

Abstract
With a diverse physiological interface to colonize, mammalian skin is the first line of defense against pathogen invasion and harbors a consortium of microbes integral in maintenance of epithelial barrier function and disease prevention. While the dynamic roles of skin bacterial residents are expansively studied, contributions of fungal constituents, the mycobiome, are largely overlooked. As a result, their influence during skin injury, such as disruption of skin integrity in burn injury and impairment of host immune defense system, is not clearly delineated. Burn patients experience a high risk of developing hard-to-treat fungal infections in comparison to other hospitalized patients. To discern the changes in the mycobiome profile and network assembly during cutaneous burn-injury, a rat scald burn model was used to survey the mycobiome in healthy (n = 30) (sham-burned) and burned (n = 24) skin over an 11-day period. The healthy skin demonstrated inter-animal heterogeneity over time, while the burned skin mycobiome transitioned toward a temporally stabile community with declining inter-animal variation starting at day 3 post-burn injury. Driven primarily by a significant increase in relative abundance of Candida, fungal species richness and abundance of the burned skin decreased, especially in days 7 and 11 post-burn. The network architecture of rat skin mycobiome displayed community reorganization toward increased network fragility and decreased stability compared to the healthy rat skin fungal network. This study provides the first account of the dynamic diversity observed in the rat skin mycobiome composition, structure, and network assembly associated with postcutaneous burn injury.
Keywords: rat skin mycobiome, deep-partial thickness burn, burned skin mycobiome, skin fungal community structure, skin fungal network assembly, Rattus norvegicus
Introduction
Mammals form a complex and poorly understood relationship with a diverse collection of microbial communities (fungi, bacteria, viruses, and archaea), called the microbiome, across a topologically diverse host-body interface with some interactions established before birth (e.g., maternal microbiome and fetal development).1–3 The ability of the mammalian skin to shield internal tissues and defend against pathogen invasion is linked largely to the microbes that evolved to coexist in dynamic equilibrium with the mammalian host as commensal skin residents.4–9 An unperturbed skin microbial community resides in homeostasis with the host ecosystem and aids in host defense via mechanisms that include nutrient competition with pathogens, host immune-response regulation, and antimicrobial agent production that can also lead to opportunistic commensal pathogenesis (e.g., Malassezia) in response to alterations of surrounding environment (e.g., impaired host-immune response, nutrient accessibility).6,10-14
Dynamics of the skin microbiome is shaped by individuality (e.g., host genetics)15,16, temporal changes (e.g., puberty versus adulthood), anatomical location (e.g., dry vs moist skin)5,17 and environmental factors (e.g., residential climate). Illness or injury to the skin perturbs the resident microbiome, called dysbiosis, and could influence the state of human health and disease sequelae (e.g., acne).18–20 Although the bacterial constituents of the skin microbiome, the bacteriome, have undergone expansive surveys,5,21–23 phylogenetic profiling of host fungal residents in the healthy and diseased state, is in its infancy.24,25 Assessing the impact of disease on the skin mycobiome is critical to understanding fungal dynamics in skin function and immunity. Burn injuries pose a serious public health threat with more than 500 000 burned individuals seeking medical treatment, resulting in 40 000 hospitalizations and 4000 deaths annually in the United States alone.26–28 A combination of factors, including burn patient demography (e.g., age), environmental factors (e.g., inhalation exposure), burn-wound severity (e.g., size), and treatment strategy (e.g., broad-spectrum antibiotics), increases the risk of developing hard-to-treat fungal infections that can persist due largely to depletion of bacterial microbiota and a weakened host immune system.29–32 A limited number of studies have surveyed the burned skin bacteriome33,34; however, the effects of burn-injury on skin mycobiome and influence of fungi in burn-wound progression and healing process is not well understood.33,35,36 Rats are commonly used in burn research to uncover intricate molecular mechanisms and complex pathophysiology of burn-injuries,37–39 while the influence of rat microbiome in burn-wound progression and healing process remains elusive37,39–43 and is an emerging area of interest.24,25,44,45 Limited culture-dependent methods have failed to reveal many fungal and bacterial residents including those that are fastidious or uncultivable but may be critical for host health and host-microbe homeostasis.46 To address these gaps in knowledge, we used amplicon sequencing of ITS2 rRNA gene marker (fITS7-ITS4 region)47,48 to survey the mycobiome of healthy (sham-burned) and deep partial-thickness burned (PTB) rat skin when subjected to burn injury over an 11-day period. In addition to the healthy and burned rat skin, the mycobiome of the rat's surrounding environment (exposome) including food, bedding, and water were surveyed to identify potential rat skin colonization sources. Compared to the healthy skin mycobiome, there was a significant enrichment of the opportunistic pathogen, Candida, and an overall depletion of species diversity and abundance in the PTB skin. Fungal shift in PTB specimens was displayed by increased temporal stability and reduced inter-animal variation among PTB skin specimens. In response to cutaneous burn injury, the fungal community network assembly transformed toward greater fragility and susceptibility to community collapse compared to the unburned skin mycobiome.
Methods
Ethics statement and animal housing
Research was conducted in compliance with the Animal Welfare Act, the implementing Animal Welfare Regulations, and the principles of the Guide for the Care and Use of Laboratory Animals, National Research Council. The facility's Institutional Animal Care and Use Committee approved all research conducted in this study. The facility where this research was conducted is fully accredited by AAALAC International. Male Sprague Dawley rats (Rattus norvegicus) of 3 to 6 months old weighed approximately 350 to 450 grams were allowed ad libitum access to food and water. Animals were randomly assigned to groups of sham-burn (n = 30) (denoted as healthy or unburned) or subjected to deep partial-thickness burn (n = 24), after 14 day acclimation period.
Induction of burn injury and specimen collection
Induction of deep partial-thickness burn was performed as previously described.49 In brief, anesthetized rats were subjected to a scald burn (99°C for 3 second) of the dorsum that comprised ∼10% of the total body surface area (TBSA).50 The study end-points for burned rats were set for days 1, 3, 7, and 11, while the end-points for sham-burned rats (treated the same as the burned skin except for burn-injury) were days 0, 1, 3, 7, and 11 post-burn injury. Before euthanasia, blood was collected via cardiac puncture for complete blood count (CBC) using ADVIA® 120 hematology system (Siemens Healthcare, Tarrytown, NY, USA). After euthanasia, the burn wound was excised, and tissue biopsies collected with a 7 millimeter biopsy punch. Biopsy punch specimens were immediately frozen in liquid nitrogen and stored at −80°C until processed for DNA extraction. To investigate the rat exposome mycobiome, environmental specimens were collected from rat's bedding (Anderson Bed-o’Cobs Corn Cob bedding, Maumee, OH, USA), food (Purina rodent diet 5001), nutritional supplement (HydroGel®), and water (chlorinated RO water) for immediate DNA extraction.
