

Abstract
Nonalcoholic steatohepatitis (NASH) is a metabolic disorder in which poor nutrition and the gut‐to‐liver interaction play a major role. We previously established that hepatic high mobility group box‐1 (HMGB1) is involved in chronic liver disease. HMGB1 increases in patients with NASH and it is expressed in intestinal epithelial cells (IEC); yet, the role of intestinal HMGB1 in the pathogenesis of NASH has not been investigated. Thus, we hypothesized that IEC‐derived HMGB1 could play a role in NASH due to local effects in the intestine that govern hepatic steatosis. Control littermates and Hmgb1 ΔIEC mice were fed for 1 or 24 weeks a control diet or a high fat, high cholesterol (CHO) and fructose‐enriched diet (HFCFD). Hepatic and intestinal injury were analyzed. Hmgb1 ΔIEC mice were protected from HFCFD‐induced NASH after 1 or 24 weeks of feeding; however, they showed extensive atypical lipid droplet accumulation and increased concentrations of triglycerides (TG) and CHO in jejunal IEC together with lower TG and other lipid classes in serum. Olive oil or CHO gavage resulted in decreased serum TG and CHO in Hmgb1 ΔIEC mice, respectively, indicating delayed and/or reduced chylomicron (CM) efflux. There was significant up‐regulation of scavenger receptor class B type 1 (SR‐B1) and down‐regulation of apolipoprotein B48 (ApoB48) proteins, suggesting decreased lipid packaging and/or CM formation that resulted in lesser hepatosteatosis. Conclusion: Ablation of Hmgb1 in IEC causes up‐regulation of SR‐B1 and down‐regulation of ApoB48, leads to lipid accumulation in jejunal IEC, decreases CM packaging and/or release, reduces serum TG, and lessens liver steatosis, therefore protecting Hmgb1 ΔIEC mice from HFCFD‐induced NASH.
We have identified that ablation of Hmgb1 in intestinal epithelial cells causes up‐regulation of SR‐B1 and down‐regulation of ApoB48, leads to lipid accumulation in jejunal intestinal epithelial cells, decreases chylomicron packaging and/or release, reduces serum triglycerides, and lessens liver steatosis, thereby protecting Hmgb1 ΔIEC mice from a high‐fat, high‐cholesterol and fructose‐induced NASH.

Abbreviations
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- ApoB48
apolipoprotein B48
- AUC
area under the curve
- CD
control diet
- CE
cholesterol ester
- CHO
cholesterol
- CM
chylomicron
- DAG
diacylglycerol
- FA
fatty acid
- FITC
fluorescein isothiocyanate
- GSH
glutathione
- H&E
hematoxylin and eosin
- HFCFD
high‐fat, high‐cholesterol and fructose‐enriched diet
- HMGB1
high mobility group box‐1
- IEC
intestinal epithelial cells
- LD
lipid droplet
- LDL
low density lipoproteins
- MTP
microsomal triglyceride transfer protein
- NAFLD
nonalcoholic fatty liver disease
- NAS
NASH activity score
- NASH
nonalcoholic steatohepatitis
- NEFA
nonesterified fatty acids
- OO
olive oil
- PA
phosphatidylethanolamine
- PBS
phosphate‐buffered saline
- PC
phosphatidylcholine
- SR‐B1
scavenger receptor class B type 1
- TG
triglyceride
- VLDL
very low density lipoprotein
- wk
week
- WT
wild type
Key features of nonalcoholic fatty liver disease (NAFLD) are steatosis and inflammation occurring in the absence of alcohol abuse. In about 20% of patients, simple fatty liver progresses to nonalcoholic steatohepatitis (NASH), identified by the presence of significant necroinflammatory activity and fibrosis. Unfortunately, NASH can progress to cirrhosis and increases the risk of developing hepatocellular carcinoma. The molecular mechanisms responsible for the onset of NAFLD and its progression to NASH are still not fully understood. Therefore, identification of targets that drive the pathogenesis of NASH would greatly aid the development of therapies to treat these patients.
The gut‐to‐liver axis plays an important role in metabolic disorders, including NAFLD.1, 2 The intestine and the liver are connected via the portal vein, allowing the passage of nutrients, antigens, and bacterial products. The composition of the diet and changes in gut permeability are important contributors for the onset and progression of NASH.3, 4 Increased intestinal absorption of fatty acids (FA) and cholesterol (CHO) from the diet overwhelm the storage capacity from the adipose tissue; as a consequence, the liver acts as a reservoir to store the excess of lipids. Intestinal epithelial cells (IEC) elicit local inflammatory responses that, together with damage to the mucosal lining of the gut, cause increased gut permeability and escalate the translocation of toxic gut‐derived products and inflammatory mediators to the liver. Abnormal gut permeability is observed in patients with NAFLD and promotes liver steatosis, inflammation, and fibrosis.5, 6, 7 Targeting gut events and/or gut‐derived mediators, which could signal to the liver, may slow down the progression of NASH.
HMGB1 is a damage‐associated molecular pattern that plays an important role in mediating inflammatory responses during tissue injury.8, 9, 10 HMGB1 is involved in alcoholic liver disease,11 fibrosis,12 hepatocellular carcinoma,13, 14 and drug‐induced liver injury,15 and has been suggested to be implicated in NAFLD. Indeed, hepatocytes’ HMGB1 expression increases in mice fed a high‐fat diet.16 Furthermore, hepatocytes exposed to lipids or undergoing cell damage release HMGB18, 10, 11 that can bind to numerous receptors on adjacent cells to evoke downstream pro‐inflammatory and pro‐fibrogenic processes.12, 17, 18 This literature suggests a role for HMGB1 in modulating the response of the liver to high lipid contents.
Moreover, following local injury, IEC secrete HMGB1 as a danger signal19 that could further act as a mediator of enteric inflammation in NAFLD.20 Notably, HMGB1 modulates gut permeability in experimental endotoxemia.21, 22 Despite this, there is little investigation on the role of IEC‐derived HMGB1, its local effects in the intestine, and the communication between the gut and the liver in the setting of NASH. It is likely that this important proinflammatory protein plays a pathogenic role in both the gut and the liver during NASH.
In this study, we hypothesized that IEC‐derived HMGB1 could play a role in NASH due to local effects in the intestine that govern hepatic steatosis. To investigate this, we generated mice with conditional ablation of Hmgb1 in IEC (Hmgb1 ΔIEC) and fed them a high‐fat, high‐cholesterol and fructose‐enriched diet (HFCFD) mimicking the western diet, which has been proven to recapitulate the symptoms of human NASH.23, 24, 25 We found that the livers from Hmgb1 ΔIEC mice were protected from NASH after both short‐term and long‐term feeding. This protection resulted from up‐regulation of scavenger receptor class B type 1 (SR‐B1) and down‐regulation of apolipoprotein B48 (ApoB48) that lead to lipid accumulation in IEC, decreased chylomicron (CM) packaging and/or release, reduced serum TG, and lessened liver steatosis.
Materials and Methods
Induction of NASH
We used a diet containing 40% Kcal from fat (mostly partially hydrogenated corn oil), 20% Kcal of fructose, and 2% of CHO (HFCFD, D16010101), or a control diet containing 10% Kcal of fat (CD, D09100304) with additional calories balanced by corn starch. Both diets were purchased from Research Diets (New Brunswick, NJ) and are a well‐established model to induce NASH.23, 24, 25 Mice were allowed free access to the diets and were sacrificed at 1 week or 24 weeks in the fed state. Supporting Fig. S1A shows a schematic of the experimental design. Blood was collected from the submandibular and portal vein under isoflurane anesthesia, mice were then sacrificed, organs harvested, images of liver and gut captured, liver and gut weight recorded, and gut length measured. Gut permeability was analyzed by gavaging mice with 40 mg/mL of fluorescein isothiocyanate (FITC)‐Dextran 4 kDa (Sigma, St. Louis, MO) and measuring the fluorescence in portal serum after 6 hours.
