MTS assay for cell viability were performed at same condition. Normalized relative luminescence by cell viability was presented in the graph. For MycCaP cells system, cells were stably transduced with Lentivirus vector pLV-mCherry-P2A-luciferase (MycCaP-luc) (Anker et al., 2018) or MYC E-box-luciferase reporter (MycCap Ebox-luc) were plated at 10000 cells per well in 96 well white-wall plate. The following day, a serial dilution of 361 were added to the cells. At 4 hr of treatment, luminescence signal was determined as above.
RNA-seq
PC3 cells were treated with 6 μM 361 or 8 μM 975 and P493–6 cells were treated with 0.1 μg/ml Tetracycline or 6 μM 975 for 24 hr in triplicates. Then cells were washed with PBS and RNA was extracted using RNAeasy Plus mini kit (Qiagen). The concentration and quality of total RNA samples was first assessed using Agilent 2100 Bioanalyzer, and a RIN (RNA Integrity Number) threshold of 9 was employed for all samples. A total amount of 50ng RNA used to prepared single-indexed strand-specific cDNA library using TruSeq RNA Access Library Prep Kit (Illumina). RNA-seq was performed by Medical Genomics in Indiana University. The resulting libraries were assessed for its quantity and size distribution using Qubit and Agilent 2100 Bioanalyzer. Two hundred pico molar pooled libraries were utilized per flowcell for clustering amplification on cBot using HiSeq 3000/4000 PE Cluster Kit and sequenced with 2×75bp paired-end configuration on HiSeq4000 (Illumina) using HiSeq 3000/4000 PE SBS Kit. A Phred quality score (Q score) was used to measure the quality of sequencing. More than 90% of the sequencing reads reached Q30 (99.9% base call accuracy). GSEA and Gene Ontology (GO) enrichment analysis were used for cellular pathway analysis.
RT-qPCR
RNA from cell lines was isolated using RNAeasy Plus mini kit (Qiagen). cDNA preparation and RT-PCR using SYBR-Green (Bio-Rad) performed on QuantStudio 6 Flex Real-Time PCR System (Applied Biosystems) were described as in previous study (Anker et al., 2018). Results were represented as fold expression. The sequences of primers used for qPCR analysis were listed in Key Resources Table.
In vivo experiments
Rapid in vivo screening and efficacy study in MycCaP allograft/xenograft prostate mouse model
FVB or NSG male mice of 6–8 weeks of age and ~25 g weight were acclimated after shipping for > 4 days. MycCaP Ebox-Luc cells (1 × 106) suspended in 100 μl BD matrigel were subcutaneously injected into flanks of mice. When the tumor size reached 150 to 200 mm3, mice were grouped with similar average tumor size in each group. Compounds were dissolved in PBS with 10% DMSO and 20% TWEEN80 (MP Biomedicals), the formulation used in the most of in vivo efficacy studies unless otherwise indicated.
In rapid screening, compounds were given i.p. for 3 days at a low dose (30 to 50 mg/kg/day) and another 3 days at a high dose (100 to 200 mg/kg/day) as tolerated by the mice (n = 4 allografts). Caliper measurement of tumor size and live imaging of bioluminescent signal in tumor were performed before and after each block of 3 days of low and high dose treatment.
For 361 efficacy study, the compound was dosed at 100 mg/kg/day for 2 days (50 mg/kg, twice daily). The treatment was paused until tumor attained the original size, and 70 mg/kg/day of 361 was given consecutively for another 9 days (n = 6 to 8 allografts). For short-term treatment experiments of 361 in both FVB and NSG mice baring MycCaP grafts, 50 mg/kg/day were given for 4 days, tumor volume were measured before and after the 4 day treatment. For tumor infiltrating lymphocytes analysis after 361 treatment, 361 (50 mg/kg/day) were administrated by i.p. for 2 days on/off for 2 rounds, and tumor and peripheral lymph nodes (LNs) were analyzed. In 361 combination with anti-PD1 therapy study, 50 mg/kg/day of 361 was given for 2 days following by two day treatment of 100 μg/day of PD-L1 antibody (BioXcell, BE0146) or IgG2a isotype control (BioXcell, BE0089) in PBS by i.p., and keep the 2 days on and off treatment of 361 and PD1 alternatively for 16 days (n = 4 to 6 mice).
For 975 efficacy study as single agent in MycCaP allografts, the compound was dissolved in 5% DMSO of corn oil, and given by i.p. at 100 mg/kg/day (once daily) consecutively for 14 days (n = 8 to 10 allografts). The systematic toxicology were evaluated under the same experimental conditions with mice (no tumor burden) treated with 975 at 100 mg/kg daily for 14 days. Animals were analyzed one week later on hematology, blood chemistry and pathology of the major organs. For the short term treatment of 975 in both FVB and NSG mice baring MycCaP grafts, 975 was dissolved in PBS with 10% DMSO and 20% TWEEN80, and given at 100 mg/kg/day (50 mg/kg, twice daily) by i.p. for three days. In 975 combination with anti-PDl therapy study, 100 mg/kg/day of 975 (50 mg/kg, twice daily) was given for 2 days following by two day treatment of 100 μg/day of PD-L1 antibody in PBS by i.p., and keep the 2 days on and off treatment of 975 and PD1 alternatively for 20 days (n = 5 to 7 mice).
Prostate cancer PDX model
Patient derived xenografts (PDX) model of prostate cancer (TM00298) was obtained from The Jackson Laboratory. About 10 mm2 size of minced tumor fragments were injected to flanks of NSG mice (male). When the grafts were measurable, 361 (55 mg/kg/day) were administrated by i.p. 3 consecutive days a week for two weeks (n = 9 or 10 grafts).
LLC1 allograft Lung cancer mouse model
LLC1 cells (1 × 106) suspended in PBS and BD matrigel (1:1) were subcutaneously injected into flanks of C57BL/6 (female) mice. After 3 days of cell inoculation, mice were randomly grouped and 100 mg/kg/day of 975 (50 mg/kg, twice daily) were treated through i.p. injectiion.
AML xenograft
The experiment of 975 combination with Ara-C in AML xenograft was performed by Developmental Therapeutics Core in Northwestern University. Briefly CB17 SCID mice were inoculated with MV-411 cells at the density of 5×106 suspended in PBS and matrigel (1:1). The mice were randomized based on tumor volume into different groups after the tumor reached ~200 to 500 mm3. The mice were treated either with vehicle, 975 (50 mg/kg/day) alone or in combination with Cytarabine (Ara-C, 20 mg/kg/day) for 3 weeks (5 days a week).
Tumor growth was monitored by the measurement of tumor size using calipers twice per week and calculated with the formula (length × width × width)/2 or by bioluminescent imaging: Luciferase-expressing tumor-bearing mice were injected i.p. with 10 μl/g body weight of 15 mg/ml D-luciferin (sodium salt, Gold Bio), and 12 min after injection, the mice were imaged with an IVIS Spectrum Imaging System (PerkinElmer). Images were analyzed and quantified using Living Image software.
Pharmacokinetic studies
Pharmacokinetic studies on 361 and 975 were performed by Sanford Burnham Prebys Medical Discovery Institute, with mice strain C57BL/6 male and CD-1 male, respectively. MYCi 361 was formulated in PBS with 10% DMSO and 20% TWEEN80 and 50 mg/kg was given at a single dose either by i.p. or p.o. routes. 975 was formulated in corn oil with 5% DMSO for i.p. and p.o. dosing (100 mg/kg and 250 mg/kg), and in 10% HPBCD with 5% Ethanol and 2% Tween80 for i.v. dosing (2 mg/kg). Each data point represents the mean value from three experimental mice.
Maximum tolerance dose (MTD) determination
MTD studies for 361 and 975 were performed by Developmental Therapeutics Core in Northwestern University. Briefly, CD-1 mice (between 6 to 15 weeks old) were acclimated up to 5 days before treatment. Three mice were used for each giving dose. Compounds were formulated in 5% DMSO of corn oil treated by p.o.. After the treatment, the clinical signs were observed 3x within 24 hr and then the next higher dose was provided as scheduled above until the concentration which caused any adverse event was found. Once the adverse event was determined, the next step would be to treat in between the last dose and the safe dose prior to it.
Hematology and blood chemistry analysis
Mouse peripheral blood was collected by cardiac puncture and placed in serum separator or dipotassium-EDTA tubes (BD Microtainer). Serum and whole blood were analyzed, the latter within 24 hr after collection, by Charles River Laboratory. Reference value ranges were used from the University of Arizona University Animal Care (https://uac.arizona.edu/clinical-pathology), and the University of Minnesota Research Animal Resources (http://www.ahc.umn.edu/rar/refvalues.html).
Flow cytometry
Single-cell dissociation from tumor tissue and lymph nodes and the following flow cytometry analysis of immune cells were performed according to the protocol described in a previous study (Anker et al., 2018). Briefly, tumor tissue was dissociated using Tumor Dissociation Kit in C Tubes (MACS Miltenyi Biotec) immediately after dissection from experimental mice. Peripheral lymph nodes were also collected and single cell suspensions were created by passing cells directly through a 70-μm filter, followed by red blood cell lysis with ACK buffer (0.15M NH4Cl, 10mM KHCO3, 0.1mM Na2-EDTA; pH 7.2–7.4; 0.2 μm filtered). Dissociated cells were treated with anti-mouse CD16/CD32 Fc block (2.4G2, BD) first. For intracellular staining, cells were resuspended in RPMI 10% FBS with 50 ng/ml PMA (Sigma), 1 μg/ml ionomycin (Cell Signaling), 1 μl/ml brefeldin A (GolgiPlug; BD), 2 μl/3 ml monensin (GolgiStop; BD), and CD107a antibody when appropriate, for 6 hr at 37 °C 5% CO2. After subsequent extracellular staining, cells were stained with LIVE/DEAD Fixable Blue Dead Cell Stain Kit (Invitrogen). FoxP3 panels were fixed and permeabilized with the FoxP3/Transcription Factor Staining Buffer Set Kit (eBioscience) before antibody incubation. All other panels were fixed in IC fixation buffer (eBioscience) before subsequent permeabilization with the Intracellular Fixation and Permeabilization Buffer Set Kit (eBioscience) and incubation with intracellular antibodies when appropriate. Samples were run on the BD FACSymphony A5 Flow Cytometer. Controls and compensation were performed using anti-rat/hamster Ig, K/negative control compensation particles set (BD) and appropriate fluorescence minus one and unstained controls. Data were analyzed using FlowJo software. A representative flow cytometry gating strategy is displayed in Supplementary Figure S6 (D: tumor, E: LNs), with initial gating on overall morphology, singlets, live cells, and CD45 positivity before proceeding with all further analyses.
Immunohistochemistry and Immunofluorescence
Tissues were fixed in 10% neutral buffered formalin for 48 hr at 4 °C and transferred to 70% ethanol before paraffin processing at the Northwestern University histology core. Paraffin sections (5 μm) were deparaffinized and rehydrated, stained with hematoxylin and eosin, or followed by antigen retrieval with citrate buffer pH6 (Dako) in press cooker for 15 min, 3% H2O2 in methanol (Sigma), blocking with 5% BSA/10% normal goat serum in PBST and incubation with primary specific antibodies (see Key Resources Table): Ki-67 (SolA15) (1:500, eBioscience, 14–5698-80), MYC (phospho T58) (1:1000, Abcam, ab185655), PD-L1 (1:200, Cell Signaling, 13684), CD3 (2GV6) (Ventana, 790–4341), B220/CD45R (1:500, BD, 550286), CD335/NKp46 (29A1.4) ( 1:200, Biolegend, 137601). For H2A.X staining in MYCi treated PC3 cells, cells were fixed in 4% paraformaldehyde (PFA), permeabilized with 0.5% Triton-X, and incubated with primary antibody Phospho-Histone H2A.X (Ser139) (20E3) (1:1000, Cell Signaling, 9718S).
Pathology assessment of the organ tissues from MYCi-treated mice for toxicity evaluation carried out by PATHOGENESIS, LLC (https://pathogenesisllc.com).
Kinases and phosphatases screen
Kinases inhibition screen for 361 was carried out with the KINOMEscan™ screening platform from DiscoverX Corporation (www.discoverx.com). In the assay, results for primary screen binding interactions are reported as Percent Control, where lower numbers indicate stronger hits in the matrix. The selectivity Score (S-score) is a quantitative measure of compound selectivity. It is calculated by dividing the number of kinases that compounds bind to by the total number of distinct kinases tested, excluding mutant variants. Phosphatases screen was performed by Reaction Biology Corporation (http://www.reactionbiology.com/webapps/site/cell-assays.aspx). 361 and control compound PTP1B inhibitor were tested in 10-dose IC50 mode with 3-fold serial dilution starting at 100 μM singly.
STD NMR
STD NMR Spectrum was recorded using a Bruker Advanced 500 spectrometer equipped with Cryo probes and processed by Bruker® Topspin software. A typical sample preparation consist of 5 μM protein and 100 μM ligand in 5% DMSO-d6 and deuterated PBS, pH 7.4 buffer (600 ml total volume) for STD NMR experiments. Protein used for STD NMR were tagless MYC (amino acids 353–439) (Cayman Chemicals) and MAX(S) (151 amino acids, purified in this study) (in HEPES – 50 mM, NaCl – 500 mM, B-mercapethonol - 10 mM, Glycerol - 5%, Imidazole - 30 mM). The ligand was tested in the presence and absence of individual proteins (MYC and MAX). The prepared solution was vortexed for 30 secs and submitted for NMR. Various sample preparations tested were Ligand + MYC, Ligand + MAX, G5 + MYC, G5 + MAX, Ligand alone, and G5 alone. STD NMR experiments were performed with a train of 50 ms Gaussian-shaped saturating pulses at 200 Hz power for 2 s with “on” resonance saturation at −2.5 ppm and “off” resonance saturation at 40 ppm. The relaxation delay was 2 s before the saturating pulses. The number of scans was 2048 and the spectral width was 10 ppm.
Experimental for 361 and 975
Synthesis of 7-hydroxy-2-(trifluoromethyl)-4H-chromen-4-one (2): To a suspension of 1-(2,4-dihydroxyphenyl)ethan-1-one (1) (5.00 g, 32.9 mmol) in trifluoroacetic anhydride (TFAA) (18.50 ml, 131.6 mmol) placed in a high-pressure tube, sodium 2,2,2-trifluoroacetate (9.84 g, 72.4 mmol) was added and the system was capped and stirred at 110 °C for 24 hr. The reaction was allowed to cool down to approximately 70 °C and was diluted with EtOAc (200 ml). The mixture was neutralized by saturated aqueous K2CO3 solution until no more bubbling was observed. The organic layer was separated and the aqueous portion was extracted with EtOAc (3 × 150 ml). The combined organic portion was washed with brine, dried over anhydrous Na2SO4, concentrated to 1/3rd volume of EtOAc and the flask was allowed to stand at room temperature for 1–2 days, obtaining a solid which was filtrated and dried under vacuum to obtain 4.1 g (54% yield) as a white-colored solid.
Synthesis of 7-hydroxy-8-iodo-2-(trifluoromethyl)-4H-chromen-4-one (3): A solution of 2 (4 g, 17.3 mmol, 1 equiv.), molecular iodine (17.6 g, 69.2 mmol), pyridine (5.6 ml, 69.2 mmol) in 110 ml of CHCl3 was stirred at room temperature for 16 hr. On completion, the reaction was quenched with 120 ml of saturated aqueous Na2S2O3 and the organic layer was separated. The aqueous portion was further extracted with CH2Cl2 (3 × 100 ml). The combined organic portion was washed with brine, dried over anhydrous Na2SO4. And concentrated under reduced pressure to yield 5.6 g (90% yield) as a pale white solid. 1H NMR (500 MHz, CDCl3) δ 8.11 (dd, J = 8.9, 1.0 Hz, 1H), 7.15 (dd, J = 8.9, 1.0 Hz, 1H), 6.77 – 6.69 (m, 1H), 6.36 (s, 1H) ppm.
Synthesis of 7-((4-chlorobenzyl)oxy)-8-iodo-2-(trifluoromethyl)-4H-chromen-4-one (4): A suspension of 3, (1 g, 2.8 mmol), p-chlorobenzyl bromide (3.4 mmol) and K2CO3 (0.8 g, 5.6 mmol) in 5 ml of acetone was heated at 60 °C for 16 hr. On completion, the reaction was filtered and the solvent was removed under reduced pressure to yield the crude residue which was suspended in H2O (50 ml). The solid was collected by filtration and dried to yield a brown-colored crude solid which was used for the next step without further purification.
Synthesis of 8-(3,5-bis(trifluoromethyl)phenyl)-7-((4-chlorobenzyl)oxy)-2-(trifluoromethyl)-4H-chromen-4-one (5): A suspension of 4 (0.85 g, 3.1 mmol), 3,5-bis(trifluoromethyl)phenylboronic acid (1.5 g, 3.1 mmol), Na2CO3 (0.7 g, 6.2 mmol) and Pd(dppf)Cl2 (0.23 g, 0.3 mmol) in 16 ml of a mixture 1:2:5 of EtOH:water:toluene was bubbled with N2 gas for 10 min. The vial was then heated at 100 °C for 2 hr. The dark solution was cool down to room temperature and diluted with EtOAc (25 ml). The solution was filtered through a celite pad and the collected organic portion was washed with saturated aqueous NH4Cl (50 ml), water (2 × 100 ml), dried over Na2SO4 and passed over a silica bed to remove the Palladium complex. The collected organic portion was evaporated and the residue was crystallized by 2-propanol to obtain 1.1 g (62% yield) as a dark-brown colored solid which was used for the next step without any further purification.
Synthesis of 6-((4-chlorobenzyl)oxy)-3-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-5-yl)-3’,5’-bis(trifluoromethyl)-[1,1’-biphenyl]-2-ol (NUCC-0198361): A solution of 5 (1.07 g, 1.9 mmol) and methylhydrazine (0.3 ml, 5.7 mmol) in 8 ml of EtOH was heated at 78 °C for 2 hr. On completion, the solution was cooled down to room temperature and concentrated. The solid residue was directly purified by silica gel chromatography (n-hexanes / ethyl acetate = 5:1 to 1:1) to obtain 0.4 g (36% yield) as a yellow-colored solid. 1H NMR (500 MHz, CDCl3) δ 7.88 (s, 2H), 7.84 (s, 1H), 7.28 – 7.16 (m, 3H), 7.11 (d, J = 8.1 Hz, 2H), 6.73 (d, J = 8.5 Hz, 1H), 6.56 (s, 1H), 5.01 (s, 2H), 3.80 (s, 3H) ppm; 13C NMR (126 MHz, CDCl3) δ 157.5, 151.5, 142.1 (d, J = 38.7 Hz), 139.4, 134.8 – 133.8 (m), 131.8, 131.7, 131.5, 131.4, 131.2, 131.1, 129.2, 128.9, 128.8, 128.3, 128.2, 123.3 (d, J = 272.2 Hz), 122.2, 121.6, 120.1, 115.9, 115.8, 109.8, 107.5, 105.5, 105.3, 69.9 ppm.
Synthesis of 8-(4-chloro-3-(trifluoromethyl)phenyl)-7-((4-chlorobenzyl)oxy)-2-(trifluoromethyl)-4H-chromen-4-one (7): A suspension of 4 (0.7 g, 3.1 mmol), 4-chloro-3-(trifluoromethyl)phenylboronic acid (1.5 g, 3.1 mmol), Na2CO3 (0.7 g, 6.2 mmol) and Pd(dppf)Cl2 (0.23 g, 0.3 mmol) in 16 ml of a mixture 1:2:5 of EtOH:water:toluene was bubbled with N2 gas for 10 min. The vial was then heated at 100 °C for 2 hr. The dark solution was cool down to room temperature and diluted with EtOAc (25 ml). The solution was filtered through a celite pad and the collected organic portion was washed with saturated aqueous NH4Cl (50 ml), water (2 × 100 ml), dried over Na2SO4 and passed over a silica bed to remove the palladium complex. The collected organic portion was evaporated and the residue was crystallized by 2-propanol to obtain 1.2 g (70% yield) as a dark-brown colored solid which was used for the next step without any further purification.
Synthesis of 4’-chloro-6-((4-chlorobenzyl)oxy)-3-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-5-yl)-3’-(trifluoromethyl)-[1,1’-biphenyl]-2-ol (NUCC-0200975): A solution of 7 (1.1 g, 2.1 mmol) and methylhydrazine (0.33 ml, 6.2 mmol) in 8 ml of EtOH was heated at 78 °C for 2 hr. On completion, the solution was cooled down to room temperature and concentrated. The solid residue was directly purified by silica gel chromatography (n-hexanes / ethyl acetate = 5:1 to 1:2) to obtain 0.5 g (43% yield) as a yellow-colored solid. 1H NMR (500 MHz, CDCl3) δ 7.77 (d, J = 2.0 Hz, 1H), 7.59 (d, J = 8.2 Hz, 1H), 7.51 (dd, J = 8.3, 2.1 Hz, 1H), 7.32 – 7.26 (m, 2H), 7.19 (d, J = 8.5 Hz, 1H), 7.16 – 7.10 (m, 2H), 6.71 (d, J = 8.6 Hz, 1H), 6.55 (s, 1H), 5.20 (s, 1H), 5.04 (s, 2H), 3.81 (s, 3H), 2.14 (s, 2H) ppm; 13C NMR (126 MHz, CDCl3) δ 157.4, 151.4, 141.7 (d, J = 39..1 Hz), 140.04, 135.1, 134.5, 133.9, 132.07, 131.8, 131.6, 131.03, 130.4 (d, J = 15.1 Hz), 128.9, 128.8, 128.6, 128.1, 122.7 (d, J = 274 Hz), 121.2 (d, J = 268.4 Hz), 116.07, 109.8, 105.4, 105.4, 105.3, 69.8, 37.8, 30.9 ppm.
Experimental for Phosphate-975
Synthesis of 8-(4-chloro-3-(trifluoromethyl)phenyl)-7-hydroxy-2-(trifluoromethyl)-4H-chromen-4-one (8): A suspension of 3 (0.5 g, 2.2 mmol), 4-chloro-3-(trifluoromethyl)phenylboronic acid (0.8 g, 2.2 mmol), Na2CO3 (0.5 g, 4.4 mmol) and Pd(dppf)Cl2 (0.2 g, 0.2 mmol) in 3.5 ml of a mixture 1:2:6 of EtOH:water:toluene was bubbled with N2 gas for 10 min. The vial was then heated at 100 °C for 2 hr. The reaction was cooled and the mixture was suspended in EtOAc (30 ml) and washed with H2O (2 × 40 ml) and the combined aqueous portion was further extracted with EtOAc (2 × 20 ml). The combined organic portion was passed over a celite plug (to remove the Pd-based compound) and evaporated to yield a crude dark-brown colored residue which was purified by silica gel chromatography (n-hexanes / ethyl acetate = 10:1 to 1:1). The product fractions were collected, evaporated and dried to yield 0.2 g (30% yield) as a light-yellow colored solid.
Synthesis of di-tert-butyl (((8-(4-chloro-3-(trifluoromethyl)phenyl)-4-oxo-2-(trifluoromethyl)-4H-chromen-7-yl)oxy)methyl) phosphate (9): A suspension of 8, (0.1 g, 0.26 mmol), di-tert-Butyl-(chloromethyl)phosphate (0.2 g, 0.78 mmol), Nal (0.02 g, 0.12 mmol) and Cs2CO3 (0.25 g, 0.78 mmol) in 0.5 ml of anhydrous DMF was heated at 60 °C for 48 hr. On completion, the reaction was purified by prep HPLC (40–95% ACN with 0.1% FA, C18, 50 ml/min) and the product (at retention time = 4.2 min) was collected and dried to obtain 45 mg (26% yield) of off-white colored solid.
Synthesis of di-tert-butyl (((4’-chloro-6-hydroxy-3’-(trifluoromethyl)-5-(3-(trifluoromethyl)-1H-pyrazol-5-yl)-[1,1’-biphenyl]-2-yl)oxy)methyl) phosphate (10): A solution of 9 (26 mg, 41 μmol) and hydrazine (65 μl, 1.2 mmol) in EtOH (0.25 ml) was stirred at 50 °C for 1 hr. On completion, the residue was diluted with ACN and purified by prep HPLC (30–95% ACN with 0.1% FA, C18, 50 ml/min) and the product 15 mg (56% yield) (at retention time = 4.3 min) was collected as a solid. 1H NMR (500 MHz, CDCl3) δ 7.69 (d, J = 1.9 Hz, 1H), 7.54 – 7.45 (m, 1H), 7.41 (d, J = 8.2 Hz, 1H), 7.11 (d, J = 8.7 Hz, 1H), 6.76 (s, 1H), 6.43 (d, J = 8.7 Hz, 1H), 5.42 (d, J = 12.5 Hz, 2H), 1.42 (s, 18H) ppm; 13C NMR (126 MHz, CDCl3) δ 153.6, 153.5, 135.8, 132.3, 130.8, 130.7, 130.3, 130.3, 127.8, 127.5, 126.4, 124.07, 121.9, 121.4, 119.2, 116.7, 110.9, 104.5, 100.6, 87.7, 87.6, 84.5, 84.4, 29.7, 29.7.
Synthesis of ((4’-chloro-6-hydroxy-3’-(trifluoromethyl)-5-(3-(trifluoromethyl)-1H-pyrazol-5-yl)-[1,1’-biphenyl]-2-yl)oxy)methyl dihydrogen phosphate (11): A solution of 10 (15 mg, 23 mmol) in anhydrous DCM (0.5 ml) was treated with TFA (54 μl, 0.7 mmol) and the reaction was stirred at room temp for 1 hr. On completion, the reaction was evaporated to dryness to yield 11 mg (90% yield) of off-white colored solid. 1H NMR (500 MHz, CD3OD) δ 7.77 (s, 1H), 7.67 (d, J = 9.3 Hz, 3H), 7.11 (d, J = 8.8 Hz, 1H), 7.03 (s, 1H), 5.51 (d, J = 9.2 Hz, 2H) ppm; 13C NMR (126 MHz, CD3OD) δ 155.7, 152.7, 136.4, 133.2, 130.9, 130.3, 130.1, 128.1, 127.5, 127.2, 124.2, 122.4, 122.05, 120.3, 117.6, 111.3, 106.5, 101.3, 87.7 ppm.
Experimental for Phosphate-361
Synthesis of 8-(3,5-bis(trifluoromethyl)phenyl)-7-hydroxy-2-(trifluoromethyl)-4H-chromen-4-one (12): A suspension of 3 (0.5 g, 1.4 mmol), 3, 5-bis(trifluoromethyl)phenylboronic acid (0.8 g, 1.4 mmol), Na2CO3 (0.3 g, 2.8 mmol) and Pd(dppf)Cl2 (0.1 g, 0.14 mmol) in 6 ml of a mixture 1:2:5 of EtOH:water:toluene was bubbled with N2 gas for 5 min. The vial was then closed and heated at 100 °C for 2 hr. The reaction was cooled and the mixture was evaporated to yield a crude solid residue. The solid was suspended in water (5 ml) and filtered. The collected solid was futher washed with water (2 × 10 ml), dried and dissolved in DCM (10 ml). The organic portion was passed through a celite plug (to remove Pd-based side product) from the reaction and the solvent was evaporated to yield 0.3 g (49% yield) as a yelllow-colored solid.
Synthesis of di-tert-butyl (((8-(3,5-bis(trifluoromethyl)phenyl)-4-oxo-2-(trifluoromethyl)-4H-chromen-7-yl)oxy)methyl) phosphate (13): A suspension of 12, (40 mg, 0.09 mmol), di-tert-butyl-(chloromethyl)phosphate (59 mg, 0.22 mmol), Nal (6.8 mg, 0.05 mmol) and Cs2CO3 (89 mg, 0.27 mmol) in 1 ml of anhydrous DMF was heated at 60 °C for 16 hr. On completion, the reaction was quenched with water (2 ml) and extracted with EtOAc (3 × 2 ml). The combined organic portion was evaporated, redissolved in MeOH (1 ml) and purified by prep HPLC (40–95% ACN with 0.1% FA, C18, 50 ml/min) and the product (at retention time = 4.2 min) was collected and dried to obtain 35 mg (57% yield) as buff-colored solid.
Synthesis of di-tert-butyl (((6-hydroxy-5-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-5-yl)-3’,5’-bis(trifluoromethyl)-[1,1’-biphenyl]-2-yl)oxy)methyl) phosphate (14): A solution of 13 (35 mg, 0.05 mmol) and methylhydrazine (14 μl, 0.26 mmol) in EtOH (0.5 ml) was stirred at 80 °C for 1 hr. On completion, the residue was diluted with ACN (to 1 ml) and purified by prep HPLC (40–95% ACN with 0.1% FA, C18, 50 ml/min) and the product (at retention time = 4.0 min) was collected and dried to obtain 7.0 mg (39% yield on the desired isomer) as a solid.
Synthesis of ((6-hydroxy-5-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-5-yl)-3’,5’-bis(trifluoromethyl)-[1,1’-biphenyl]-2-yl)oxy)methyl dihydrogen phosphate (15): A solution of 14 (7 mg, 0.01 mmol) in anhydrous DCM (0.5 ml) was treated with TFA (23 μl, 0.3 mmol) and the reaction was stirred at room temp for 1 hr. On completion, the reaction was evaporated to dryness to yield 5.3 mg (90% yield) of buff-colored solid. 1H NMR (500 MHz, CD3OD) δ 8.01 – 7.85 (m, 4H), 7.33 (d, J = 8.7 Hz, 1H), 7.06 (d, J = 8.7 Hz, 1H), 6.63 (s, 1H), 5.61 (d, J = 12.2 Hz, 2H), 3.81 (s, 3H). 13C NMR (126 MHz, CD3OD) δ 157.7, 157.4, 154.9, 152.2, 141.04, 139.9, 135.3, 131.2, 130.2, 129.9, 123.8, 121.7, 116.9, 115.4, 113.2, 111.7, 105.3, 104.2, 86.9, 86.9, 35.8 ppm.
Experimental for Biotinylated-361
Synthesis of tert-butyl (3-((8-iodo-4-oxo-2-(trifluoromethyl)-4H-chromen-7-yl)oxy)propyl)carbamate (16): A suspension of 3 (0.3 g, 0.9 mmol), tert-butyl (3-bromopropyl)carbamate (0.4 g, 1.7 mmol) and K2CO3 (0.2 g, 1.7 mmol) in 8 ml of acetone was heated at 60 °C for 18 hr. The reaction was diluted with quenched by adding water (10 ml) and extracted with EtOAc (10 ml × 3). Combined organic portions were dried over Na2SO4, filtrated, concentrated under reduced pressure and purified by silica gel chromatography (n-hexanes / ethyl acetate = 10:1 to 1:1). The product fractions were collected, evaporated and dried to yield 0.4 g (86% yield) as a light-yellow colored oil.
Synthesis of tert-butyl (3-((8-tert-butyl (3-((8-(3,5-bis(trifluoromethyl)phenyl)-4-oxo-2-(trifluoromethyl)-4H-chromen-7-yl)oxy)propyl)carbamate (17): A suspension of 16 (0.25 g, 0.5 mmol), 3,5-bis(trifluoromethyl)phenyl)boronic acid (0.16 g, 0.6 mmol), Na2CO3 (0.11 g, 1.0 mmol) and Pd(dppf)Cl2 (0.03 g, 0.04 mmol) in 3.6 ml of a mixture 1:2:6 of EtOH:water:toluene was bubbled with N2 gas for 10 min. The vial was then heated at 100 °C for 3 hr. The reaction was cooled and the mixture was suspended in EtOAc (30 ml) and washed with H2O (2 × 40 ml) and the combined aqueous portion was further extracted with EtOAc (2 × 20 ml). The combined organic portion was passed over a celite plug (to remove the Pd-based compound) and evaporated to yield a crude dark-brown colored residue which was purified by silica gel chromatography (n-hexanes / ethyl acetate = 10:1 to 1:1). The product fractions were collected, evaporated and dried to yield 0.28 g (76% yield) as a light-orange colored oil.
Synthesis of tert-butyl (3-((6-hydroxy-5-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-5-yl)-3’,5’-bis(trifluoromethyl)-[1,1’-biphenyl]-2-yl)oxy)propyl)carbamate (18): A solution of 17 (0.28 g, 0.47 mmol) and methylhydrazine (68 ml, 1.41 mmol) in EtOH (5 ml) was stirred at 80 °C for 45 min. On completion, the residue was concentrated under reduced pressure and purified by silica gel chromatography (n-hexanes / ethyl acetate = 10:1 to 1:2). The product fractions were collected, evaporated and dried to yield 0.13 g (43% yield) as a light-yellow colored oil.
Synthesis of N-(3-((6-hydroxy-5-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-5-yl)-3’,5’-bis(trifluoromethyl)-[1,1’-biphenyl]-2-yl)oxy)propyl)-1-(5-((4S)-2-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)pentanamido)-3,6,9,12,15,18-hexaoxahenicosan-21-amide (19): Firstly, a solution of compound 18 (15 mg, 24 μmol) in 0.5 ml solution of dichloromethane:TFA (10:1) stirred at room temperature for 4h. The crude reaction was concentrated under reduced pressure and dissolved in 300 ml of freshly distilled DMF. After that, this solution was added over a solution of Biotin-PEG6-COOH (12 mg, 23 mmol), EDC (4.4 mg, 23 μmol), HOBt (3.5 mg, 23 mmol) and Et3N (16 ml, 110 mmol) in freshly distilled DMF at 0 °C. The resulting solution stirred for 16 hr from 0 °C to room temperature. The reaction was filtrated and directly purified by preparative HPLC (water / acetonitrile = 95:5 to 1:1). The product fractions were collected, evaporated and dried to yield 13 mg (38% yield) as a colorless oil. 1H NMR (500 MHz, Chloroform-d) δ 7.97 (d, J = 11.1 Hz, 2H), 7.83 (d, J = 8.3 Hz, 1H), 7.20 (d, J = 8.5 Hz, 1H), 6.82 – 6.65 (m, 3H), 6.56 (s, 1H), 5.15 (s, 1H), 4.44 (dd, J = 7.8, 5.0 Hz, 1H), 4.24 (dd, J = 8.0, 4.7 Hz, 1H), 4.02 (t, J = 6.3 Hz, 2H), 3.84 (s, 2H), 3.66 (dt, J = 11.8, 5.6 Hz, 2H), 3.63 – 3.41 (m, 24H), 3.34 (dq, J = 7.2, 4.3 Hz, 2H), 3.23 (q, J = 6.5 Hz, 2H), 3.09 (dt, J = 7.1, 3.7 Hz, 1H), 2.86 (dd, J = 12.6, 5.3 Hz, 1H), 2.67 (d, J = 12.8 Hz, 1H), 2.37 (q, J = 5.5 Hz, 2H), 2.14 (td, J = 7.3, 2.7 Hz, 2H), 1.99 – 1.76 (m, 5H), 1.74 – 1.50 (m, 4H), 1.45 – 1.31 (m, 2H) ppm. HRMS (ESI+): calcd: 1088.3995; found: 1088.3975 (−1.86 ppm).
Experimental for Biotinylated-975
Synthesis of tert-butyl (3-((8-(4-chloro-3-(trifluoromethyl)phenyl)-4-oxo-2-(trifluoromethyl)-4H-chromen-7-yl)oxy)propyl)carbamate (20): A suspension of 16 (0.10 g, 0.19 mmol), (4-chloro-3-(trifluoromethyl)phenyl)boronic acid (0.06 g, 0.24 mmol), Na2CO3 (0.04 g, 0.39 mmol) and Pd(dppf)Cl2 (0.01 g, 0.01 mmol) in 3.6 ml of a mixture 1:2:6 of EtOH:water:toluene was bubbled with N2 gas for 10 min. The vial was then heated at 100 °C for 3 hr. The reaction was cooled and the mixture was suspended in EtOAc (30 ml) and washed with H2O (2 × 40 ml) and the combined aqueous portion was further extracted with EtOAc (2 × 20 ml). The combined organic portion was passed over a celite plug (to remove the Pd-based compound) and evaporated to yield a crude dark-brown colored residue which was purified by silica gel chromatography (n-hexanes / ethyl acetate = 10:1 to 1:1). The product fractions were collected, evaporated and dried to yield 0.11 g (98% yield) as a light-orange colored oil.
Synthesis of tert-butyl (3-((6-hydroxy-5-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-5-yl)-3’,5’-bis(trifluoromethyl)-[1,1’-biphenyl]-2-yl)oxy)propyl)carbamate (21): A solution of 20 (0.11 g, 0.19 mmol) and methylhydrazine (28 ml, 0.58 mmol) in EtOH (2 ml) was stirred at 80 °C for 45 min. On completion, the residue was concentrated under reduced pressure and purified by silica gel chromatography (n-hexanes / ethyl acetate = 10:1 to 1:2). The product fractions were collected, evaporated and dried to yield 0.06 g (52% yield) as a light-yellow colored oil.
Synthesis of N-(2-((6-hydroxy-5-(1-methyl-3-(trifluoromethyl)-1H-pyrazol-5-yl)-3’,5’-bis(trifluoromethyl)-[1,1’-biphenyl]-2-yl)oxy)ethyl)-1-(5-((4S)-2-oxohexahydro-1H-thieno[3,4-d]imidazol-4-yl)pentanamido)-3,6,9,12-tetraoxapentadecan-15-amide (22): Firstly, a solution of compound 21 (17 mg, 29 μmol) in 0.5 ml solution of dichloromethane:TFA (10:1) stirred at room temperature for 2h. The crude reaction was concentrated under reduced pressure and solved in 300 μl of freshly distilled DMF. After that, this solution was added over a solution of Biotin-PEG4-COOH (13 mg, 26 μmol), EDC (5 mg, 26 μmol), HOBt (4 mg, 26 μmol) and Et3N (13 μl, 130 μmol) in freshly distilled DMF at 0 °C. The resulting solution stirred for 16 hr from 0 °C to room temperature. The reaction was filtrated and directly purified by preparative HPLC (water / acetonitrile = 95:5 to 1:1). The product fractions were collected, evaporated and dried to yield 18 mg (71% yield) as a colorless oil. 1H NMR (500 MHz, CDCl3) δ 7.76 (d, J = 1.8 Hz, 1H), 7.62 – 7.55 (m, 2H), 7.17 (d, J = 8.5 Hz, 1H), 6.67 – 6.57 (m, 3H), 6.52 (s, 1H), 5.92 (s, 1H), 5.00 (d, J = 5.4 Hz, 1H), 4.42 (dd, J = 7.9, 4.9 Hz, 1H), 4.21 (dd, J = 8.1, 4.8 Hz, 1H), 4.00 (t, J = 6.0 Hz, 2H), 3.81 (s, 2H), 3.70 – 3.62 (m, 2H), 3.56 (dd, J = 10.1, 1.7 Hz, 11H), 3.54 – 3.43 (m, 4H), 3.34 (q, J = 5.3 Hz, 2H), 3.23 (q, J = 6.3 Hz, 2H), 3.07 (td, J = 7.3, 4.4 Hz, 1H), 2.89 – 2.81 (m, 1H), 2.65 (d, J = 12.8 Hz, 1H), 2.41 – 2.30 (m, 2H), 2.12 (t, J = 7.4 Hz, 2H), 1.85 (p, J = 6.3 Hz, 2H), 1.70 – 1.51 (m, 3H), 1.36 (dt, J = 15.2, 7.5 Hz, 2H). HRMS (ESI+): calcd: 966.3208; found: 966.3187 (−2.14 ppm).
NMR spectra
Please see Data S1.
Quantification and Statistical Analysis
Statistical analyses were performed by GraphPad Prism 7 software. Data are presented as mean ± standard error of the mean (S.E.M.). Number of technical replicates or independent biological repeats is indicated in the figure legends. Unpaired two tailed Student’s t-test and two-way ANOVA with post-hoc Sidak were utilized as appropriate. FP competition experiments were fit using the “One site - Fit Ki” analysis in Prism 7. Half-life of MYC protein calculated by “one phase decay” analysis in Prism. Correlations were analyzed by Pearson’s correlation coefficient (r) and p value. Mice survival data was analyzed by survival curve comparison in Prism. No samples, mice or data points were excluded from the analyses. For all analyses, results were considered statistically significant with *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001.
Data and Code Availability
Raw RNA-seq data were deposited into the National Center for Biotechnology Information (NCBI)’s Gene Expression Ominibus, GEO: GSE135877. The mass spectrometry proteomics data have been deposited to the PeptideAtlas, Identifier: PASS01427.
Key Resources Table
Supplementary Material
Scheme 1:

(a) TFAA, Sodium 2,2,2-trifluoroacetate, 110 °C, 24 hr; (b) Iodine, pyridine, CHCl3, room temp. overnight; (c) p-Chlorobenzyl bromide, K2CO3, acetone, 60 °C, overnight; (d) Aryl boronic acid, Pd(dppf)2Cl2, Na2CO3, toluene, EtOH, H2O, 100 °C, 2 hr; (e) Methylhydrazine, EtOH, 78 °C, 2 hr.
Scheme 2:

(a) 4-chloro-3-(trifluoromethyl)phenyl boronic acid, Pd(dppf)2Cl2, Na2CO3, toluene, EtOH, H2O, 100 °C, 2 hr; (b) di-tert-Butyl-(chloromethyl)phosphate, Cs2CO3, Nal, DMF, 60 °C, 48 hr; (c) Hydrazine 60%, EtOH, 50 °C, 1 hr; (d) TFA, DCM, room temp. 1 hr.
Scheme 3:

(a) 3,5-Bis(trifluoromethyl)phenyl boronic acid, Pd(dppf)2Cl2, Na2CO3, toluene, EtOH, H2O, 100 °C, 2 hr; (b) di-tert-Butyl-(chloromethyl)phosphate, Cs2CO3, Nal, DMF, 60 °C, 16 hr; (c) Methylhydrazine 60%, EtOH, 80 °C, 1 hr; (d) TFA, DCM, room temp. 1 hr.
Scheme 4:

(a) tert-butyl (3-bromopropyl)carbamate, K2CO3, acetone; (b) (3,5-bis(trifluoromethyl)phenyl)boronic acid, Pd(dppf)2Cl2, Na2CO3, toluene, EtOH, H2O, 100 °C, 3 hr; (c) Methylhydrazine 60%, EtOH, 80 °C, 45 min; (d) 1st .TFA, DCM, room temp. 2 hr. 2nd EDC, HOBt, Et3N, Biotin-PEG6-COOH, DMF.
Scheme 5:

(a) (3,5-bis(trifluoromethyl)phenyl)boronic acid, Pd(dppf)2Cl2, Na2CO3, toluene, EtOH, H2O, 100 °C, 3 hr; (b) Methylhydrazine 60%, EtOH, 80 °C, 45 min; (c) 1st .TFA, DCM, room temp. 2 hr. 2nd EDC, HOBt, Et3N, Biotin-PEG4-COOH, DMF
Highlights.
Development of small molecule MYC inhibitors (MYCi) that engage MYC in cells
MYCi disrupt MYC/MAX complexes, promote MYC T58 phosphorylation and MYC degradation
MYCi show favorable PK, in vivo efficacy and tolerability in mouse tumor models
MYCi treatment synergizes with anti-PD1 immunotherapy
Han et al. develop inhibitors that disrupt MYC/MAX heterodimerization, enhance MYC degradation, and impair MYC-driven gene expression. One compound exhibits potent anti-tumor efficacy in mice with good tolerability, increases tumor immune cell infiltration, and sensitizes tumors to anti-PD1 immunotherapy.
Significance.
MYC is a highly sought after target for cancer therapy. By integrating pharmacophore-based in silico screening of a large compound library with a rapid in vivo screen in mice, we identified a series of MYC inhibitors showing significant in vivo anti-tumor efficacy with favorable pharmacokinetic profiles. We showed that lead optimization is feasible, generating compound 975 with improved tolerability. Furthermore, treatment with the inhibitors remodeled the tumor immune microenvironment, thereby sensitizing otherwise refractory tumors to immune checkpoint blockade therapy. Our studies demonstrate a potential pathway for the development of viable MYC inhibitors for mechanistic studies and therapeutic interventions.
Acknowledgements
We thank Dr. Edward V. Prochownik for constructs and compound JKY-2-169, Dr. Shideng Bao for MYC mutant constructs, Dr. Chi V. Dang for P493-6 B cells, and Dr. John M. Sedivy for Rat-1 fibroblast cells. We thank the NUseq and the Robert H. Lurie Comprehensive Cancer Center Flow Cytometry cores of Northwestern University. We thank Dr. Andrew Mazar and Dr. Nicolette Zielinski for helpful discussions on the project design and Lisa Hurley for the technical assistance. This work was supported by grants from the National Cancer Institute: R01CA123484, RO1CA196270 and P50CA180995; and by the NewCures Biomedical Accelerator of Northwestern University. Part of the work was supported by the H-Foundation Multi-PI Basic Science Synergy Award made possible by a gift from the H Foundation to the Robert H. Lurie Comprehensive Cancer Center. A part of this work was performed by the Northwestern University ChemCore and the Developmental Therapeutics Core, which are funded by Cancer Center Support Grant P30CA060553 from the National Cancer Institute awarded to the Robert H. Lurie Comprehensive Cancer Center, and the Chicago Biomedical Consortium with support from the Searle Funds at the Chicago Community Trust.
Footnotes
Declaration of interests
H.H., A.D.J., J.I., R.K.M. G.E.S. and S.A.A. are co-inventors on patent applications covering the methods and assays to identify and characterize MYC inhibitors and derivatives. All other authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agrawal P, Yu K, Salomon AR, and Sedivy JM (2010). Proteomic profiling of Myc-associated proteins. Cell Cycle 9, 4908–4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anker JF, Naseem AF, Mok H, Schaeffer AJ, Abdulkadir SA, and Thumbikat P (2018). Multi-faceted immunomodulatory and tissue-tropic clinical bacterial isolate potentiates prostate cancer immunotherapy. Nat Commun 9, 1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell JB, and Holloway GA (2010). New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem 53, 2719–2740. [DOI] [PubMed] [Google Scholar]
- Berg T, Cohen SB, Desharnais J, Sonderegger C, Maslyar DJ, Goldberg J, Boger DL, and Vogt PK (2002). Small-molecule antagonists of Myc/Max dimerization inhibit Myc-induced transformation of chicken embryo fibroblasts. Proc Natl Acad Sci U S A 99, 3830–3835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwell TK, Kretzner L, Blackwood EM, Eisenman RN, and Weintraub H (1990). Sequence-specific DNA binding by the c-Myc protein. Science 250, 1149–1151. [DOI] [PubMed] [Google Scholar]
- Bretones G, Delgado MD, and Leon J (2015). Myc and cell cycle control. Biochim Biophys Acta 1849, 506–516. [DOI] [PubMed] [Google Scholar]
- Carabet LA, Rennie PS, and Cherkasov A (2018). Therapeutic Inhibition of Myc in Cancer. Structural Bases and Computer-Aided Drug Discovery Approaches. Int J Mol Sci 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey SC, Baylot V, and Felsher DW (2018). The MYC oncogene is a global regulator of the immune response. Blood 131, 2007–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey SC, Tong L, Li Y, Do R, Walz S, Fitzgerald KN, Gouw AM, Baylot V, Gutgemann I, Eilers M, et al. (2016). MYC regulates the antitumor immune response through CD47 and PD-L1. Science 352, 227–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimmperman P, Baranauskiene L, Jachimoviciute S, Jachno J, Torresan J, Michailoviene V, Matuliene J, Sereikaite J, Bumelis V, and Matulis D (2008). A quantitative model of thermal stabilization and destabilization of proteins by ligands. Biophys J 95, 3222–3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clausen DM, Guo J, Parise RA, Beumer JH, Egorin MJ, Lazo JS, Prochownik EV, and Eiseman JL (2010). In vitro cytotoxicity and in vivo efficacy, pharmacokinetics, and metabolism of 10074-G5, a novel small-molecule inhibitor of c-Myc/Max dimerization. J Pharmacol Exp Ther 335, 715–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV (2012). MYC on the path to cancer. Cell 149, 22–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV (2013). MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb Perspect Med 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang CV, O’Donnell KA, Zeller KI, Nguyen T, Osthus RC, and Li F (2006). The c-Myc target gene network. Semin Cancer Biol 16, 253–264. [DOI] [PubMed] [Google Scholar]
- Dosset M, Vargas TR, Lagrange A, Boidot R, Vegran F, Roussey A, Chalmin F, Dondaine L, Paul C, Lauret Marie-Joseph E, et al. (2018). PD-1/PD-L1 pathway: an adaptive immune resistance mechanism to immunogenic chemotherapy in colorectal cancer. Oncoimmunology 7, e1433981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzikiewicz-Krawczyk A, Kok K, Slezak-Prochazka I, Robertus JL, Bruining J, Tayari MM, Rutgers B, de Jong D, Koerts J, Seitz A, et al. (2017). ZDHHC11 and ZDHHC11B are critical novel components of the oncogenic MYC-miR-150-MYB network in Burkitt lymphoma. Leukemia 31, 1470–1473. [DOI] [PubMed] [Google Scholar]
- Evan GI, and Vousden KH (2001). Proliferation, cell cycle and apoptosis in cancer. Nature 411, 342–348. [DOI] [PubMed] [Google Scholar]
- Ewing RM, Chu P, Elisma F, Li H, Taylor P, Climie S, McBroom-Cerajewski L, Robinson MD, O’Connor L, Li M, et al. (2007). Large-scale mapping of human protein-protein interactions by mass spectrometry. Mol Syst Biol 3, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X, Zhou W, Wu Q, Huang Z, Shi Y, Yang K, Chen C, Xie Q, Mack SC, Wang X, et al. (2017). Deubiquitinase USP13 maintains glioblastoma stem cells by antagonizing FBXL14-mediated Myc ubiquitination. J Exp Med 214, 245–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farrell AS, and Sears RC (2014). MYC degradation. Cold Spring Harb Perspect Med 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, et al. (2010). Selective inhibition of BET bromodomains. Nature 468, 1067–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher S, and Prochownik EV (2015). Small-molecule inhibitors of the Myc oncoprotein. Biochim Biophys Acta 1849, 525–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follis AV, Hammoudeh DI, Wang H, Prochownik EV, and Metallo SJ (2008). Structural rationale for the coupled binding and unfolding of the c-Myc oncoprotein by small molecules. Chem Biol 15, 1149–1155. [DOI] [PubMed] [Google Scholar]
- Galaktionov K, Chen X, and Beach D (1996). Cdc25 cell-cycle phosphatase as a target of c-myc. Nature 382, 511–517. [DOI] [PubMed] [Google Scholar]
- Guo J, Parise RA, Joseph E, Egorin MJ, Lazo JS, Prochownik EV, and Eiseman JL (2009). Efficacy, pharmacokinetics, tisssue distribution, and metabolism of the Myc-Max disruptor, 10058-F4 [Z,E]-5-[4-ethylbenzylidine]-2-thioxothiazolidin-4-one, in mice. Cancer Chemother Pharmacol 63, 615–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammoudeh DI, Follis AV, Prochownik EV, and Metallo SJ (2009). Multiple independent binding sites for small-molecule inhibitors on the oncoprotein c-Myc. J Am Chem Soc 131, 7390–7401. [DOI] [PubMed] [Google Scholar]
- Hart JR, Garner AL, Yu J, Ito Y, Sun M, Ueno L, Rhee JK, Baksh MM, Stefan E, Hartl M, et al. (2014). Inhibitor of MYC identified in a Krohnke pyridine library. Proc Natl Acad Sci U S A 111, 12556–12561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M, Arvanitis C, Chu K, Dewey W, Leonhardt E, Trinh M, Sundberg CD, Bishop JM, and Felsher DW (2002). Sustained loss of a neoplastic phenotype by brief inactivation of MYC. Science 297, 102–104. [DOI] [PubMed] [Google Scholar]
- Jiang H, Bower KE, Beuscher A.E.t., Zhou B, Bobkov AA, Olson AJ, and Vogt PK (2009). Stabilizers of the Max homodimer identified in virtual ligand screening inhibit Myc function. Mol Pharmacol 76, 491–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung KY, Wang H, Teriete P, Yap JL, Chen L, Lanning ME, Hu A, Lambert LJ, Holien T, Sundan A, et al. (2015). Perturbation of the c-Myc-Max protein-protein interaction via synthetic alpha-helix mimetics. J Med Chem 58, 3002–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalkat M, Resetca D, Lourenco C, Chan PK, Wei Y, Shiah YJ, Vitkin N, Tong Y, Sunnerhagen M, Done SJ, et al. (2018). MYC Protein Interactome Profiling Reveals Functionally Distinct Regions that Cooperate to Drive Tumorigenesis. Mol Cell 72, 836–848 e837. [DOI] [PubMed] [Google Scholar]
- Kapeli K, and Hurlin PJ (2011). Differential regulation of N-Myc and c-Myc synthesis, degradation, and transcriptional activity by the Ras/mitogen-activated protein kinase pathway. J Biol Chem 286, 38498–38508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim R, Tse G, Putti T, Scolyer R, and Lee S (2004). The significance of the Wnt pathway in the pathology of human cancers. Pathology 36, 120–128. [DOI] [PubMed] [Google Scholar]
- Kepp O, Senovilla L, Vitale I, Vacchelli E, Adjemian S, Agostinis P, Apetoh L, Aranda F, Barnaba V, Bloy N, et al. (2014). Consensus guidelines for the detection of immunogenic cell death. Oncoimmunology 3, e955691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiessling A, Sperl B, Hollis A, Eick D, and Berg T (2006). Selective inhibition of c-Myc/Max dimerization and DNA binding by small molecules. Chem Biol 13, 745–751. [DOI] [PubMed] [Google Scholar]
- Koch HB, Zhang R, Verdoodt B, Bailey A, Zhang CD, Yates JR 3rd, Menssen A, and Hermeking H (2007). Large-scale identification of c-MYC-associated proteins using a combined TAP/MudPIT approach. Cell Cycle 6, 205–217. [DOI] [PubMed] [Google Scholar]
- Kortlever RM, Sodir NM, Wilson CH, Burkhart DL, Pellegrinet L, Brown Swigart L, Littlewood TD, and Evan GI (2017). Myc Cooperates with Ras by Programming Inflammation and Immune Suppression. Cell 171, 1301–1315 e1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress TR, Sabo A, and Amati B (2015). MYC: connecting selective transcriptional control to global RNA production. Nat Rev Cancer 15, 593–607. [DOI] [PubMed] [Google Scholar]
- Krovat EM, Fruhwirth KH, and Langer T (2005). Pharmacophore identification, in silico screening, and virtual library design for inhibitors of the human factor Xa. J Chem Inf Model 45, 146–159. [DOI] [PubMed] [Google Scholar]
- Lu Y, Hu Z, Mangala LS, Stine ZE, Hu X, Jiang D, Xiang Y, Zhang Y, Pradeep S, Rodriguez-Aguayo C, et al. (2018). MYC Targeted Long Noncoding RNA DANCR Promotes Cancer in Part by Reducing p21 Levels. Cancer Res 78, 64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez Molina D, Jafari R, Ignatushchenko M, Seki T, Larsson EA, Dan C, Sreekumar L, Cao Y, and Nordlund P (2013). Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science 341, 84–87. [DOI] [PubMed] [Google Scholar]
- Mathivanan S, Periaswamy B, Gandhi TK, Kandasamy K, Suresh S, Mohmood R, Ramachandra YL, and Pandey A (2006). An evaluation of human protein-protein interaction data in the public domain. BMC Bioinformatics 7 Suppl 5, S19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeown MR, and Bradner JE (2014). Therapeutic strategies to inhibit MYC. Cold Spring Harb Perspect Med 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer N, and Penn LZ (2008). Reflecting on 25 years with MYC. Nat Rev Cancer 8, 976–990. [DOI] [PubMed] [Google Scholar]
- Mimura K, Teh JL, Okayama H, Shiraishi K, Kua LF, Koh V, Smoot DT, Ashktorab H, Oike T, Suzuki Y, et al. (2018). PD-L1 expression is mainly regulated by interferon gamma associated with JAK-STAT pathway in gastric cancer. Cancer Sci 109, 43–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mo H, and Henriksson M (2006). Identification of small molecules that induce apoptosis in a Myc-dependent manner and inhibit Myc-driven transformation. Proc Natl Acad Sci U S A 103, 6344–6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustata G, Follis AV, Hammoudeh DI, Metallo SJ, Wang H, Prochownik EV, Lazo JS, and Bahar I (2009). Discovery of novel Myc-Max heterodimer disruptors with a three-dimensional pharmacophore model. J Med Chem 52, 1247–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardoll DM (2012). The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12, 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfirschke C, Engblom C, Rickelt S, Cortez-Retamozo V, Garris C, Pucci F, Yamazaki T, Poirier-Colame V, Newton A, Redouane Y, et al. (2016). Immunogenic Chemotherapy Sensitizes Tumors to Checkpoint Blockade Therapy. Immunity 44, 343–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Posternak V, and Cole MD (2016). Strategically targeting MYC in cancer. F1000Res 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seal J, Lamotte Y, Donche F, Bouillot A, Mirguet O, Gellibert F, Nicodeme E, Krysa G, Kirilovsky J, Beinke S, et al. (2012). Identification of a novel series of BET family bromodomain inhibitors: binding mode and profile of I-BET151 (GSK1210151A). Bioorg Med Chem Lett 22, 2968–2972. [DOI] [PubMed] [Google Scholar]
- Shachaf CM, and Felsher DW (2005). Tumor dormancy and MYC inactivation: pushing cancer to the brink of normalcy. Cancer Res 65, 4471–4474. [DOI] [PubMed] [Google Scholar]
- Shi J, Stover JS, Whitby LR, Vogt PK, and Boger DL (2009). Small molecule inhibitors of Myc/Max dimerization and Myc-induced cell transformation. Bioorg Med Chem Lett 19, 6038–6041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soucek L, Whitfield J, Martins CP, Finch AJ, Murphy DJ, Sodir NM, Karnezis AN, Swigart LB, Nasi S, and Evan GI (2008). Modelling Myc inhibition as a cancer therapy. Nature 455, 679–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sterling T, and Irwin JJ (2015). ZINC 15--Ligand Discovery for Everyone. J Chem Inf Model 55, 2324–2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trumpp A, Refaeli Y, Oskarsson T, Gasser S, Murphy M, Martin GR, and Bishop JM (2001). c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature 414, 768–773. [DOI] [PubMed] [Google Scholar]
- Unno K, Roh M, Yoo YA, Al-Shraideh Y, Wang L, Nonn L, and Abdulkadir SA (2017). Modeling African American prostate adenocarcinoma by inducing defined genetic alterations in organoids. Oncotarget 8, 51264–51276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Hammoudeh DI, Follis AV, Reese BE, Lazo JS, Metallo SJ, and Prochownik EV (2007). Improved low molecular weight Myc-Max inhibitors. Mol Cancer Ther 6, 2399–2408. [DOI] [PubMed] [Google Scholar]
- Watson PA, Ellwood-Yen K, King JC, Wongvipat J, Lebeau MM, and Sawyers CL (2005). Context-dependent hormone-refractory progression revealed through characterization of a novel murine prostate cancer cell line. Cancer Res 65, 11565–11571. [DOI] [PubMed] [Google Scholar]
- Welcker M, Orian A, Jin J, Grim JE, Harper JW, Eisenman RN, and Clurman BE (2004). The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci U S A 101, 9085–9090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, and Pan W (2010). GSK3: a multifaceted kinase in Wnt signaling. Trends Biochem Sci 35, 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Shi J, Yamamoto N, Moss JA, Vogt PK, and Janda KD (2006). A credit-card library approach for disrupting protein-protein interactions. Bioorg Med Chem 14, 2660–2673. [DOI] [PubMed] [Google Scholar]
- Yap JL, Wang H, Hu A, Chauhan J, Jung KY, Gharavi RB, Prochownik EV, and Fletcher S (2013). Pharmacophore identification of c-Myc inhibitor 10074-G5. Bioorg Med Chem Lett 23, 370–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildiz I, Ertan T, Bolelli K, Temiz-Arpaci O, Yalcin I, and Aki E (2008). QSAR and pharmacophore analysis on amides against drug-resistant S. aureus. Sar Qsar Environ Res 19, 101–113. [DOI] [PubMed] [Google Scholar]
- Yin X, Giap C, Lazo JS, and Prochownik EV (2003). Low molecular weight inhibitors of Myc-Max interaction and function. Oncogene 22, 6151–6159. [DOI] [PubMed] [Google Scholar]
- Zhou Z, He C, and Wang J (2015). Regulation mechanism of Fbxw7-related signaling pathways (Review). Oncol Rep 34, 2215–2224. [DOI] [PubMed] [Google Scholar]
- Zou J, Zhuang M, Yu X, Li N, Mao R, Wang Z, Wang J, Wang X, Zhou H, Zhang Lv et al. (2018). MYC inhibition increases PD-L1 expression induced by IFN-gamma in hepatocellular carcinoma cells. Mol Immunol 101, 203–209. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw RNA-seq data were deposited into the National Center for Biotechnology Information (NCBI)’s Gene Expression Ominibus, GEO: GSE135877. The mass spectrometry proteomics data have been deposited to the PeptideAtlas, Identifier: PASS01427.