Genomic DNA isolation, ITS2 rRNA gene marker amplification and sequencing
Burned and sham-burned skin biopsies were subjected to bead-based cell lysis and genomic DNA extraction using BioFire Platinum Path IT 1-2-3 DNA/RNA Extraction Kit (BioFire Defense, Murray, UT, USA), per manufacturer's instructions. Purified DNA was assessed for quality (260/280) and quantity (ng/ml) using UV-spectrophotometer (NanoDrop™ 2000, Thermo Fisher Scientific, Waltham, MA, USA) and Qubit® fluorimeter (Life Technologies™, Carlsbad, CA, USA), respectively. To reduce length-based polymerase chain reaction (PCR) bias and increase specificity for fungi, qualified DNA extracts underwent PCR amplification targeting fungal internal transcribed spacer 2 (ITS2) rRNA gene marker (forward fITS7 primer: 5′-GTGARTCATCGAATCTTTG-3′47; reverse ITS4 primer: 5′-TCCTCCGCTTATTGATATGC-3′48) described by Ihmark et al.47 as annealing to the 5.8S and LSU rRNA genes flanking the ITS2 region. This was followed by indexing of target ITS2 rRNA gene marker region with Illumina adapters. For primer-dimer removal, ITS2 rRNA amplicons underwent magnetic-bead purification using the Agencourt® AMPure® XP kit (Beckman Coulter Genomics, Indianapolis, IN, USA). Integrity of ITS2 rRNA amplicon libraries was examined using gel electrophoresis with Agilent D1000 screentape and Agilent 2200 TapeStation system (Agilent Technologies, Santa Clara, CA, USA). DNA concentration of ITS2 rRNA gene marker amplicon libraries was calculated using Qubit® fluorimeter. Indexing of ITS2 rRNA gene amplicon libraries was conducted using the Illumina manufacturer's instructions. Bioanalyzer 2100 system (Agilent Technologies) and Qubit® fluorimeter were used to assess quality and quantity of indexed amplicon libraries, respectively. The ITS2 rRNA gene marker amplicon libraries were pooled at equimolar concentration with 20–30% PhiX spike-in control. The Illumina MiSeq platform (two 300-bp paired-end [PE] chemistry) with MiSeq Reagent V3 chemistry (600 cycle, MS-102-3003) was employed for amplicon sequencing.
Data availability
Sequenced reads were submitted to the NCBI Sequence Read Archive and are accessible under BioProject PRJNA476563.
Computational bioinformatics analyses
Generated FASTQ files from fungal ITS2 rRNA gene marker amplicon sequencing were preprocessed using a custom pipeline workflow available in GitHub.51 In brief, demultiplexed paired-end (PE) reads were merged using PEAR52 and quality filtered using FASTQC.53 High-quality sequenced reads were subjected to the PIPITS pipeline54 for ITS2 processing using the ITS2 reference database.55 The extracted ITS2 target region was clustered into operational taxonomic units (OTUs) at 97% sequence homology threshold (genus level) using VSEARCH,56 and chimera was removed using UNITE UCHIME (2017 version) reference data set. Taxonomy was assigned to OTUs using RDP classifier.55,57 Fungal ITS2 rRNA samples were rarefied to the fraction depth of most depauperate specimen, 20000 subsampling per sample. Alpha-diversity estimation of fungal skin communities were conducted using Chao1, Shannon diversity index, Faith's phylogenetic distance (PD),58,59 Simpson diversity index, number of observed species (richness), and Good's coverage measures with QIIME alpha_diversity.py script.60 Beta-diversity was calculated with Bray-Curtis index,61 using the QIIME beta_diversity.py script.60 Correlational study of rat skin mycobiome in relation with rat skin metadata was performed for postoperative day (POD) and CBC measurements (File S1). Statistical analyses were conducted in R62 and Python (www.python.prg), as applicable. Statistical significance across groups (sham-burned vs. burned skin) was measured using PERMANOVA, PERMDISP, and ANOSIM using QIIME scripts. Fungal skin community differences were measured with 1000 permutations and P-values corrected using false discovery rate (FDR).63 Kruskal-Wallis test was applied for cross-comparison testing of taxonomic assignments across sample groups. Core mycobiome profiling was performed using QIIME compute_core_microbiome.py script at threshold of 100% (maximum fraction of core) and 50% (minimum fraction of core) skin fungal resident membership.
Inference of skin fungal ecological network in health and disease
Sparse InversE Covariance Estimation for Ecological Association Inference (SPIEC-EASI) pipeline64 was used to construct fungal networks and investigate changes in assembly of the skin mycobiome network following burn injury.64,65 For inverse covariance estimation, neighborhood selection approach of Mein-shausen and Buhlmann (MB) method was selected,66 and optimal sparsity parameters were based on the stability approach and regularization selection (StARS).67 The skin mycobiome network data was visualized and analyzed using R package igraph (http://igraph.org/r) and seqtime (hallucigenia-sparsa/seqtime). OTUs present as singleton (n < 2) in each sample and present in fewer than 18% of specimen groups (minimal occurrence cutoff possible for this study) of healthy (2 out of 11 specimens) or burned (5 out of 23 specimens) skin group were removed. For network assembly comparison of healthy versus burned skin, edge and node degree, assortativity, transitivity, average path length, and modularity was investigated.
Results
Mammalian microbial diversity is not limited to the bacteriome; fungal residents can also play an integral role in maintaining human health, microbial community stability, and disease development.12,24,68–71 Of the 30 studied healthy skin specimens, 11 were positive for ITS2 rRNA gene marker (36.6%) in the healthy skin community, while of the 24 studied burned skin specimens, 23 were positive (95.8%) for presence of fungal ITS2 rRNA gene marker in the PTB skin community. Fungal rRNA gene marker was detected in specimens collected from rat's exposome. Only one healthy skin specimen from day 7 and one from day11 tested positive for fungi rRNA gene marker preventing a definitive statistical conclusion about fungal community of healthy skin community at those specific time points.
Within-community (alpha diversity) analysis of rat skin mycobiome
Alpha diversity rarefied accumulation curves for Good's coverage estimate exhibited curves that were stabilizing, indicating appropriate representation of majority of the target ecosystems (Fig. S1).72 Overall, the healthy rat skin ecosystem's alpha-diversity measures of species richness, evenness, abundance, and phylogenetic diversity were greater compared to PTB skin or the exposome ecosystems (Fig. 1). When temporal changes were considered, PTB skin mycobiome exhibited a drastic decline in species richness, evenness, and diversity by POD 7 and 11, while the healthy skin mycobiome appeared to decline at POD 3 but exhibited a rise by POD 11.