Mice
The Hmgb1fl/fl mice were donated by Dr. Timothy R. Billiar (University of Pittsburgh, Pittsburgh, PA). The Hmgb1loxP allele was created by inserting loxP sites within introns 1 and 2 flanking exon 2 of Hmgb1. 26 The Hmgb1fl/fl mice were bred with Villin (Vil1)‐Cre mice (B6.Cg‐Tg[Vil1‐cre]997Gum/J, Stock No. 004586; Jackson Laboratory, Bar Harbor, ME) to generate IEC‐specific (Hmgb1 ΔIEC) mice. Control littermates (Hmgb1fl/fl−/− Vil‐Cre+ and Hmgb1fl/fl+/+ Vil‐Cre‐), referred to as wild type (WT) on the text for simplicity purposes only, were used in all experiments. First, each Cre mouse line that had not been crossed to any fl/fl mouse line was used to control for Cre‐induced recombination at cryptic lox sites in the genome; and second, each fl/fl mouse line that had not been crossed to any Cre mouse line was used to control for the effects of introducing loxP sites into the intron regions of the gene of interest. All loxP‐flanked (floxed) strains were rederived using C57BL/6J oocytes donors. All mice lacked a liver or intestinal phenotype in the absence of any treatment.
Study Approval
All animals received humane care according to the criteria outlined in the Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences and published by the National Institutes of Health. Housing and husbandry conditions were approved by our Institutional Animal Care and Use Committee office prior to initiating the studies. All in vivo experiments were carried out according to the ARRIVE guidelines.
Statistical Analysis
Data are expressed as mean ±SEM. Data were analyzed for normal distribution and then subject to either an unpaired t test or Wilcoxon test if normally or not normally distributed, respectively. The metabolomics and lipidomics data were normalized to the respective injection standards in positive or negative mode. Data were then subject to generalized linear model analysis in R software, including an interaction term to compare the effects of both variables in the study. A pairwise t test was then performed, and metabolites with a P < 0.05 were plotted in a heatmap with hierarchical clustering.
Results
Hmgb1 ΔIEC Mice Are Protected From HFCFD‐Induced Hepatic Steatosis and Inflammation After 1 Week
To determine the possible role of IEC‐derived HMGB1 in the early stages of NASH, we ablated Hmgb1 in IEC and then fed these mice with the CD and HFCFD for 1 week. Hmgb1 was successfully deleted from IEC as shown on immunohistochemistry for HMGB1 in the jejunum (Fig. 1A) and throughout the length of the small and large intestine (Supporting Fig. S2). WT mice fed the HFCFD for 1 week displayed signs of NAFLD/early NASH such as microvesicular and macrovesicular hepatic steatosis, localized primarily to zones 1 and 2, and inflammation, resulting in a NASH activity score (NAS) of 2.48 ± 1.06. Mild hepatocyte ballooning degeneration and ductular reaction were also observed in some WT mice fed the HFCFD; however, these changes were not significant at 1 week. In addition, there was no fibrosis in these mice (Fig. 1B,C). The liver‐to‐body weight ratio was increased in WT mice fed the HFCFD for 1 week, alongside decreased serum TG, CHO, and nonesterified fatty acids (NEFA), and a trend of increased liver TG, CHO and NEFA, corroborating the steatosis observed by histology. As commonly observed in patients with NAFLD,27 alanine aminotransferase (ALT) and aspartate aminotransferase (AST) activities were higher in WT mice fed the HFCFD than those fed the CD (Fig. 1D‐F). Hmgb1 ΔIEC mice fed the HFCFD for 1 week were protected from hepatic steatosis and inflammation. They displayed no hepatocyte ballooning degeneration, ductular reaction or fibrosis, overall resulting in a NAS of 0.05 ± 0.02 (Fig. 1B,C). The increase in body weight was higher than that in WT mice, which may be explained by accumulation of fat in other organs including the intestine and adipose tissue (Fig. 1D). There was less liver injury as revealed by lower ALT activity (Fig. 1F). No macroscopic differences in adipose tissue mass or phenotype were observed between genotypes (data not shown). These data indicate that mice lacking Hmgb1 in IEC are protected from HFCFD‐induced NASH at 1 week. Next, we investigated how this protection is conferred.
Figure 1.
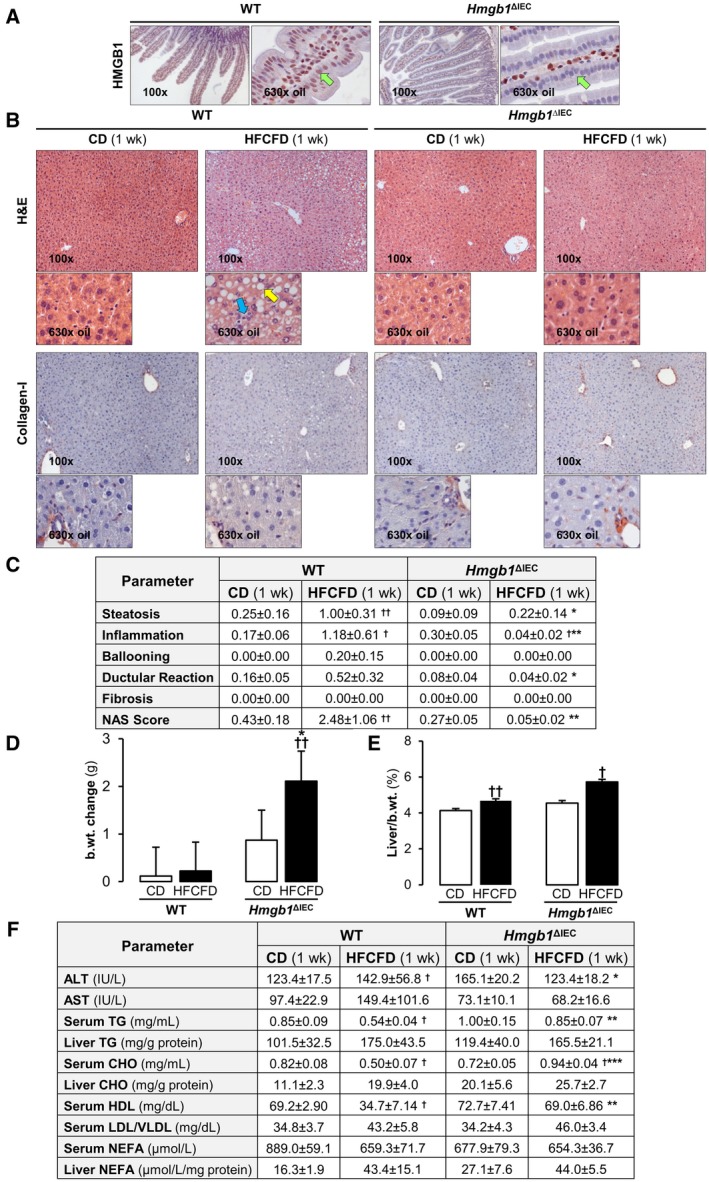
Hmgb1 ΔIEC mice are protected from HFCFD‐induced hepatic steatosis and inflammation after 1 week. WT and Hmgb1 ΔIEC mice were fed the CD or HFCFD for 1 week. (A) Immunostaining for HMGB1 (red) in the jejunum (green arrows: IEC). (B) Hematoxylin and eosin (H&E) staining (top) and collagen‐I immunostaining of the liver (yellow arrow: steatosis; blue arrow: inflammation). (C) Pathology scores. (D) Change in the mouse body weight. (E) Liver‐to‐body weight ratio. (F) Serum ALT and AST activities; serum TG, CHO, HDL, LDL/VLDL, and NEFA levels; liver TG, CHO, and NEFA concentration. Data are expressed as mean ± SEM (n ≥ 8 mice/group). † P < 0.05 and †† P < 0.01 for HFCFD versus CD; *P < 0.05, **P < 0.01, and ***P < 0.001 for Hmgb1 ΔIEC versus WT mice. Abbreviations: b.wt., body weight; wk, week.
