

Abstract
Fuchs Endothelial Corneal Dystrophy (FECD) is an age-related genetically complex disease characterized by increased oxidative DNA damage and progressive degeneration of corneal endothelial cells (HCEnCs). FECD has a greater incidence and advanced phenotype in women, suggesting a possible role of hormones in the sex-driven differences seen in the disease pathogenesis. In this study, catechol estrogen (4-OHE2), the byproduct of estrogen metabolism, induced genotoxic estrogen-DNA adducts formation, macromolecular DNA damage, and apoptotic cell death in HCEnCs; these findings were potentiated by menadione (MN)-mediated reactive oxygen species (ROS). Expression of NQO1, a key enzyme that neutralizes reactive estrogen metabolites, was downregulated in FECD, indicating HCEnC susceptibility to reactive estrogen metabolism in FECD. NQO1 deficiency in vitro exacerbated the estrogen-DNA adduct formation and loss of cell viability, which was rescued by the supplementation of N-acetylcysteine, a ROS scavenger. Notably, overexpression of NQO1 in HCEnCs treated with MN and 4-OHE2 quenched the ROS formation, thereby reducing the DNA damage and endothelial cell loss. This study signifies a pivotal role for NQO1 in mitigating the macromolecular oxidative DNA damage arising from the interplay between intracellular ROS and impaired endogenous estrogen metabolism in post-mitotic ocular tissue cells. A dysfunctional Nrf2-NQO1 axis in FECD renders HCEnCs susceptible to catechol estrogens and estrogen-DNA adducts formation. This novel study highlights the potential role of NQO1-mediated estrogen metabolite genotoxicity in explaining the higher incidence of FECD in females.
Keywords: Corneal endothelium, NQO1, catechol estrogen, estrogen-DNA adducts, Menadione, Fuchs endothelial corneal dystrophy
Graphical Abstract
1. Introduction
Corneal endothelium plays a central role in the maintenance of corneal transparency and forms a monolayer of regular hexagonal cells in the innermost layer of the cornea lacking proliferative capacity [1–3]. Fuchs Endothelial Corneal Dystrophy (FECD) is the most common cause of endogenous corneal endothelial degeneration and is a late-onset oxidative stress disorder characterized by gradual loss of human corneal endothelial cells (HCEnCs). Hallmark features of FECD pathogenesis include morphological alterations of CEnCs due to loss of junctional contacts, endothelial cellular apoptosis, [4, 5] and concomitant deposition of extracellular matrix in the form of mound-shaped excrescences known as guttae [6]. FECD is estimated to affect 4% of the U.S. population above 40 years of age, majorly females, and currently corneal transplantation is the only treatment available, making it the leading cause accounting for 36% of 46,900 corneal transplants carried out in 2016 [7]. Lack of pharmacological treatments for FECD heightens the need for studies driven towards understanding the molecular mechanisms and sex-driven differences in the disease pathogenesis.
FECD is an age-related disorder with higher incidence in women compared to men (3-4:1 ratio) [8–12]. Various reports also show that a higher percentage of women (9-11%) develops central corneal guttae, relative to men (3.5-7%) [9, 13]. In addition to age, female sex is considered a significant risk factor for advanced FECD development [14]. A study comprised of 1241 subjects showed that a higher percentage of women developed more advanced FECD compared to men [14]. Similarly, it has been reported that smoking and diabetes have a more pronounced effect on development of FECD in female compared to male cohorts [14]. Furthermore, women comprised 77% of patients undergoing corneal transplantation in the observational case series spanning 30 years, pointing to the predominance of advanced FECD in women [12, 15]. Nevertheless, there is a lack of knowledge of why women are drastically more affected by FECD than men.
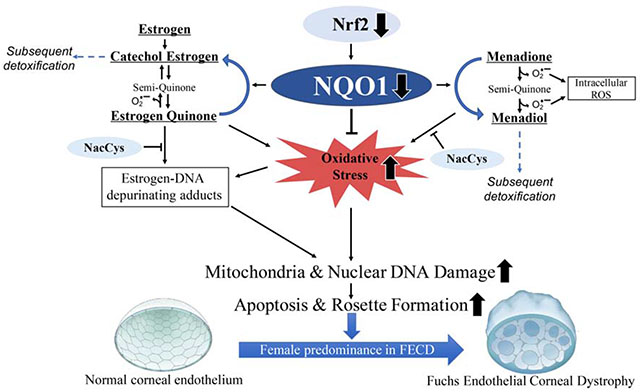
FECD is a genetically heterogenous disease and intronic trinucleotide CTG repeat expansion in TCF4 gene (> 40 CTG repeats) is the major genetic marker significantly associated with FECD [16]. Regardless of the genetic mutation, susceptibility to oxidative stress is key to FECD pathogenesis. Post-mitotically arrested CEnCs are rich in mitochondria and are metabolically active owing to their pump and barrier function, and hence are highly prone to oxidative stress and the ensuing molecular damage. Previous studies, including from our laboratory, have demonstrated that acquired oxidative DNA damage [17–19] and pro-oxidant milieu are characteristic features of FECD. Reduced expression of peroxiredoxins and Nrf2 (nuclear factor erythroid 2-related factor 2), a key redox-sensitive transcription factor that activates antioxidant defense-related genes, in FECD ex vivo specimens account for the oxidant-antioxidant imbalance in FECD [20–24]. NQO1 (NAD(P)H:quinone oxidoreductase 1; DT-Diaphorase) is a highly inducible and cytoprotective flavoprotein that catalyzes the reduction of endogenous and exogenous quinones, quinoneimines and nitroaromatic compounds, thereby limiting the formation of ROS and free radicals in the cellular environment [25]. Nrf2 transcription factor activates NQO1 expression by binding to antioxidant response element (ARE) sequence in the NQO1 upstream promoter region [23, 26, 27]. We have established an in vitro FECD model by inducing endogenous oxidative stress with menadione (MN), forming pathognomonic rosettes that resemble the acellular centers around guttae observed during CEnC apoptosis [23]. Menadione is an exogenous quinone that generates intracellular mitochondrial superoxide and an unstable semiquinone radical, thereby increasing intracellular ROS [17]. NQO1 is involved in MN detoxification into a stable quinone - menadiol, thereby making MN a useful pro-oxidant substrate to obtain deeper understanding of the role of this enzyme in the oxidant-antioxidant imbalance as seen in FECD (Fig. 1A) [17].
Fig. 1.

Decreased protein expression of NQO1 and COMT in FECD specimens and increased DNA damage and cytotoxicity induced by catechol estrogens A. Schematic diagram of estrogen metabolism pathway with corresponding enzymes and NQO1-mediated MN detoxification. B, C. Representative western blots of NQO1 and COMT (s-COMT) in normal (n=11 for NQO1 and n=6 for COMT) and FECD (n=21 for NQO1, n=16 for COMT) specimens. ß-actin is used as normalizing control. Two bands in COMT blot represent membrane (M) and soluble (S) forms. Corresponding densitometry of each enzyme levels in normal (black) and FECD (grey) specimens is indicated below as bar graphs. * p <0.05, ** p<0.01, two-tailed Mann-Whitney U test. D. Western blot analysis of baseline NQO1 levels in HCEnC-SV-67F-16, FECD-SV-73F-74 and FECD-SV-61F-18 cell lines. E. Dose–response analysis of cellular viability of 4-OHE2-treated HCEnC-SV-67F-16, FECD-SV-73F-74 and -61F-18 cells using the Cell-Titer Glo assay. F. Cellular morphology of HCEnC-SV-67F-16, FECD-SV-73F-74 and -61F-18 cells treated with 30 μM 4-OHE2 for 24 h by phase contrast microscopy. G. LA-qPCR analysis of mtDNA amplification and lesions in HCEnC-SV-67F-16 (white bar), FECD-SV-73F-74 (black) and FECD-SV-61F-18 (grey) cells treated with 30 μM 4-OHE2 for indicated time points. Y-axis denotes the ratio of intensity of large mitochondrial fragment divided by short mitochondrial fragment amplification. The short mitochondrial fragment serves as copy number control. n=3, data are mean ± SE, * indicates P < 0.05 by two-way ANOVA compared to untreated sample. H. Amplification of a nuclear encoded ß-globin gene by LA-qPCR in 30 μM 4-OHE2 treated HCEnC-SV-67F-16, FECD-SV-73F-74 and -61F-18 cells. Dotted line indicate the untreated samples to which the treated samples are normalized. Error bars represent SEM. Data expressed as mean values ± SE. (*p < 0.05, **p<0.01, Student’s t-test and Two-way ANOVA with Tukey post tests for comparison of multiple data sets).
Endogenous estrogen quinones are also known to be NQO1 substrates, wherein NQO1 catalyzes the reduction of estrogen quinones (E2-3,4,Q) by converting them back to catechol estrogen (4-hydroxyestradiol; 4-OHE2) [28], thereby diverting the quinones from binding to DNA and generating depurinating estrogen-DNA adducts. Further, catechol estrogens are converted to less reactive and non-toxic methoxy estrogen conjugates (4-OCH3E2) by a deactivating enzyme, catechol—O-methyl transferase (COMT) (Fig. 1A) [29]. Imbalance in the endogenous estrogen metabolism and redox cycling between estrogen metabolites resulting in accumulation of depurinating DNA adducts (Fig. 1A) is widely implicated in breast and prostate cancers and lymphoma [30].
The effect of impaired estrogen metabolism in FECD pathogenesis is poorly understood. In the current work, we have examined the effect of catechol estrogens on HCEnCs and the role of NQO1 in mitigating the DNA damage mediated by intracellular reactive oxygen species (ROS) and estrogen metabolites in FECD pathogenesis. Combination of ROS and catechol estrogen induced the accumulation of depurinating estrogen-DNA adducts in HCEnCs. Loss of NQO1 further triggered the accumulation of estrogen-DNA adducts and increased the propensity for cell loss, which was reversed by the addition of ROS scavenger N-acetylcysteine (NAC). We report a significant loss of NQO1 in FECD patient specimens and this decrease was further corroborated in FECD patient-derived cell lines exhibiting oxidative DNA damage. Overexpression of NQO1 significantly decreased the ROS levels and DNA damage induced by MN and 4-OHE2 stressors, highlighting the cytoprotective role of NQO1. This novel study provides mechanistic insights into the role of oxidative stress and estrogen genotoxicity mediated by NQO1 in FECD pathogenesis. Collectively, these findings indicate that NQO1, a key component of cellular antioxidant defense, is a potential therapeutic target for FECD.
2. Materials and methods
2.1. Human tissue
This study was conducted according to the Declaration of Helsinki and approved by the Massachusetts Eye and Ear Institutional Review Board. Written and informed consent was obtained from patients undergoing surgical treatment for FECD. Corneal endothelial tissue from FECD patients (n=22, age range 47-83 yrs) was isolated by endothelial keratoplasty (EK) performed at Price Vision Group (Indianapolis, IN), and Massachusetts Eye and Ear Infirmary (Boston, MA). After surgical removal, the tissue was immediately placed in the storage medium (Optisol-GS; Bausch & Lomb) at 4°C. Normal donor corneas (n=11, age range 32–75 yrs) were purchased from Northeast Pennsylvania Lions Eye Bank (Bethlehem, PA), Eversight (Ann Arbor, MI) and Lions VisionGift (Portland, OR) according to the criteria previously reported [24, 31]. These criteria include utilizing corneas with CEnC counts ≥1500 cells/mm2. Corneas were not accepted if death-to-preservation period was >24 h, there was presence of corneal guttae or any other endothelial abnormality seen on the specular biomicroscopy. Both normal and FECD corneas were stored in Optisol-GS after procurement to rule out any effects of storage conditions on protein expression.
2.2. Cell culture
HCEnC-21T cells were generated by immortalization using retrovirus transfection containing pBABE-puro-hTERT [32]. NQO1+/+ and NQO1−/− cells were generated from HCEnC-21T cells using the CRISPR-Cas9 system as previously reported [33]. HCEnC-SV-67F-16 (67 yr old female donor with 15/16 CTG repeats), FECD-SV-73F-74 (73 year old female FECD patient with 16/74 CTG repeats) and FECD-SV-61F-18 (61 year old female FECD patient with 18/18 CTG repeats) cell lines were derived by immortalization using SV40 T Antigen Cell Immortalization Kit (Alstem Cell Advancements, Richmond, CA) as per the methodology described previously [34]. Cells were cultured in Chen’s medium [35] containing OptiMEM-I; Thermo Fisher Scientific, Waltham, MA), 8% fetal bovine serum (HyClone, Rockford, IL), 5 ng/mL epidermal growth factor (EMD Millipore, Billerica, MA), 100 mg/mL bovine pituitary extract (Thermo Fisher Scientific), 200 mg/L calcium chloride (Sigma-Aldrich, St. Louis, MO), 0.08% chondroitin sulfate (Sigma-Aldrich), 50 mg/mL gentamicin (Thermo Fisher Scientific), and 1:100 diluted antibiotic/antimycotic solution (Sigma-Aldrich). Sub-culturing of HCEnCs was performed using 0.05% trypsin (Thermo Fisher Scientific) for 5 min at 37°C.
For treatments with 4-OHE2, HCEnCs were seeded in estrogen-free medium (phenol red-free OptiMEM with charcoal stripped FBS, 200 mg/L calcium chloride, 0.08% chondroitin sulfate) and treated with the desired concentration of MN and 4-OHE2. For cell viability assays, HCEnCs were treated with 50 μM MN and 10 μM 4-OHE2 in estrogen free medium for 24 h, while for western blotting, cells were harvested at 8 h. For estrogen metabolite analysis, the medium was changed to estrogen-free medium and incubated for 48 h, and treated either with or without 8.5 μM MN (Sigma-Aldrich), 3.75 μM 4-OHE2 (Steraloids, Newport, RI), or 25 μM NAC (Sigma-Aldrich) for 24 h. Following the treatments, the media were harvested, supplemented with 2 mM ascorbic acid (Sigma-Aldrich) to prevent possible decomposition of the compounds and processed for estrogen metabolite analysis [36]. Media from untreated HCEnC-21T and NQO1+/+ cells were used as controls.
2.3. Western blot analysis
Whole cell extracts of tissue specimens were prepared by lysis using the ReadyPrep Sequential Extraction Kit Reagent 3 and 1 mM tributyl phosphine (Biorad, Hercules, CA). Proteins were loaded onto 10% Bis-Tris NuPAGE gels (Thermo Fisher Scientific). Peptides were transferred to a polyvinylidene difluoride membrane (EMD Millipore) and non-specific binding sites were blocked with 5% non-fat dry milk in Tris Buffered Saline plus 0.1% Tween 20 (TBS-T) for 1 h. Membranes were incubated overnight at 4°C with anti-COMT (AB5873-I, EMD Millipore) diluted 1:1000 and goat polyclonal anti-NQO1 (ab2346, Abcam, Cambridge, MA) diluted 1:1000. Mouse anti-β-actin (Sigma-Aldrich) diluted 1:4000 was used to normalize protein loading. Blots were rinsed with TBS-T, and exposed for 1 h to horseradish peroxidase—conjugated goat anti-rabbit IgG at 1:1000 for COMT blots,HRP-conjugated donkey anti-goat IgG at 1:1000 for NQO1 blots and HRP-conjugated donkey anti-mouse IgG at 1:4000 for β-actin blots, in blocking buffer. After washing with TBS-T, antibody binding was detected with SuperSignal west pico plus chemiluminescent substrate (Thermo Fisher Scientific). Densitometry was analyzed with ImageJ software (developed by Wayne Rasband, National Institutes of Health, Bethesda, MD), and protein content was normalized relative to β-actin content. Experiments were repeated a minimum of three times. Results were averaged and SEM values were calculated. 95% confidence interval of the difference in means of two populations is indicated.
2.4. Transfection of NQO1 plasmid to CEnCs
HCEnC-21T cells were seeded in 12-well plate and transiently transfected with myc-tagged NQO1 plasmid (pCMV6-NQO1-myc, Origene, Rockville, MD) and control plasmid (pCMV6-myc) at 1.5 μg/well with 3 μL Lipofectamine 2000 (Thermo Fisher Scientific) in OptiMEM. 24 h post-transfection, cells were treated with either 10 μM MN, 20 μM 4-OHE2 or in combination for 8 h. Cells were collected and lysed in RIPA buffer supplemented with reducing agent and HALT protease/inhibitor cocktail (Thermo Fisher Scientific). Protein samples were loaded onto 10% Bis-Tris NuPAGE gels, transferred and blotted with NQO1 antibody (Abcam, ab2346) to detect both NQO1-myc and endogenous NQO1.
2.5. Detection of DNA damage using long-extension QPCR from HCEnCs
Long amplicon-qPCR (LA-qPCR) analysis for mtDNA and nDNA was performed as previously described [37]. For LA-qPCR analysis, HCEnCs were plated in estrogen-free medium for 24 h before incubation with 20-30 μM 4-OHE2 and 10 μM MN in phenol red-free OptiMEM for varying time points (3, 6, 12 and 24 h). Total genomic DNA was isolated treated endothelial cell lines using Genomic tip-20/G and QIAamp DNA mini kit (Qiagen, Hilden, Germany). The following primers were used for amplifying human mtDNA and nDNA: Mitochondrial large fragment (8.9 kb): FP: 5999 5’-TCT AAG CCT CCT TAT TCG AGC CGA-3’; RP: 14841 5’-TTT CAT CAT GCG GAG ATG TTG GAT GG-3’; Mitochondrial short fragment (221 bp): FP: 14620 5’-CCC CAC AAA CCC CAT TAC TAA ACC CA -3’; RP: 14841 5’-TTT CAT CAT GCG GAG ATG TTG GAT GG-3’. Nuclear β-globin gene (13.5 kb): FP: 48510 5’-CGA GTA AGA GAC CAT TGT GGC AG-3’; RP: 62007 5’-GCA CTG GCT TAG GAG TTG GAC T-3’. The thermocycler (Biorad T100; Biorad) program included an initial denaturation at 94°C for 1 min, 26 cycles for β-globin (16 and 14 cycles for large and small mitochondrial target) at 94°C for 15 s, annealing at 64°C for 25-30 s, and extension at 68°C for 15 min for β-globin (9 min for large mitochondrial target and 30 s for small mitochondrial target). DNA lesion frequencies were calculated as described [17]. Two-way analysis of variance (ANOVA) was utilized for statistical analysis, with P values corresponding to <0.05 (*) as significant.
2.6. Cellular viability and morphology
Cell numbers were measured using an automatic cell counter (Countess; Thermo Fisher Scientific) and trypan blue dye exclusion. Phase-contrast microscopy (Leica DM IL LED) was employed to visualize cell morphology. Cell viability was determined by the Cell Titer Glo reagent (Promega, Madison, WI) according to the manufacturer’s protocol. The luminescence was determined by a luminometer (Turner Biosystems, Sunnyvale, CA).
2.7. Flow cytometry
HCEnC-21T cells were seeded in a 6-well plate and cultured for 24 h. Cells were treated with/without MN (50 μM) or 4-OHE2 (10 μM), and incubated for 16 h in estrogen-free medium. Cells were then trypsinized, washed in cold PBS, and incubated in the dark with 3 μL of Annexin-V/FITC (Ann) (Thermo Fisher Scientific) and 2 μL of propidium iodide (PI, Sigma-Aldrich) in a total volume of 50 μL for 30 min at room temperature. Cells were re-suspended in 2 mL of Isoflow sheath fluid (Beckman Coulter, Fullerton, CA) and 20,000 live cells were acquired on a LSR II (BD Biosciences) with DIVA software (BD Biosciences) gated for PI and Annexin V with unstained cells as a negative control. Data were then analyzed with Summit 4.3 (Beckman Coulter, Fullerton, CA).
2.8. Determination of mitochondrial membrane potential (ΔΨm)
Mitochondrial membrane potential was determined as described in MitoProbeTM JC-1 Assay Kit (Cat # M34152, Thermo Fisher Scientific). Briefly, approximately 1 X 106 cells/mL were suspended in 1 mL warm Chen’s media, and further incubated with 1 μL of 50 mM CCCP for 5 min at 37°C, followed by addition of 2 μM JC-1 stain with an incubation time of 20-30 min at 37°C. Cells were washed with 500 μL PBS followed by analysis on BD LSR II flow cytometer with 488 excitation using emission filters appropriate for Alexa Fluor 488 dye and R-phycoerythrin. The live gate was applied to FITC histogram according to gate P2 and the percentage of cells was analyzed in this gate in control and CCCP groups for NQO1+/+ and NQO1−/− cells.
2.9. Detection of intracellular ROS production
ROS production was measured using DCFDA cellular ROS detection assay kit (Abcam, Cambridge MA) according to the manufacturer manual. Post-treatment in 96-well plate, HCEnCs were stained with 25 μM 5-(and-6)-carboxy-2’, 7’-dichlorodihydrofluorescein diacetate (carboxy-H2DCFDA) for 45 min at 37°C. DCFDA solution was then removed and the fluorescence (Ex/Em=485/535 nm) was immediately analyzed by using a fluorescence microplate reader (BioTek-Synergy 2; BioTek Instruments, Inc., Winooski, VT).
2.10. Estrogen Metabolite Analysis
The cell culture medium (50 mL) was adjusted to pH 7.5 and passed drop-wise through pre-conditioned Varian Certify II cartridges. Once all media passed, the cartridge was washed with 0.3 mL of conditioning buffer, 0.1 M potassium phosphate buffer (pH 7.5). Finally, the analytes were eluted with 1 mL of elution buffer, comprised of methanol: acetonitrile: water: trifluoroacetic acid (8:1:1:0.1), evaporated to 200 μL and then lyophilized. The residue was re-suspended in 70 μL of 50% methanol in water and filtered through a 5000-MW cut-off filter (Millipore, Bedford, MA) before analysis by ultraperformance liquid chromatography/tandem mass spectrometry (UPLC-MS/MS).
All of the samples were analyzed on a Waters Acquity UPLC equipped with a MicroMass QuattroMicro triple stage quadrupole mass spectrometer (Waters, Milford, MA). The 10-μL injections were carried out on a Waters Acquity UPLC BEHC18 column (1.7 μm, 10 × 100 mm). The instrument was operated in positive electrospray ionization mode. All aspects of system operation, data acquisition and processing were controlled using QuanLynx v4.2 software (Waters). The column was eluted starting with 20% acetonitrile in water (0.1% formic acid) for 4 min at a flow rate of 150 μL/min, then to 55% acetonitrile in 10 min. Ionization was achieved using the following settings: capillary voltage 3 kV; cone voltage 15-40 V; source block temperature 120°C; desolvation temperature 200°C, with a nitrogen flow of 700 L/h. Five-point calibration curves were run for each standard and triplicate samples were analyzed for each data point as described previously [38].
3. Results
3.1. Reduced expression of deactivating estrogen metabolic enzymes NQO1 and COMT in FECD specimens
The catechol estrogen (4-OHE2) undergoes oxidative metabolism to form E2-3,4-quinones (E2-3,4-Q) that readily react with DNA producing genotoxic adducts. The highly reactive estrogen quinones are detoxified by various mechanisms including NQO1-mediated conversion of E2-3,4-Q back to 4-OHE2 which are further methylated by COMT or undergo glutathione-mediated conjugation making them less reactive (Fig. 1A, schematic). Supporting the decreased Nrf2 levels in FECD [23], we noted a 6.2-fold decrease in its downstream target NQO1 in human FECD specimens as compared to normal samples (p <0.01, 95% confidence interval (CI) of the difference between the means [0.9, 2.85], Fig. 1B). Additionally, we also observed a 2.8-fold reduction in S-COMT (soluble form) protein levels in FECD specimens (p <0.05, 95% CI [0.13, 0.58], Fig. 1C). COMT exists in two distinct forms in the cell: soluble protein in the cytoplasm (S-COMT) and bound to membranes (M-COMT) and the soluble form is known to account for 90% of the enzyme activity [39]. These observations suggest that downregulation of detoxifying enzymes in the estrogen metabolism pathway in FECD specimens can potentially drive the pathway towards the generation of depurinating estrogen-DNA adducts.
3.2. Reduced NQO1 expression in FECD patient-derived immortalized cell lines
Next, we assessed the NQO1 levels in immortalized CEnC lines derived from normal and FECD ex vivo specimens previously generated in our laboratory: HCEnC-SV-67F-16, FECD-SV-73F-74 (CTG repeats > 40) and FECD-SV-61F-18 (CTG repeats < 40) [34]. Western blot analysis of these cell lines revealed a significant reduction of NQO1 in FECD cell lines. Interestingly, the extent of reduction was variable in the two FECD cell lines. NQO1 protein was nearly absent in FECD-SV-61F-18 cells lacking TCF4 repeat expansion, while was reduced to a lesser extent (3-fold) (p<0.05, 95% CI [0.45, 1.68]) in FECD-SV-73F-74 cells (Fig. 1D). Overall, NQO1 is downregulated in vitro in FECD cell lines, supporting the findings noted in ex vivo specimens.
3.3. Catechol estrogen induces cytotoxicity and DNA damage in FECD cell lines
In order to investigate the effect of catechol estrogen metabolites in vitro, we assessed the dose-response effect of 4-OHE2 on the cell viability of the above described cell lines. Cells were treated with varying concentrations of 4-OHE2 for 24 h. Treatment with 4-OHE2 elicited dose-dependent cytotoxicity in both normal and FECD cells (Fig. 1E). Significant differential cytotoxicity was noted between HCEnC and FECD cell lines starting from 30 μM 4-OHE2, which was exacerbated at higher doses (Fig. 1E, black and grey bars). At the higher doses (40 and 50 μM), FECD cells were even more susceptible (41.2 ± 5.7 and 26.1 ± 3 % in FECD-SV-73F-74; 18.7 ± 3.3 and 8.7 ± 1.7 % in FECD-SV-61F-18) to 4-OHE2 than normal cells (63.4 ± 2.1 and 46.3 ± 4 %), (p < 0.01 respectively, Fig. 1E). Interestingly, FECD cell line SV-61F-18 lacking NQO1 (Fig. 1D) was more susceptible compared to SV-73F-74 (Fig. 1D). The cytotoxicity in the cell lines upon 4-OHE2 treatment was supported by cellular morphology changes including a reduction in cell size and disruption of cellular junctions indicative of apoptosis at the higher doses (≥ 30 μM) (Fig. 1F) as determined by phase contrast microscopy.
Catechol estrogen 4-OHE2 is known to induce single strand DNA (ssDNA) breaks in hamster kidney and hydroxyl radical-mediated 8-hydroxylation of guanine bases of DNA (8-OHdG) in vitro and in hamsters in vivo [40]. Accumulation of nuclear (nDNA) and mitochondrial DNA (mtDNA) damage is a hallmark feature of FECD pathogenesis [17]. We next assessed the 4-OHE2-mediated nDNA and mtDNA damage by LA-qPCR [41]. As templates for PCR, we used total genomic DNA isolated from normal and two FECD cell lines that showed differential cytotoxicity with 30 μM 4-OHE2 and non-treated cells at each time-point served as corresponding controls. The relative amplification of small mtDNA fragment was employed as an estimate of mtDNA copy number (Supplemental Fig. 1A). Interestingly, mtDNA damage occurred as early as 6 h post treatment, which was sustained after 12 and 24 h in FECD-61F-18 cells, while significant mtDNA damage in FECD-SV-73F-74 cells was noted much later at 24 h (Fig. 1G, black and grey bars). However, 4-OHE2-induced nDNA damage in the FECD cell lines occurred only at the later time point of 24 h, where the reduction in amplification of ß-globin (by 31 and 57%, 0.3 and 0.49 lesions/10kb) was seen in both FECD cell lines but not in HCEnC-SV cells (p < 0.05, Fig. 1H). Taken together, mtDNA of FECD cell lines was more susceptible to 4-OHE2-induced DNA damage than nDNA.
3.4. Menadione downregulates NQO1 protein levels in HCEnC-21T cells
We have previously established an in vitro oxidative stress-induced FECD model using the pro-oxidant MN to investigate the pathogenesis of FECD in HCEnCs [17]. As FECD is an oxidative stress-induced disease that results in progressive loss of HCEnCs due to apoptosis, we investigated the interplay between MN-induced intracellular oxidative stress and estrogen metabolites in HCEnC-21T cells, an immortalized normal CEnC line generated in our laboratory [32]. We previously showed that MN-induced DNA damage (25 and 50 μM MN) in HCEnCs leads to rosette formation, corresponding to findings in ex vivo FECD specimens [17]. Cellular morphology analysis revealed rosette-like clusters with clear acellular centers (white asterisks) and spindle-like cells (black arrowheads) surrounding them with MN alone and co-treatment with MN and 4-OHE2 in CEnC hexagonal monolayers (Fig. 2A). Menadione is a known substrate of NQO1 and is catalytically reduced to menadiol by 2-electron transfer [25]. To study the effect of MN-induced oxidative stress on detoxifying estrogen metabolic enzymes such as NQO1 and COMT, HCEnC-21T cells were treated with increasing concentrations of MN. The cells showed a dose-dependent decrease in NQO1 protein levels with 2.3-fold decrease with 35 μM MN (p< 0.05, 95% CI [0.25, 0.88], Fig. 2B) and a 5.3 fold decrease with 50 μM MN (p < 0.001, 95% CI [0.78, 0.84], Fig. 2B). Treatment with 10 μM 4-OHE2 alone did not alter the NQO1 levels (Fig. 2C). Cotreatment with 10 μM 4-OHE2 and 50 μM MN decreased NQO1 levels (2.1-fold; p < 0.05, 95% CI [−0.4, 1.23], Fig. 2C) although not significantly different from only MN treatment. However, MN and 4-OHE2 treatments both individually or in combination did not cause significant difference in COMT levels compared to non-treated cells. Overall, oxidative stress induced by MN, modeling the changes seen in FECD, decreases NQO1 protein levels corresponding to its lower expression in FECD tissues relative to normal specimens.
Fig. 2.

Cotreatment with MN and 4-OHE2 decreased NQO1 protein levels, accumulated estrogen-DNA adducts and increased apoptosis in normal HCEnC-21T cells. A. Analysis of cell morphology using phase-contrast microscopy of hexagonal monolayers of HCEnC-21T cells treated with 50 μM MN, 10 μM 4-OHE2 and co-treated with both. Rosette-like clusters with clear acellular centers (white asterisks) and spindle-like cells (black arrowheads) were observed in treated cells. B. Representative western blots of NQO1 protein expression in HCEnC-21T cells treated with increasing dose of MN (8.5, 17, 35, 50 μM) for 8 h. β-actin is used as a normalizing control. Relative expression of NQO1 protein is indicated below as bar graphs. C. Western blot analysis of NQO1 and COMT levels in HCEnC-21T cells treated with either 50 μM MN and 10 μM 4-OHE2 alone or co-treated with both for 8 h. Two bands in COMT blot represent membrane (M) and soluble (S) forms. D. Assessment of cellular viability in HCEnC-21T cells treated with either 50 μM MN and 10 μM 4-OHE2 alone or co-treated with both for 24 h. E, F. Estimation of percent apoptotic cells in MN and 4-OHE2 treated cells by FACS stained with Annexin-V (Ann) and propidium iodide (PI). Representative image of flow cytometry analysis gated for vital (Ann−/PI−), early apoptotic (Ann+/PI−), late apoptotic (Ann+/PI+), and necrotic (Ann−/PI+) cell populations in HCEnC-21T cells are shown. G. Determination of intracellular ROS production by DCFDA assay in HCEnC-21T cells treated with 50 μM MN and 10 μM 4-OHE2 for 5h. H-J. Quantification of estrogen-DNA adducts, 4-OCH3E2 and quinone conjugates in HCEnC-21T cells treated with either 3.75 μM 4-OHE2 alone or co-treated with 8.5 μM MN and 3.75 μM 4-OHE2 by UPLC-MS/MS. Error bars represent SEM. Data expressed as mean values ± SE. (*p < 0.05, **p < 0.01, ***p < 0.001, One-way ANOVA with Tukey post tests for comparison of multiple data sets).
3.5. Combined exposure to MN and estrogen metabolites leads to increased ROS and apoptosis in HCEnC-21T cells
Treatment with 50 μM MN resulted in a significant decrease in cellular viability in HCEnC-21T cells (66.8 ± 1.8 %, p < 0.01, Fig. 2D), as previously noted [17]. Cell viability was further lowered when 4-OHE2 and 50 μM MN were combined (8 ± 2.7 %, p < 0.01, Fig. 2D) indicating an additive effect. To evaluate the role of 4-OHE2 in oxidative-stress-induced apoptosis, HCEnC-21T cells were treated with 50 μM MN and 10 μM 4-OHE2 for 16 h. Combined treatment led to a greater increase in late apoptotic cells represented in the PI+ (Propidium iodide) and Ann+ (Annexin V) population (75 ± 5%, p < 0.01) determined by flow cytometry compared to the cells treated with MN alone (53 ± 2.1%, p < 0.01, Fig. 2E, F). The loss of cell viability correlated with the increase in intracellular ROS production upon MN and 4-OHE2 treatments (Fig. 2G). Intracellular ROS significantly increased upon MN treatment while 4-OHE2 alone did not induce ROS. Combination of MN and 4-OHE2 led to drastic increase in ROS generation compared to MN alone (Fig. 2G). These data indicate the augmented susceptibility to apoptotic cell death caused by the combination of reactive catechol estrogen and MN-induced oxidative stress.
3.6. Menadione-induced ROS and estrogen metabolites increase the accumulation of depurinating estrogen-DNA adducts in HCEnC-21T cells
Since impaired estrogen homeostasis increases the formation of catechol estrogen quinones and downstream depurinating estrogen-DNA adducts [42, 43], we investigated the effect of MN and 4-OHE2 treatments on the generation of these compounds in HCEnCs. We used moderately cytotoxic doses of MN and 4-OHE2 for these experiments for efficient estrogen metabolite analysis. Optimal dose and course of treatment for the analysis were determined by the analysis of survival plots of MN- and 4-OHE2-mediated cytotoxicity. The 50% killing dose (LC50) for MN and 4-OHE2 was estimated to be 34 and 15 μM, respectively (Supplemental Fig. 1B, C). Since the combination of 4-OHE2 and MN induced further cytotoxicity than each compound alone, co-treatment of 8.5 μM MN and 3.75 μM 4-OHE2, an LC25 of each, was used for these studies. The culture supernatant of the cells treated under various conditions was taken up for estrogen metabolite analysis by UPLC/MS-MS. We detected a statistically significant increase in depurinating estrogen-DNA adducts when cells were treated with 4-OHE2 compared to untreated cells where levels were barely detectable (p < 0.05; Fig. 2H, Supplemental Fig. 2A). Formation of estrogen-DNA adducts drastically increased upon addition of MN along with 4-OHE2 compared to only 4-OHE2 treated cells (112% increase, p < 0.01, 95% CI [0.4, 7.5], Fig. 2H). Levels of less reactive or inactive metabolites 4-OCH3E2 and quinone conjugates, catalyzed by COMT and glutathione respectively, significantly increased upon treatment with 4-OHE2 (p < 0.01, Fig. 2I, J, Supplemental Fig. 2B, C) compared to untreated cells. This is expected, as supplementation of 4-OHE2 would also lead to activation of the protective pathways to detoxify the reactive metabolites along with the competitive oxidation to form catechol quinones. However, addition of MN and 4-OHE2 showed a statistically significant decrease in 4-OCH3E2 compared to 4-OHE2 alone treated cells (33 % decrease, p < 0.01, 95% CI [1668, 6603], Fig. 2I). This suggests an oxidant-antioxidant imbalance in the pathway such that the generation of depurinating adducts dominates over the protective inactivation of the catechol estrogens mediated by COMT and NQO1. However, levels of quinone conjugates were not significantly changed with cotreatment with MN and 4-OHE2 indicating glutathione-based conjugation pathway is unaffected upon treatment (29% decrease, p = 0.13, Fig. 2J). Collectively, combining MN-induced oxidative stress and catechol metabolites drive the estrogen metabolism pathway towards generating genotoxic estrogen-DNA adducts in HCEnCs.
3.7. Deletion of NQO1 increases estrogen metabolite levels, depurinating DNA adducts and cytotoxicity induced by MN and 4-OHE2
The reduced expression of NQO1 in ex vivo FECD specimens and also upon cotreatment with both MN and 4-OHE2 in vitro prompted us to understand its role in the cross talk between oxidative stress and impaired estrogen metabolism. Previously in our laboratory, NQO1−/− cells were generated employing CRISPR-Cas9 mediated excision along with a corresponding wild-type NQO1+/+ cell line [33]. Loss of NQO1 resulted in morphological changes such as fibroblast-like elongation unlike the regular hexagonal monolayer formation seen in NQO1+/+ cells (Fig. 3A). Similar to FECD cell lines, NQO1−/− cells showed increased susceptibility to 4-OHE2 treatment in a dose-dependent manner (Fig. 3B, grey bar). Even at lower doses starting from 5 μM, NQO1+/+ cells had reduced cell viability compared to wild-type cells (Fig. 3B). Furthermore, NQO1−/− cells showed a significant decrease in cell viability compared to NQO1+/+ cells when treated with either 50 μM MN (25.5 ± 2.0% and 45.2 ± 3.8%, p < 0.01) or 10 μM 4-OHE2 (79.7 ± 0.1 and 82.7 ± 0.5 %, p < 0.01) (Fig. 3C). Co-treatment of MN and 4-OHE2 resulted in a drastic reduction in cell viability in both cell lines (Fig. 3C).
Fig. 3.

Deletion of NQO1 resulted in increased susceptibility to 4-OHE2, greater generation of estrogen-DNA adducts and DNA damage compared to NQO1+/+ cells. A. Comparative cellular morphology analysis of NQO1+/+ and NQO1−/− cells by phase-contrast microscopy. White arrowheads indicate the elongated processes in NQO1−/− cells. B. Percent cellular viability was determined in NQO1+/+ (black bar) and NQO1−/− cells (grey bar) treated with increasing doses of 4-OHE2 for 24 h. C. Comparison of percent cellular viability between NQO1+/+ (black bar) and NQO1−/− cells (grey bar) treated with either 50 μM MN, 10 μM 4-OHE2 alone or co-treated with both. D. Assessing the morphological differences in NQO1+/+ and NQO1−/− cells post treatment with either 30 μM 4-OHE2 alone or both 10 μM MN and 20 μM 4-OHE2 for 24 h. E. Detection of nDNA damage by ß-globin amplification and DNA lesions using LA-qPCR analysis in NQO1+/+ cells treated with 30 μM 4-OHE2 (black bar) and 20 μM 4-OHE2 and 10 μM MN (white bar); NQO1−/− cells treated with 30 μM 4-OHE2 (dark grey bar) and 20 μM 4-OHE2 and 10 μM MN (light grey bar). Dotted line indicate the untreated samples to which the treated samples are normalized. F. Estimation of mtDNA damage by relative amplification of large fragment by small fragment using LA-qPCR analysis in NQO1+/+ cells treated with 30 μM 4-OHE2 (black bar) and 20 μM 4-OHE2 and 10 μM MN (white bar); NQO1−/− cells treated with 30 μM 4-OHE2 (dark grey bar) and 20 μM 4-OHE2 and 10 μM MN (light grey bar). Dotted line indicate the untreated samples to which the treated samples are normalized. The statistical signficance is tested against the corresponding untreated sample. G. Determination of ΔΨm levels in NQO1+/+ (black) and NQO1−/− cells (grey) (n=3) at baseline and upon treatment with 50 mM CCCP. Bar graph showing the percentage of cells in gate P2 upon flow cytometry analysis for three different experiments. H-I. Quantification of estrogen-DNA adducts, 4-OCH3E2 and quinone conjugates in NQO1+/+ (black bar) and NQO1−/− cells (grey bar) treated with 3.75 μM 4-OHE2 alone, co-treated with 8.5 μM MN and 3.75 μM 4-OHE2 and co-treated with 8.5 μM MN, 3.75 μM 4-OHE2 and supplemented with 25 μM NAC for 24 h by UPLC-MS/MS. Error bars represent SEM. Data expressed as mean values ± SE. (*p < 0.05, **p < 0.01, ***p < 0.001, Two-way ANOVA with Tukey post tests for comparison of multiple data sets).
Next, we sought to delineate and compare the extent of mtDNA and nDNA damage occurring in NQO1+/+ and NQO1−/− cells upon treatment with 4-OHE2 and MN. The treatment with 4-OHE2 alone and cotreatment with both 4-OHE2 and MN caused differential cellular morphological changes in NQO1+/+ and NQO1−/− cells (Fig. 3D). As 50 μM MN and 10 μM 4-OHE2 treatments caused severe loss of cell viability, we chose lower doses of MN (10 μM) and 4-OHE2 (20 μM) for their combined treatment for DNA damage studies. Upon treatment with 30 μM 4-OHE2, we detected reduced amplification of nuclear encoded ß-globin gene starting from 6 h post treatment in NQO1−/− cells (Fig. 3E, dark grey bar). Similarly, mtDNA damage also ensued from 6 h in NQO1−/− cells (Fig. 3F, dark grey bar). Co-treatment of 20 μM 4-OHE2 and 10 μM MN resulted in a severe reduction in relative mtDNA amplification in NQO1−/− cells from as early as 3 h that persisted up to 24 h (Fig. 3F, light grey bar). In NQO1+/+ cells, mtDNA damage was noted only by 12 h (Fig. 3F, white bar) however by 24 h, the extent of damage in NQO1+/+ and NQO1−/− cells is comparable (data not shown). Similarly, reduced amplification of ß-globin gene was noted at both 6 and 12 h post treatment in NQO1−/− cells compared to NQO1+/+ cells (Fig. 3E light grey and white bars). Supporting the increased susceptibility of NQO1−/− cells to mtDNA damage, mitochondrial membrane potential (ΔΨm) was significantly reduced in NQO1−/− cells when treated with a classic mitochondrial membrane depolarizer CCCP (carbonyl cyanide m-chlorophenyl hydrazone) compared to wild-type cells (Fig. 3G).
Next, we sought to investigate the levels of estrogen metabolites in NQO1−/− cells. Although NQO1−/− cells treated with 4-OHE2 showed marginal increase in estrogen-DNA adducts compared to NQO1+/+ cells (Fig. 3H), with 4-OHE2 and MN cotreatment, NQO1−/− cells showed significantly higher levels of depurinating estrogen-DNA adducts compared to NQO1+/+ cells (p < 0.01, Fig. 3H). Upon 4-OHE2 treatment, NQO1−/− cells also showed significantly higher levels of less reactive 4-OCH3E2 and quinone conjugates compared to NQO1+/+ cells (53% increase, p < 0.05, 95% CI [6112, 22697], 313% increase, p < 0.01, 95% CI [66.6, 121.36], Fig. 3I, J) indicating the activation of detoxification pathways. However, upon combining MN and 4-OHE2, the extent of increase of these inactive metabolites was significantly lower compared to NQO1+/+ cells. These observations corroborate with the increase in estrogen-DNA adducts in NQO1−/−cells treated with MN and 4-OHE2 indicative of increased levels of the unstable estrogen quinones in the absence of NQO1. These observations further underscore the protective role of NQO1 in converting E2-3,4-Q to less reactive forms to prevent the formation of estrogen-DNA adducts. Therefore, downregulation of NQO1 in FECD further drives the estrogen metabolic pathway towards the generation of DNA adducts.
N-acetylcysteine (NAC), a free radical scavenger and antioxidant, is an aminothiol and precursor of intracellular glutathione (GSH) [44]. Zahid et al., have shown that NAC is one of the best inhibitors of the formation of depurinating adducts. In reaction mixtures containing E2-3,4-Q or enzyme-activated 4-OHE2 and DNA, DNA adduct formation was reduced due to reaction of the E2-3,4-Q with NAC instead of DNA [45]. Thus, since NAC has been shown to be cytoprotective in estrogen metabolic pathways, we tested its effect on NQO1−/− cells exposed to both catechol estrogen and MN-induced oxidative stress. In our study, the addition of NAC decreased the accumulation of estrogen-DNA adducts (71% decrease, p < 0.01, 95% CI [4.5, 97.5], Fig. 3H) supporting our hypothesis. We also observed reduced levels of methoxy conjugates (71% decrease, p < 0.01, 95% CI [11263, 24848], Fig. 3I) and increased quinone conjugates (194% increase, p < 0.01, 95% CI [15, 214.9], Fig. 3J) in NQO1−/− cells. This is expected as NAC is known to bind to E2-3,4-Q forming conjugates as part of its detoxification process. Therefore, supplementation of NAC reduced the effect of oxidative stress contributed by both MN and estrogen metabolites in NQO1−/− cells.
3.8. Overexpression of NQO1 reduces MN and estrogen metabolite induced ROS and DNA damage
To ascertain the central contribution of NQO1 in alleviating the molecular defects of ROS and estrogen metabolites in CEnCs, we tested the effect of overexpression of NQO1 on MN and 4-OHE2 treated HCEnC-21T cells. The strong CMV6 promoter drove the expression of C-terminal myc-tagged NQO1 in a plasmid background (pCMV6-NQO1-myc). Empty vector backbone lacking NQO1 (pCMV6-myc) served as the control. Western blot analysis using anti-NQO1 antibody confirmed the evident overexpression of NQO1-myc in HCEnC-21T cells treated with 20 μM MN, 10 μM 4-OHE2 or both in comparison to the endogenous NQO1 (Fig. 4A). Elevating the levels of NQO1 significantly increased the cell viability of HCEnC-21T cells treated with 20 μM 4-OHE2 and 10 μM MN for 12 h (Fig. 4B). Supporting this data, plasmid-mediated overexpression of NQO1 significantly inhibited the MN and 4-OHE2 induced intracellular ROS generation at an early time point of 3 h post treatment (Fig. 4C). Furthermore, increased NQO1 protein levels also significantly rescued both mtDNA and nDNA damage induced by 4-OHE2 + MN treatment at 12 h (Fig. 4D). These observations strongly indicate that overexpression of NQO1 drastically reduced the ROS generation, thereby decreasing the extent of mtDNA and nDNA damage consequently leading to rescue of cell viability. Collectively, these data underscore NQO1 as an important factor of cellular defense to combat ROS and maintain balanced estrogen metabolism. Downregulation of NQO1 in FECD is one of the key contributing factors leading to ROS formation and imbalanced estrogen metabolism, thereby causing formation of estrogen-DNA adducts and DNA damage that ultimately results in endothelial cell death.
Fig. 4.

Overexpression of NQO1 reduces ROS formation and DNA damage induced by MN and 4-OHE2. A. Western blot analysis of plasmid expressed NQO1-myc and endogenous NQO1 levels in HCEnC-21T cells transfected with pCMV6-myc and pCMV6-NQO1-myc followed by treatment with 50 μM MN and 10 μM 4-OHE2. The bands corresponding to NQO1-myc and endogenous NQO1 are indicated. ß-actin serves as a normalizing control. B. Assessment of cell viability of HCEnC-21T cells transfected with pCMV6-myc (black) and pCMV6-NQO1-myc (grey) and treated with 10 μM MN and 20 μM 4-OHE2 for 24 h by Cell-titer glo assay. C. Determination of intracellular ROS production in HCEnCs transfected with vector (black) and NQO1 plasmid (grey) by DCFDA assay 5 h post treatment with 10 μM MN and 20 μM 4-OHE2. Statistical significance tested using Student’s t test. D. Assessment of mtDNA (left) and nDNA (right) damage by LA-qPCR analysis of HCEnC-21T cells transfected with vector (black) and NQO1 plasmid (grey) and treated with 10 μM MN and 20 μM 4-OHE2 for 12 h. (* p< 0.05, Student’s t test).
4. Discussion
Females are reported to have increased incidence and advanced FECD disease development [8–11]. Although a possible hormonal role has been postulated to account for this, no studies have been done to explain the mechanisms of FECD predominance in females [46]. In this study, we demonstrate that NQO1 downregulation seen in FECD renders HCEnCs susceptible to catechol estrogen and estrogen-DNA adducts formation. This is the first study that provides a mechanistic insight into the interplay between ROS-mediated oxidative stress and estrogen metabolites, mediated by NQO1 in FECD pathogenesis.
Maintaining balanced estrogen metabolism requires a tight interplay between estrogen activating enzymes, such as CYP1B1, and deactivating enzymes, such as COMT and NQO1. Under a normal cellular scenario, the formation of estrogen quinones is low, and consequently the formation of genotoxic DNA adducts leading to HCEnCs apoptosis is low. Diminished expression of detoxification enzymes and/or upregulation of enzymes of the oxidation pathway can disrupt this homeostasis. For example, low COMT activity, which can be due to presence of low activity genetic polymorphisms [47–49], or low NQO1 activity are contributory factors in the increased formation of estrogen–DNA adducts [36, 50, 51]. A concomitant decrease in NQO1 and COMT along with increase in CYP1B1 is associated with elevated risk of breast and ovarian cancers [42, 52]. In this study, we showed that diminished expression of the detoxification enzyme NQO1 leads to increased formation of estrogen-DNA adducts and mtDNA and nDNA damage in FECD. Overexpression of NQO1 was able to significantly diminish the ROS production and DNA damage arising from MN and catechol estrogen treatment. Studies from our laboratory provide the first line of evidence on this etiological factor in FECD, suggesting that estrogen metabolites play a notable role in endothelial cell death in FECD by inducing increased estrogen-DNA adducts and DNA damage in FECD endothelium that in turn leads to subsequent induction of apoptosis of endothelial cells. In a parallel study from our laboratory, we observed that physiological stressor UVA caused an increase in the expression of estrogen-oxidizing enzyme CYP1B1 preferentially in female mice leading to increased accumulation of estrogen-DNA adducts (Liu et al., PNAS, in Press), concordant with the imbalance in levels of estrogen metabolizing enzymes in FECD. A recent genome-wide association study (GWAS) identified sex-specific association between FECD and several genetic factors, where LAMC1 increased the risk of FECD in women and TCF4 increased the risk of FECD in men [53]. LAMC1 is a cell adhesion protein in the basement membranes such as DM, while TCF4 is a basic helix-loop-helix transcription factor, involved in cellular growth and differentiation. The mechanism by which these alleles confer sex-specific risk in developing FECD is still unknown. Further studies are needed to investigate the correlation between the genetic factors and estrogen metabolite toxicity in rendering females to be more susceptible to develop FECD. However, it has been shown that estrogen-DNA adducts form to a higher extent in men with prostate cancer compared to men without cancer [54], indicating that impaired estrogen metabolism may also increase the risk of FECD in genetically susceptible male patients. In support of this, we observed decreased NQO1 levels in both male and female FECD patient specimens.
NQO1 mediates the protective function of antioxidants Vitamin C and butylated hydroxyanisol against oxidative DNA damage and estrogen-mediated mammary tumorigenesis [55]. Similarly, the antioxidants sulforaphane and resveratrol also induce protective enzymes including NQO1 and COMT to reverse estrogen-induced damage [56,57]. Further, NQO1 is involved in the detoxification of dopamine hydroquinones [58] resulting from the oxidative metabolism of dopamine and oxidative stress that leads to the degeneration of dopinamergic post-mitotic neurons in Parkinson’s disease (PD) [59]. Activation of the Nrf2/ARE signal mitigates the 6-hydroxydopamine-induced neurotoxicity in human neuroblastoma cell lines [60]. In our study, plasmid-mediated NQO1 overexpression as well as supplementation with antioxidant NAC significantly inhibited the ROS formation, DNA damage and cell death induced by MN and estrogen metabolites. NAC is known to directly react with estrogen quinones to form non-toxic conjugates, thereby preventing the binding of quinones to DNA and forming mutagenic adducts [61]. While NQO1 is an important link between the effect of ROS and estrogen metabolite-mediated damage in FECD, lack of complete rescue of cell viability with NQO1 overexpression suggests potential concerted contribution of other factors.
NQO1 levels are reduced in ex vivo FECD specimens, MN treated HCEnCs and in FECD patient-derived cell lines, highlighting its importance in maintaining the oxidant-antioxidant balance in functional corneal endothelium. NQO1−/− cells showed an increased propensity for DNA damage and cell loss induced by MN and 4-OHE2. Furthermore, NQO1-null mice show increased susceptibility to MN toxicity compared to wild-type mice corroborating our in vitro findings [62]. Altered NQO1 expression is reported in various pathologies including cancer and strikingly in neurodegenerative diseases like Alzheimer’s (AD) and PD [59, 63, 64]. In both AD and PD, NQO1 levels differ in an age-dependent manner in which its expression increases in the earlier stages in accordance with its inducible cytoprotective function in response to oxidative stress. However, the expression progressively declines with age in the pathological scenario [64, 65]. Similarly, NQO1 levels are reduced in FECD ex vivo specimens that represent late stage of the disease progression. Supprting this, we have previously reported increased MN-induced endothelial-mesenchymal transition (EnMT) in NQO1cells, a characteristic feature of FECD [33]. A recent study has shown that Akt kinase phosphorylates NQO1 and couples it to E3 ligase Parkin leading to NQO1 ubiquination and subsequent proteasomal degradation [65]. Interestingly, we recently demonstrated increased activation of Parkin-mediated mitophagy in ex vivo FECD specimens as well as MN-induced oxidative stress [31].
In this study, we noted reduced ΔΨm and early mtDNA damage in NQO1−/− cells compared to wild-type cells, suggesting a possible function for NQO1 in mitochondria, an organelle highly prone to ROS-mediated oxidative stress. There is very limited literature on the direct role of NQO1 in mitochondrial function. Although NQO1 is primarily considered a cytosolic protein [66], a recent study detected dioxin-induced NQO1 expression in the microsomal, mitochondrial and cytosolic fractions of various mouse organs [67]. In another study, increased NQO1 was shown to improve the activity of mitochondrial electron chain complexes and offer protection against mitochondrial toxins [68].
NQO1 is a multifunctional cytosolic flavoenzyme whose role in the reduction of quinones is well studied. Apart from its enzymatic function, NQO1 is known to play a cytoprotective role by binding and stabilizing p53, a factor activated in FECD [69], thereby preventing its proteasomal degradation [25, 70]. It would also be interesting to investigate whether NQO1 polymorphisms potentially increase the genetic predisposition to FECD. Clear absence of NQO1 protein expression in a FECD patient derived cell line lacking TCF4 repeat expansion is also noteworthy in this context. Notably, cigarette smoking is known to alter NQO1 expression owing to the generation of free radicals; and smoking has also been identified as a risk factor for FECD [71]. Taken together, this study highlights a possible role of NQO1 and estrogen metabolism in the pathogenesis of FECD, thereby offering novel portals for potential therapeutic intervention.
5. Conclusions
NQO1, an important estrogen quinone-detoxifying enzyme regulated by Nrf2 transcription factor, is downregulated in FECD specimens, FECD patient derived cells lines and in the in vitro MN-mediated oxidative stress model in HCEnCs. Menadione-mediated ROS and catechol estrogens induced estrogen-DNA adduct formation and apoptosis in HCEnCs. Loss of NQO1 enzyme further aggravated the estrogen-DNA adducts formation and loss of cell viability. Notably, NQO1−/− cells show increased susceptibility to mtDNA damage and reduced MMP. Addition of NAC, an effective scavenger of ROS, rescued the estrogen-DNA adduct formation in NQO1−/− cells. Overexpression of NQO1 in HCEnCs treated with MN and 4-OHE2 quenched the intracellular ROS formation, reduced DNA damage and rescued cell viability, underscoring the cytoprotective role of NQO1. Overall, our findings indicate that NQO1 downregulation seen in FECD leads to increased susceptibility of HCEnCs to catechol estrogen, estrogen-DNA adducts formation, and mtDNA and nDNA damage. This novel study highlights the potential role of impaired estrogen metabolism in FECD development and provides a potential target for therapeutic intervention.
Supplementary Material
Fig. 1. A. Bar graph plotting the relative amplification of small mtDNA in 30 μM 4-OHE2 treated HCEnC-SV-67F-16, FECD-SV-73F-74 and -61F-18 cells. Dotted line indicate the untreated samples to which the treated samples are normalized. B, C. Survival plots of the percent cell viability of HCEnC-21T cells treated with varying doses of MN and 4-OHE2 determined by Cell Titer Glo assay.
Fig. 2. A-C. Comparison of the levels of estrogen-DNA adducts, 4-973 OCH3E2 and quinone conjugates in untreated HCEnC-21T cells versus. treated with either 8.5 μM MN alone, 3.75 μM 4-OHE2 alone or co-treated with 8.5 μM MN and 3.75 μM 4-OHE2 by UPLC-MS/MS.
Highlights.
Loss of NQO1 points to role of reactive estrogen metabolism in FECD pathogenesis
Catechol estrogens induce DNA damage and apoptosis in corneal endothelium
Loss of NQO1 in vitro aggravates ROS-induced estrogen-DNA adduct formation
Overexpression of NQO1 reduces DNA damage and endothelial cell loss in FECD
Acknowledgments
This work was supported by NIH/NEI Grant R01EY20581 (UVJ), Core Grant P30EY003790, Alcon Research Award (UVJ), Japan Eye Bank Association Overseas Grant 2018 (TM) and American Heart Association Postdoctoral Fellowship (18POST34030385) (VK).
Abbreviations
- BSA
bovine serum albumin
- FECD
Fuchs endothelial corneal dystrophy
- HCEnC
human corneal endothelial cell
- MN
menadione
- NAC
N-acetylcysteine
- NQO1
NAD(P)H-quinone oxireductase
- Nrf2
nuclear factor erythroid 2-related factor
- PD
Parkinson’s disease
- AD
Alzheimer’s disease
- TBS
tris-buffered saline
- PI
propidium iodide
- ROS
reactive oxygen species
- SEM
standard error of the mean
- RT-PCR
Real-time reverse transcription–polymerase chain reaction.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure
The authors declare that they have no conflict of interest.
References
- [1].Joyce NC, Proliferative capacity of the corneal endothelium, Progress in retinal and eye research 22(3) (2003) 359–389. [DOI] [PubMed] [Google Scholar]
- [2].Peh GS, Toh K-P, Wu F-Y, Tan DT, Mehta JS, Cultivation of human corneal endothelial cells isolated from paired donor corneas, PloS one 6(12) (2011) e28310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Senoo T, Joyce NC, Cell cycle kinetics in corneal endothelium from old and young donors, Investigative ophthalmology & visual science 41(3) (2000) 660–667. [PubMed] [Google Scholar]
- [4].Borderie VM, Baudrimont M, Vallee A, Ereau TL, Gray F, Laroche L, Corneal endothelial cell apoptosis in patients with Fuchs’ dystrophy, Invest Ophthalmol Vis Sci 41(9) (2000) 2501–5. [PubMed] [Google Scholar]
- [5].Li QJ, Ashraf MF, Shen DF, Green WR, Stark WJ, Chan CC, O’Brien TP, The role of apoptosis in the pathogenesis of Fuchs endothelial dystrophy of the cornea, Arch Ophthalmol 119(11) (2001) 1597–604. [DOI] [PubMed] [Google Scholar]
- [6].Son H-S, Villarreal G, Meng H, Eberhart CG, Jun AS, On the origin of ‘guttae’, British Journal of Ophthalmology 98(9) (2014) 1308–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gain P, Jullienne R, He Z, et al. , Global survey of corneal transplantation and eye banking, JAMA Ophthalmology 134(2) (2016) 167–173. [DOI] [PubMed] [Google Scholar]
- [8].Yeh P, Colby K, Corneal Endothelial Dystrophies, in: Foster C, Azar D, Dohlman C (Eds.), Smolin and Thoft’s The Cornea, Lippincott Williams & Wilkins, Philadelphia, 2004. [Google Scholar]
- [9].Wilson SE, Bourne WM, Fuchs’ dystrophy, Cornea 7(1) (1988) 2–18. [PubMed] [Google Scholar]
- [10].Krachmer JH, Purcell JJ Jr., Young CW, Bucher KD, Corneal endothelial dystrophy. A study of 64 families, Arch Ophthalmol 96(11) (1978) 2036–9. [DOI] [PubMed] [Google Scholar]
- [11].Rosenblum P, Stark WJ, Maumenee IH, Hirst LW, Maumenee AE, Hereditary Fuchs’ Dystrophy, Am J Ophthalmol 90(4) (1980) 455–62. [DOI] [PubMed] [Google Scholar]
- [12].Afshari NA, Pittard AB, Siddiqui A, Klintworth GK, Clinical study of Fuchs corneal endothelial dystrophy leading to penetrating keratoplasty: a 30-year experience, Archives of ophthalmology 124(6) (2006) 777–780. [DOI] [PubMed] [Google Scholar]
- [13].Zoega GM, Fujisawa A, Sasaki H, Kubota A, Sasaki K, Kitagawa K, Jonasson F, Prevalence and risk factors for cornea guttata in the Reykjavik Eye Study, Ophthalmology 113(4) (2006) 565–569. [DOI] [PubMed] [Google Scholar]
- [14].Zhang X, Igo RP Jr., Fondran J, Mootha VV, Oliva M, Hammersmith K, Sugar A, Lass JH, Iyengar SK, Fuchs’ G Genetics Multi-Center Study, Association of smoking and other risk factors with Fuchs’ endothelial corneal dystrophy severity and corneal thickness, Invest Ophthalmol Vis Sci 54(8) (2013) 5829–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chan SWS, Yucel Y, Gupta N, New trends in corneal transplants at the University of Toronto, Can J Ophthalmol 53(6) (2018) 580–587. [DOI] [PubMed] [Google Scholar]
- [16].Wieben ED, Aleff RA, Tosakulwong N, Butz ML, Highsmith WE, Edwards AO, Baratz KH, A common trinucleotide repeat expansion within the transcription factor 4 (TCF4, E2-2) gene predicts Fuchs corneal dystrophy, PLoS One 7(11) (2012) e49083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Halilovic A, Schmedt T, Benischke AS, Hamill C, Chen Y, Santos JH, Jurkunas UV, Menadione-Induced DNA Damage Leads to Mitochondrial Dysfunction and Fragmentation During Rosette Formation in Fuchs Endothelial Corneal Dystrophy, Antioxid Redox Signal 24(18) (2016) 1072–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Czarny P, Kasprzak E, Wielgorski M, Udziela M, Markiewicz B, Blasiak J, Szaflik J, Szaflik JP, DNA damage and repair in Fuchs endothelial corneal dystrophy, Molecular Biology Reports 40(4) (2013) 2977–2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Czarny P, Seda A, Wielgorski M, Binczyk E, Markiewicz B, Kasprzak E, Jimenez-Garcia MP, Grabska-Liberek I, Pawlowska E, Blasiak J, Szaflik J, Szaflik JP, Mutagenesis of mitochondrial DNA in Fuchs endothelial corneal dystrophy, Mutat Res 760 (2014) 42–7. [DOI] [PubMed] [Google Scholar]
- [20].Liu C, Vojnovic D, Kochevar IE, Jurkunas UV, UV-A Irradiation Activates Nrf2-Regulated Antioxidant Defense and Induces p53/Caspase3-Dependent Apoptosis in Corneal Endothelial Cells, Invest Ophthalmol Vis Sci 57(4) (2016) 2319–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Liu C, Chen Y, Kochevar IE, Jurkunas UV, Decreased DJ-1 leads to impaired Nrf2-regulated antioxidant defense and increased UV-A-induced apoptosis in corneal endothelial cells, Invest Ophthalmol Vis Sci 55(9) (2014) 5551–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bitar MS, Liu C, Ziaei A, Chen Y, Schmedt T, Jurkunas UV, Decline in DJ-1 and decreased nuclear translocation of Nrf2 in Fuchs endothelial corneal dystrophy, Invest Ophthalmol Vis Sci 53(9) (2012) 5806–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Jurkunas UV, Bitar MS, Funaki T, Azizi B, Evidence of oxidative stress in the pathogenesis of fuchs endothelial corneal dystrophy, Am J Pathol 177(5) (2010) 2278–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Jurkunas UV, Rawe I, Bitar MS, Zhu C, Harris DL, Colby K, Joyce NC, Decreased expression of peroxiredoxins in Fuchs’ endothelial dystrophy, Invest Ophthalmol Vis Sci 49(7) (2008) 2956–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Dinkova-Kostova AT, Talalay P, NAD (P) H: quinone acceptor oxidoreductase 1 (NQO1), a multifunctional antioxidant enzyme and exceptionally versatile cytoprotector, Archives of biochemistry and biophysics 501(1) (2010) 116–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Favreau LV, Pickett CB, The rat quinone reductase antioxidant response element. Identification of the nucleotide sequence required for basal and inducible activity and detection of antioxidant response element-binding proteins in hepatoma and non-hepatoma cell lines, J Biol Chem 270(41) (1995) 24468–74. [DOI] [PubMed] [Google Scholar]
- [27].Nioi P, McMahon M, Itoh K, Yamamoto M, Hayes JD, Identification of a novel Nrf2-regulated antioxidant response element (ARE) in the mouse NAD(P)H:quinone oxidoreductase 1 gene: reassessment of the ARE consensus sequence, Biochem J 374(Pt 2) (2003) 337–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Gaikwad NW, Rogan EG, Cavalieri EL, Evidence from ESI-MS for NQO1-catalyzed reduction of estrogen ortho-quinones, Free Radic Biol Med 43(9) (2007) 1289–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].He X, Chen MG, Lin GX, Ma Q, Arsenic induces NAD (P) H-quinone oxidoreductase I by disrupting the Nrf2 · Keap1 · Cul3 complex and recruiting Nrf2 · Maf to the antioxidant response element enhancer, Journal of Biological Chemistry 281(33) (2006) 23620–23631. [DOI] [PubMed] [Google Scholar]
- [30].Cavalieri EL, Rogan EG, Unbalanced metabolism of endogenous estrogens in the etiology and prevention of human cancer, The Journal of steroid biochemistry and molecular biology 125(3-5) (2011) 169–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Miyai T, Vasanth S, Melangath G, Deshpande N, Kumar V, Benischke AS, Chen Y, Price MO, Price FW Jr., Jurkunas UV, Activation of PINK1-Parkin-Mediated Mitophagy Degrades Mitochondrial Quality Control Proteins in Fuchs Endothelial Corneal Dystrophy, Am J Pathol (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Schmedt T, Chen Y, Nguyen TT, Li S, Bonanno JA, Jurkunas UV, Telomerase immortalization of human corneal endothelial cells yields functional hexagonal monolayers, PLoS One 7(12) (2012) e51427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Katikireddy KR, White TL, Miyajima T, Vasanth S, Raoof D, Chen Y, Price MO, Price FW, Jurkunas UV, NQO1 downregulation potentiates menadione-induced endothelial-mesenchymal transition during rosette formation in Fuchs endothelial corneal dystrophy, Free Radic Biol Med 116 (2018) 19–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Benischke AS, Vasanth S, Miyai T, Katikireddy KR, White T, Chen Y, Halilovic A, Price M, Price F Jr., Liton PB, Jurkunas UV, Activation of mitophagy leads to decline in Mfn2 and loss of mitochondrial mass in Fuchs endothelial corneal dystrophy, Sci Rep 7(1) (2017) 6656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zhu C, Joyce NC, Proliferative Response of Corneal Endothelial Cells from Young and Older Donors, Investigative Ophthalmology & Visual Science 45(6) (2004) 1743–1751. [DOI] [PubMed] [Google Scholar]
- [36].Lu F, Zahid M, Saeed M, Cavalieri EL, Rogan EG, Estrogen metabolism and formation of estrogen-DNA adducts in estradiol-treated MCF-10F cells. The effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin induction and catechol-O-methyltransferase inhibition, J Steroid Biochem Mol Biol 105(1–5) (2007) 150–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Santos JH, Mandavilli BS, Van Houten B, Measuring oxidative mtDNA damage and repair using quantitative PCR, Mitochondrial DNA, Springer; 2002, pp. 159–176. [DOI] [PubMed] [Google Scholar]
- [38].Mondal B, Chen H, Wen W, Cavalieri EL, Rogan EG, Zahid M, Modulation of Cellular Response to Arsenic Trioxide Toxicity by Resveratrol, ACS Omega 3(5) (2018) 5511–5515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Dawling S, Roodi N, Mernaugh RL, Wang X, Parl FF, Catechol-O-methyltransferase (COMT)-mediated metabolism of catechol estrogens: comparison of wild-type and variant COMT isoforms, Cancer Res 61(18) (2001) 6716–22. [PubMed] [Google Scholar]
- [40].Liehr JG, Ricci MJ, 4-Hydroxylation of estrogens as marker of human mammary tumors, Proceedings of the National Academy of Sciences 93(8) (1996) 3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kovalenko OA, Santos JH, Analysis of Oxidative Damage by Gene-Specific Quantitative PCR, Current Protocols in Human Genetics 62(1) (2009) 19.1.1–19.1.13. [DOI] [PubMed] [Google Scholar]
- [42].Zahid M, Beseler CL, Hall JB, LeVan T, Cavalieri EL, Rogan EG, Unbalanced estrogen metabolism in ovarian cancer, Int J Cancer 134(10) (2014) 2414–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zahid M, Saeed M, Lu F, Gaikwad N, Rogan E, Cavalieri E, Inhibition of catechol-O-methyltransferase increases estrogen-DNA adduct formation, Free Radic Biol Med 43(11) (2007) 1534–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Sun S-Y, N-acetylcysteine, reactive oxygen species and beyond, Cancer biology & therapy 9(2) (2010) 109–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Zahid M, Gaikwad NW, Rogan EG, Cavalieri EL, Inhibition of depurinating estrogen-DNA adduct formation by natural compounds, Chem Res Toxicol 20(12) (2007) 1947–53. [DOI] [PubMed] [Google Scholar]
- [46].Adamis AP, Filatov V, Tripathi BJ, Fuchs’ endothelial dystrophy of the cornea, Survey of ophthalmology 38(2) (1993) 149–168. [DOI] [PubMed] [Google Scholar]
- [47].Lavigne JA, Helzlsouer KJ, Huang H-Y, Strickland PT, Bell DA, Selmin O, Watson MA, Hoffman S, Comstock GW, Yager JD, An association between the allele coding for a low activity variant of catechol-O-methyltransferase and the risk for breast cancer, Cancer Research 57(24) (1997) 5493–5497. [PubMed] [Google Scholar]
- [48].Huang C-S, Chern H-D, Chang K-J, Cheng C-W, Hsu S-M, Shen C-Y, Breast cancer risk associated with genotype polymorphism of the estrogen-metabolizing genes CYP17, CYP1A1, and COMT: a multigenic study on cancer susceptibility, Cancer research 59(19) (1999) 4870–4875. [PubMed] [Google Scholar]
- [49].Yim D-S, Park SK, Yoo K-Y, Yoon K-S, Chung HH, Kang HJ, Ahn S-H, Noh D-Y, Choe K-J, Jang I-J, Relationship between the Val158Met polymorphism of catechol O-methyl transferase and breast cancer, Pharmacogenetics and Genomics 11(4) (2001) 279–286. [DOI] [PubMed] [Google Scholar]
- [50].Cavalieri EL, Kumar S, Todorovic R, Higginbotham S, Badawi AF, Rogan EG, Imbalance of estrogen homeostasis in kidney and liver of hamsters treated with estradiol: implications for estrogen-induced initiation of renal tumors, Chemical research in toxicology 14(8) (2001) 1041–1050. [DOI] [PubMed] [Google Scholar]
- [51].Rogan EG, Badawi AF, Devanesan PD, Meza JL, Edney JA, West WW, Higginbotham SM, Cavalieri EL, Relative imbalances in estrogen metabolism and conjugation in breast tissue of women with carcinoma: potential biomarkers of susceptibility to cancer, Carcinogenesis 24(4) (2003) 697–702. [DOI] [PubMed] [Google Scholar]
- [52].Singh S, Chakravarti D, Edney JA, Hollins RR, Johnson PJ, West WW, Higginbotham SM, Cavalieri EL, Rogan EG, Relative imbalances in the expression of estrogen-metabolizing enzymes in the breast tissue of women with breast carcinoma, Oncology reports 14(4) (2005) 1091–1096. [PubMed] [Google Scholar]
- [53].Afshari NA, Igo RP Jr., Morris NJ, Stambolian D, Sharma S, Pulagam VL, Dunn S, Stamler JF, Truitt BJ, Rimmler J, Kuot A, Croasdale CR, Qin X, Burdon KP, Riazuddin SA, Mills R, Klebe S, Minear MA, Zhao J, Balajonda E, Rosenwasser GO, Baratz KH, Mootha VV, Patel SV, Gregory SG, Bailey-Wilson JE, Price MO, Price FW Jr., Craig JE, Fingert JH, Gottsch JD, Aldave AJ, Klintworth GK, Lass JH, Li YJ, Iyengar SK, Genome-wide association study identifies three novel loci in Fuchs endothelial corneal dystrophy, Nat Commun 8 (2017) 14898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Cavalieri EL, Rogan EG, Zahid M, Critical depurinating DNA adducts: Estrogen adducts in the etiology and prevention of cancer and dopamine adducts in the etiology and prevention of Parkinson’s disease, Int J Cancer 141(6) (2017) 1078–1090. [DOI] [PubMed] [Google Scholar]
- [55].Singh B, Bhat NK, Bhat HK, Induction of NAD(P)H-quinone oxidoreductase 1 by antioxidants in female ACI rats is associated with decrease in oxidative DNA damage and inhibition of estrogen-induced breast cancer, Carcinogenesis 33(1) (2012) 156–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Yang L, Zahid M, Liao Y, Rogan EG, Cavalieri EL, Davidson NE, Yager JD, Visvanathan K, Groopman JD, Kensler TW, Reduced formation of depurinating estrogen-DNA adducts by sulforaphane or KEAP1 disruption in human mammary epithelial MCF-10A cells, Carcinogenesis 34(11) (2013) 2587–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Zahid M, Gaikwad NW, Ali MF, Lu F, Saeed M, Yang L, Rogan EG, Cavalieri EL, Prevention of estrogen-DNA adduct formation in MCF-10F cells by resveratrol, Free Radic Biol Med 45(2) (2008) 136–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Segura-Aguilar J, Lind C, On the mechanism of the Mn3(+)-induced neurotoxicity of dopamine:prevention of quinone-derived oxygen toxicity by DT diaphorase and superoxide dismutase, Chem Biol Interact 72(3) (1989) 309–24. [DOI] [PubMed] [Google Scholar]
- [59].van Muiswinkel FL, de Vos RA, Bol JG, Andringa G, Jansen Steur EN, Ross D, Siegel D, Drukarch B, Expression of NAD(P)H:quinone oxidoreductase in the normal and Parkinsonian substantia nigra, Neurobiol Aging 25(9) (2004) 1253–62. [DOI] [PubMed] [Google Scholar]
- [60].Kim DW, Lee KT, Kwon J, Lee HJ, Lee D, Mar W, Neuroprotection against 6-OHDA-induced oxidative stress and apoptosis in SH-SY5Y cells by 5,7-Dihydroxychromone: Activation of the Nrf2/ARE pathway, Life Sci 130 (2015) 25–30. [DOI] [PubMed] [Google Scholar]
- [61].Zahid M, Saeed M, Ali MF, Rogan EG, Cavalieri EL, N-acetylcysteine blocks formation of cancer-initiating estrogen-DNA adducts in cells, Free Radic Biol Med 49(3) (2010) 392–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Radjendirane V, Joseph P, Lee YH, Kimura S, Klein-Szanto AJ, Gonzalez FJ, Jaiswal AK, Disruption of the DT diaphorase (NQO1) gene in mice leads to increased menadione toxicity, J Biol Chem 273(13) (1998) 7382–9. [DOI] [PubMed] [Google Scholar]
- [63].Oh E-T, Park HJ, Implications of NQO1 in cancer therapy, BMB reports 48(11) (2015) 609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Chhetri J, King AE, Gueven N, Alzheimer’s Disease and NQO1: Is there a Link?, Current Alzheimer Research 15(1) (2018) 56–66. [DOI] [PubMed] [Google Scholar]
- [65].Luo S, Su Kang S, Wang ZH, Liu X, Day JX, Wu Z, Peng J, Xiang D, Springer W, Ye K, Akt Phosphorylates NQO1 and Triggers its Degradation, Abolishing its Antioxidative Activities in Parkinson’s Disease, J Neurosci (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Winski SL, Koutalos Y, Bentley DL, Ross D, Subcellular Localization of NAD(P)H:quinone Oxidoreductase 1 in Human Cancer Cells, Cancer Research 62(5) (2002) 1420–1424. [PubMed] [Google Scholar]
- [67].Dong H, Shertzer HG, Genter MB, Gonzalez FJ, Vasiliou V, Jefcoate C, Nebert DW, Mitochondrial targeting of mouse NQO1 and CYP1B1 proteins, Biochemical and biophysical research communications 435(4) (2013) 727–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Kim J, Kim SK, Kim HK, Mattson MP, Hyun DH, Mitochondrial function in human neuroblastoma cells is up-regulated and protected by NQO1, a plasma membrane redox enzyme, PLoS One 8(7) (2013) e69030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Azizi B, Ziaei A, Fuchsluger T, Schmedt T, Chen Y, Jurkunas UV, p53-regulated increase in oxidative-stress--induced apoptosis in Fuchs endothelial corneal dystrophy: a native tissue model, Invest Ophthalmol Vis Sci 52(13) (2011) 9291–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Asher G, Lotem J, Kama R, Sachs L, Shaul Y, NQO1 stabilizes p53 through a distinct pathway, Proceedings of the National Academy of Sciences 99(5) (2002) 3099–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Zhang X, Igo RP, Fondran J, Mootha VV, Oliva M, Hammersmith K, Sugar A, Lass JH, Iyengar SK, Association of smoking and other risk factors with Fuchs’ endothelial corneal dystrophy severity and corneal thickness, Investigative ophthalmology & visual science 54(8) (2013) 5829–5835. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. 1. A. Bar graph plotting the relative amplification of small mtDNA in 30 μM 4-OHE2 treated HCEnC-SV-67F-16, FECD-SV-73F-74 and -61F-18 cells. Dotted line indicate the untreated samples to which the treated samples are normalized. B, C. Survival plots of the percent cell viability of HCEnC-21T cells treated with varying doses of MN and 4-OHE2 determined by Cell Titer Glo assay.
Fig. 2. A-C. Comparison of the levels of estrogen-DNA adducts, 4-973 OCH3E2 and quinone conjugates in untreated HCEnC-21T cells versus. treated with either 8.5 μM MN alone, 3.75 μM 4-OHE2 alone or co-treated with 8.5 μM MN and 3.75 μM 4-OHE2 by UPLC-MS/MS.