Figure 1.
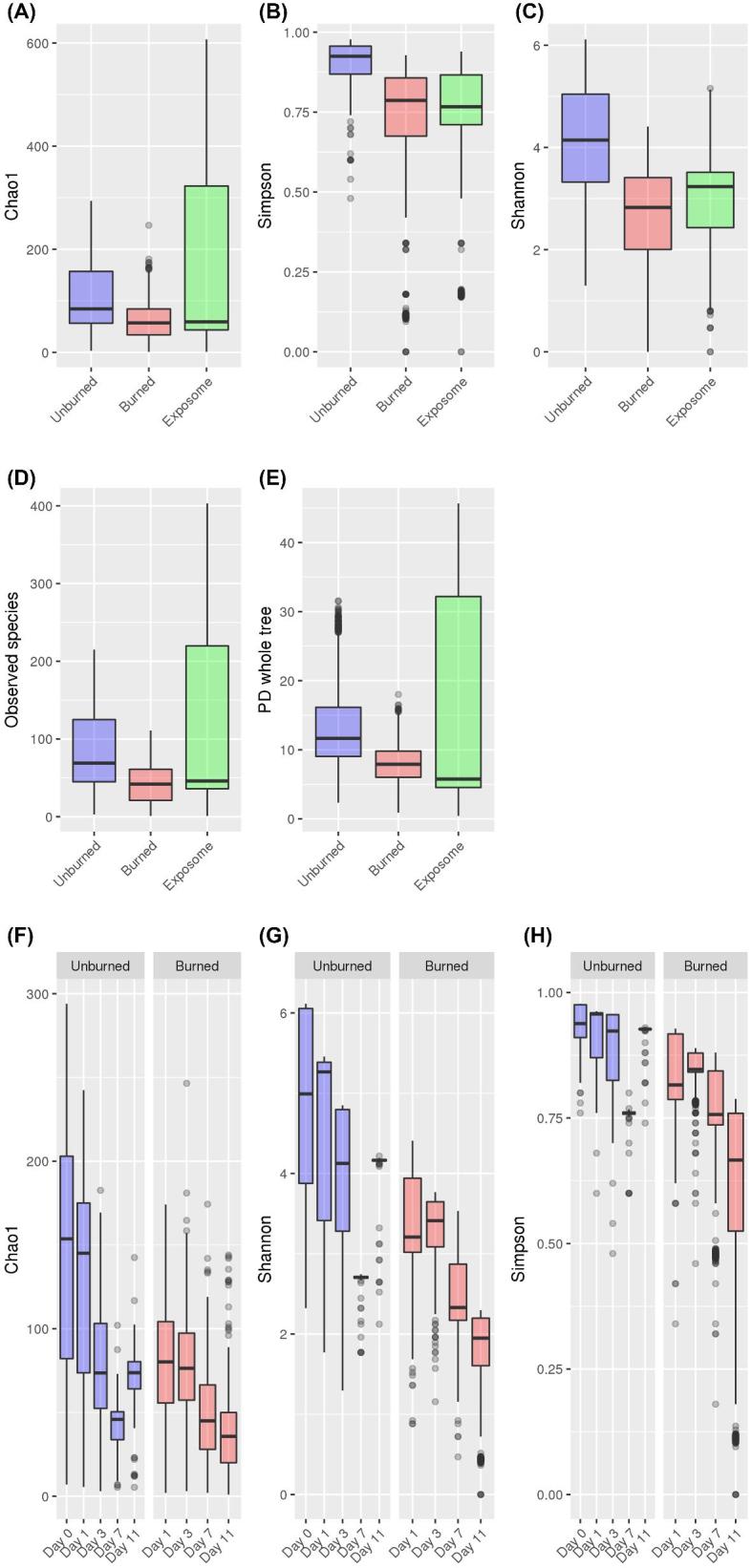
Within-community (alpha) diversity longitudinal analysis of healthy and burned rat skin and exposome mycobiome. (A) With the greatest variability observed in the rat exposome, the species richness estimate of healthy (unburned) skin was slightly higher than the PTB skin community. (B) Fungal species abundance was highest in the healthy rat skin when compared to the mycobiome of the burned skin and the exposome. (C) Species evenness of the healthy skin mycobiome was the highest relative to the burned skin and the exposome. (D) The healthy rat skin mycobiome harbored the most number of unique OTUs per community (richness) than the burned skin. Greatest variability was observed in the rat exposome. (E) Phylogenetic diversity of the PTB skin mycobiome was reduced compared to the healthy skin with greatest variability observed in the rat exposome mycobiome. (F) Species richness estimate generated from alpha diversity rarefactions declined over time in both healthy and PTB skin mycobiome; while healthy skin specimen showed a rise by POD 11, species richness of PTB skin continued to decline. (G) Fungal species evenness showed a pattern of decline in the PTB skin mycobiome starting at POD 3, while the healthy skin specimen showed increased evenness by POD 11. (H) PTB skin showed a decreasing pattern of species abundance that continued through POD 11.
Comparative analysis of fungal communities’ taxa composition and abundance between healthy and burned skin
The phyla profile of fungi in burned versus healthy rat skin were comparable in composition and abundance, with notable differences observed at the genus level. The two major phyla identified in the healthy rat skin mycobiome were Ascomycota (71.3%) and Basidiomycota (17.3%) (Fig. 2A). Ascomycota was dominated by members of order Capnodiales (14.7%) and family Pleosporaceae (11.0%), whereas Basidiomycota was dominated by Wallemia (3.4%) genus in the healthy skin mycobiome (Fig. 2B). Similarly Ascomycota (73.3%) and Basidiomycota (18.1%) were the dominating phyla in the PTB skin (Fig. 2C). However, Ascomycota was mainly composed of Candida (24.8%) and Wallemia (2.5%) was the dominant genus of Basidiomycota in the PTB skin mycobiome (Fig. 2D). Comparable to the rat skin, the rat exposome consisted mainly of members of Ascomycota followed by Basidiomycota (Fig. S2A). Capnodiales (37.4%) was the major component of Ascomycota, while Panellus (3.1%) genus predominated Basidiomycota in the rat exposome (Fig. S2B). When fungal relative abundance in PTB skin community was examined with respect to temporal changes, Capnodiales (18.0% to 10.3%) and Nectriaceae (7.0% to 1.8%) abundance declined from POD 1 through 11, while Candida was enriched between POD 1 through 11 (13.5% to 48%) (Fig. 2D). Environmental specimens, in particular water and food, exhibited a substantially higher abundance of Capnodiales than in the rat skin mycobiome (Fig. S2B).
Figure 2.

Phylogenetic profile of rat skin mycobiome. (A) Phyla distribution in healthy rat skin mycobiome was dominated by Ascomycota and Basidiomycota. (B) Healthy rat skin mycobiome showed a degree of heterogeneity in genera distribution that was associated with temporal changes over 11-day study period. (C) Phyla profiling of compromised PTB skin mycobiome was mainly composed of Ascomycota and Basidiomycota. (D) Genera distribution exhibited variations associated with temporal changes including an increase in Candida and a decline in members of Nectriaceae, Capnodiales (Cladosporium and Cercospora), and Wallemia in relative abundance.
Pairwise comparisons (beta diversity) of healthy and burned rat skin mycobiome
Temporal stability of burned and healthy fungal skin specimens assessed over time using Bray-Curtis dissimilarity index resulted in two main clusters (Fig. 3A) separating PTB skin specimens collected on POD 7 and 11 from majority of other specimens (Fig. 3B). Partial-thickness burn specimens belonging to PODs 1 and 3 exhibited greater inter-animal heterogeneity and were scattered across the formed clusters. In contrast, PODs 7 and 11 specimens showed a drastic reduction in temporal instability and inter-animal mycobiome variation compared to all other specimens (Fig. 3B, D). Scattered distance matrix profiles of healthy skin fungal specimens in different PODs showed that the variable composition of healthy skin mycobiome was not associated with temporal changes. Comparison of rat skin mycobiome taxa profile relative to CBC measurements of rat blood exhibited an association between elevated monocyte levels (%) and enrichment of Candida (File S2, Fig. 3E). Significance analysis of differentially distributed fungi in the healthy and PTB rat skin revealed a significant enrichment of members of Candida, Fusarium, and Talaromyces genera in the PTB skin, while members of Onygenales and Plectosphaerellaceae were enriched in the healthy skin compared to the PTB skin mycobiome (FDR P < .05).
Figure 3.

Beta-diversity analysis of fungal communities in rat skin and exposome mycobiome. (A) Bray-Curtis analysis demonstrated a grouping of fungal specimens that separate a group of PTB specimens from remaining specimens. The rat exposome specimens were scattered between burned and unburned rat skin fungal communities. (B) When temporal changes are considered, majority of PTB of POD 7 and 11 skin specimens were separated into a cluster, while PTB specimens of POD 1 and 3 were scattered between the two clustering groups. The healthy skin specimens exhibited inter-animal variation across time. (C) Monocyte levels (%) obtained from CBC analysis of rat blood showed elevated monocyte values associated mostly with the PTB skin specimens of POD 7 and 11. Only a subgroup of rat skin specimens (27 rats) were randomly selected for undergoing CBC analysis. (D) Comparison distance plot of mycobiome sequence abundance (Bray-Curtis) over time for healthy and PTB rat skin specimens (Monte Carlo permutations = 2000) demonstrated a fungal shift between POD 3 and POD 7 in PTB skin relative to healthy skin community. (E) Significant enrichment of Candida in specimens exhibiting elevated monocyte levels (%) (FDR P = .03).
The core mycobiome of rat skin community
To identify the core mycobiome, the fungal community was assessed for 100% and 50% community membership in the skin mycobiome (File S3). In the context of this study, the core was considered taxa present in all of the target fungal group (100% frequency) and prevalent or common members were those identified in ≥50% of the target fungal group. Core mycobiome analysis (100% persistence) of healthy skin identified members of Ascomycota of order Capnodiales, whereas prevalent members (50% membership) consisted mostly of members of Capnodiales, Pleosporales, and Saccharomycetes. In addition to Capnodiales, core mycobiome (100% persistence) of PTB skin group was comprised of members of Nectriaceae, Candida, and Thermomyces.
Fungal network assembly in healthy versus burned skin
A considerable reduction in the number of nodes (taxa) and edges (links) was observed in the PTB skin compared to the healthy skin fungal network assembly (Fig. 4A, B, Table 1). Compared to healthy skin, the clustering strength (modularity) of the skin network assembly and number or partitioning of network clusters (potential niche) increased in the PTB skin (Table 1). Relative to the healthy skin, the average path length between the taxa in the PTB mycobiome increased and formed a fungal network lacking a core network structure (Fig. 4B). Estimated probability that close taxa were connected (network transitivity) demonstrated that burned skin fungal network formed more complex network structures than those depicted in the healthy skin. Healthy skin harbored fewer clusters than the PTB skin network (Table 1). However, there were a greater number of OTUs associated with most clusters in the healthy skin than the burned skin network. Compared to the healthy skin, the predicted average degree distribution of taxa in PTB skin network assembly declined (Fig. S3A), while the degree of homophily (assortativity) increased between associated taxa (Table 1). Comparison of network stability using attack robustness estimate revealed higher degree of network stability in the healthy skin than the PTB skin mycobiome, which depicted increased fragility or vulnerability towards community collapse (Fig. S3B).
Figure 4.

Rat skin mycobiome network structure analysis in healthy and burned skin. (A) The healthy skin mycobiome network displayed a large connected network component that was dominated by members of Ascomycota. (B) The PTB skin fungal network assembly exhibited one large connected component along with smaller disconnected networks (e.g., singleton, dyad, and triad). Each node is colored by phyla and edges between nodes represent predicted interactions.
Table 1.
Comparison of rat skin fungal network assembly in healthy versus burned skin.
| Group ID | Edge degree (total) | Degree distribution (average) | Assortativity (r, homophily degree) | Transitivity (T, global) | Average path length (mean distance) | Modularity (Q, strength of cluster separation) |
|---|---|---|---|---|---|---|
| Healthy skin | 486 | 3.811765 | 0.1741892 | 0.2491857 | 4.72725 | 0.5933399 |
| PTB skin | 246 | 2.877193 | 0.3231011 | 0.07147221 | 7.295518 | 0.8063653 |
Discussion
Community structure and temporal stability differs in healthy versus burned skin mycobiome
In the absence of a human burn-wound cutaneous mycobiome study, we are unable to perform a comprehensive comparison of burned rat skin to a human burned skin. Compared to the human healthy skin, the rat skin specimens were rarely colonized by members of Basidiomycota of Malassezia genus, which are abundant members of human sebaceous skin (e.g., dorsum).25,71 Instead, the healthy and burned rat dorsum were dominated by members of Ascomycota; fungi classified as members of Onygenales and Sordariomycetes were significantly enriched in the healthy skin compared to the PTB skin and part of the healthy skin core mycobiome. Similar phyla proportions have been reported in mycobiome of human chronic diabetic foot ulcers (DFU),73 and dominance of Ascomycota members has been described in the arms and lower extremities (e.g., popliteal crease) of healthy human skin.71 While differential distribution of fungi across different human body sites has been reported,71,74 fungal distribution associated with spatial diversity of the rat skin is not yet explored, and we have only identified the rat dorsal mycobiome as the common experimental site for burn and wound-healing research. Influence of temporal and spatial differences of the microbiome structure on community function and the host's cutaneous or systemic immune response remains largely unknown and an emerging area of research.15,36,75,76 Furthermore, the extent to which microbiome density and composition of chronic wounds differs from acute wounds, specifically in burn-wounds, has not been explored.
The PTB rat skin microbiome reported a higher percentage of fungi-positive skin specimens (>90%) than past fungal survey of more than 900 human chronic wounds (23%).44 Analogous to a fungal survey of the human chronic ulcer wound,44 we observed a significant enrichment of Candida in burn-wounds compared to the healthy rat skin mycobiome (File S2). In fact, the relative abundance of Candida was substantially increased from POD 1 (13.5%) through 11 (48%) post-burn injury. The core mycobiome of the burned rat skin (100% persistence) carried a diverse assortment of fungi compared to the healthy skin, signaling a shift in fungal community structure (File S3). This shift is accompanied by a decline in temporal instability, inter-animal heterogeneity (Fig. 3B) and expansion of the core community from only members of Capnodiales to the addition of other fungi including members of Candida and Nectriaceae (File S3). It is possible that changes in the skin bacterial dynamics (e.g., depletion) and impairment of host-immune response during burn injury facilitated the changes in composition and abundance of the dysbiotic PTB skin mycobiome, compared to the healthy skin.
Within-community fungal dversity declines in burned skin mycobiome
Human microbiome studies have largely exhibited an inverse relationship between microbial alpha diversity and disease severity;33,36,77 similarly, the PTB rat skin mycobiome exhibited reduced alpha diversity profile compared to the healthy skin. The transient decline observed in alpha diversity of the healthy rat skin mycobiome could be influenced by mechanical (e.g., shaving) or chemical stress (e.g., hair depilation) introduced to the healthy (sham-burned) rat skin. While pretreatment of animal skin prior to induction of burn-injury is a common practice (e.g., rat, pig, and mice) to remove dense hair and achieve the desired burn depth; the extent to which pretreatment of the animal skin itself influences the burn-wound outcome and disease progression has not been investigated. The sham-burned rat skin specimens harbored a higher level of species richness and abundance compared to PTB skin mycobiome. The availability of one fungi positive skin specimen for POD 7 and 11 in healthy skin group of our study coupled with low rate of fungi positive human volunteers44 highlights the need to sample a larger healthy population for future mycobiome studies and gain better insight into the mycobiome of healthy ecosystems to account for possible lack of detectable fungi in healthy groups in future studies.
Reduction in temporal instability in burned skin mycobiome
The healthy rat skin mycobiome demonstrated greater degree of inter-animal variation compared to the PTB skin specimens. Comparable to past microbiome studies of diseased versus healthy host,10,33,77–79 increased temporal stability and reduced inter-animal mycobiome variation (Bray-Curtis index) (Fig. 3B) in the burned (dysbiotic) rat skin mycobiome was observed as a shift toward single species dominance (e.g., Candida) by POD 7 in an immunocompromised host with disrupted skin ecosystem. Physiological and environmental differences likely play an important role in the prevalence and abundance of resident microbes in different hosts80–84 and manipulation of such factors could lead to a remodeling of host resident microbe structure and its potential function. Possible influences of different wound therapies (e.g., antimicrobials) on skin mycobiome composition and abundance or its implications on skin barrier integrity and clinical outcome remains poorly understood. Potential benefits of reintroducing microbial heterogeneity or temporal instability to a dysbiotic burned skin microbiome by exploitation of the microbiome structure and introduction of bio-therapeutic agents (e.g., microbial transplantation) has been suggested but remains to be tested.33,85 Follow-up studies to investigate the intricate and complex cross-kingdom interactions (e.g., pathway landscape) with the mycobiome and potential implications on the health of an ecosystem and differential modulation of the host immune response post-burn injury are needed as a first step towards development of alternative therapies.
Reorganization of burned skin mycobiome network assembly
Comparison of healthy versus PTB rat skin fungal network assembly revealed reorganization of PTB fungal community into a fragile network that was more niche-based with increased susceptibility to collapse after cutaneous burn injury. Similar observations of increased network fragility and reduced complexity are reported for mice gut microbiome post antibiotics treatment86 or in human skin microbiome in megacities compared to rural areas caused by changes in climate and socioeconomic status.87 The fungal network derived from healthy rat skin mycobiome implied that healthy skin clusters exercised more association among each other than compared to the PTB skin fungal network. Comparable trends in network structure is reported for healthy human scalp demonstrating increased network stability and connectedness compared to individuals with seborrheic dermatitis and dandruff.88 Inferring microbial interactions among different species in a community and unraveling their influence on the health of the resident ecosystem and influence on host factors (e.g., immune system) is of great importance in ecology and medicine for microbiome remodeling research.
An impaired host immune system and application of broad-spectrum antibiotics as standard of care in burn-injury patients provides the ideal condition for overgrowth of opportunistic fungi that might otherwise be commensal in an immune-competent host.89 Considering that fungi are linked with clinical complications of human skin diseases (e.g., seborrheic dermatitis), injuries (e.g., burn wounds), and disease outcomes (e.g., diabetic ulcers),18,73,89–92 understanding the role of skin fungal residents in host immune regulation and host-microbiome homeostasis is necessary for improvement of therapeutic strategies for skin disorders and injuries. The longitudinal aspect of our rat skin burn study established the dynamics of fungal diversity and abundance in the perturbed rat skin mycobiome in response to cutaneous thermal trauma. With respect to the rat's immune response to cutaneous burn injury, elevated blood monocyte levels (%) were observed, especially in specimens of PODs 7 and 11 post-burn injury (Fig. 3C). These circulating monocytes differentiate to macrophages at injury site and are vital in tissue remodeling of burn-wound and host immune defense.93 To our knowledge, this is the first fungal survey of cutaneous mycobiome of burn wounds in an effort to unravel the diversity and abundance of rat skin resident fungi pre- and post-burn injury, not previously possible through culture-based approaches. As a major etiological agent of burn-wound infections (e.g., Candida and Aspergillus), further studies addressing the influence of the mycobiome's composition and abundance in disease progression after cutaneous burn-injury with and without burn-wound standard of care treatment will be necessary for clinical improvement of burn-wound outcome and severity.
Supplementary Material
Acknowledgments
The work was supported by the Intramural Program [G_001-2016-USAISR], Combat Casualty Care Research Directorate, US Army Medical Research and Materiel Command and the Naval Medical Research Center's Advanced Medical Development Program [N3239815MHX040]. We would like to thank the expertise of Mr. Michael T. Strosaker in development of computational pipelines (available in Github), workstation setup, and data-related consultations. We thank and acknowledge efforts of Drs. Zhao Lai and Brian Hermann in conducting the amplicon sequencing portion of the project. Sequencing was conducted with assistance of Dr. Brian Hermann at the UTSA Genomics Core [NIH grant G12MD007591, and NSF grant DBI-1337513] and with the assistance of Dr. Zhao Lai, Mrs. Dawn Garcia, and Korri Weldon in the Greehey Children's Cancer Research Institute's Genome Sequencing Facility supported by UT Health San Antonio [NIH-NCI P30 CA054174] (Cancer Center Support Grant).
DOD disclaimer
The opinions or assertions contained herein are the private views of the authors and are not to be construed as official or as reflecting the views of the Department of the Army or the Department of Defense.
Declaration of interest
The authors report no conflicts of interest and the authors are solely responsible for the writing and content of this paper.
References
- 1. Neuman H, Koren O. The pregnancy microbiome. Nestle Nutr Inst Workshop Ser. 2017; 88: 1–9. [DOI] [PubMed] [Google Scholar]
- 2. Cho I, Blaser MJ. The human microbiome: at the interface of health and disease. Nat Rev Genet. 2012; 13: 260–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kaminska D, Gajecka M. Is the role of human female reproductive tract microbiota underestimated? Benef Microbes. 2017; 8: 327–343. [DOI] [PubMed] [Google Scholar]
- 4. Oh J, Byrd AL, Deming C et al.. Biogeography and individuality shape function in the human skin metagenome. Nature. 2014; 514: 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Grice EA, Kong HH, Conlan S et al.. Topographical and temporal diversity of the human skin microbiome. Science. 2009; 324: 1190–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rosenthal M, Goldberg D, Aiello A, Larson E, Foxman B. Skin microbiota: microbial community structure and its potential association with health and disease. Infect Genet Evol. 2011; 11: 839–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Guani-Guerra E, Santos-Mendoza T, Lugo-Reyes SO, Teran LM. Antimicrobial peptides: general overview and clinical implications in human health and disease. Clinical immunology. 2010; 135: 1–11. [DOI] [PubMed] [Google Scholar]
- 8. Zackular JP, Baxter NT, Iverson KD et al.. The gut microbiome modulates colon tumorigenesis. MBio. 2013; 4: e00692–00613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kang D, Shi B, Erfe MC, Craft N, Li H. Vitamin B12 modulates the transcriptome of the skin microbiota in acne pathogenesis. Sci Transl Med. 2015; 7: 293ra103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Belizario JE, Napolitano M. Human microbiomes and their roles in dysbiosis, common diseases, and novel therapeutic approaches. Front Microbiol. 2015; 6: 1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Grice EA. The skin microbiome: potential for novel diagnostic and therapeutic approaches to cutaneous disease. Semin Cutan Med Surg. 2014; 33: 98–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Harada K, Saito M, Sugita T, Tsuboi R. Malassezia species and their associated skin diseases. J Dermatol. 2015; 42: 250–257. [DOI] [PubMed] [Google Scholar]
- 13. Smeekens SP, Huttenhower C, Riza A et al.. Skin microbiome imbalance in patients with STAT1/STAT3 defects impairs innate host defense responses. J Innate Immun. 2014; 6: 253–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mayer FL, Wilson D, Hube B. Candida albicans pathogenicity mechanisms. Virulence. 2013; 4: 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Naik S, Bouladoux N, Wilhelm C et al.. Compartmentalized control of skin immunity by resident commensals. Science. 2012; 337: 1115–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Napflin K, Schmid-Hempel P. Immune response and gut microbial community structure in bumblebees after microbiota transplants. Proc Biol Sci. 2016; 283: 20160312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Grice EA, Kong HH, Renaud G et al.. A diversity profile of the human skin microbiota. Genome Res. 2008; 18: 1043–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gaitanis G, Magiatis P, Hantschke M, Bassukas ID, Velegraki A. The Malassezia genus in skin and systemic diseases. Clin Microbiol Rev. 2012; 25: 106–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sikorska H, Smoragiewicz W. Role of probiotics in the prevention and treatment of meticillin-resistant Staphylococcus aureus infections. Int J Antimicrob Agents. 2013; 42: 475–481. [DOI] [PubMed] [Google Scholar]
- 20. Liu J, Yan R, Zhong Q et al.. The diversity and host interactions of Propionibacterium acnes bacteriophages on human skin. ISME J. 2015; 9: 2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Grice EA, Segre JA. The skin microbiome. Nat Rev Microbiol. 2011; 9: 244–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Edmonds-Wilson SL, Nurinova NI, Zapka CA, Fierer N, Wilson M. Review of human hand microbiome research. Journal of dermatological science. 2015; 80: 3–12. [DOI] [PubMed] [Google Scholar]
- 23. Byrd AL, Belkaid Y, Segre JA. The human skin microbiome. Nat Rev Microbiol. 2018; 16: 143–155. [DOI] [PubMed] [Google Scholar]
- 24. Tipton L, Muller CL, Kurtz ZD et al.. Fungi stabilize connectivity in the lung and skin microbial ecosystems. Microbiome. 2018; 6: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jo JH, Kennedy EA, Kong HH. Topographical and physiological differences of the skin mycobiome in health and disease. Virulence. 2017; 8: 324–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nielson CB, Duethman NC, Howard JM, Moncure M, Wood JG. Burns: Pathophysiology of systemic complications and current management. J Burn Care Res. 2017; 38: e469–e481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rowan MP, Cancio LC, Elster EA et al.. Burn wound healing and treatment: review and advancements. Critical Care. 2015; 19: 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Colohan SM. Predicting prognosis in thermal burns with associated inhalational injury: a systematic review of prognostic factors in adult burn victims. J Burn Care Res. 2010; 31: 529–539. [DOI] [PubMed] [Google Scholar]
- 29. Luo G, Peng Y, Yuan Z, Cheng W, Wu J, Fitzgerald M. Yeast from burn patients at a major burn centre of China. Burns. 2011; 37: 299–303. [DOI] [PubMed] [Google Scholar]
- 30. Moore EC, Padiglione AA, Wasiak J, Paul E, Cleland H. Candida in burns: risk factors and outcomes. J Burn Care Res. 2010; 31: 257–263. [DOI] [PubMed] [Google Scholar]
- 31. Jarvis WR. Epidemiology of nosocomial fungal infections, with emphasis on Candida species. Clin Infect Dis. 1995; 20: 1526–1530. [DOI] [PubMed] [Google Scholar]
- 32. Ballard J, Edelman L, Saffle J et al.. Positive fungal cultures in burn patients: a multicenter review. J Burn Care Res. 2008; 29: 213–221. [DOI] [PubMed] [Google Scholar]
- 33. Plichta JK, Gao X, Lin H et al.. Cutaneous burn injury promotes shifts in the bacterial Mmcrobiome in autologous donor skin: implications for skin grafting outcomes. Shock. 2017; 48: 441–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu SH, Huang YC, Chen LY, Yu SC, Yu HY, Chuang SS. The skin microbiome of wound scars and unaffected skin in patients with moderate to severe burns in the subacute phase. Wound Repair Regen. 2018; 26: 182–191. [DOI] [PubMed] [Google Scholar]
- 35. Plichta JK, Holmes CJ, Nienhouse V et al.. Cutaneous burn injury modulates urinary antimicrobial peptide responses and the urinary microbiome. Crit Care Med. 2017; 45: e543–e551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Earley ZM, Akhtar S, Green SJ et al.. Burn injury alters the intestinal microbiome and increases gut permeability and bacterial translocation. PLoS One. 2015; 10: e0129996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Walker HL, Mason AD Jr.. A standard animal burn. J Trauma. 1968; 8: 1049–1051. [DOI] [PubMed] [Google Scholar]
- 38. Abdullahi A, Amini-Nik S, Jeschke MG. Animal models in burn research. Cell Mol Life Sci. 2014; 71: 3241–3255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Walker HL, Mason AD Jr., Raulston GL. Surface infection with Pseudomonas aeruginosa. Ann Surg. 1964; 160: 297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sayeed MM. Inflammatory/cardiovascular-metabolic responses in a rat model of burn injury with superimposed infection. Shock. 2005; 24: 40–44. [DOI] [PubMed] [Google Scholar]
- 41. Eloy R, Cornillac AM. Wound healing of burns in rats treated with a new amino acid copolymer membrane. Burns. 1992; 18: 405–411. [DOI] [PubMed] [Google Scholar]
- 42. Campelo AP, Campelo MW, Britto GA, Ayala AP, Guimaraes SB, Vasconcelos PR. An optimized animal model for partial and total skin thickness burns studies. Acta Cir Bras. 2011; 26: 38–42. [DOI] [PubMed] [Google Scholar]
- 43. Cai EZ, Ang CH, Raju A et al.. Creation of consistent burn wounds: a rat model. Arch Plas Srg. 2014; 41: 317–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dowd SE, Delton Hanson J, Rees E et al.. Survey of fungi and yeast in polymicrobial infections in chronic wounds. J Wound Care. 2011; 20: 40–47. [DOI] [PubMed] [Google Scholar]
- 45. Ward TL, Dominguez-Bello MG, Heisel T, Al-Ghalith G, Knights D, Gale CA. Development of the human mycobiome over the first mMonth of life and across body sites. mSystems. 2018; 3: pii: e00140-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Myles IA, Reckhow JD, Williams KW, Sastalla I, Frank KM, Datta SK. A method for culturing Gram-negative skin microbiota. BMC Microbiol. 2016; 16: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ihrmark K, Bodeker IT, Cruz-Martinez K et al.. New primers to amplify the fungal ITS2 region–evaluation by 454-sequencing of artificial and natural communities. FEMS Microbiol Ecol. 2012; 82: 666–677. [DOI] [PubMed] [Google Scholar]
- 48. White TJ BT, Lee S, Taylor J Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfland DH, Sninsky JJ, White TJ, eds. PCR Protocols: A Guide to Methods and Applications. Cambridge, MA: Academic Press, 1990: 313–322. [Google Scholar]
- 49. Brandenburg KS, Weaver AJ Jr., Qian L et al.. Development of Pseudomonas aeruginosa biofilms in partial-thickness burn wounds using a Sprague-Dawley rat model. J Burn Care Res. 2019; 40: 44–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gilpin DA. Calculation of a new Meeh constant and experimental determination of burn size. Burns. 1996; 22: 607–611. [DOI] [PubMed] [Google Scholar]
- 51. Strosaker MT. Microbiome-analysis-pipeline. 2017; https://github.com/mstrosaker/microbiome-analysis-pipeline/releases/tag/v0.1. [Google Scholar]
- 52. Zhang J, Kobert K, Flouri T, Stamatakis A. PEAR: a fast and accurate illumina paired-end read merger. Bioinformatics. 2014; 30: 614–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Brown J, Pirrung M, McCue LA. FQC Dashboard: integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics. 2017. doi: 10.1093/bioinformatics/btx373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gweon HS, Oliver A, Taylor J et al.. PIPITS: an automated pipeline for analyses of fungal internal transcribed spacer sequences from the Illumina sequencing platform. Methods Ecol Evol. 2015; 6: 973–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ankenbrand MJ, Keller A, Wolf M, Schultz J, Forster F. ITS2 Database V: Twice as Much. Mol Biol Evol. 2015; 32: 3030–3032. [DOI] [PubMed] [Google Scholar]
- 56. Rognes T, Flouri T, Nichols B, Quince C, Mahe F. VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016; 4: e2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wang Q, Garrity GM, Tiedje JM, Cole JR. Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microbiol. 2007; 73: 5261–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Faith DP. The role of the phylogenetic diversity measure, PD, in bio-informatics: getting the definition right. Evol Bioinform Online. 2007; 2: 277–283. [PMC free article] [PubMed] [Google Scholar]
- 59. Faith DP, Baker AM. Phylogenetic diversity (PD) and biodiversity conservation: some bioinformatics challenges. Evol Bioinform Online. 2007; 2: 121–128. [PMC free article] [PubMed] [Google Scholar]
- 60. Caporaso JG, Kuczynski J, Stombaugh J et al.. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010; 7: 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Bray JR, Curtis JT. An ordination of the upland forest communities of Southern Wisconsin. Ecological Monographs. 1957; 27: 325–349. [Google Scholar]
- 62. Team R. RStudio: Integrated development for R. 2015; http://www.rstudio.com/. [Google Scholar]
- 63. Benjamini Y, Drai D, Elmer G, Kafkafi N, Golani I. Controlling the false discovery rate in behavior genetics research. Behav Brain Res. 2001; 125: 279–284. [DOI] [PubMed] [Google Scholar]
- 64. Kurtz ZD, Muller CL, Miraldi ER, Littman DR, Blaser MJ, Bonneau RA. Sparse and compositionally robust inference of microbial ecological networks. PLoS Comput Biol. 2015; 11: e1004226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhao T, Liu H, Roeder K, Lafferty J, Wasserman L. The huge package for high-dimensional undirected graph estimation in R. J Mach Learn Res. 2012; 13: 1059–1062. [PMC free article] [PubMed] [Google Scholar]
- 66. Meinshausen NB, Peter. High-dimensional graphs and variable selection with the lasso. Ann Stat. 2006; 34: 1436–1462. [Google Scholar]
- 67. Liu H, Roeder K, Wasserman L. Stability approach to regularization selection (StARS) for high dimensional graphical models. Adv Neural Inf Process Syst. 2010; 24: 1432–1440. [PMC free article] [PubMed] [Google Scholar]
- 68. Rizzetto L, De Filippo C, Cavalieri D. Richness and diversity of mammalian fungal communities shape innate and adaptive immunity in health and disease. Eur J Immunol. 2014; 44: 3166–3181. [DOI] [PubMed] [Google Scholar]
- 69. Peleg AY, Hogan DA, Mylonakis E. Medically important bacterial-fungal interactions. Nat Rev Microbiol. 2010; 8: 340–349. [DOI] [PubMed] [Google Scholar]
- 70. Chehoud C, Albenberg LG, Judge C et al.. Fungal signature in the gut microbiota of pediatric patients with inflammatory bowel disease. Inflamm Bowel Dis. 2015; 21: 1948–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Findley K, Oh J, Yang J et al.. Topographic diversity of fungal and bacterial communities in human skin. Nature. 2013; 498: 367–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hughes JB, Hellmann JJ, Ricketts TH, Bohannan BJ. Counting the uncountable: statistical approaches to estimating microbial diversity. Appl Environ Microbiol. 2001; 67: 4399–4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kalan L, Loesche M, Hodkinson BP et al.. Redefining the chronic-wound microbiome: fungal communities are prevalent, dynamic, and associated with delayed healing. MBio. 2016; 7: pii: e01058-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sugita T, Yamazaki T, Makimura K et al.. Comprehensive analysis of the skin fungal microbiota of astronauts during a half-year stay at the International Space Station. Med Mycol. 2016; 54: 232–239. [DOI] [PubMed] [Google Scholar]
- 75. Valvis SM, Waithman J, Wood FM, Fear MW, Fear VS. The immune response to skin trauma is dependent on the etiology of injury in a mouse model of burn and excision. J Invest Dermatol. 2015; 135: 2119–2128. [DOI] [PubMed] [Google Scholar]
- 76. Kuethe JW, Armocida SM, Midura EF et al.. Fecal microbiota transplant restores mucosal integrity in a murine model of burn injury. Shock. 2016; 45: 647–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Loesche M, Gardner SE, Kalan L et al.. Temporal stability in chronic wound microbiota is associated with poor healing. J Invest Dermatol. 2017; 137: 237–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Huang G, Sun K, Yin S et al.. Burn injury leads to increase in relative abundance of opportunistic pathogens in the rat gastrointestinal microbiome. Front Microbiol. 2017; 8: 1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Shreiner AB, Kao JY, Young VB. The gut microbiome in health and in disease. Curr Opin Gastroenterol. 2015; 31: 69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Zhang M, Jiang Z, Li D et al.. Oral antibiotic treatment induces skin microbiota dysbiosis and influences wound healing. Microb Ecol. 2015; 69: 415–421. [DOI] [PubMed] [Google Scholar]
- 81. Devine DA, Marsh PD, Meade J. Modulation of host responses by oral commensal bacteria. J Oral Microbiol. 2015; 7: 26941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Cho I, Yamanishi S, Cox L et al.. Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature. 2012; 488: 621–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Manus MB, Yu JJ, Park LP et al.. Environmental influences on the skin microbiome of humans and cattle in rural Madagascar. Evol Med Public Health. 2017; 2017: 144–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Dreno B, Bettoli V, Araviiskaia E, Sanchez Viera M, Bouloc A. The influence of exposome on acne. J Eur Acad Dermatol Venereol. 2018; 32: 812–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Myles IA, Williams KW, Reckhow JD et al.. Transplantation of human skin microbiota in models of atopic dermatitis. JCI Insight. 2016; 1: pii: 86955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Ruiz VE, Battaglia T, Kurtz ZD et al.. A single early-in-life macrolide course has lasting effects on murine microbial network topology and immunity. Nat Commun. 2017; 8: 518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kim HJ, Kim H, Kim JJ et al.. Fragile skin microbiomes in megacities are assembled by a predominantly niche-based process. Sci Adv. 2018; 4: e1701581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Park T, Kim HJ, Myeong NR et al.. Collapse of human scalp microbiome network in dandruff and seborrhoeic dermatitis. Exp Dermatol. 2017; 26: 835–838. [DOI] [PubMed] [Google Scholar]
- 89. Struck MF, Gille J. Fungal infections in burns: a comprehensive review. Ann Burns Fire Disasters. 2013; 26: 147–153. [PMC free article] [PubMed] [Google Scholar]
- 90. Glatz M, Bosshard PP, Hoetzenecker W, Schmid-Grendelmeier P. The role of Malassezia spp. in atopic dermatitis. J Clin Med. 2015; 4: 1217–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Javad G, Taheri Sarvtin M, Hedayati MT, Hajheydari Z, Yazdani J, Shokohi T. Evaluation of Candida colonization and specific humoral responses against Candida albicans in patients with atopic dermatitis. BioMed Res Int. 2015; 2015: 849206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Seebacher C, Bouchara JP, Mignon B. Updates on the epidemiology of dermatophyte infections. Mycopathologia. 2008; 166: 335–352. [DOI] [PubMed] [Google Scholar]
- 93. Suda S, Williams H, Medbury HJ, Holland AJ. A review of monocytes and monocyte-derived cells in hypertrophic scarring post burn. J Burn Care Res. 2016; 37: 265–272. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequenced reads were submitted to the NCBI Sequence Read Archive and are accessible under BioProject PRJNA476563.