Hmgb1 ΔIEC Mice Display HFCFD‐Induced Atypical Lipid Droplet Accumulation in the Jejunum After 1 Week
Jejunal inflammation, either intraepithelial or in the lamina propria, was similar between groups. The duodenum, jejunum, ileum, and colon of WT mice fed the HFCFD remained healthy and showed similar weight and length than when fed the CD (Fig. 2A,B). A small number of WT mice displayed lipid droplets (LD) in the jejunum, but the frequency and the overall area of LD per sample was low (Fig. 2B). Strikingly, Hmgb1 ΔIEC mice fed the HFCFD displayed extensive LD accumulation in the jejunal IEC, which was confirmed by BODIPY (Thermo Fisher Scientific, Waltham, MA) FA staining. The LD were both microvesicular and macrovesicular and localized primarily to the IEC at the tip of the villi, with fewer being found near the crypts and some in the lamina propria (Fig. 2A,B). LD were not apparent in the IEC of any other region of the intestine, reflecting that lipid absorption occurs primarily in the jejunum28 (Supporting Fig. S3). Increased gut permeability was observed in both groups of mice fed the HFCFD compared with CD diet, correlating with increased HMGB1 in portal serum, however, there were minimal differences between genotypes (Fig. 2C,D). Importantly, while TG and CHO were increased in IEC from CD‐fed Hmgb1 ΔIEC, the HFCFD further increased TG, CHO, and NEFA compared with WT mice (Fig. 2D). These data indicate that deletion of Hmgb1 from IEC leads to HFCFD‐induced accumulation of TG and CHO‐containing LD in IEC in the jejunum from mice after 1 week, paralleling the decreased lipid accumulation and inflammation in the liver. We next asked whether this accumulation of LD in IEC and the related protection from NASH was conferred long term.
Figure 2.
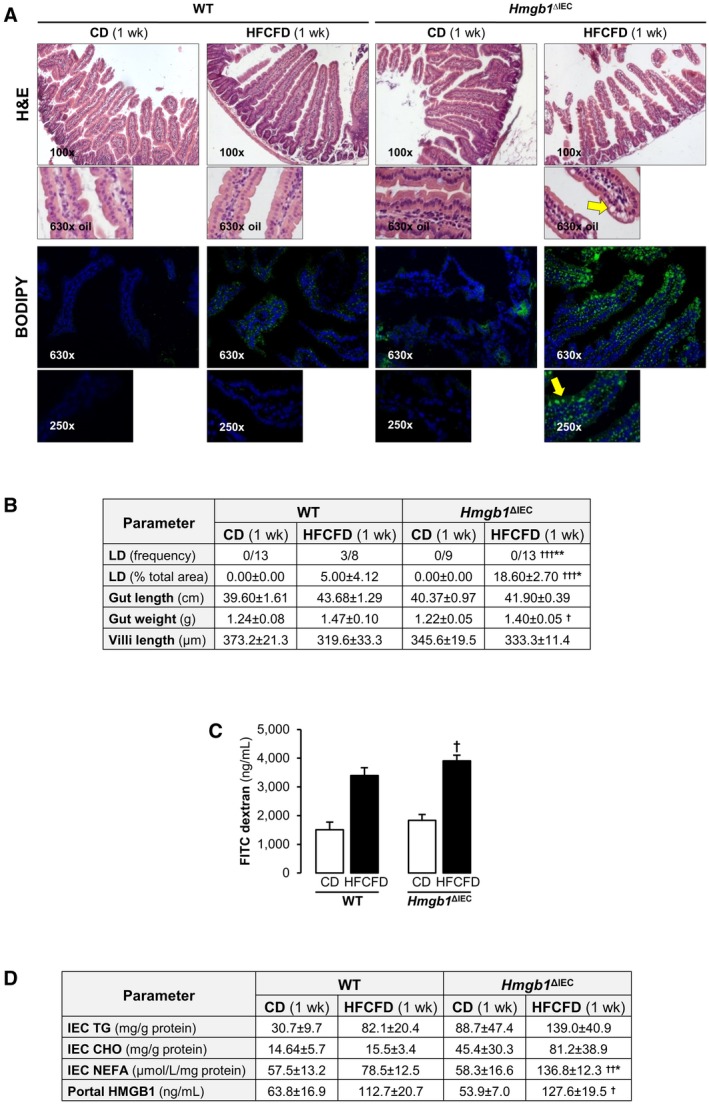
Hmgb1 ΔIEC mice display HFCFD‐induced atypical LD accumulation in the jejunum after 1 week. WT and Hmgb1 ΔIEC mice were fed the CD or HFCFD for 1 week. (A) H&E staining (top) and BODIPY FA staining (bottom) of the jejunum (yellow arrows: LD). (B) Pathology scores. (C) FITC‐Dextran concentration in portal serum indicative of gut permeability. (D) IEC TG, CHO, and NEFA and HMGB1 levels in portal serum. Data are expressed as mean ± SEM except for LD frequency, which is shown as the number of cases per group (n ≥ 8 mice/group). † P < 0.05, †† P < 0.01, and ††† P < 0.001 for HFCFD versus CD; *P < 0.05 and **P < 0.01 for Hmgb1 ΔIEC versus WT mice.
Hmgb1 ΔIEC Mice Are Protected From HFCFD‐Induced NASH and Display Atypical LD Accumulation in the Jejunum After 24 Weeks
WT mice fed the HFCFD for 24 weeks developed symptoms of advanced NASH with an overall NAS of 7.42 ± 0.66. Macrovesicular steatosis localized primarily to zone 1 and microvesicular steatosis to zone 3. Inflammation and hepatocyte ballooning degeneration were observed across all zones. Chicken‐wire fibrosis, which is observed in human NASH, as well as thickening of fibers around the portal triad, was present (Fig. 3A,B). Increased body‐weight change and liver‐to‐body weight ratio were observed compared with mice fed the CD, supporting the development of a NASH phenotype (Fig. 3C,D). Mice on CD also displayed hepatic steatosis and mild inflammation, likely due to the high fat‐derived calories of the CD; however, the NAS (1.58 ± 0.19) was not representative of NASH (Fig. 3A,B). Low density lipoprotein (LDL) particles are CHO‐rich, whereas very low density lipoprotein (VLDL) particles are TG‐rich. The LDL‐to‐VLDL ratio was elevated by the NASH diet (Fig. 3E). Hmgb1 ΔIEC mice fed the HFCFD for 24 weeks were protected from NASH, as shown by the NAS (5.86 ± 0.49), due to reduced steatosis, significantly less ballooned hepatocytes, and chicken‐wire fibrosis together with thinner scars (Fig. 3A,B). Nonetheless, inflammation was comparable between both genotypes. There were no clear differences in the number of neutrophils, Kupffer cells, and T cells in the livers from each group of mice regardless of genotype (Supporting Fig. S4). A slight reduction in body‐weight change and liver‐to‐body weight ratio was observed in the HFCFD‐fed Hmgb1 ΔIEC mice (Fig. 3C,D). There was less liver injury as shown by lower ALT and AST activities and serum TG compared with the HFCFD‐fed WT mice (Fig. 3E). Intrahepatic HMGB1 did not contribute to these effects, as there was no difference in the expression in both HFCFD‐fed groups of mice (Supporting Fig. S4).
Figure 3.
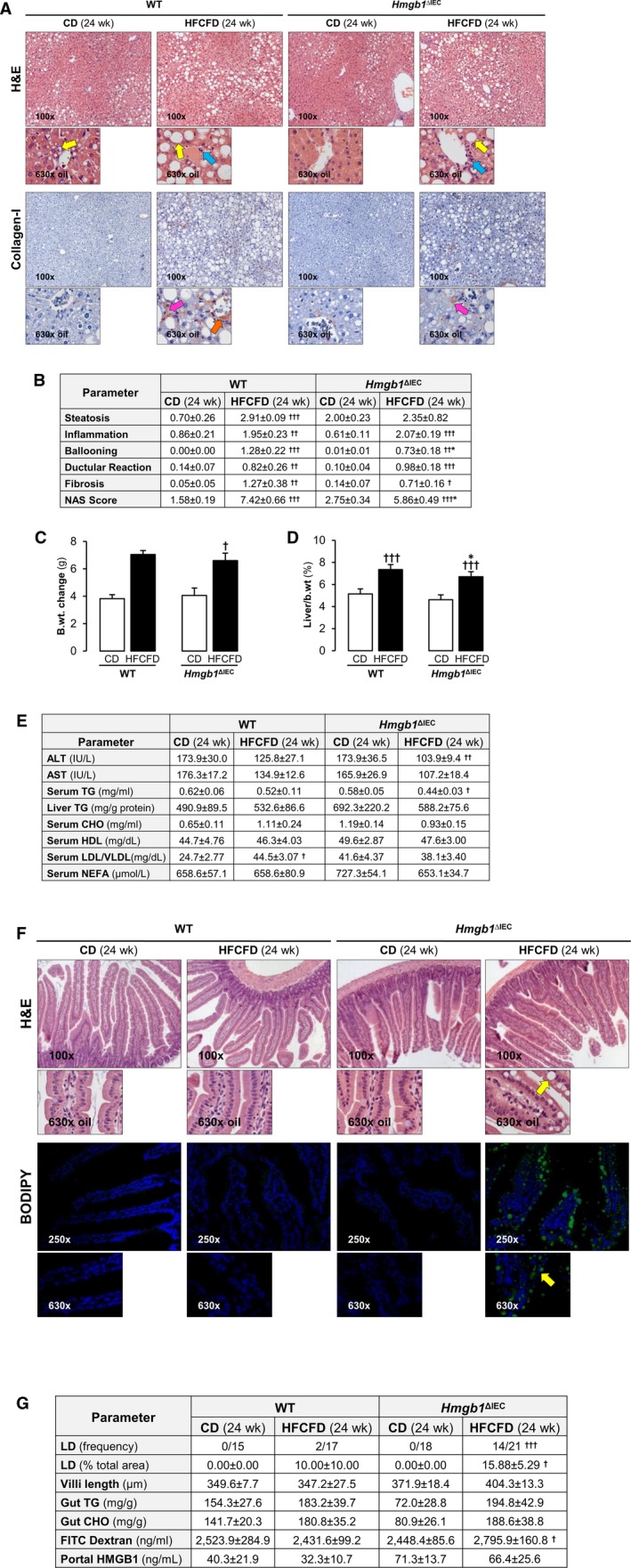
Hmgb1 ΔIEC mice are protected from HFCFD‐induced NASH and display atypical LD accumulation in the jejunum after 24 weeks. WT and Hmgb1 ΔIEC mice were fed the CD or HFCFD for 24 weeks. (A) H&E staining (top) and collagen‐I immunostaining (bottom) of the liver (yellow arrows: steatosis; blue arrows: inflammation; pink arrows: chicken‐wire fibrosis; orange arrow: thickening of portal triad fibers). (B) Pathology scores. (C) Change in body weight. (D) Liver‐to‐body weight ratio. (E) Serum ALT and AST activities; serum TG, CHO, HDL, LDL/VLDL, and NEFA levels; and Liver TG concentration. (F) H&E staining (top) and BODIPY FA stain (bottom) of the jejunum (yellow arrows: LD). (G) LD frequency and percentage, villi length, FITC dextran indicative of gut permeability, and HMGB1 levels. Intestinal TG and CHO normalized to protein concentration. Data are expressed as mean ± SEM except for LD frequency, which is shown as the number of cases per group (n ≥ 15 mice/group). † P < 0.05, †† P < 0.01, and ††† P < 0.001 for HFCFD versus CD; *P < 0.05 for Hmgb1 ΔIEC versus WT mice. Abbreviation: b.wt., body weight.
We next looked into whether these mice had the same intestinal phenotype after long‐term feeding. The duodenum, jejunum, ileum, and colon of WT mice fed the HFCFD for 24 weeks displayed no notable signs of injury and only a small amount of LD (Fig. 3F,G and Supporting Fig. S5). The frequency and quantity of LD accumulation in the IEC of Hmgb1 ΔIEC mice fed the HFCFD for 24 weeks replicated what was observed at 1 week, with similar localization (Fig. 3F,G). Gut permeability was higher in Hmgb1 ΔIEC mice when fed the HFCFD than the CD (Fig. 3G). Although counterintuitive, HMGB1 in portal serum increased in HFCFD‐fed Hmgb1 ΔIEC compared with WT mice (Fig. 3F). Taken together, these results demonstrate that mice lacking Hmgb1 in IEC are protected from HFCFD‐induced NASH after 1 week, which is sustained after long‐term feeding for 24 weeks, and parallels increased LD accumulation in jejunal IEC in these mice. Thus, the accumulation of LD in the intestine and the decreased steatosis in the liver from Hmgb1 ΔIEC mice confers protection from NASH. We next asked whether alterations in lipid and/or metabolic profiles contributed to this protection.
Hmgb1 ΔIEC Mice Fed the HFCFD Display Altered Lipid Profile in Portal Serum After 24 Weeks
Untargeted lipidomics was performed in the portal serum from WT and Hmgb1 ΔIEC mice fed the CD or HFCFD for 24 weeks. Figure 4A depicts a heatmap of the lipid classes, showing significant changes between the groups. The lipidomics analysis revealed differences in clustering between Hmgb1 ΔIEC mice and WT mice fed the HFCFD, with an overall decrease in the lipid pools in Hmgb1 ΔIEC mice. Specifically, there was a reduction in a large number of TGs, phosphatidylcholines (PCs), lysoPCs, phosphatidylethanolamines (PEs), lysoPE, plasmanylPE, plasmenaylPEs, phosphatidylinositols, some sphingomyelins, and sphingosine. Only acylcarnitine 18:1 and ceramide d17:1/18:1 were increased in HFCFD‐fed Hmgb1 ΔIEC mice. Overall, the absence of HMGB1 in the IEC leads to a reduction in the amount and/or subclasses of lipids in the portal blood.
Figure 4.
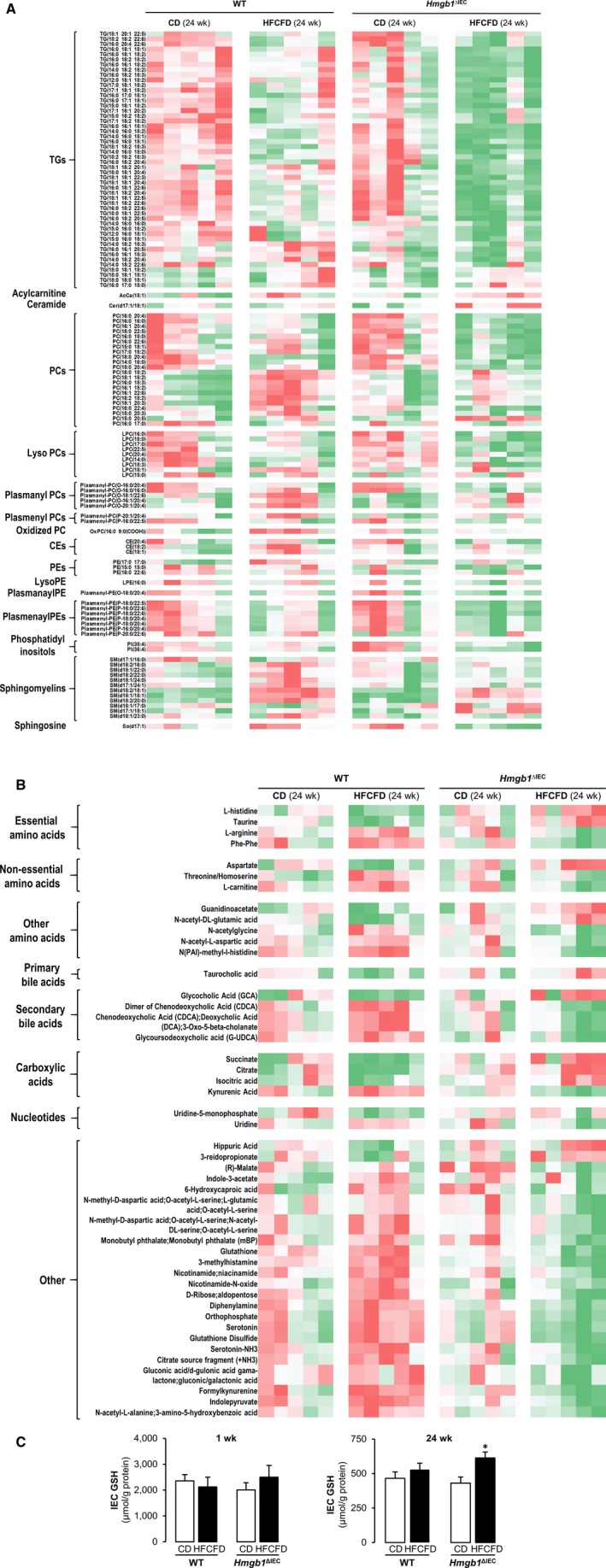
Hmgb1 ΔIEC mice fed the HFCFD display altered lipid and metabolic profiles in portal serum after 24 weeks. WT and Hmgb1 ΔIEC mice were fed the CD or HFCFD for 24 weeks. Portal serum was collected following sacrifice, and untargeted lipidomics and metabolomics analysis were performed. Peak intensities were normalized to internal standards and subject to generalized linear model analysis; species with significant (P < 0.05) differences in peak intensity between genotype or diet are plotted in a heatmap with hierarchical clustering; intermediates are separated by lipid classes or molecule type. Lipidomics data are plotted in (A) and metabolomics in (B) (green: low; red: high). (C) GSH levels in IEC at 1 week and 24 weeks of feeding the CD or HFCFD. Data are expressed as means ± SEM (n ≥ 5 mice/group). *P < 0.05 for Hmgb1 ΔIEC versus WT mice.
Hmgb1 ΔIEC Mice Fed the HFCFD Display Altered Metabolic Profile in Portal Serum After 24 Weeks
Next, metabolomics analysis was performed in portal serum from WT and Hmgb1 ΔIEC mice fed the CD or HFCFD (Fig. 4B). We found that essential amino acids such as taurine and l‐histidine were increased in Hmgb1 ΔIEC mice, whereas l‐arginine and phenylalanine were decreased. There were significant increases in aspartate, a non‐essential amino acid, other amino acids (guanidinoacetate and N‐acetyl‐dl‐glutamic acid), and carboxylic acids (succinate, citrate, and isocitrate). There was an increase in the primary bile acid taurocholic acid and in the secondary bile acid glycocholic acid, and a decrease in other secondary bile acids in the serum of HFCFD‐fed Hmgb1 ΔIEC mice compared with all other groups. Notably, glutathione disulphide (GSSG), an indicator of oxidative stress, was only up‐regulated in HFCFD‐fed WT mice but was low in Hmgb1 ΔIEC mice fed the HFCFD. The latter mice showed higher glutathione (GSH) levels. Hence, lack of HMGB1 in the IEC alters key metabolites, which could possibly account for the protection against NASH.
Hmgb1 ΔIEC Mice Have Delayed and/or Reduced Chylomicron Release
We next asked whether a defect in lipid packaging and/or CM release was responsible for the atypical LD accumulation in HFCFD‐fed Hmgb1 ΔIEC mice. To this end, we analyzed CM release by injecting WT and Hmgb1 ΔIEC mice with Tyloxapol, to block lipoprotein lipase, and gavaged them with either 200 µL phosphate‐buffered saline (PBS), OO, or 2% CHO dissolved in OO. Serum TG and CHO were analyzed at baseline and over 6 hours to observe the amount of gavaged OO or CHO secreted into the systemic blood. Hmgb1 ΔIEC mice had reduced serum TG over time compared to WT mice, as reflected by lower area under the curve (AUC) (Fig. 5A top). Similarly, when the mice were gavaged with CHO, serum CHO was decreased in Hmgb1 ΔIEC mice, as shown by the reduced AUC (Fig. 5A bottom). Consistently, TG and CHO in IEC were increased in Hmgb1 ΔIEC mice to a greater extent than in WT, after gavage with OO or CHO, respectively (Fig. 5B). The jejunum histology from mice gavaged with OO, but not with CHO, reflected this finding, as there was increased frequency and percentage of area occupied by LD in Hmgb1 ΔIEC compared with WT mice (Fig. 5C,D). These data suggest that CM release is reduced in Hmgb1 ΔIEC, resulting in LD accumulation in IEC, reduced serum lipids, and protection from NASH. We next investigated the mechanism by which the defect in lipid absorption, packaging, and/or CM release was being affected.
Figure 5.
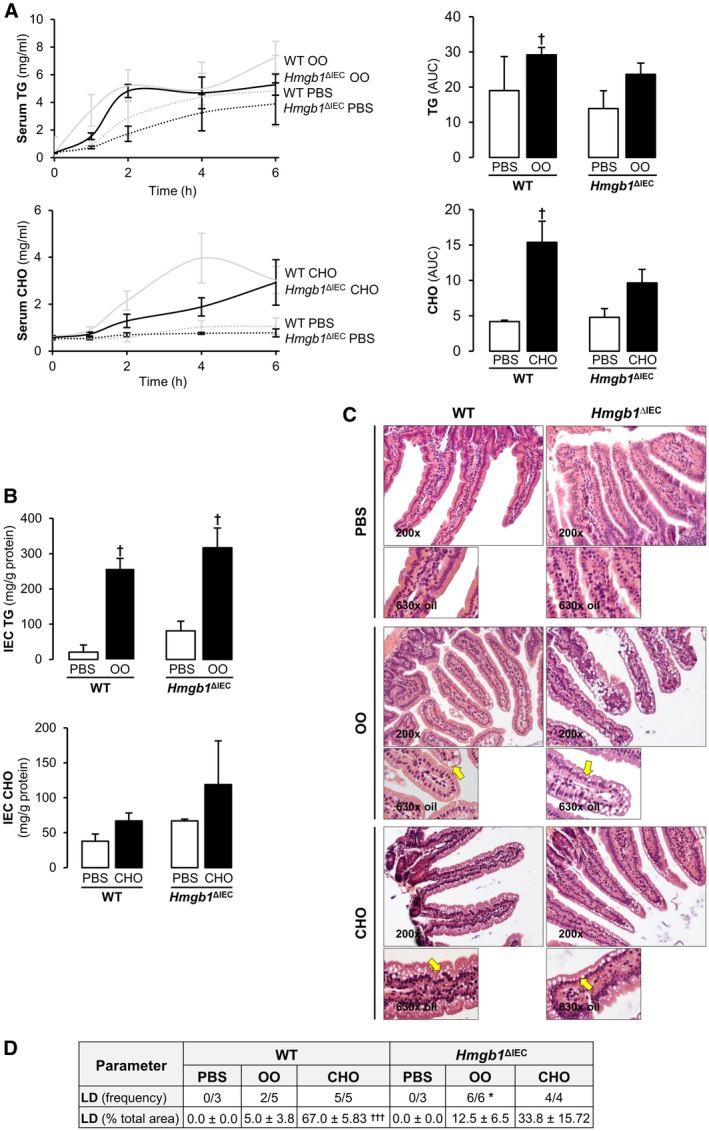
Hmgb1 ΔIEC mice have delayed and/or reduced CM release. WT and Hmgb1 ΔIEC mice were fasted overnight and serum was drawn to record baseline levels of TG and CHO. Then, mice were injected intraperitoneally with 300 mg/mL of Tyloxapol, and 5 minutes later they were gavaged with 200 µL of OO, 2% CHO, or PBS alone. Serum samples were drawn at 1, 2, 4, and 6 hours after gavage. Mice were sacrificed at 6 hours, tissues harvested, and IEC isolated for histological and biochemical analyses. (A) Time‐course analysis and matching AUC of serum TG and CHO. (B) IEC TG and CHO concentration. (C) H&E staining of the jejunum (yellow arrows: steatosis). (D) LD frequency and percentage of total area. Data are expressed as means ± SEM except for LD frequency, which is shown as the number of cases per group (n ≥ 3 mice/group). † P < 0.05 and ††† P < 0.001 for OO or CHO versus PBS; *P < 0.05 for Hmgb1 ΔIEC versus WT mice.
Hmgb1 ΔIEC Mice on the HFCFD Have Increased SR‐B1 and Decreased ApoB48 Protein Expression in IEC
Proteins required for efficient lipid transport from the diet are CD36 (cluster of differentiation 36), FABP2 (fatty acid binding protein), NPC1L1 (Niemann‐Pick C1‐like 1) and SR‐B1, and packaging is facilitated by MOGAT2 (2‐acylglycerol O‐acyltransferase 2), DGAT (diacylglycerol O‐acyltransferase 1), ACAT2 (acetyl‐CoA acetyltransferase 2), and MTP (microsomal triglyceride transfer protein). Once inside the IEC, CE and TG are assembled with ApoB48, through interaction with MTP in the endoplasmic reticulum. Thereafter, CM are exported to the cis‐Golgi by pre‐CM transport vesicles, before being targeted to the enterocyte basolateral side to enter the systemic circulation via the thoracic duct.29, 30 Thus, we analyzed the expression of these proteins by western blot (Fig. 6) and/or quantitative real‐time polymerase chain reaction (Supporting Fig. S6) at 1 week of feeding the HFCFD. Of all the proteins analyzed, there was a significant increase in SR‐B1 and a decrease in ApoB48 protein along with a minor decrease in Mttp messenger RNA in IEC from HFCFD‐fed Hmgb1 ΔIEC compared with WT mice (Fig. 6A and Supporting Fig. S6). These results indicate that Hmgb1 deletion from IEC increases SR‐B1, which facilitates the selective uptake of CHO, and lowers the ApoB48 protein, which disrupts the packaging of TG and CHO into CM, resulting in accumulation of TG and CHO in IEC and a reduction in the amount and/or subclasses of lipids in serum, thereby protecting these mice from NASH (Fig. 6B, working model). These data highlight an important and previously unknown role for intestinal HMGB1 in lipid transport.
Figure 6.
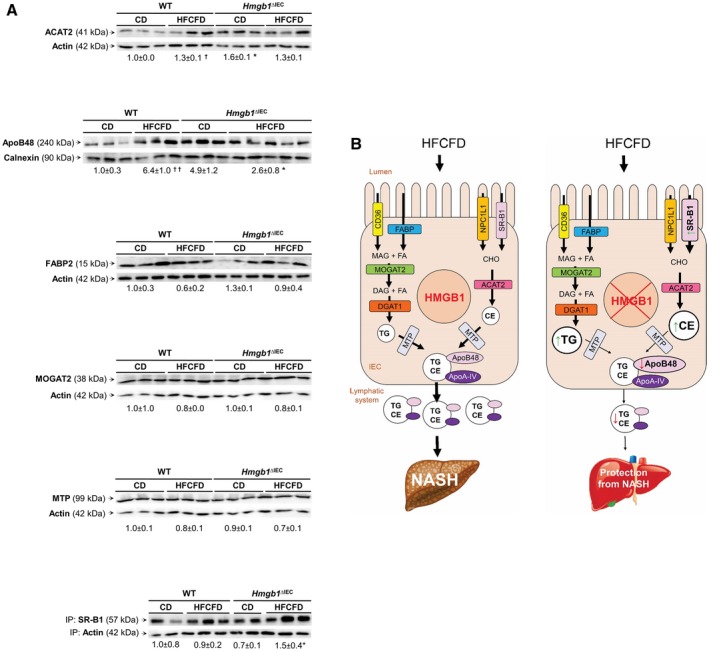
Hmgb1 ΔIEC mice on the HFCFD have increased SR‐B1 and decreased ApoB48 protein expression in IEC. WT and Hmgb1 ΔIEC mice were fed the CD or HFCFD for 1 week, and IEC were isolated. (A) Western blot analysis for ACAT2, ApoB48, FABP2, MOGAT2, MTP, and immunoprecipitation followed by western blot for SR‐B1 normalized to actin or calnexin. Data are expressed as means ± SEM (n ≥ 3 mice/group). † P < 0.05 for HFCFD versus CD; *P < 0.05 for Hmgb1 ΔIEC versus WT mice. (B) Proposed mechanism by which HMGB1 is delaying and/or reducing CM release: In Hmgb1ΔIEC mice fed the HFCFD, SR‐B1 is increased and ApoB48 expression is significantly decreased, whereas Mttp (encoding MTP) is only slightly down‐regulated. This contributes to increased absorption of CHO and hinders the packaging of TG and CHO to form CM, thereby diminishing CM efflux. This results in lower lipid classes in serum and hence less hepatosteatosis. Abbreviations: IP, immunoprecipitation; MAG, monoacylglycerol.
Discussion
HMGB1 participates in chronic liver disease,31 primarily due to its proinflammatory role; however, until now, the precise mechanism by which HMGB1 acts in NASH was unknown. Because HMGB1 increases in patients with NASH,32 it is highly expressed in IEC and intestinal immune cells, and the gut‐to‐liver axis plays a key role in metabolic disorders such as NASH, hence, we hypothesized that IEC‐derived HMGB1 could play a role in NASH due to local effects in the intestine that govern hepatic steatosis.
To investigate this, we generated a conditional null mouse of Hmgb1 in IEC, the cells responsible for the absorption of dietary nutrients and transport into the circulation, as well as for acting as a barrier to prevent the passage of dangerous pathogens or molecules to the liver. To induce NASH, we used a HFCFD, shown to depict key features of human NASH.23, 24, 25 In this study, we recapitulated the early and the well‐established NASH phenotype after 1 week and 24 weeks, respectively, of feeding mice this diet.
Overall, our data suggest that IEC‐derived HMGB1 is involved in the pathogenesis of NASH, primarily because of its local effects in the intestine that, as a consequence, lessen hepatosteatosis. First, we found that HMGB1 plays a role in the initiation and/or progression of NAFLD to NASH, as deletion of the gene from IEC was protective from HFCFD‐induced liver steatosis and inflammation in mice as early as 1 week. Second, we observed atypical LD accumulation in jejunal IEC from Hmgb1 ΔIEC mice. This phenotype has been previously observed in mice and in patients with defects in lipid absorption, packaging, and/or CM secretion.33, 34 In severe cases, this causes malnourishment, as nutrients are unable to reach vital organs35; however, in our study, this led to a modest decrease in the amount of lipids reaching the liver.
Lipidomics analysis revealed that numerous TGs and other lipid classes (i.e., PCs, lysoPCs, PEs, lysoPEs, plamanylPE, plasmenaylPEs, phosphatidylinositols, sphingomyelins, and sphingosine) were reduced in the serum of HFCFD‐fed Hmgb1 ΔIEC mice. Serum PCs are significantly increased in patients with NASH.36 Importantly, it has been described that treatments to reduce the PC‐to‐PE ratio in the liver protect against the development of hepatic insulin resistance.37 LysoPC is a death effector in the lipoapoptosis of hepatocytes.38 In NASH, the diacylglycerol (DAG) that is normally consumed to produce PEs by the CDP‐ethanolamine pathway is instead converted to TGs, promoting LD formation.39 Sphingomyelins produce DAG and have a role in cell apoptosis by hydrolyzing into ceramide.40 When lipid rafts are depleted of sphingomyelins, this leads to insulin sensitivity.41 Despite these findings that may have protected the livers from HFCFD‐fed Hmgb1 ΔIEC mice, we also found that acylcarnitine (18:1) and ceramide (17:1/18:1) were elevated. Although these mice presented less hepatosteatosis, mitochondrial function eventually failed with progression to NASH, leading to increased acylcarnitine.42 Likewise, disruption in phospholipid metabolism may have occurred as in human NASH.43
Our findings lead us to postulate that Hmgb1 deletion could cause a defect in lipid absorption, packaging, and/or CM release, leading to accumulation of lipids within IEC and, as a result, decreased levels in serum. HFCFD‐fed Hmgb1 ΔIEC mice presented increased SR‐B1 protein expression, which facilitated CHO uptake; however, they had inefficient transport of CHO or TG from the intestine into the serum compared with WT mice. ApoB48, responsible for the packaging of TG and CHO into CM, was significantly down‐regulated. This, in turn, reduced CM formation and/or release, lowering serum lipids.
These results are, to a certain degree, unexpected, as HMGB1 has a well‐established role in inflammation, the key reason for its involvement in other liver disorders as previously described by us,11, 12 which formed the basis for our preliminary hypothesis. Despite our initial prediction that HMGB1 could be involved in NASH development due to its modulation of liver and gut inflammation, we in fact found little change in portal blood HMGB1 and gut permeability between WT and Hmgb1 ΔIEC mice; hence, it was unlikely that intestinal HMGB1 was a key signal in the portal blood driving NASH. Indeed, it is possible that IEC‐derived HMGB1 was not secreted to the portal blood. The source of portal blood HMGB1 may have been local immune cells that increased its expression with disease progression or the presence of more immune cells. It appears that removal of IEC‐derived HMGB1 reduces hepatic lipid accumulation and inflammation in the early stages of NASH, neither per se nor due to increased translocation of dangerous products from the gut as anticipated, but primarily because of a decreased availability of lipids for storage. Accordingly, HMGB1 had a dominant local effect in the intestine. We also found that the protection conferred in Hmgb1 ΔIEC mice in the later stages of NASH had a negligible effect on inflammation, indicating that the observed fibrosis was driven mostly by lipid‐induced hepatocyte damage and/or ballooning degeneration rather than by inflammation. Likewise, Hmgb1 deletion was protective by modulating hepatic lipid content.
In addition, key metabolites were found to be increased (i.e., l‐lysine and taurine) or decreased (i.e., l‐arginine and phenylalanine) in HFCFD‐fed Hmgb1 ΔIEC mice. Lysine levels are essential to maintaining carnitine homeostasis, which is involved in long‐chain fatty acid oxidation.44 Taurine is a biosynthetic precursor to the bile salts sodium taurochenodeoxycholate and sodium taurocholate. Taurine reduces the secretion of apolipoprotein B100 and lipids in HepG2 cells,45 has a blood CHO‐lowering effect in young overweight adults,46 and acts as a glycation inhibitor.47 All of these may have contributed to the protective phenotype in HFCFD‐fed Hmgb1 ΔIEC mice.
Moreover, the primary bile acid taurocholic acid and the secondary bile acid glycocholic acid were increased in HFCFD‐fed Hmgb1 ΔIEC mice, which are involved in regulating CHO homeostasis. Therefore, these intermediates may participate in protecting the livers of Hmgb1 ΔIEC mice from the effects of the HFCFD. Although speculative, the lack of HMGB1 may have conveyed these changes to the secondary bile acid pool by changing the composition of the gut microbiota. Dysbiosis in itself can alter lipid metabolism by affecting the synthesis of short‐chain FAs,5, 48 the activity of lipoprotein lipase,49 gut permeability, and intestinal inflammation. Further investigation into the interaction of HMGB1 with the microbiome in metabolic disorders would be worth pursuing.
An interesting observation in this study is that GSSG levels were decreased and GSH elevated in IEC over time, suggesting that Hmgb1 ΔIEC mice may be protected from HFCFD‐induced oxidative stress, despite not observing much prevention of increased gut permeability in addition to displaying atypical LD accumulation. Indeed, it has been described that blockade of HMGB1 protects the intestine, where it not only relieved the oxidative stress, but also the gut barrier dysfunction and microbiome changes in a rodent model of acute necrotizing pancreatitis.50
In conclusion, our study suggests that ablation of Hmgb1 in IEC has a local effect in the intestine that results in up‐regulation of SR‐B1 and down‐regulation of ApoB48, leads to lipid accumulation in IEC, decreases CM formation and/or release, reduces serum TG, and lowers liver steatosis, thereby protecting Hmgb1 ΔIEC mice from HFCFD‐induced NASH. Further studies are needed to dissect how HMGB1 modulates SR‐B1 and ApoB48 protein expression and to identify potential targets for NASH treatment.
Supporting information
Acknowledgment
The authors thank Dr. Timothy R. Billiar (University of Pittsburgh, Pittsburgh, PA) for donating the Hmgb1fl/fl mice. They also thank all past and current members from the Nieto Laboratory for their helpful comments, support, and suggestions throughout the course of this project. Histology and fluorescent imaging services were provided by the Research Resources Center Histology Core (University of Illinois at Chicago).
Supported by the National Institute of Diabetes and Digestive and Kidney Diseases (R01 DK111677), and Chicago Biomedical Consortium.
Potential conflict of interest: Nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Compare D, Coccoli P, Rocco A, Nardone OM, De Maria S, Carteni M, et al. Gut–liver axis: the impact of gut microbiota on non alcoholic fatty liver disease. Nutr Metab Cardiovasc Dis 2012;22:471‐476. [DOI] [PubMed] [Google Scholar]
- 2. Vanni E, Bugianesi E. The gut‐liver axis in nonalcoholic fatty liver disease: another pathway to insulin resistance? Hepatology 2009;49:1790‐1792. [DOI] [PubMed] [Google Scholar]
- 3. Mouzaki M, Allard JP. The role of nutrients in the development, progression, and treatment of nonalcoholic fatty liver disease. J Clin Gastroenterol 2012;46:457‐467. [DOI] [PubMed] [Google Scholar]
- 4. Goodwin M, Herath C, Jia Z, Leung C, Coughlan MT, Forbes J, et al. Advanced glycation end products augment experimental hepatic fibrosis. J Gastroenterol Hepatol 2013;28:369‐376. [DOI] [PubMed] [Google Scholar]
- 5. Leung C, Rivera L, Furness JB, Angus PW. The role of the gut microbiota in NAFLD. Nat Rev Gastroenterol Hepatol 2016;13:412‐425. [DOI] [PubMed] [Google Scholar]
- 6. Rahman K, Desai C, Iyer SS, Thorn NE, Kumar P, Liu Y, et al. Loss of junctional adhesion molecule a promotes severe steatohepatitis in mice on a diet high in saturated fat, fructose, and cholesterol. Gastroenterology 2016;151:733‐746.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Federico A, Dallio M, Godos J, Loguercio C, Salomone F. Targeting gut‐liver axis for the treatment of nonalcoholic steatohepatitis: translational and clinical evidence. Transl Res 2016;167:116‐124. [DOI] [PubMed] [Google Scholar]
- 8. Li L, Chen L, Hu L, Liu Y, Sun HY, Tang J, et al. Nuclear factor high‐mobility group box1 mediating the activation of toll‐like receptor 4 signaling in hepatocytes in the early stage of nonalcoholic fatty liver disease in mice. Hepatology 2011;54:1620‐1630. [DOI] [PubMed] [Google Scholar]
- 9. Willingham SB, Allen IC, Bergstralh DT, Brickey WJ, Huang MT, Taxman DJ, et al. NLRP3 (NALP3, Cryopyrin) facilitates in vivo caspase‐1 activation, necrosis, and HMGB1 release via inflammasome‐dependent and ‐independent pathways. J Immunol 2009;183:2008‐2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Arriazu E, Ge X, Leung TM, Magdaleno F, Lopategi A, Lu Y, et al. Signalling via the osteopontin and high mobility group box‐1 axis drives the fibrogenic response to liver injury. Gut 2017;66:1123‐1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ge X, Antoine DJ, Lu Y, Arriazu E, Leung TM, Klepper AL, et al. High mobility group box‐1 (HMGB1) participates in the pathogenesis of alcoholic liver disease (ALD). J Biol Chem 2014;289:22672‐22691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ge X, Arriazu E, Magdaleno F, Antoine DJ, Dela Cruz R, Theise N, et al. High mobility group box‐1 drives fibrosis progression signaling via the receptor for advanced glycation end products in mice. Hepatology 2018;68:2380‐2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Masuda K, Ono A, Aikata H, Kawaoka T, Nelson Hayes C, Teraoka Y, et al. Serum HMGB1 concentrations at 4 weeks is a useful predictor of extreme poor prognosis for advanced hepatocellular carcinoma treated with sorafenib and hepatic arterial infusion chemotherapy. J Gastroenterol 2018;53:107‐118. [DOI] [PubMed] [Google Scholar]
- 14. Dong YD, Cui L, Peng CH, Cheng DF, Han BS, Huang F. Expression and clinical significance of HMGB1 in human liver cancer: knockdown inhibits tumor growth and metastasis in vitro and in vivo. Oncol Rep 2013;29:87‐94. [DOI] [PubMed] [Google Scholar]
- 15. Antoine DJ, Jenkins RE, Dear JW, Williams DP, McGill MR, Sharpe MR, et al. Molecular forms of HMGB1 and keratin‐18 as mechanistic biomarkers for mode of cell death and prognosis during clinical acetaminophen hepatotoxicity. J Hepatol 2012;56:1070‐1079. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16. Chen X, Ling Y, Wei Y, Tang J, Ren Y, Zhang B, et al. Dual regulation of HMGB1 by combined JNK1/2‐ATF2 axis with miR‐200 family in nonalcoholic steatohepatitis in mice. FASEB J 2018;32:2722‐2734. [DOI] [PubMed] [Google Scholar]
- 17. Yu M, Li JH, Yang LH, Obar R, Newman W, Mason J, et al. HMGB1 signals through toll‐like receptor 2. Shock 2004;21:14. [Google Scholar]
- 18. Chen GY, Tang J, Zheng P, Liu Y. CD24 and Siglec‐10 selectively repress tissue damage‐induced immune responses. Science 2009;323:1722‐1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Liu S, Stolz DB, Sappington PL, Macias CA, Killeen ME, Tenhunen JJ, et al. HMGB1 is secreted by immunostimulated enterocytes and contributes to cytomix‐induced hyperpermeability of Caco‐2 monolayers. Am J Physiol Cell Physiol 2006;290:C990‐C999. [DOI] [PubMed] [Google Scholar]
- 20. Chandrashekaran V, Seth RK, Dattaroy D, Alhasson F, Ziolenka J, Carson J, et al. HMGB1‐RAGE pathway drives peroxynitrite signaling‐induced IBD‐like inflammation in murine nonalcoholic fatty liver disease. Redox Biol 2017;13:8‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sappington PL, Yang R, Yang H, Tracey KJ, Delude RL, Fink MP. HMGB1 B box increases the permeability of Caco‐2 enterocytic monolayers and impairs intestinal barrier function in mice. Gastroenterology 2002;123:790‐802. [DOI] [PubMed] [Google Scholar]
- 22. Yang R, Miki K, Oksala N, Nakao A, Lindgren L, Killeen ME, et al. Bile high‐mobility group box 1 contributes to gut barrier dysfunction in experimental endotoxemia. Am J Physiol Regul Integr Comp Physiol 2009;297:R362‐R369. [DOI] [PubMed] [Google Scholar]
- 23. Sanches SC, Ramalho LN, Augusto MJ, da Silva DM, Ramalho FS. Nonalcoholic steatohepatitis: a search for factual animal models. Biomed Res Int 2015;2015:574832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lau JK, Zhang X, Yu J. Animal models of non‐alcoholic fatty liver disease: current perspectives and recent advances. J Pathol 2017;241:36‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ibrahim SH, Hirsova P, Malhi H, Gores GJ. Animal models of nonalcoholic steatohepatitis: eat, delete, and inflame. Dig Dis Sci 2016;61:1325‐1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Huang H, Nace GW, McDonald KA, Tai S, Klune JR, Rosborough BR, et al. Hepatocyte‐specific high‐mobility group box 1 deletion worsens the injury in liver ischemia/reperfusion: a role for intracellular high‐mobility group box 1 in cellular protection. Hepatology 2014;59:1984‐1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Neuschwander‐Tetri BA. Handbook of Liver Disease. Third edition Amsterdam, Netherlands: Elsevier; 2012. [Google Scholar]
- 28. Booth CC, Read AE, Jones E. Studies on the site of fat absorption. 1: The sites of absorption of increasing doses of I‐labelled triolein in the rat. Gut 1961;2:23‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hesse D, Jaschke A, Chung B, Schurmann A. Trans‐golgi proteins participate in the control of lipid droplet and chylomicron formation. Biosci Rep 2013;33:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Levy E. Insights from human congenital disorders of intestinal lipid metabolism. J Lipid Res 2015;56:945‐962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gaskell H, Ge X, Nieto N. High‐mobility group box‐1 and liver disease. Hepatol Commun 2018;2:1005‐1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yates KP, Deppe R, Comerford M, Masuoka H, Cummings OW, Tonascia J, et al. Serum high mobility group box 1 protein levels are not associated with either histological severity or treatment response in children and adults with nonalcoholic fatty liver disease. PLoS One 2017;12:e0185813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang Y, Iordanov H, Swietlicki EA, Wang L, Fritsch C, Coleman T, et al. Targeted intestinal overexpression of the immediate early gene tis7 in transgenic mice increases triglyceride absorption and adiposity. J Biol Chem 2005;280:34764‐34775. [DOI] [PubMed] [Google Scholar]
- 34. Nakano T, Inoue I, Koyama I, Kanazawa K, Nakamura K, Narisawa S, et al. Disruption of the murine intestinal alkaline phosphatase gene Akp3 impairs lipid transcytosis and induces visceral fat accumulation and hepatic steatosis. Am J Physiol Gastrointest Liver Physiol 2007;292:G1439‐G1449. [DOI] [PubMed] [Google Scholar]
- 35. Shoulders CC, Brett DJ, Bayliss JD, Narcisi TM, Jarmuz A, Grantham TT, et al. Abetalipoproteinemia is caused by defects of the gene encoding the 97 kDa subunit of a microsomal triglyceride transfer protein. Hum Mol Genet 1993;2:2109‐2116. [DOI] [PubMed] [Google Scholar]
- 36. Tiwari‐Heckler S, Gan‐Schreier H, Stremmel W, Chamulitrat W, Pathil A. Circulating phospholipid patterns in NAFLD patients associated with a combination of metabolic risk factors. Nutrients 2018;10:649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. van der Veen JN, Lingrell S, McCloskey N, LeBlond ND, Galleguillos D, Zhao YY, et al. A role for phosphatidylcholine and phosphatidylethanolamine in hepatic insulin signaling. FASEB J 2019;33:5045‐5057. [DOI] [PubMed] [Google Scholar]
- 38. Han MS, Park SY, Shinzawa K, Kim S, Chung KW, Lee JH, et al. Lysophosphatidylcholine as a death effector in the lipoapoptosis of hepatocytes. J Lipid Res 2008;49:84‐97. [DOI] [PubMed] [Google Scholar]
- 39. Fullerton MD, Hakimuddin F, Bonen A, Bakovic M. The development of a metabolic disease phenotype in CTP:phosphoethanolamine cytidylyltransferase‐deficient mice. J Biol Chem 2009;284:25704‐25713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Green DR. Apoptosis and sphingomyelin hydrolysis. The flip side. J Cell Biol 2000;150:F5‐F7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gaillet RP. Open question to those responsible for the measles‐mumps‐rubella vaccination program. Rev Med Suisse Romande 1988;108:711‐716. [PubMed] [Google Scholar]
- 42. Peng KY, Watt MJ, Rensen S, Greve JW, Huynh K, Jayawardana KS, et al. Mitochondrial dysfunction‐related lipid changes occur in nonalcoholic fatty liver disease progression. J Lipid Res 2018;59:1977‐1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ma DW, Arendt BM, Hillyer LM, Fung SK, McGilvray I, Guindi M, et al. Plasma phospholipids and fatty acid composition differ between liver biopsy‐proven nonalcoholic fatty liver disease and healthy subjects. Nutr Diabetes 2016;6:e220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hoppel C. The role of carnitine in normal and altered fatty acid metabolism. Am J Kidney Dis 2003;41:S4‐S12. [DOI] [PubMed] [Google Scholar]
- 45. Yanagita T, Han SY, Hu Y, Nagao K, Kitajima H, Murakami S. Taurine reduces the secretion of apolipoprotein B100 and lipids in HepG2 cells. Lipids Health Dis 2008;7:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhang M, Bi LF, Fang JH, Su XL, Da GL, Kuwamori T, et al. Beneficial effects of taurine on serum lipids in overweight or obese non‐diabetic subjects. Amino Acids 2004;26:267‐271. [DOI] [PubMed] [Google Scholar]
- 47. Huang JS, Chuang LY, Guh JY, Yang YL, Hsu MS. Effect of taurine on advanced glycation end products‐induced hypertrophy in renal tubular epithelial cells. Toxicol Appl Pharmacol 2008;233:220‐226. [DOI] [PubMed] [Google Scholar]
- 48. Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity‐associated gut microbiome with increased capacity for energy harvest. Nature 2006;444:1027‐1031. [DOI] [PubMed] [Google Scholar]
- 49. Marra F, Svegliati‐Baroni G. Lipotoxicity and the gut‐liver axis in NASH pathogenesis. J Hepatol 2018;68:280‐295. [DOI] [PubMed] [Google Scholar]
- 50. Huang L, Zhang D, Han W, Guo C. High‐mobility group box‐1 inhibition stabilizes intestinal permeability through tight junctions in experimental acute necrotizing pancreatitis. Inflamm Res 2019;68:677‐689. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials