

Abstract
Hyperoxidized albumin promotes inflammation and modulates several immune cells in severe alcoholic hepatitis (SAH). Platelets mediate inflammation by interacting with immune cells, endothelium, and other cells. The role of hyperoxidized albumin in platelet activation and alteration of platelet phenotype/functions is not known. Quantitative platelet proteomics performed in 10 patients with SAH was compared with 10 patients with alcoholic cirrhosis and 10 healthy controls, respectively. Dysregulated pathways were identified and validated in a separate cohort (n = 40). Healthy platelets were exposed to patient plasma or purified albumin or ex vivo modified albumin (human‐mercaptalbumin, humannonmercaptalbumin‐1, and human nonmercaptalbumin 2) in the presence or absence of CD36 blockade, and platelet secretome was analyzed. Two hundred and two up‐regulated proteins linked to platelet activation, complement regulation, lipid transportation, and 321 down‐regulated proteins related to platelet hemostasis and coagulation (fold change ± 1.5, P < 0.01) were identified. Blood transcription module enrichment showed an inflammatory phenotype of SAH platelet. Increased level of platelet factor‐4, P‐selectin, and soluble cluster of differentiation‐40 ligand correlated with severity (Model for End‐Stage Liver Disease score, r > 0.3, P < 0.05) in SAH. Transcripts linked to platelet activation (increased) and granular secretions (decreased in SAH) correlated with disease severity. SNARE (soluble‐N‐ethylmaleimide‐sensitive‐factor‐activating‐protein‐receptor) complex proteins (SNAP‐23 [synaptosomal‐associated protein 23] and VAMP‐8 [vesicle‐associated membrane protein 3]) were down‐regulated in SAH platelets (P < 0.05). In vitro stimulation of healthy platelets showed enhanced activation with patient plasma, or purified albumin‐treatment blocking of CD36 blunted this effect (P < 0.05). Ex vivo modified albumin (primarily nonmercaptalbumin–human nonmercaptalbumin 2 [HNA2; 1 mg/mL]) showed high activation and aggregation and intracellular reactive oxygen species production in healthy platelets (P < 0.05), which significantly reduced after CD36 neutralization. Platelet secretome showed reduced inflammatory mediators and increased repair proteins. Conclusion: Hyperoxidized albumin triggers platelet activation (possibly through the CD36 receptor), promotes inflammation and oxidative stress, and contributes to disease severity in patients with SAH.
Hyperoxidized albumin activates immune cells; however, its contribution in activation of platelets and change in proteome, which correlates with outcomes in SAH, is unknown. Platelets of patients with SAH are hyperactivated, facilitate oxidative stress and systemic inflammation, and have dysregulated granule secretion due to alteration in the expression of SNARE proteins. Oxidized albumin human nonmercaptoalbumin‐2 (HNA2) causes platelet activation and promotes inflammation and oxidative stress through the CD36 receptor–mediated redox pathway. Neutralization or blockade of platelet CD36 receptor and/or removal of HNA2 could serve as an attractive therapeutic strategy for reducing systemic inflammation and oxidative stress in patients with SAH.

Abbreviations
- AOPP
advance oxidative protein product
- DEP
differentially expressed protein
- EGTA
ethylene glycol tetraacetic acid
- FC
fold change
- Gp2b/3a
glycoprotein integrin αIIbβ
- HC
healthy control
- HMA
humanmercaptalbumin
- HNA1
humannonmercaptalbumin‐1
- HNA2
human nonmercaptalbumin 2
- MELD
Model for End‐Stage Liver Disease
- mRNA
messenger RNA
- PAC‐1
procaspase‐activating compound‐1
- PRP
platelet‐rich plasma
- ROS
reactive oxygen species
- SAH
severe alcoholic hepatitis
- SNAP‐23
synaptosomal‐associated protein 23
- SNARE
soluble N‐ethylmaleimide‐sensitive factor activating protein receptor
- VAMP‐3
vesicle‐associated membrane protein 3
Severe alcoholic hepatitis (SAH) is linked with poor prognosis and high short‐term mortality.1 In patients with SAH, episodes of variceal and nonvariceal bleeding contribute to high morbidity and mortality.2 Thrombocytopenia and altered function of platelets are common in patients with liver cirrhosis.3 Thrombocytopenia is pronounced in patients with alcoholism and is linked to increased platelet apoptosis, decrease in thrombopoietin levels, and/or consumption of platelets by splenic sequestration.4, 5 This mediates hemodynamic instability and leads to progression of severity of liver. Patients with cirrhosis are in a hypercoagulable state,6 and thrombosis is common in alcoholics due to an increase in gut permeability.7 Hyperactivation of platelets in patients with alcoholism results in higher oxidative stress.8 Platelet activation generate reactive oxygen species (ROS),9 express CD40L, and releases its soluble form (sCD40L), which acts as an inflammatory mediator.10 Increase in CD40L promotes platelet‐leucocyte aggregation.11 However, the phenotype of platelets, proteins carried by them, and their function are not clearly understood in SAH. Furthermore, the contribution of platelets in inflammation and stress in alcoholic liver disease is also elusive.12
Platelets mediate inflammation as they interact with immune cells, endothelium, and other cells.13 Furthermore, increase in advance oxidative protein products (AOPPs) induce activation of platelets through scavenger receptors CD36.14 In SAH, synthesis and function of albumin decreases.15 Change in circulating pro‐coagulant and anticoagulant protein levels predisposes patients with SAH to both bleeding, as well as thrombotic complications.16 Previously we have shown hypo‐albuminemia with increase in oxidative modification and bilirubin binding in SAH.17 We also demonstrated how oxidative modification in albumin activates neutrophils of SAH.18 This was complemented by a recent work demonstrating how oxidative albumin (human nonmercaptalbumin HNA1 [humannonmercaptalbumin‐1] and HNA2) trigger peripheral leukocytes and induce systemic inflammation in liver failure.19 This evidence suggests that oxidized albumin in SAH may contribute to platelet activation and systemic inflammation.
To understand, proteomic profile of platelet was studied and validated. Causality of platelet dysfunction was determined by incubating healthy platelets with purified albumin (patients with SAH) or ex vivo oxidized albumin (human nonmercaptalbumin HNA1 and HNA2; concentration as in patients with SAH) in the presence or absence of CD36 receptor blockade. The secretome of such platelets was also analyzed. Our results show that oxidized albumin contributes to platelet dysfunction, and promotes inflammation and oxidative stress through CD36 receptor signaling in SAH.
Patients and Methods
Patients
Eighty patients with biopsy‐proven SAH, who were admitted to the Department of Hepatology, Institute of Liver and Biliary Sciences (New Delhi, India) between September 2015 and January 2018 were enrolled. Thirty of these patients were excluded (as detailed in Supporting Fig. S1). Of the remaining 50 patients with SAH, 10 were included in the discovery cohort (platelet proteomics). The results were validated in the validation cohort of 40 patients with SAH, and 20 patients with alcoholic cirrhosis were included as disease control. SAH was diagnosed based on histological criteria and a Maddrey’s discriminant function of >32.20 Alcoholic cirrhosis was diagnosed on previous history of chronic heavy alcohol intake (>1‐month alcohol restraint) and with a combination of clinical, biochemical, endoscopic, and radiological criteria.21 Additionally, healthy controls (HC) with no history of present or previous illness were enrolled. Baseline blood samples were drawn and stored at −80°C. The study protocol was approved by the ethical committee (IEC/IRB No. 37/M‐3) of the Institute of Liver and Biliary Sciences.
Discovery Cohort
Platelet proteomics of 10 patients with SAH and age/sex‐matched 10 HC was performed as detailed in the Supporting Methods.
Validation Cohort
Differentially expressed proteins (DEPs) linked to platelet functions were validated in 40 patients with SAH and 20 patients with alcoholic cirrhosis (Supporting Methods).
Pathway Enrichment and Correlation Analyses
DEPs were enriched for Gene Ontology/Kyoto Encyclopedia of Genes and Genomes classification22 and blood transcription module analysis,23 as detailed in the Supporting Methods.
Albumin Purification and Characterization
Circulating albumin was purified and subjected to mass spectrometry,18 as detailed in the Supporting Methods.
Ex Vivo Preparation of Oxidized Albumin Forms
Albumin purified from HC was subjected to oxidative modification by incubating it with cystine (17 mM) at 37°C for 24 hours to obtain HNA1. For HNA2, purified albumin was incubated with H2O2 (45 mM) for 1 hour at room temperature19 (Supporting Methods).
Platelet Activation Markers and sCD40L Assay
Plasma‐citrated blood samples were centrifuged for 15 minutes at 3,000g, and levels of PF‐4 (E‐EL‐H2216; Elabscience, Houston, TX), P‐selectin (E‐EL‐H0917; Elabscience), and sCD40L (E‐EL‐H0035; Elabscience) were measured.
Platelet Preparation and Flow‐Cytometry Analysis
Whole blood from patients and healthy donors was collected into tubes containing 3.8% sodium citrate (9:1) and was centrifuged at 150g for 15 minutes to obtain platelet‐rich plasma (PRP).7 Washed platelets were obtained from PRP. To avoid leukocyte contamination, only the top 75% of the PRP was collected. The purity of platelets was checked by platelet‐specific markers (CD45‐ve and CD61+) by flow cytometry, as described previously.24 To perform the ex vivo tests, platelets from HC were diluted with Tyrode’s buffer to obtain the same number of platelets as that of the patient. Platelet aggregation, intracellular calcium flux (362561; BioLegend, San Diego, CA), anti‐procaspase‐activating compound‐1 (PAC‐1) (362803; BioLegend), P‐selectin (304903; BioLegend), and CD40L (310809; BioLegend) expression analyses were performed.7 Intracellular ROS were measured (109244‐58‐8; Cayman Chemical Co., Ann Arbor, MI)25 (Supporting Methods).
Western Blot Analysis
Expression of Gp2b/3a (glycoprotein integrin αIIbβ) (53417; Santa Cruz Biotechnology, Dallas, TX), SNAP‐23 (E‐AB 33501; Elabscience), Syntaxin‐11 (E‐AB‐11626; Elabscience), Rab21 (E‐AB‐16748; Elabscience), Rab27b (EPP18496; Elabscience), VAMP‐8 (EPP19134; Elabscience), and Munc13‐4 (GTX108105; Elabscience) was studied in platelets.
Blocking/Neutralizing Assay
Platelets were pretreated with anti‐CD36 (AB17044; Lucerna‐Chem‐AG, Luzern, Switzerland) blocking antibody (1 μg/mL for 30 minutes at room temperature), which specifically blocks an epitope in CD36 receptor and a respective isotype control before exposure with oxidized albumin fractions (HMA [humanmercaptalbumin], HNA1, and HNA2, all at 1 mg/mL) to healthy platelets14 (Supporting Methods).
Secretome Analysis
Total protein in the supernatant of platelets before and after neutralization assay was analyzed using high‐resolution mass spectrometry (Supporting Methods).
Statistical Analysis
Results are represented as means with SD unless indicated otherwise. Statistical significance was seen at P < 0.05 (GraphPad 6/SPSS 20). For comparison of continuous variables between two groups, the unpaired (two‐tailed) Student t test and Mann‐Whitney U test were used. Spearmen correlation analysis was performed between platelet activation markers with the disease severity. Variability between discovery and validation cohort was assessed using the Bland‐Altman test (Supporting Fig. S2).
Results
Demographics of the Study Cohort
The study cohort included 50 patients with SAH (mean age 45 ± 8 years and 92.5% male). These were compared with 20 matched HC. Clinical characteristics of the discovery cohort were similar to the validation cohort. Baseline neutrophil count and severity indices were higher in patients with SAH as compared with other groups (P < 0.01; Table 1 and Supporting Fig. S3).
Table 1.
Baseline Clinical Parameters of All Patients (Demographic Profile)
| Discovery Cohort | Validation Cohort | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| SAH (n = 10) | HC (n = 10) | SAH (n = 40) | AC (n = 20) | HC (n = 20) | P Value | P Value | P Value | P Value | |
| A | B | C | D | E | A vs. B | A vs. C | A vs. D | C vs. E | |
| Gender (male %) | 100 | 80 | 100 | 100 | 90 | — | — | — | — |
| Age (years) | 44 ± 8 | 32 ± 10 | 45 ± 8 | 46 ± 9 | 38 ± 9 | 0.21 | 0.71 | 0.62 | 0.68 |
| Height (cm) | 167.5 ± 4.1 | 170.1 ± 4.0 | 169.5 ± 7.1 | 171.5 ± 6.1 | 170.1 ± 4.0 | 0.85 | 1.41 | 0.55 | 0.51 |
| Weight (kg) | 70.6 ± 16.6 | 68.5 ± 12.8 | 72.2 ± 17.1 | 75 ± 16.5 | 71.1 ± 11.1 | 0.46 | 1.13 | 0.75 | 0.88 |
| BMI (kg/m2) | 24.5 ± 3.4 | 21.8 ± 2.7 | 25.5 ± 4.7 | 24.5 ± 5.6 | 22.3 ± 2.3 | 0.05 | 0.71 | 0.08 | <0.05 |
| Jaundice duration (days) | 24 ± 20 | — | 25 ± 21 | — | — | — | 0.71 | — | — |
| Baseline Laboratory Parameters of All Patients | |||||||||
| Total bilirubin (mg/dL) | 6.0 ± 3.0 | 0.6 ± 0.4 | 5.0 ± 4.0 | 2.3 ± 3.0 | 0.7 ± 0.3 | <0.001 | 0.71 | <0.001 | <0.01 |
| Direct bilirubin (mg/dL) | 1.64 ± 1.8 | 0.1 ± 0.1 | 1.74 ± 1.7 | — | 0.1 ± 0.1 | — | 0.07 | — | <0.01 |
| AST (IU/L) | 80.4 ± 47.6 | 20.0 ± 8.0 | 76.6 ± 49.2 | 34 ± 41.2 | 18.0 ± 10.0 | <0.001 | 2.67 | <0.001 | <0.01 |
| ALT (IU/L) | 42.0 ± 32.7 | 16.0 ± 5.1 | 42.0 ± 32.7 | 40 ± 31.5 | 16.0 ± 5.1 | <0.01 | 0.35 | <0.01 | <0.01 |
| Albumin (g/dL) | 2.7 ± 0.5 | 4.1 ± 1.0 | 2.9 ± 0.7 | 4.0 ± 0.9 | 4.1 ± 1.0 | <0.001 | 0.14 | <0.001 | <0.05 |
| PT (seconds) | 24.2 ± 8.1 | 11.6 ± 0.6 | 23.4 ± 9.1 | 18.1 ± 5.2 | 11.6 ± 0.6 | <0.001 | 0.57 | <0.001 | <0.01 |
| INR | 2.3 ± 0.6 | 0.9 ± 0.3 | 2.2 ± 0.7 | 1.1 ± 0.2 | 0.9 ± 0.3 | <0.001 | 0.07 | <0.001 | <0.01 |
| Hb (g/dL) | 9.2 ± 1.7 | 12.9 ± 0.5 | 9.4 ± 1.6 | 11 ± 1.5 | 12.9 ± 0.5 | <0.001 | 0.14 | <0.001 | <0.05 |
| TLC (cells/µL) | 6.3 ± 2.7 | 5.6 ± 1.7 | 6.1 ± 2.9 | 5.9 ± 1.6 | 5.6 ± 1.7 | <0.05 | 0.07 | <0.05 | <0.05 |
| Platelet count (cells/µL) | 65.5 ± 15.5 | 190.0 ± 20.0 | 63.4 ± 12.5 | 160 ± 31.5 | 190.0 ± 20.0 | <0.01 | 1.48 | <0.01 | <0.01 |
| Neutrophil % of TLC | 88.4 ± 44.6 | 55.0 ± 12.0 | 83.4 ± 40.4 | 60 ± 20 | 55.0 ± 12.0 | <0.01 | 3.54 | <0.01 | <0.01 |
| Baseline Severity Parameters of All Patients | |||||||||
| Child‐Turcotte‐Pugh score | 16 ± 4 | — | 18 ± 4 | 12 ± 4 | — | — | 1.41 | — | — |
| MELD score | 23 ± 8 | — | 25 ± 7 | 16 ± 4 | — | — | 1.57 | — | — |
| MELD plus sodium score | 75 ± 46 | — | 76 ± 56 | 22 ± 10 | — | — | 0.61 | — | — |
| Discriminant function score | 9 ± 3 | — | 10 ± 2 | 6 ± 2 | — | — | 0.78 | — | — |
| Age bilirubin INR creatinine score | 8.2 ± 2.3 | — | 7.8 ± 2.7 | 6 ± 1 | — | — | 0.55 | — | — |
For continuous variables, descriptive statistics are presented as mean ± SD, whereas for categorical variables, statistics are presented as number (percentage).
Abbreviations: AC, alcoholic cirrhosis; BMI, body mass index; ALT, alanine aminotransferase; AST, aspartate aminotransferase; Hb, hemoglobin; INR, international normalized ratio; PT, prothrombin time; TLC, total leukocyte count.
Evaluation of Platelet Proteome and Phenotype in SAH
A total of 202 proteins were up‐regulated (fold change [FC] ≥ +1.5 fold) and 321 proteins were down‐regulated (FC ≤ −1.5 fold; P < 0.05) in patients with SAH when compared with HC (Fig. 1A and Supporting Table S1). Most of the up‐regulated proteins were associated with complement activation, inflammation, and platelet activation (Fig. 1B, Supporting Fig. S4, and Supporting Table S2). Down‐regulated proteins were linked to platelet and neutrophil degranulation, vesicular transport, and others (Fig. 1B, Supporting Fig. S4, and Supporting Table S2). The total DEPs in SAH were enriched on the blood transcription module (BTM) space23 for phenotype assessment. The up‐regulated proteins were enriched for monocytes (>5%), recruitment of neutrophils (>8%), and inflammation modules (>35%) (Supporting Fig. S5 and Supporting Table S3). Down‐regulated proteins were enriched for blood coagulation (>30%), respiratory electron transport chain (>30%), and protein folding (>40%) (Supporting Fig. S5 and Supporting Table S3). Furthermore, DEPs linked to platelet phenotype/functions were further validated by in vivo and in vitro analyses (Fig. 1C). Results suggest that platelets of patients with SAH transport a relatively higher percentage of inflammatory mediators and are depleted in coagulation, protein folding, and repair proteins.
Figure 1.
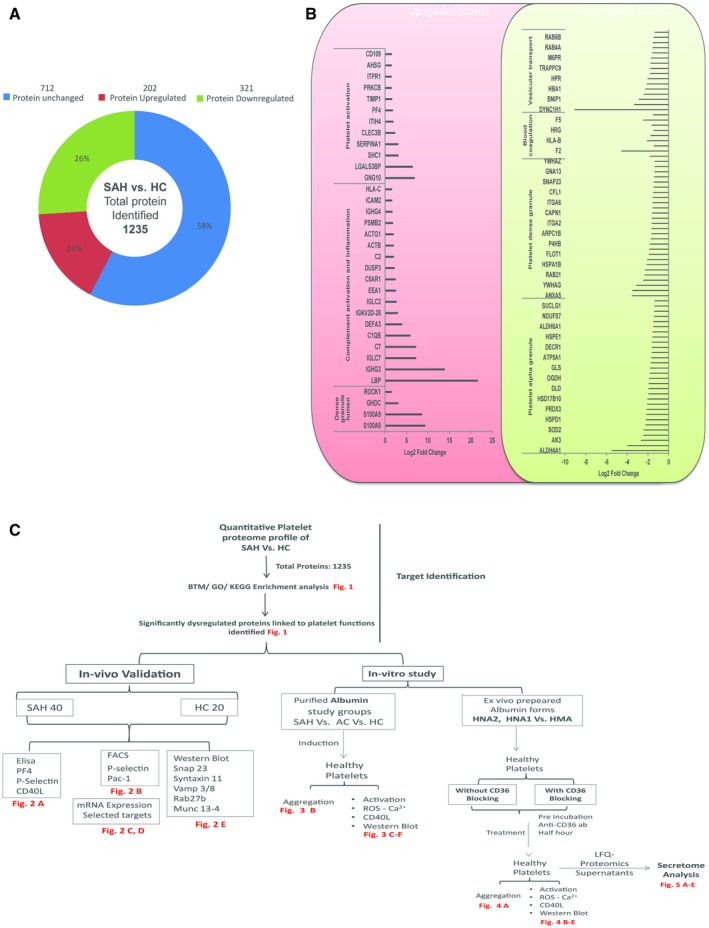
Quantitative platelet proteome profile and phenotype in patients with SAH. (A) Quantitative platelet proteomic in patients with SAH (n = 10) versus HC (n = 10): up‐regulated protein > 1.5‐fold and down‐regulated proteins < 0.65‐fold. (B) Classification of up‐regulated and down‐regulated proteins based on protein function. Representative log2 FC of up‐regulated and down‐regulated platelet proteins in patients with SAH associated with different platelet functions. (C) Flowchart and summary of the experiments performed with the number of samples studied. Abbreviations: AC, alcoholic cirrhosis; ELISA, enzyme‐linked immunosorbent assay; FACS, fluorescence‐activated cell sorting; GO, Gene Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; LFQ, Label‐Free Quantification.
SAH Platelets Show Increased Activation/Aggregation and Impaired Granules Secretion: Validation Phase
In our study, the markers of platelet activation, cytosolic Ca2+ flux, and aggregation were significantly (P < 0.05) higher in SAH platelets (P < 0.05; Fig. 2A) and was validated by their messenger RNA (mRNA) expression (P < 0.05; Fig. 2B). Interestingly, mRNA linked to granule secretion, coagulation, and homeostasis were down‐regulated (P < 0.05; Fig. 2B, Supporting Fig. S6, and Supporting Table S4). The result of flow cytometry analysis showed a significant increase in the expression of PAC‐1, P‐selectin, and calcium release in SAH platelets as compared with healthy platelets. This observation was validated by analyzing plasma levels of PF‐4, P‐selectin, and sCD40L (known indicators of platelet activation) in patients with SAH and HC.8 Patients with SAH showed higher plasma levels of PF‐4, P‐selectin, and sCD40L (Fig. 2C). Results of the enzyme‐linked immunosorbent assay experiment were concordant with the flow experiment and suggest that platelets in SAH express a higher level of activation markers as compared with healthy platelets. In addition, circulating levels of PF‐4, P‐selectin, and sCD40L documented direct correlation with the Model for End‐Stage Liver Disease (MELD) score of patients with SAH (r > 0.5; P < 0.05; Fig. 2C). Expression of SNARE complex proteins SNAP‐23, VAMP‐3, Rab‐27b, Syntaxin‐11, and Munc‐13‐4 protein were decreased in SAH platelets (P < 0.05; Fig. 2D). These results suggest that SAH platelets are hyperactivated and have dysregulated granule secretory capacity. Moreover, platelet functions such as activation and inflammation correlate directly with the severity in patients with SAH.
Figure 2.
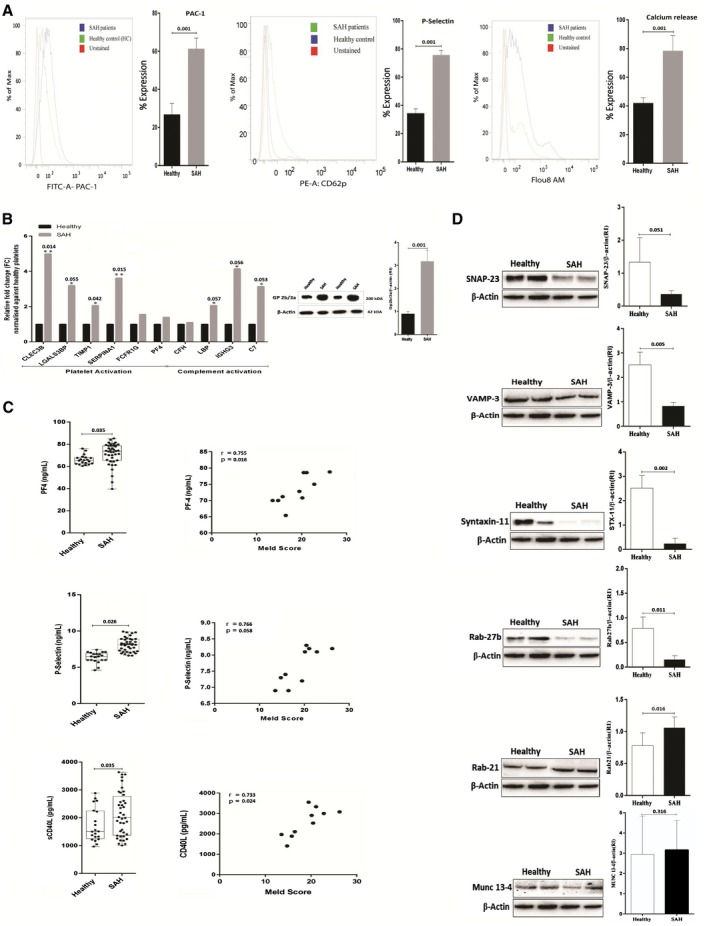
Validation of platelet functionality in SAH. (A) Platelet activation/aggregation in SAH histogram shows the percent expression of PAC‐1, P‐selectin (CD62p), and intracellular Ca+2(secretion) in SAH and HC. (B) Increased expression (relative change) of various mRNA in SAH platelets linked to platelet and complement activation. Protein expression of Gp2b/3a/β‐actin ratio in platelets of patients with SAH. Results expressed as mean ± SEM (*P < 0.05, **P < 0.01 for SAH versus HC). (C) Patients with SAH showed higher plasma levels of PF‐4 (65.85 ± 0.9457 versus 81.13 ± 6.595 ng/mL, P < 0.02), P‐selectin (6.360 ± 0.1639 versus 10.118 ± 2.1492 ng/mL, P < 0.0001), and soluble cluster of differentiation 40 ligand (1,726 ± 134.4 versus 2,528 ± 1.525.4 pg/mL, P < 0.05). Circulating levels of PF‐4, P‐selectin, and sCD40L correlated with the MELD score in SAH (r > 0.5, P < 0.05). (D) Representative western blot of SNARE complex proteins in platelets isolated from SAH versus HC (n = 5). Results are expressed as mean ± SEM (*P < 0.05, **P < 0.01, ***P < 0.001 as indicated).
Oxidized Albumin Promotes Activation and Induces Oxidative Stress in Healthy Platelets
Results of our study show that platelets in SAH are hyperactivated. To establish the cause for platelet hyperactivation, healthy platelets were exposed to purified albumin from patients with SAH, and its activation capacity was compared with known platelet activators.7, 26 Expression of PAC‐1 and P‐selectin was markedly higher in platelets exposed to purified albumin as compared with other platelet activators (Fig. 3A). Platelet aggregation/activation was significantly higher in healthy platelets exposed to purified albumin from SAH as compared with purified albumin from other study groups (P < 0.05; Fig. 3B‐D). Concomitantly, the expression of CD40L and production of ROS in healthy platelets was increased when exposed to purified albumin from SAH (Fig. 3E). In addition, purified SAH albumin significantly increased SNAP‐23 and Syntaxin‐11 expression (P < 0.05, Fig. 3F). Together, these findings suggest that purified SAH albumin induces aggregation/activation, inflammation, and oxidative stress in healthy platelets.
Figure 3.
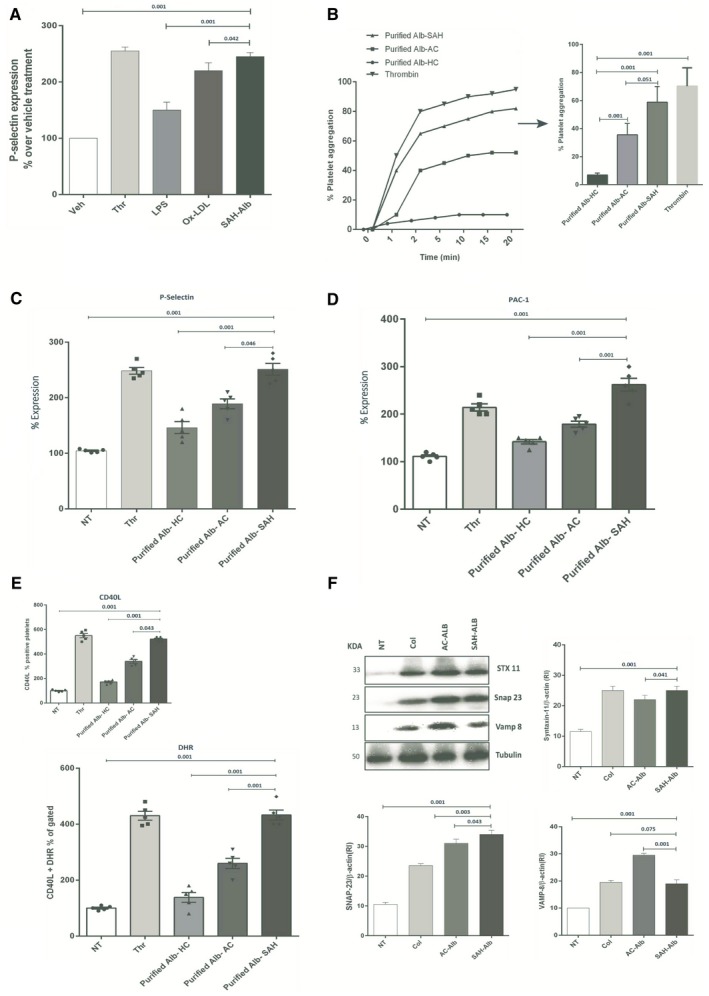
Purified albumin induces activation and ROS production in healthy platelets. (A) SAH albumin increases the expression of P‐selectin compared with lipopolysaccharide and oxidized low‐density lipoprotein. (B) Platelets were incubated with purified albumin‐HC (Alb‐HC, 1 mg/mL), purified albumin‐AC (Alb‐AC, 1 mg/mL), and purified albumin‐SAH (Alb‐SAH, 1 mg/mL). Platelet aggregation was measured after 30 minutes according to the final protocol at 37°C. A representative recording and bar graph is shown in mean values of three independent experiments, expressed as a percentage of maximal platelet aggregation. (C,D) Histogram showing the up‐regulation of P‐selectin, PAC‐1, and intracellular Ca2+ in platelets exposed to purified Alb‐SAH (1 mg/mL) versus others (percent expression). (E) Increased CD40L and ROS production in platelets exposed to Alb‐SAH compared with others (results shown as percentage of vehicle treatment; n = 3). (F) Representative western blot expression of SNARE complex protein Syntaxin‐11, SNAP‐23, and VAMP‐8 levels in healthy platelets incubated with purified Alb‐HC, Alb‐AC, or Alb‐SAH (1 mg/mL). All values are shown as mean ± SEM (*P < 0.05, **P < 0.01, ***P < 0.001 as indicated). Abbreviations: DHR, Dihydrorhodamine; KD, kilodalton.
Oxidized Albumin Mediated Increase in Platelet Activation and Oxidative Stress Was Reduced Following CD36 Neutralization
Recently, we showed markedly high oxidized albumin and advanced oxidative protein product (AOPP) levels in SAH.17, 18 Furthermore, AOPPs induce platelet activation through receptor CD36.14 To check which form of oxidized albumin induces platelet activation, oxidative stress and CD36 neutralization reverse the observed effects. Healthy platelets were first pretreated with or without CD36 blocking antibody and then with equal amounts (1 mg/mL) of ex vivo prepared albumin isoforms HMA, HNA1, and HNA2. Platelets exposed to HNA2 showed more increased aggregation than HNA1 or HMA (P < 0.05; Fig. 4A). Similarly, CD40L and ROS production was higher in HNA2‐exposed platelets, which was attenuated with CD36 neutralization (P < 0.05; Fig. 4B). A dose‐dependent reduction in CD40L and ROS was seen by ethylene glycol tetraacetic acid (EGTA), a chemical inhibitor, following HNA2 exposure (P < 0.05; Fig. 4C). Exposure to HNA2 significantly increased platelet activation compared with HNA1 or controls, which got ameliorated under CD36 neutralization (P < 0.05; Fig. 4D). In our study, the expression of SNAP‐23 was significantly increased following HNA2 exposure, and CD36 neutralization was able to reduce the expression (P < 0.05; Fig. 4E). These results suggest that majorly HNA2 (oxidized albumin form) is involved in the induction of platelet activation and mediates inflammation/oxidative stress through CD36 receptor signaling.
Figure 4.

HNA2 induces platelet aggregation and activation. (A) Platelets were incubated with ex vivo prepared HMA (1 mg/mL), HNA1 (1 mg/mL), and HNA2 (1 mg/mL), and aggregation was induced with collagen (0.2 μg/mL). Representative reading and bar graph indicate increased aggregation in HNA2‐treated platelets as compared with HNA1 and HMA. Data are expressed as percent of normalized platelet aggregation (n = 3). (B) CD40L and ROS production in healthy platelets was significantly increased when exposed to HNA2 (1 mg/mL). Pretreatment with anti‐CD36 blocking antibody (1 μg/mL) significantly reverses the effect, particularly in HNA2‐exposed platelets. (C) EGTA decreases HNA2‐induced platelet expression of CD40L and ROS production in a dose‐dependent manner. Platelets were pre‐incubated with EGTA, and activation was induced with HNA2 (1 mg/mL). (D) Histogram represents the percent expression of platelets exposed to HNA2 (1 mg/mL) showing increased expression of P‐selectin, PAC‐1, and intracellular calcium as compared with HNA1 (n = 5). (E) Expression of SNARE complex proteins (Syntaxin‐11, SNAP‐23, and VAMP‐8) increases in healthy platelets exposed to HNA2 (1 mg/mL) as compared with HNA1 and HMA. Interestingly, anti‐CD36 blocking antibody (1 μg/mL) reduced the effect of HNA2 over others. Results expressed as mean ± SEM (*P < 0.05, **P < 0.01, ***P < 0.001).
CD36 Neutralization Attenuates HNA2 Effects and Reduces Inflammatory Platelet Secretions
To validate the beneficial effect of CD36 neutralization, the secretome of CD36‐neutralized platelets were compared with controls (CD36 active) platelets. We quantitated 581 proteins in the secretome of the platelets (Fig. 5A and Supporting Table S5). The secretome heat map of HNA2: HNA2 + CD36 exposure was distinct (Fig. 5B). CD36 neutralization induced/increased 174 proteins in HMA‐exposed platelets, 187 proteins in HNA1‐exposed platelets, and 213 proteins in HNA2‐exposed platelets (FC > 1.5, P < 0.05). CD36 neutralization inhibited/decreased 102 proteins in HMA, 151 proteins in HNA1, and 39 proteins in HNA2 exposure (FC > −1.5; P < 0.05; Fig. 5B). Venny analysis identified 153 proteins that are specific to HNA2, which were induced following CD36 neutralization (Fig. 5B lower panel), and 23 HNA2‐specific proteins that were inhibited following CD36 neutralization (Fig. 5B lower panel). Pathway analysis of the 153 HNA2‐specific proteins showed significant increase in protein processing, amino‐acid metabolism, and glycolysis (P < 0.05; Fig. 5C and Supporting Table S6). Down‐regulated 23 HNA2‐specific proteins were enriched for platelet degranulation/activation, in response to elevated Ca2+ and glycolysis (Fig. 5C and Supporting Table S7). In addition, secretome analysis of HMA‐exposed and HNA‐1‐exposed platelets were analyzed, and differentially expressed proteins were subjected to pathway analysis. The up‐regulated proteins showed significant increase in pathways associated with platelet scavenging receptors. In contrast, down‐regulated proteins exhibit a decrease in peroxisome and amino acid metabolism–related pathways (Supporting Fig. S8). The results of the secretome analysis were in agreement with our previous results and validated that HNA2‐specific changes in platelets such as activation/aggregation, response to cytosolic Ca2+, and degranulation were reduced under CD36 neutralization. Thus, CD36 neutralization could prevent healthy platelets from HNA2‐mediated damage.
Figure 5.
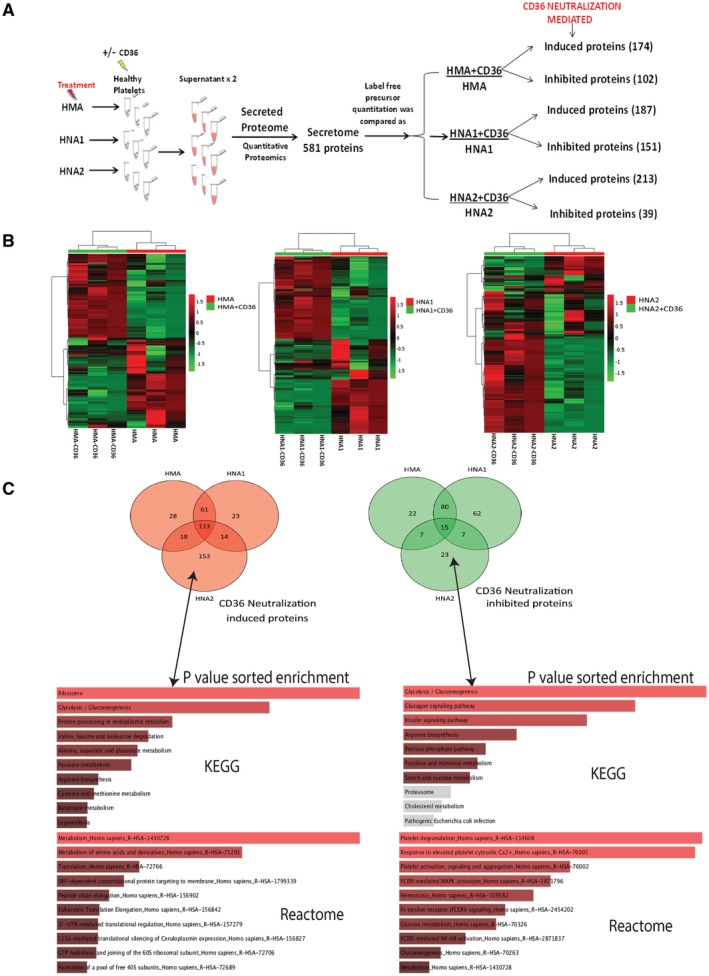
Secretome analysis of platelets with CD36 blockade. (A) Experimental flow diagram of secretome analysis using high‐resolution mass spectrometry. (B) Heat map representation of secretome for HMA+CD36:HMA; HNA+CD36:HNA1; and HNA2+CD36:HNA2 represents distinct secretome in HNA2 group comparison. Red Venn represents proteins that are induced/increased, and green Venn represents proteins that get reduced/inhibited with CD36 neutralization (P < 0.05). (C) Pathway overrepresentation analysis (Kyoto Encyclopedia of Genes and Genomes/Reactome) of HNA2‐specific 153 protein‐induced and 23 HNA2‐specific proteins inhibited under CD36 neutralization (P < 0.05, C‐score > 1).
Dysregulated Platelet Functions Inversely Correlate With the Severity of SAH
The mRNAs for platelet activation directly correlated (r > 0.3, P < 0.05), whereas the mRNA for platelet alpha/dense granules’ vesicular secretion inversely correlated, with the severity scores (r > −0.3; P < 0.05; Supporting Table S8), suggesting that platelet mRNA expression could also be used for prediction of outcomes in SAH.
Discussion
The results of this study show that the platelets in patients with severe alcoholism are hyperactivated and have impaired granule secretion. The oxidized albumin circulating in patients with SAH induces platelet oxidative stress and impairs granule secretion. We demonstrate that the irreversible form of oxidized albumin HNA2 is the prominent form of oxidized albumin playing a dominant role in platelet activation and oxidative stress. In addition, the results of our study show that CD36 neutralization significantly reduces the observed inflammatory and oxidative changes in platelets.
One of the important outcomes of the present study is that platelet proteins associated with activation and inflammation were up‐regulated, and proteins linked to coagulation (granular secretions) were down‐regulated. This was also supported by previous studies.27 In pathophysiological conditions like diabetes, advanced liver disease and chronic kidney disease, platelet activation is a result of endothelial damage and calcium dysregulation.28, 29 We observed higher platelet activation with higher expression of aggregation marker GP2b/3a and intracellular Ca2+ secretion as compared with controls. This corroborates with our result that platelet activation and inflammation markers correlate directly with the severity of SAH. BTM enrichment of platelet proteome also confirmed the inflammatory nature of SAH platelets and showed that these platelets are deprived of coagulation and regeneration capacity. In totality, platelets in SAH are activated, show inflammatory prototype, and may have thrombotic implications.30, 31, 32
Another key finding of our study is that platelets in patients with SAH have decreased degranulation capacity. Activated platelets release alpha/dense granules, which play a major role in hemostasis, leucocyte recruitment, repair, and regeneration.9 Granule release is the key function of platelets, which controls the vascular integrity and regulates hemostasis and inflammation.33 Granule secretion requires fusion of plasma membrane t‐SNAREs (Syntaxin and SNAP‐23) with vesicular membrane v‐SNARES (VAMP‐8, VAMP‐3) in the presence of MUNC13‐4, RAB27b, along with other small guanosine triphosphatases.25 In our study, the expression of SNARE complex proteins SNAP‐23, VAMP‐3, Rab‐27b, Syntaxin‐11, and Munc‐13‐4 were down‐regulated in SAH platelets, suggesting reduced capabilities for granular secretions, which may also correlate with the pathophysiology of SAH. In addition, significant reduction in the SNARE complex proteins in SAH platelets may highlight immature and dysregulated platelet formation at the megakaryocyte level in patients with SAH.
In the current study, we provide experimental evidence that hyperoxidized albumin is one of the inflammatory proteins that promotes platelet activation through the platelet CD36 receptor–mediated redox pathway, proposing also a prothrombotic role of oxidized albumin (Fig. 6F). Platelets express both types of class B scavenger receptors: SR‐BI and CD36. CD36 is a major glycoprotein present in platelets and recognizes a number of ligands that include oxidized proteins/lipoproteins.34 Recently, our group showed that oxidized albumin induces neutrophil activation.18 Furthermore, the ability of AOPP to activate platelets and trigger ROS production through CD36 receptor signaling is also documented.14 These observations led to the hypothesis that oxidized albumin through the CD36 receptor contributes to platelet activation and dysregulated SNARE complex protein expression. To validate, healthy platelets were first treated with or without anti‐CD36 blocking antibody followed by purified albumin exposure from the study groups. It was clear that albumin purified from SAH plasma was more potent in the activation/aggregation of platelets. SAH‐purified albumin resulted in a higher level of inflammatory mediators, ROS generation, and expression of proteins linked to SNARE complex. These derangements were markedly reduced after CD36 neutralization, indicating CD36‐dependent activation of platelets (Supporting Fig. S9). These findings may help to delineate the mechanisms of inflammation and disease progression in patients with SAH.
Figure 6.
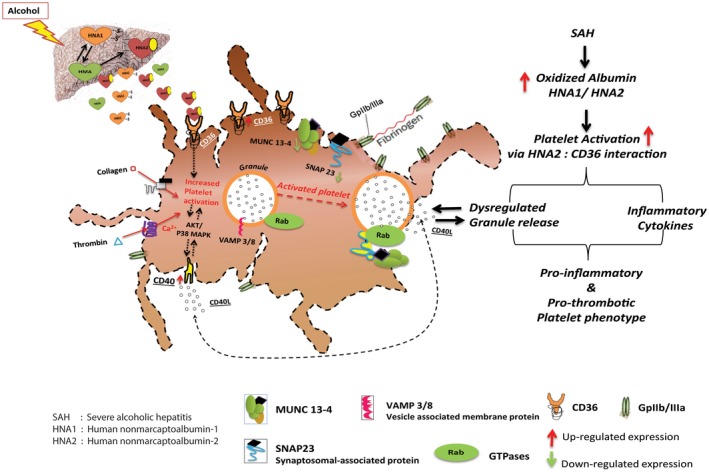
Paradigm of platelet activation in SAH. Proposed proinflammatory/prothrombotic activities of oxidized albumin and CD36 receptor. In SAH platelets, HNA2 promotes inflammatory and oxidative activation through CD36 receptor–mediated autocrine signaling much more than HNA1. In brief, HNA2 interacts with CD36 and harbors the SNARE complex proteins, which are then used quickly for autocrine activation and reloading the membrane CD36 levels. These phenomena result in intracellular calcium mobilization, ROS production, and CD40L secretion. By blocking CD36, HNA2‐mediated activation of platelet through CD36 is attenuated and the SNARE complex proteins are available for normal degranulation of repair and regenerative proteins.
Recently, reversibly oxidized albumin isoform (HNA1) was shown to induce leucocyte in advanced liver diseases.19 In contrast, we showed how Fenton‐modified albumin (HNA2, in vitro) was able to induce neutrophil oxidative burst. This probable reason for differential activation could be variable expression scavenger receptor CD36. Because the expression of CD36 is highest (Supporting Fig. S10) in platelets, these cells are more prone to oxidative damage by HNA2. Furthermore, blocking CD36 expression in platelets may inhibit its hyperactivation. This was successfully documented in our study, in which we show that blocking of CD36 significantly reduced activation markers, levels of CD40L, and ROS even after HNA2 exposure on platelets; this suggests that HNA2 possibly works through the CD36 pathway. Furthermore, a dose‐dependent decrease in activation/aggregation, inflammation, and oxidative stress with EGTA (chemical chelator of AOPP) also confirms that among the albumin isoforms, HNA2 specifically induces platelet activation response (Fig. 5D). Furthermore, significant reduction of SNAP‐23 expression following CD36 neutralization also suggests a prominent role of HNA2 not only in the activation but also in the modulation of SNARE proteins. These observations suggest that inhibition of the CD36‐HNA2 axis may decrease inflammatory activation and boost the granular secretion capacity of platelets in SAH.
Secretome analysis of the platelets exposed to HMA, HNA1, and HNA2 in the presence and absence of CD36 blockade showed a significant decrease in proteins linked to platelet activation aggregation (cytosolic Ca2+) and degranulation in the supernatant of CD36‐neutralized HNA2 counterpart platelets (Fig. 5B). This indicates that CD36 neutralization would reduce the pro‐oxidative and pro‐inflammatory effect of HNA2 in platelets (Fig. 6) In addition, exposure of HMA and HNA1 showed the increase in scavenging receptors in platelets. This observation was supported by previous studies that showed how CD36 knockout mice were protected from steatosis and inflammation.35
In conclusion, our study shows that the predominantly oxidized circulating albumin (HNA2) potentially increases platelet activation in patients with SAH, which may further facilitate vicious cycle of inflammation and oxidative stress in these patients through CD36 hyperactivation and autocrine signaling. This phenomenon was attenuated by blocking the CD36 receptor on platelets, suggesting that anti‐CD36 could be an attractive therapeutic option.
Supporting information
Supported by the Science and Engineering Research Board (DST‐SERB) (EMR/2016/004829).
Potential conflict of interest: Nothing to report.
Contributor Information
Jaswinder Singh Maras, Email: jassi2param@gmail.com, Email: sksarin@ilbs.in, Email: shivsarin@gmail.com.
Shiv K. Sarin, Email: sksarin@ilbs.in, Email: shivsarin@gmail.com.
References
Author names in bold designate shared co‐first authorship.
- 1. Hughes E, Hopkins LJ, Parker R. Survival from alcoholic hepatitis has not improved over time. PLoS ONE 2018;13:e0192393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Qamar AA, Grace ND, Groszmann RJ, Garcia‐Tsao G, Bosch J, Burroughs AK, et al. Incidence, prevalence, and clinical significance of abnormal hematologic indices in compensated cirrhosis. Clin Gastroenterol Hepatol 2009;7:689‐695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Witters P, Freson K, Verslype C, Peerlinck K, Hoylaerts M, Nevens F, et al. Review article: blood platelet number and function in chronic liver disease and cirrhosis. Aliment Pharmacol Ther 2008;27:1017‐1029. [DOI] [PubMed] [Google Scholar]
- 4. Afdhal N, McHutchison J, Brown R, Jacobson I, Manns M, Poordad F, et al. Thrombocytopenia associated with chronic liver disease. J Hepatol 2008;48:1000‐1007. [DOI] [PubMed] [Google Scholar]
- 5. Liu L, Chen M, Zhao L, Zhao Q, Hu R, Zhu J, et al. Ethanol induces platelet apoptosis. Alcohol Clin Exp Res 2017;41:291‐298. [DOI] [PubMed] [Google Scholar]
- 6. Northup PG, Sundaram V, Fallon MB, Reddy KR, Balogun RA, Sanyal AJ, et al. Hypercoagulation and thrombophilia in liver disease. J Thromb Haemost 2008;6:2‐9. [DOI] [PubMed] [Google Scholar]
- 7. Raparelli V, Basili S, Carnevale R, Napoleone L, Del Ben M, Nocella C, et al. Low‐grade endotoxemia and platelet activation in cirrhosis. Hepatology 2017;65:571‐581. [DOI] [PubMed] [Google Scholar]
- 8. Basili S, Raparelli V, Riggio O, Merli M, Carnevale R, Angelico F, et al. NADPH oxidase‐mediated platelet isoprostane over‐production in cirrhotic patients: implication for platelet activation. Liver Int 2011;31:1533‐1540. [DOI] [PubMed] [Google Scholar]
- 9. Ferroni P, Vazzana N, Riondino S, Cuccurullo C, Guadagni F, Davi G. Platelet function in health and disease: from molecular mechanisms, redox considerations to novel therapeutic opportunities. Antioxid Redox Signal 2012;17:1447‐1485. [DOI] [PubMed] [Google Scholar]
- 10. Henn V, Slupsky JR, Grafe M, Anagnostopoulos I, Forster R, Muller‐Berghaus G, et al. CD40 ligand on activated platelets triggers an inflammatory reaction of endothelial cells. Nature 1998;391:591‐594. [DOI] [PubMed] [Google Scholar]
- 11. Stoy S, Patel VC, Sturgeon JP, Manakkat Vijay GK, Lisman T, et al. Platelet‐leucocyte aggregation is augmented in cirrhosis and further increased by platelet transfusion. Aliment Pharmacol Ther 2018;47:1375‐1386. [DOI] [PubMed] [Google Scholar]
- 12. Lisman T, Luyendyk JP. Platelets as modulators of liver diseases. Semin Thromb Hemost 2018;44:114‐125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dovizio M, Alberti S, Guillem‐Llobat P, Patrignani P. Role of platelets in inflammation and cancer: novel therapeutic strategies. Basic Clin Pharmacol Toxicol 2014;114:118‐127. [DOI] [PubMed] [Google Scholar]
- 14. Pasterk L, Lemesch S, Leber B, Trieb M, Curcic S, Stadlbauer V, et al. Oxidized plasma albumin promotes platelet‐endothelial crosstalk and endothelial tissue factor expression. Sci Rep 2016;6:22104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Garcia‐Martinez R, Caraceni P, Bernardi M, Gines P, Arroyo V, Jalan R. Albumin: pathophysiologic basis of its role in the treatment of cirrhosis and its complications. Hepatology 2013;58:1836‐1846. [DOI] [PubMed] [Google Scholar]
- 16. Violi F, Ferro D. Clotting activation and hyperfibrinolysis in cirrhosis: implication for bleeding and thrombosis. Semin Thromb Hemost 2013;39:426‐433. [DOI] [PubMed] [Google Scholar]
- 17. Das S, Hussain MS, Maras JS, Kumar J, Shasthry SM, Nayak S, et al. Modification patterns of urinary albumin correlates with serum albumin and outcome in severe alcoholic hepatitis. J Clin Gastroenterol 2018. [DOI] [PubMed] [Google Scholar]
- 18. Das S, Maras JS, Hussain MS, Sharma S, David P, Sukriti S, et al. Hyperoxidized albumin modulates neutrophils to induce oxidative stress and inflammation in severe alcoholic hepatitis. Hepatology 2017;65:631‐646. [DOI] [PubMed] [Google Scholar]
- 19. Alcaraz‐Quiles J, Casulleras M, Oettl K, Titos E, Flores‐Costa R, Duran‐Guell M, et al. Oxidized albumin triggers a cytokine storm in leukocytes through p38 MAP kinase: role in systemic inflammation in decompensated cirrhosis. Hepatology 2018;68:1937‐1952. [DOI] [PubMed] [Google Scholar]
- 20. Thursz MR, Richardson P, Allison M, Austin A, Bowers M, Day CP, et al. Prednisolone or pentoxifylline for alcoholic hepatitis. N Engl J Med 2015;372:1619‐1628. [DOI] [PubMed] [Google Scholar]
- 21. Kumar Acharya S, Kumar Sharma P, Singh R, Kumar Mohanty S, Madan K, Kumar Jha J, et al. Hepatitis E virus (HEV) infection in patients with cirrhosis is associated with rapid decompensation and death. J Hepatol 2007;46:387‐394. [DOI] [PubMed] [Google Scholar]
- 22. Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 2016;44:W90‐W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li S, Sullivan NL, Rouphael N, Yu T, Banton S, Maddur MS, et al. Metabolic phenotypes of response to vaccination in humans. Cell 2017;169:862‐877, e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lee H, Chae S, Park J, Bae J, Go EB, Kim SJ, et al. Comprehensive proteome profiling of platelet identified a protein profile predictive of responses to an antiplatelet agent sarpogrelate. Mol Cell Proteomics 2016;15:3461‐3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ghasemzadeh M, Hosseini E. Platelet granule release is associated with reactive oxygen species generation during platelet storage: a direct link between platelet pro‐inflammatory and oxidation states. Thromb Res 2017;156:101‐104. [DOI] [PubMed] [Google Scholar]
- 26. Magwenzi S, Woodward C, Wraith KS, Aburima A, Raslan Z, Jones H, et al. Oxidized LDL activates blood platelets through CD36/NOX2‐mediated inhibition of the cGMP/protein kinase G signaling cascade. Blood 2015;125:2693‐2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cevik O, Baykal AT, Sener A. Platelets proteomic profiles of acute ischemic stroke patients. PLoS ONE 2016;11:e0158287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Vairappan B. Endothelial dysfunction in cirrhosis: role of inflammation and oxidative stress. World J Hepatol 2015;7:443‐459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wu H. Calcium signaling in platelet activation. Sheng Li Ke Xue Jin Zhan 2012;43:417‐421. [PubMed] [Google Scholar]
- 30. Ogasawara F, Fusegawa H, Haruki Y, Shiraishi K, Watanabe N, Matsuzaki S. Platelet activation in patients with alcoholic liver disease. Tokai J Exp Clin Med 2005;30:41‐48. [PubMed] [Google Scholar]
- 31. Pluta A, Gutkowski K, Hartleb M. Coagulopathy in liver diseases. Adv Med Sci 2010;55:16‐21. [DOI] [PubMed] [Google Scholar]
- 32. Arshad F, Lisman T, Porte RJ. Hypercoagulability as a contributor to thrombotic complications in the liver transplant recipient. Liver Int 2013;33:820‐827. [DOI] [PubMed] [Google Scholar]
- 33. Iwakiri Y, Groszmann RJ. Vascular endothelial dysfunction in cirrhosis. J Hepatol 2007;46:927‐934. [DOI] [PubMed] [Google Scholar]
- 34. Valiyaveettil M, Podrez EA. Platelet hyperreactivity, scavenger receptors and atherothrombosis. J Thromb Haemost 2009;7(Suppl 1):218‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Clugston RD, Yuen JJ, Hu Y, Abumrad NA, Berk PD, Goldberg IJ, et al. CD36‐deficient mice are resistant to alcohol‐ and high‐carbohydrate‐induced hepatic steatosis. J Lipid Res 2014;55:239‐246. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials