

Abstract
Several recent studies have reported that reactive oxygen species (ROS), superoxide anion and hydrogen peroxide (H2O2), play important roles in various cellular signaling networks. NADPH oxidase (Nox) isozymes have been shown to mediate receptor-mediated ROS generation for physiological signaling processes involved in cell growth, differentiation, apoptosis, and fibrosis. Detectable intracellular levels of ROS can be induced by the electron leakage from mitochondrial respiratory chain as well as by activation of cytochrome p450, glucose oxidase and xanthine oxidase, leading to oxidative stress. The up-regulation and the hyper-activation of NADPH oxidases (Nox) also likely contribute to oxidative stress in pathophysiologic stages. Elevation of the renal ROS level through hyperglycemia-mediated Nox activation results in the oxidative stress which induces a damage to kidney tissues, causing to diabetic nephropathy (DN). Nox inhibitors are currently being developed as the therapeutics of DN. In this review, we summarize Nox-mediated ROS generation and development of Nox inhibitors for therapeutics of DN treatment.
Keywords: NADPH oxidase, Oxidative stress, Signal transduction, Kidney, Diabetic nephropathy, Nox inhibitor
INTRODUCTION
Complications of diabetes mellitus (DM) occur in the eyes (retinopathy) (Heng et al., 2013; Gorin et al., 2015; Khan et al., 2017), nerves (neuropathy) (Said, 2007; Vinik et al., 2013) and kidneys (nephropathy) (Thomas et al., 2006; Caramori et al., 2013; Forbes and Cooper, 2013; Thomas et al., 2015). Among these complications, diabetic nephropathy (DN) is the leading cause of renal failure in patients with diabetes mellitus which ultimately results in end-stage renal disease (ESRD) (Molitch et al., 2004; Thomas et al., 2015; Papadopoulou-Marketou et al., 2017). In the initial stage of diabetic nephropathy (DN), commonly observed signs include albuminuria mainly resulting from reduction of the glomerular filtration rate (GFR) (Jha et al., 2016). Increasing albuminuria, reduction of GFR or mesangial expansion, glomerulosclerosis, podocyte apoptosis, and tubulointerstitial fibrosis are well-established features seen in DN progression (Jha et al., 2016). Generally, DN can be divided into multiple stages; (1) increased GFR (hyperfiltration) phase with increased volume of the kidney; (2) normal filtration phase with thickening of the glomerular basement membrane (GBM); (3) microalbuminuria (>30 mg/day); (4) macro-albuminuria (>300 mg/day); (5) uremia with ESRD (Rocco and Berns, 2009; Palatini, 2012; Reidy et al., 2014; Lv et al., 2015). In addition to functional changes, structural changes are also observed; glomerular basement membrane (GBM) thickening, mesangial expansion and overexpression of ECM proteins, tubulointerstitial fibrosis and glomeruolsclerosis (Mason and Wahab, 2003; Tervaert et al., 2010; Caramori et al., 2013). Several lines of evidence strongly suggest that oxidative stress serves as an etiological factor in the initiation and progression of DN (Jha et al., 2016). In particular, renal oxidative stress is known to be closely associated with the activation or the overexpression of various NADPH oxidase (Nox) isozymes (Jha et al., 2016). This review will discuss about the function of Nox isozymes in pathology of DN and recent on-going development of Nox inhibitors for treatment of DN.
STRUCTURE AND REGULATION OF NOX ISOZYMES
The gp91phox (renamed as Nox2) was the first identified isozyme, and it was in phagocytic cells. Subsequently, six additional homologues have been isolated in non-phagocytic cells (Nox1, Nox3, Nox4, Nox5, Duox1 and Duox2) (Bae et al., 2011). All Nox isozymes are single polypeptide enzymes and share common features. Nox isozymes are structurally divided into two functional domains. The long carboxyl terminal region of Nox contains NADPH- and FAD-binding sites homologous to that of ferredoxin-NADP+ reductase (FNR) (Segal et al., 1992; Sumimoto et al., 1992). The six transmembrane α-helical domains also found in the carboxyl terminal region bearing tandem heme groups homologous to that in ferric reductase (Fre) (Fig. 1A) (Finegold et al., 1996). The sequential electron transfer from NADPH to FAD and heme groups stimulates the reduction of oxygen molecules leading to generation of reactive oxygen species (ROS) such as superoxide anion and hydrogen peroxide. Among the seven Nox isozymes in human, Nox1, Nox3, and Nox4 have similar structures with Nox2 requiring p22phox integral protein for the activation and formation of Nox complex. Nox5 and Duox1/2 isozymes do not require p22phox integral protein, but contain the EF hand domain responsible for binding of intracellular calcium (Fig. 1B) (Bae et al., 2011).
Fig. 1.

NADPH Oxidase (Nox) structure and Nox isozymes. (A) General structure of Nox isozyme. Nox contains six transmembrane domains with two heme binding sites and long COOH-terminal region for NADPH binding site and FAD-binding site as prosthestic group. (B) Regulation of Nox isozymes. Nox2-p22phox dimer associates p47phox through interaction of SH3 of p47phox with PRR of p22phox. The PRR domain of p47phox serves as the binding site for p67phox. Subsequently, the p67phox protein recruites p40phox and Rac protein. Nox1 also binds to p22phox and requires Nox organizer 1 (NoxO1), Nox activator1 (NoxA1), and Rac protein. Nox4 requires only p22phox for the activity. Nox5 and Duox 1/2 isozymes contain Ca2+-binding EF hand domain at N-terminus region, indicating that these isozymes are regulated by intracellular calcium mobilization.
Nox2 isozyme requires one integral protein, p22phox, multiple cytosolic proteins (p47phox, p40phox, and p67phox) and a small GTPase Rac (Bokoch and Zhao, 2006; Bedard and Krause, 2007; Bae et al., 2011; Jha et al., 2016) for its activity. The formation of Nox2 complex is mediated by sophisticated protein-protein interactions (Fig. 1B). Nox2 tightly associates with the integral protein p22phox whose proline-rich region (PRR) functions as the binding site for the Src homology domain 3 (SH3) of p47phox. This interaction is key to subsequent formation of active Nox2 complex. The p47phox also contains phox (PX) domain in the NH3-terminal region, two SH3 domains in the central region, and auto-inhibitory region (AIR) and PRR in the COOH-terminal region. In the resting stage, AIR interacts with tandem SH3 domains to keep intramolecular folding consistent with inactive state. Various serine/threonine kinases including PKC, PKA, MAPK, and Akt phosphorylate the COOH-terminal region of p47phox to interrupt intramolecular SH3-AIR interaction and then to induce the opening of SH3 domain for interaction with PRR of p22phox (Sumimoto et al., 1996; Groemping et al., 2003; Bedard and Krause, 2007). Subsequently, the PRR domain of p47phox serves as the binding site for p67phox (Bae et al., 2011). The p67phox protein contains two SH3 domains in its COOH-terminal region that interacts with the PRR domain of p47phox and four tetratricopeptide repeats (TPR) in the NH3-terminal region that functions as the binding site of small G-protein Rac. The p67phox protein also contains the PB1 domain between the two SH3 domains which mediates molecular interaction with the PB1 domain of p40phox. Receptor-mediated Nox2 complex formation induces electron transfers among NADPH, FAD and heme group, leading to generation of superoxide anions.
Nox1 was identified as a homologue of Nox2 from non-phagocytic colon epithelium (Suh et al., 1999; Bae et al., 2011). Nox1 also binds to p22phox and requires Nox organizer 1 (NoxO1) and Nox activator1 (NoxA1) as the homologues and functional counterparts of p47phox and p67phox of Nox2 complex respectively as well as the small G protein Rac (Fig. 1B) (Geiszt et al., 2003; Kawahara et al., 2005; Cheng et al., 2006). NoxO1 contains two SH3 domains in the central region for interaction with the PRR domain of p22phox and one PRR domain in the COOH-terminal region for binding to the SH3 domain of NoxA1. It has generally been assumed that the Nox1 complex is constitutively active because a domain homologous to the AIR domain of p47phox is missing in NoxO1 (Sumimoto, 2008; Dutta and Rittinger, 2010). However, it has been reported that Nox1 activity is transiently increased in response to various agonists including EGF (Lassegue et al., 2001; Park et al., 2006; Schroder et al., 2007; Choi et al., 2008; Lee et al., 2009, 2013), suggesting that an additional mechanism is in operation for the regulation of Nox1 activity. Joo et al. (2016) reported a molecular regulatory process in which Casitas B–lineage lymphoma (Cbl) E3 ligase is recruited to Nox1 complex through the interaction between NoxO1 and growth receptor–bound protein 2 (Grb2) leading to the disassembly of Nox1 complex and a subsequent reduction in ROS generation upon epidermal growth factor (EGF) stimulation.
The primary structure of Nox3 shares 56% homology with that of Nox2 (Kikuchi et al., 2000; Jha et al., 2016). Nox3 also requires p22phox and NoxO1 for the activation (Fig. 1B). However, the molecular mechanism for Nox3 complex formation remains to be elucidated (Banfi et al., 2004a; Cheng et al., 2004; Ueno et al., 2005; Ueyama et al., 2006; Bedard and Krause, 2007). Nox4 was initially identified from kidney and requires only p22phox for the activity (Fig. 1B). However, no cytosolic protein regulating Nox4 activity has been reported. Many reports suggested that Nox4 seems to be constitutively active and Nox4 activity is dependent largely on its expression level in cells. However, Nox4 isozyme activity is regulated by receptor-mediated cell signaling networks, suggesting that unknown protein or proteins regulate Nox4 complex formation and stimulates its activity (Bedard and Krause, 2007; Leto et al., 2009; Block and Gorin, 2012; Lassegue et al., 2012; Lambeth and Neish, 2014). Poldip2 (polymerase delta-interacting proteins2), a p22phox interacting protein, has been identified as a positive regulator for Nox4 activity in SMC migration (Lyle et al., 2009). However, molecular mechanisms by which poldip2 associates and interacts with Nox4-p22phox complex remain to be determined.
Unlike Nox1-4 isozymes, Nox5 does not require p22phox or cytosolic accessory proteins for activation. Given that Nox5 isozyme contains long intracellular NH3-terminus region containing Ca2+-binding EF hand domain, Nox5 isozyme is likely regulated by intracellular calcium mobilization (Fig. 1B) (Banfi et al., 2001, 2004b). As in Nox5, the EF-hand domain is localized in the NH3-terminus region of Duox 1/2 isozymes and functions as the binding site of intracellular calcium (Fig. 1B) (Ameziane-El-Hassani et al., 2005; Bae et al., 2011). Recently, we reported that testosterone binds to GPRC6A located in plasma membrane and subsequently induces the activation of Gq protein, IP3 generation, and [Ca2+]i mobilization ultimately leading to Duox1 activation (Ko et al., 2014).
EXPRESSION OF NOX ISOZYMES IN KIDNEY CELLS
It is now clear that multiple Nox isozymes including Nox1, Nox2, Nox4 and Nox5 (in case of human genomes) and their accessory proteins are expressed in kidney tissues (Fig. 2). Together, these isozymes represent one of the major sources of ROS in normal and pathological states of kidney (Jha et al., 2014; Holterman et al., 2015; Cha et al., 2017). The pathological states of kidney in particular includes diabetic nephropathy and fibrosis in which the major function of kidney, elimination of metabolic wastes in blood through glomerular filtration, is severely compromised (Jha et al., 2016). The renal vessels are composed of vascular smooth muscle cells (VSMCs) and endothelia cells (ECs) expressing Nox1 and Nox4 which are responsible for ROS generation in response to angiotensin II (Fig. 2) (Wingler et al., 2001). Glomerulus plays an important role in maintaining kidney function and is mainly composed of mesangial cells and podocytes. Many reports have shown that upregulation of Nox1, Nox2, Nox4, and Nox5 and the regulatory subunits p22phox, p47phox and p67phox contributes to mesangial cell hypertrophy, tissue expansion, ECM protein accumulation and podocyte apoptosis in glomerulus (Jha et al., 2016). Proximal tubule cells express Nox1, Nox2, Nox4 and Nox5 isozymes (Yang et al., 2012; Yu et al., 2014; Holterman et al., 2015), and macula densa expresses Nox2 and Nox4 as well as p47phox, p67phox and p22phox (Fig. 2) (Holterman et al., 2015).
Fig. 2.

Distribution of Nox isozymes and its components in the kidney.
ACTIVATION OF NOX IN DIABETIC NEPHROPATHY
High glucose (HG)-induced kidney damages are associated with several metabolic risk factors (Fig. 3). Activation of renin-angiotensin-aldosterone pathway, activation of PKC, production of advanced glycation end products (AGEs) and TGFβ-induced fibrosis have been reported as high risk factors for the initiation and progression of DN. A high HG level induces increased generation of advanced glycation end products (AGEs) which serve as ligands for cellular receptor RAGE, transducing signals for inflammation (Schmidt et al., 1995; Hofmann et al., 1999). AGEs from ECM fragments including protein and lipid species thereby lead to structural changes in kidney tissues (Silbiger et al., 1993; Forbes et al., 2003). Activation of signaling networks by AGE stimulates nuclear factor-kB (NF-kB) activity which induces the generation of proinflammatory cytokines including interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) (Neumann et al., 1999). AGEs are also considered as potent stimulators for VEGF expression and PKC activation, leading to upregulation of TGFβ1 expression, which in turn is causally linked to expression of ECM protein for mesangial expansion (Osicka et al., 2000; Lal et al., 2002; Singh et al., 2014). Activation of TGFβ1 also stimulates Nox4 expression (Fig. 3) (Jiang et al., 2014). Importantly, knockdown of Nox4 expression suppressed TGFβ1-dependent fibrotic markers including fibronectin, collagen I, and α-smooth muscle actin (Jiang et al., 2014). Moreover, Nox inhibitor treatment inhibited TGF-β-induced Smad2/3 activation and then suppressed expression of marker proteins for fibrosis, indicating that Nox-dependent ROS generation may have critical roles in TGF-β-mediated fibrosis in kidney tissues (Jiang et al., 2014).
Fig. 3.

Activation of Nox in diabetic nephropathy. High glucose (HG), transforming growth factor β (TGF β), angiotensin II (Ang II) and advanced glycation end products (AGEs) stimulate Nox activity or its expression, leading to overproduction of ROS. Nox-derived ROS regulate Akt/PKB, p38, and caspase 3 activation leading to podocyte apoptosis. ROS induce inflammation in renal cells by increasing MCP by NF-κB activation. Additionally, the activation of protein kinase C (PKC) and AMP-activated kinase (AMPK) are involved in Nox derived ROS dependent events in the progression of DN.
Activation of protein kinase C (PKC) is involved in hyperglycemia-dependent nephropathy. HG-RAGE-phospholipase Cβ (PLC) pathway and angiotensin-angiotensin receptor II-PLCβ pathway can stimulate DAG generation and intracellular calcium mobilization through hydrolysis of phosphatidylinositol 4,5, bisphosphate (PIP2). Elevated levels of intracellular calcium and DAG activate renal PKC affecting vascular functions through inducing changes in endothelial permeability, vasoconstriction, extracellular matrix (ECM) maintenance, cytokine activation and leukocyte adhesion (Fig. 3). Of interest, PKC inhibition ameliorates structural and functional abnormalities in the kidney in db/db mice (Koya et al., 2000). Importantly, HG-induced translocation of PKCα to renal membranes stimulates Nox2 activation. In type I diabetic mice, treatment of PKCα inhibitor (Ro-32-0432) attenuates Nox-dependent superoxide production. Renal extracellular matrix accumulation of fibronectin and collagen IV is decreased by Nox inhibitor apocynin (Thallas-Bonke et al., 2008). These reports together suggest that Nox2 activation by PKCα is the key downstream event of the AGE-receptor signaling in diabetic nephropathy.
Recently, oxidative stress has come to be recognized as a key player in the pathogenesis of DN. Most ROS produced intracellularly originate from the mitochondrial respiratory chain, and they certainly can bring damages in renal tissues when mitochondria become dysfunctional. Relatively small amounts (1–2%) of total O2 consumed during mitochondrial respiration are normally converted to ROS (Chance et al., 1979; Kudin et al., 2004). However, the formation of ROS is increased from dysfunctional mitochondria in hyperglycemia stage. ROS accumulation can lead to oxidative stresses in the cell, leading to damages of cellular components including protein and DNA. Ultimately, the oxidative stress leads to renal fibrosis and decline in renal function (Badal and Danesh, 2014). ROS mediate activation of a number of pathways including PKC, NF-kB, hexosamine synthesis and AGE (Nishikawa et al., 2000).
As has been described above, ROS can also be produced by activities of Nox isozymes. Many reports have indicated that HG, angiotensin II or TGFβ stimulate Nox4-derived ROS generation, resulting in fibrotic response in mesangial cells, podocytes and tubular cells (Jha et al., 2016). The pathogenic process leading to fibrosis has been studied with Nox4-deficient mice (Jha et al., 2014; Gorin et al., 2015). Interestingly, treatment of Nox inhibitors (GKT137831 or APX-115) resulted in resistance to DN progression as evidenced by reduction in ECM accumulation and albuminuria. In addition, inflammation events in Nox4 deficient renal cells were accompanied by reduced levels of NF-kB activity and subsequently of chemokines and MCP-1 and in renal tubular cell or podocyte in response to HG stimulation (Xu et al., 2010; Jha et al., 2014).
DEVELOPMENT OF NOX INHIBITOR FOR THE TREATMENT OF DN
Oxidative stress plays an important role in DN, and clinical trials for therapeutic intervention for reduction of such oxidative stress are ongoing. That Nox isozymes are sources of oxidative stress for kidney is now well-established. Therefore, it is tempting to speculate that Nox isozymes will be viable therapeutic targets for DN treatment. Nox isozymes are expressed in various renal cells; Nox4 in EC, Nox1 in SMC, Nox1, Nox2, Nox4, and Nox5 in glomeruli, Nox4 and Nox1 in tubule cells. Of importance, studies with diabetic nephropathy animal models with Nox gene mutation suggest that pan-Nox inhibitors are more effective rather than isozyme-specific inhibitors.
GKT137831 (Setanaxib)
Pyrazolopyridine compound, a dual inhibitor of Nox1 and Nox4 was developed by Genkyotex. GKT136901 was the lead compound identified from a high-throughput screening (Table 1) (Laleu et al., 2010). GKT137831 compound as a Nox1/4 inhibitor was subsequently obtained from a structure-activity relationship study of GKT136901. GKT137831 has been applied to various disease models. These preclinical studies showed that GKT137831 have therapeutic potential in fibrotic and inflammatory diseases of the liver (Aoyama et al., 2012), lung (Green et al., 2012) and other organs as well as kidney. According to the preclinical data, GKT137831 attenuated various parameters of glomerular injury, such as ECM accumulation, glomerular structural changes and albuminuria in streptozotocin-induced diabetic ApoE−/− mice (Jha et al., 2014). In another study, application of GKT137831 significantly reduced mesangial matrix expansion, urinary albumin excretion, and podocyte loss in OVE mice as a type I diabetes model (Gorin et al., 2015). GKT137831 is not only a dual inhibitor for the Nox1 and Nox4 isozymes but also a partial inhibitor of Nox5 (Gaggini et al., 2011; Jha et al., 2016). GKT137831 is now enrolled in a phase 2 clinical trial in primary biliary cholangitis (PBC, a fibrotic orphan disease) and type I diabetes and kidney disease (DKD).
Table 1.
NADPH oxidase inhibitors
| Compound | Mechanism of action | Chemical name | Chemical structure | Target | References |
|---|---|---|---|---|---|
| GKT137981 | Dual Nox1/4 inhibitor | 2-(2-Chlorophenyl)-4-(3-dimethylamino)phenyl)-5-methyl-1H-pyrazolo[4,3-c] pyridine-3,6(2H,5H)-dione |
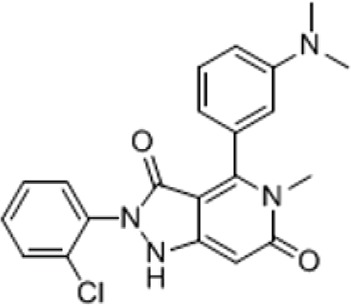
|
Liver fibrosis, kidney fibrosis, lung fibrosis | Laleu et al., 2010; Gaggini et al., 2011; Aoyama et al., 2012; Green et al., 2012; Jha et al., 2014; Gorin et al., 2015; Jha et al., 2016 |
| APX-115 | Pan-Nox inhibitor | 3-Phenyl-4-propyl-1-(pyridin-2-yl)-1H-pyrazol-5-olhydrochloride |
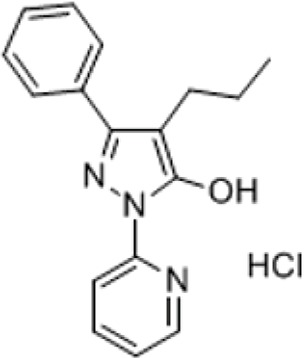
|
Diabetic Nephropathy | Cha et al., 2017; Kwon et al., 2017; Dorotea et al., 2018 |
APX-115
APX-115 is first-in-class pan Nox inhibitor developed by Aptabio (Table 1). The Apx-115 compound protects against diabetic nephropathy in type II diabetic model db/db mice. Specifically, treatment of APX-115 significantly improved insulin resistance in diabetic mice. Oxidative stress as measured by plasma 8-isoprostane level was decreased by the APX-115 treatment. APX-115 also suppressed urinary albumin excretion and preserved creatinine clearance in db/db mice. In diabetic kidneys, APX-115 significantly suppressed mesangial expansion. Comparative study showed that APX-115 is more effective than dual Nox1/4 inhibitor GKT137831 (Cha et al., 2017). Another study compared APX-115 and Losartan (renin-angiotensin system inhibitor) in Streptozotocin (STZ)-induced type I diabetic mouse. APX-115 inhibited renal oxidative stress and prevented albuminuria, glomerular hypertrophy, tubular injury, podocyte injury, fibrosis and inflammation in diabetic kidneys as effectively as losartan alone which is currently used as the standard treatment against kidney injuries in diabetic patients (Kwon et al., 2017). Moreover, treatment with APX-115 in STZ-induced diabetic rat was also as effective as losartan (Dorotea et al., 2018). APX-115 is more effective than GKT137831 based on inflammation or fibrosis. This suggests that pan-Nox inhibitors are more effective therapeutics for DN treatment than Nox isozyme-specific inhibitors. This is not surprising given that multiple Nox isozymes are expressed in overlapping manners in multiple cell types of kidney, which all appear to be affected by ROS one way or another. Of note, APX-115 treatment decreased expression of all Nox isozyme expression at the protein level in the kidney. Pan-Nox inhibition might therefore be a promising therapy for diabetic nephropathy.
CONCLUSION
Currently, no effective therapy exists for treatment of DN. Based on the damage ROS brings to kidney during pathogenesis of DN, Nox isozymes have emerged as promising targets. It should be noted that chronic hyperglycemia in diabetes stimulates the expression and activation of Nox isozymes in renal tissues leading to a sustained oxidative stress. There are various pathways that may contribute to stimulation of Nox-dependent ROS generation. Therefore, Nox isozymes in renal tissues should be effective therapeutic targets for DN treatment. Now, two Nox inhibitors (GKT137831 and APX-115) are enrolled in clinical phase II for DN patients. These Nox inhibitors may represent best hope to date to many DN patients with no other recourse.
Acknowledgments
This work was supported by Aging project (2017M3A9D 8062955 to YSB) and Bio-SPC (2018M3A9G1075771 to YSB) funded by the National Research Foundation of Korea (NRF) and Ministry of Science and ICT.
REFERENCES
- Ameziane-El-Hassani R, Morand S, Boucher JL, Frapart YM, Apostolou D, Agnandji D, Gnidehou S, Ohayon R, Noel-Hudson MS, Francon J, Lalaoui K, Virion A, Dupuy C. Dual oxidase-2 has an intrinsic Ca2+-dependent H2O2-generating activity. J. Biol. Chem. 2005;280:30046–30054. doi: 10.1074/jbc.M500516200. [DOI] [PubMed] [Google Scholar]
- Aoyama T, Paik YH, Watanabe S, Laleu B, Gaggini F, Fioraso-Cartier L, Molango S, Heitz F, Merlot C, Szyndralewiez C, Page P, Brenner DA. Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent. Hepatology. 2012;56:2316–2327. doi: 10.1002/hep.25938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badal SS, Danesh FR. New insights into molecular mechanisms of diabetic kidney disease. Am. J. Kidney Dis. 2014;63:S63–S83. doi: 10.1053/j.ajkd.2013.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae YS, Oh H, Rhee SG, Yoo YD. Regulation of reactive oxygen species generation in cell signaling. Mol. Cells. 2011;32:491–509. doi: 10.1007/s10059-011-0276-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banfi B, Malgrange B, Knisz J, Steger K, Dubois-Dauphin M, Krause KH. NOX3, a superoxide-generating NADPH oxidase of the inner ear. J. Biol. Chem. 2004a;279:46065–46072. doi: 10.1074/jbc.M403046200. [DOI] [PubMed] [Google Scholar]
- Banfi B, Molnar G, Maturana A, Steger K, Hegedus B, Demaurex N, Krause KH. A Ca(2+)-activated NADPH oxidase in testis, spleen, and lymph nodes. J. Biol. Chem. 2001;276:37594–37601. doi: 10.1074/jbc.M103034200. [DOI] [PubMed] [Google Scholar]
- Banfi B, Tirone F, Durussel I, Knisz J, Moskwa P, Molnar GZ, Krause KH, Cox JA. Mechanism of Ca2+ activation of the NADPH oxidase 5 (NOX5) J. Biol. Chem. 2004b;279:18583–18591. doi: 10.1074/jbc.M310268200. [DOI] [PubMed] [Google Scholar]
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Block K, Gorin Y. Aiding and abetting roles of NOX oxidases in cellular transformation. Nat. Rev. Cancer. 2012;12:627–637. doi: 10.1038/nrc3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokoch GM, Zhao T. Regulation of the phagocyte NADPH oxidase by Rac GTPase. Antioxid. Redox Signal. 2006;8:1533–1548. doi: 10.1089/ars.2006.8.1533. [DOI] [PubMed] [Google Scholar]
- Caramori ML, Parks A, Mauer M. Renal lesions predict progression of diabetic nephropathy in type 1 diabetes. J. Am. Soc. Nephrol. 2013;24:1175–1181. doi: 10.1681/ASN.2012070739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha JJ, Min HS, Kim KT, Kim JE, Ghee JY, Kim HW, Lee JE, Han JY, Lee G, Ha HJ, Bae YS, Lee SR, Moon SH, Lee SC, Kim G, Kang YS, Cha DR. APX-115, a first-in-class pan-NADPH oxidase (Nox) inhibitor, protects db/db mice from renal injury. Lab. Invest. 2017;97:419–431. doi: 10.1038/labinvest.2017.2. [DOI] [PubMed] [Google Scholar]
- Chance B, Sies H, Boveris A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979;59:527–605. doi: 10.1152/physrev.1979.59.3.527. [DOI] [PubMed] [Google Scholar]
- Cheng G, Diebold BA, Hughes Y, Lambeth JD. Nox1-dependent reactive oxygen generation is regulated by Rac1. J. Biol. Chem. 2006;281:17718–17726. doi: 10.1074/jbc.M512751200. [DOI] [PubMed] [Google Scholar]
- Cheng G, Ritsick D, Lambeth JD. Nox3 regulation by NOXO1, p47phox, and p67phox. J. Biol. Chem. 2004;279:34250–34255. doi: 10.1074/jbc.M400660200. [DOI] [PubMed] [Google Scholar]
- Choi H, Leto TL, Hunyady L, Catt KJ, Bae YS, Rhee SG. Mechanism of angiotensin II-induced superoxide production in cells reconstituted with angiotensin type 1 receptor and the components of NADPH oxidase. J. Biol. Chem. 2008;283:255–267. doi: 10.1074/jbc.M708000200. [DOI] [PubMed] [Google Scholar]
- Dorotea D, Kwon G, Lee JH, Saunders E, Bae YS, Moon SH, Lee SJ, Cha DR, Ha H. A pan-NADPH oxidase inhibitor ameliorates kidney injury in type 1 diabetic rats. Pharmacology. 2018;102:180–189. doi: 10.1159/000491398. [DOI] [PubMed] [Google Scholar]
- Dutta S, Rittinger K. Regulation of NOXO1 activity through reversible interactions with p22(phox) and NOXA1. PLoS ONE. 2010;5:e10478. doi: 10.1371/journal.pone.0010478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finegold AA, Shatwell KP, Segal AW, Klausner RD, Dancis A. Intramembrane bis-heme motif for transmembrane electron transport conserved in a yeast iron reductase and the human NADPH oxidase. J. Biol. Chem. 1996;271:31021–31024. doi: 10.1074/jbc.271.49.31021. [DOI] [PubMed] [Google Scholar]
- Forbes JM, Cooper ME. Mechanisms of diabetic complications. Physiol. Rev. 2013;93:137–188. doi: 10.1152/physrev.00045.2011. [DOI] [PubMed] [Google Scholar]
- Forbes JM, Cooper ME, Oldfield MD, Thomas MC. Role of advanced glycation end products in diabetic nephropathy. J. Am. Soc. Nephrol. 2003;14:S254–S258. doi: 10.1097/01.ASN.0000077413.41276.17. [DOI] [PubMed] [Google Scholar]
- Gaggini F, Laleu B, Orchard M, Fioraso-Cartier L, Cagnon L, Houngninou-Molango S, Gradia A, Duboux G, Merlot C, Heitz F, Szyndralewiez C, Page P. Design, synthesis and biological activity of original pyrazolo-pyrido-diazepine, -pyrazine and -oxazine dione derivatives as novel dual Nox4/Nox1 inhibitors. Bioorg. Med. Chem. 2011;19:6989–6999. doi: 10.1016/j.bmc.2011.10.016. [DOI] [PubMed] [Google Scholar]
- Geiszt M, Lekstrom K, Witta J, Leto TL. Proteins homologous to p47phox and p67phox support superoxide production by NAD(P)H oxidase 1 in colon epithelial cells. J. Biol. Chem. 2003;278:20006–20012. doi: 10.1074/jbc.M301289200. [DOI] [PubMed] [Google Scholar]
- Gorin Y, Cavaglieri RC, Khazim K, Lee DY, Bruno F, Thakur S, Fanti P, Szyndralewiez C, Barnes JL, Block K, Abboud HE. Targeting NADPH oxidase with a novel dual Nox1/Nox4 inhibitor attenuates renal pathology in type 1 diabetes. Am. J. Physiol. Renal Physiol. 2015;308:F1276–F1287. doi: 10.1152/ajprenal.00396.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DE, Murphy TC, Kang BY, Kleinhenz JM, Szyndralewiez C, Page P, Sutliff RL, Hart CM. The Nox4 inhibitor GKT137831 attenuates hypoxia-induced pulmonary vascular cell proliferation. Am. J. Respir. Cell Mol. Biol. 2012;47:718–726. doi: 10.1165/rcmb.2011-0418OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groemping Y, Lapouge K, Smerdon SJ, Rittinger K. Molecular basis of phosphorylation-induced activation of the NADPH oxidase. Cell. 2003;113:343–355. doi: 10.1016/S0092-8674(03)00314-3. [DOI] [PubMed] [Google Scholar]
- Heng LZ, Comyn O, Peto T, Tadros C, Ng E, Sivaprasad S, Hykin PG. Diabetic retinopathy: pathogenesis, clinical grading, management and future developments. Diabet. Med. 2013;30:640–650. doi: 10.1111/dme.12089. [DOI] [PubMed] [Google Scholar]
- Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, Neurath MF, Slattery T, Beach D, McClary J, Nagashima M, Morser J, Stern D, Schmidt AM. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97:889–901. doi: 10.1016/S0092-8674(00)80801-6. [DOI] [PubMed] [Google Scholar]
- Holterman CE, Read NC, Kennedy CR. Nox and renal disease. Clin. Sci. (Lond.) 2015;128:465–481. doi: 10.1042/CS20140361. [DOI] [PubMed] [Google Scholar]
- Jha JC, Banal C, Chow BS, Cooper ME, Jandeleit-Dahm K. Diabetes and kidney disease: role of oxidative stress. Antioxid. Redox Signal. 2016;25:657–684. doi: 10.1089/ars.2016.6664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha JC, Gray SP, Barit D, Okabe J, El-Osta A, Namikoshi T, Thallas-Bonke V, Wingler K, Szyndralewiez C, Heitz F, Touyz RM, Cooper ME, Schmidt HH, Jandeleit-Dahm KA. Genetic targeting or pharmacologic inhibition of NADPH oxidase nox4 provides renoprotection in long-term diabetic nephropathy. J. Am. Soc. Nephrol. 2014;25:1237–1254. doi: 10.1681/ASN.2013070810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang F, Liu GS, Dusting GJ, Chan EC. NADPH oxidase-dependent redox signaling in TGF-beta-mediated fibrotic responses. Redox. Biol. 2014;2:267–272. doi: 10.1016/j.redox.2014.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo JH, Oh H, Kim M, An EJ, Kim RK, Lee SY, Kang DH, Kang SW, Keun Park C, Kim H, Lee SJ, Lee D, Seol JH, Bae YS. NADPH oxidase 1 activity and ROS generation are regulated by Grb2/Cbl-mediated proteasomal degradation of NoxO1 in colon cancer cells. Cancer Res. 2016;76:855–865. doi: 10.1158/0008-5472.CAN-15-1512. [DOI] [PubMed] [Google Scholar]
- Kawahara T, Ritsick D, Cheng G, Lambeth JD. Point mutations in the proline-rich region of p22phox are dominant inhibitors of Nox1- and Nox2-dependent reactive oxygen generation. J. Biol. Chem. 2005;280:31859–31869. doi: 10.1074/jbc.M501882200. [DOI] [PubMed] [Google Scholar]
- Khan A, Petropoulos IN, Ponirakis G, Malik RA. Visual complications in diabetes mellitus: beyond retinopathy. Diabet. Med. 2017;34:478–484. doi: 10.1111/dme.13296. [DOI] [PubMed] [Google Scholar]
- Kikuchi H, Hikage M, Miyashita H, Fukumoto M. NADPH oxidase subunit, gp91(phox) homologue, preferentially expressed in human colon epithelial cells. Gene. 2000;254:237–243. doi: 10.1016/S0378-1119(00)00258-4. [DOI] [PubMed] [Google Scholar]
- Ko E, Choi H, Kim B, Kim M, Park KN, Bae IH, Sung YK, Lee TR, Shin DW, Bae YS. Testosterone stimulates Duox1 activity through GPRC6A in skin keratinocytes. J. Biol. Chem. 2014;289:28835–28845. doi: 10.1074/jbc.M114.583450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koya D, Haneda M, Nakagawa H, Isshiki K, Sato H, Maeda S, Sugimoto T, Yasuda H, Kashiwagi A, Ways DK, King GL, Kikkawa R. Amelioration of accelerated diabetic mesangial expansion by treatment with a PKC beta inhibitor in diabetic db/db mice, a rodent model for type 2 diabetes. FASEB J. 2000;14:439–447. doi: 10.1096/fasebj.14.3.439. [DOI] [PubMed] [Google Scholar]
- Kudin AP, Bimpong-Buta NY, Vielhaber S, Elger CE, Kunz WS. Characterization of superoxide-producing sites in isolated brain mitochondria. J. Biol. Chem. 2004;279:4127–4135. doi: 10.1074/jbc.M310341200. [DOI] [PubMed] [Google Scholar]
- Kwon G, Uddin MJ, Lee G, Jiang S, Cho A, Lee JH, Lee SR, Bae YS, Moon SH, Lee SJ, Cha DR, Ha H. A novel pan-Nox inhibitor, APX-115, protects kidney injury in streptozotocin-induced diabetic mice: possible role of peroxisomal and mitochondrial biogenesis. Oncotarget. 2017;8:74217–74232. doi: 10.18632/oncotarget.18540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lal MA, Brismar H, Eklof AC, Aperia A. Role of oxidative stress in advanced glycation end product-induced mesangial cell activation. Kidney Int. 2002;61:2006–2014. doi: 10.1046/j.1523-1755.2002.00367.x. [DOI] [PubMed] [Google Scholar]
- Laleu B, Gaggini F, Orchard M, Fioraso-Cartier L, Cagnon L, Houngninou-Molango S, Gradia A, Duboux G, Merlot C, Heitz F, Szyndralewiez C, Page P. First in class, potent, and orally bioavailable NADPH oxidase isoform 4 (Nox4) inhibitors for the treatment of idiopathic pulmonary fibrosis. J. Med. Chem. 2010;53:7715–7730. doi: 10.1021/jm100773e. [DOI] [PubMed] [Google Scholar]
- Lambeth JD, Neish AS. Nox enzymes and new thinking on reactive oxygen: a double-edged sword revisited. Annu. Rev. Pathol. 2014;9:119–145. doi: 10.1146/annurev-pathol-012513-104651. [DOI] [PubMed] [Google Scholar]
- Lassegue B, San Martin A, Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ. Res. 2012;110:1364–1390. doi: 10.1161/CIRCRESAHA.111.243972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassegue B, Sorescu D, Szocs K, Yin Q, Akers M, Zhang Y, Grant SL, Lambeth JD, Griendling KK. Novel gp91(phox) homologues in vascular smooth muscle cells: nox1 mediates angiotensin II-induced superoxide formation and redoxsensitive signaling pathways. Circ. Res. 2001;88:888–894. doi: 10.1161/hh0901.090299. [DOI] [PubMed] [Google Scholar]
- Lee JH, Joo JH, Kim J, Lim HJ, Kim S, Curtiss L, Seong JK, Cui W, Yabe-Nishimura C, Bae YS. Interaction of NADPH oxidase 1 with Toll-like receptor 2 induces migration of smooth muscle cells. Cardiovasc. Res. 2013;99:483–493. doi: 10.1093/cvr/cvt107. [DOI] [PubMed] [Google Scholar]
- Lee MY, San Martin A, Mehta PK, Dikalova AE, Garrido AM, Datla SR, Lyons E, Krause KH, Banfi B, Lambeth JD, Lassegue B, Griendling KK. Mechanisms of vascular smooth muscle NADPH oxidase 1 (Nox1) contribution to injury-induced neointimal formation. Arterioscler. Thromb. Vasc. Biol. 2009;29:480–487. doi: 10.1161/ATVBAHA.108.181925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leto TL, Morand S, Hurt D, Ueyama T. Targeting and regulation of reactive oxygen species generation by Nox family NADPH oxidases. Antioxid. Redox Signal. 2009;11:2607–2619. doi: 10.1089/ars.2009.2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lv M, Chen Z, Hu G, Li Q. Therapeutic strategies of diabetic nephropathy: recent progress and future perspectives. Drug Discov. Today. 2015;20:332–346. doi: 10.1016/j.drudis.2014.10.007. [DOI] [PubMed] [Google Scholar]
- Lyle AN, Deshpande NN, Taniyama Y, Seidel-Rogol B, Pounkova L, Du P, Papaharalambus C, Lassegue B, Griendling KK. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ. Res. 2009;105:249–259. doi: 10.1161/CIRCRESAHA.109.193722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason RM, Wahab NA. Extracellular matrix metabolism in diabetic nephropathy. J. Am. Soc. Nephrol. 2003;14:1358–1373. doi: 10.1097/01.ASN.0000065640.77499.D7. [DOI] [PubMed] [Google Scholar]
- Molitch ME, DeFronzo RA, Franz MJ, Keane WF, Mogensen CE, Parving HH, Steffes MW, American Diabetes A. Nephropathy in diabetes. Diabetes Care. 2004;27:S79–S83. doi: 10.2337/diacare.27.2007.S79. [DOI] [PubMed] [Google Scholar]
- Neumann A, Schinzel R, Palm D, Riederer P, Munch G. High molecular weight hyaluronic acid inhibits advanced glycation endproduct-induced NF-kappaB activation and cytokine expression. FEBS Lett. 1999;453:283–287. doi: 10.1016/S0014-5793(99)00731-0. [DOI] [PubMed] [Google Scholar]
- Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, Yorek MA, Beebe D, Oates PJ, Hammes HP, Giardino I, Brownlee M. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature. 2000;404:787–790. doi: 10.1038/35008121. [DOI] [PubMed] [Google Scholar]
- Osicka TM, Yu Y, Panagiotopoulos S, Clavant SP, Kiriazis Z, Pike RN, Pratt LM, Russo LM, Kemp BE, Comper WD, Jerums G. Prevention of albuminuria by aminoguanidine or ramipril in streptozotocin-induced diabetic rats is associated with the normalization of glomerular protein kinase C. Diabetes. 2000;49:87–93. doi: 10.2337/diabetes.49.1.87. [DOI] [PubMed] [Google Scholar]
- Palatini P. Glomerular hyperfiltration: a marker of early renal damage in pre-diabetes and pre-hypertension. Nephrol. Dial. Transplant. 2012;27:1708–1714. doi: 10.1093/ndt/gfs037. [DOI] [PubMed] [Google Scholar]
- Papadopoulou-Marketou N, Chrousos GP, Kanaka-Gantenbein C. Diabetic nephropathy in type 1 diabetes: a review of early natural history, pathogenesis, and diagnosis. Diabetes Metab. Res. Rev. 2017;33 doi: 10.1002/dmrr.2841. [DOI] [PubMed] [Google Scholar]
- Park HS, Park D, Bae YS. Molecular interaction of NADPH oxidase 1 with betaPix and nox organizer 1. Biochem. Biophys. Res. Commun. 2006;339:985–990. doi: 10.1016/j.bbrc.2005.11.108. [DOI] [PubMed] [Google Scholar]
- Reidy K, Kang HM, Hostetter T, Susztak K. Molecular mechanisms of diabetic kidney disease. J. Clin. Invest. 2014;124:2333–2340. doi: 10.1172/JCI72271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocco MV, Berns JS. KDOQI in the era of global guidelines. Am. J. Kidney Dis. 2009;54:781–787. doi: 10.1053/j.ajkd.2009.08.001. [DOI] [PubMed] [Google Scholar]
- Said G. Diabetic neuropathy--a review. Nat. Clin. Pract. Neurol. 2007;3:331–340. doi: 10.1038/ncpneuro0504. [DOI] [PubMed] [Google Scholar]
- Schmidt AM, Hori O, Chen JX, Li JF, Crandall J, Zhang J, Cao R, Yan SD, Brett J, Stern D. Advanced glycation endproducts interacting with their endothelial receptor induce expression of vascular cell adhesion molecule-1 (VCAM-1) in cultured human endothelial cells and in mice. A potential mechanism for the accelerated vasculopathy of diabetes. J. Clin. Invest. 1995;96:1395–1403. doi: 10.1172/JCI118175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K, Helmcke I, Palfi K, Krause KH, Busse R, Brandes RP. Nox1 mediates basic fibroblast growth factor-induced migration of vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2007;27:1736–1743. doi: 10.1161/ATVBAHA.107.142117. [DOI] [PubMed] [Google Scholar]
- Segal AW, West I, Wientjes F, Nugent JH, Chavan AJ, Haley B, Garcia RC, Rosen H, Scrace G. Cytochrome b-245 is a flavocytochrome containing FAD and the NADPH-binding site of the microbicidal oxidase of phagocytes. Biochem. J. 1992;284:781–788. doi: 10.1042/bj2840781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silbiger S, Crowley S, Shan Z, Brownlee M, Satriano J, Schlondorff D. Nonenzymatic glycation of mesangial matrix and prolonged exposure of mesangial matrix to elevated glucose reduces collagen synthesis and proteoglycan charge. Kidney Int. 1993;43:853–864. doi: 10.1038/ki.1993.120. [DOI] [PubMed] [Google Scholar]
- Singh VP, Bali A, Singh N, Jaggi AS. Advanced glycation end products and diabetic complications. Korean J. Physiol. Pharmacol. 2014;18:1–14. doi: 10.4196/kjpp.2014.18.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh YA, Arnold RS, Lassegue B, Shi J, Xu X, Sorescu D, Chung AB, Griendling KK, Lambeth JD. Cell transformation by the superoxide-generating oxidase Mox1. Nature. 1999;401:79–82. doi: 10.1038/43459. [DOI] [PubMed] [Google Scholar]
- Sumimoto H. Structure, regulation and evolution of Nox-family NADPH oxidases that produce reactive oxygen species. FEBS J. 2008;275:3984. doi: 10.1111/j.1742-4658.2008.06488.x. [DOI] [PubMed] [Google Scholar]
- Sumimoto H, Hata K, Mizuki K, Ito T, Kage Y, Sakaki Y, Fukumaki Y, Nakamura M, Takeshige K. Assembly and activation of the phagocyte NADPH oxidase. Specific interaction of the N-terminal Src homology 3 domain of p47phox with p22phox is required for activation of the NADPH oxidase. J. Biol. Chem. 1996;271:22152–22158. doi: 10.1074/jbc.271.36.22152. [DOI] [PubMed] [Google Scholar]
- Sumimoto H, Sakamoto N, Nozaki M, Sakaki Y, Takeshige K, Minakami S. Cytochrome b558, a component of the phagocyte NADPH oxidase, is a flavoprotein. Biochem. Biophys. Res. Commun. 1992;186:1368–1375. doi: 10.1016/S0006-291X(05)81557-8. [DOI] [PubMed] [Google Scholar]
- Tervaert TW, Mooyaart AL, Amann K, Cohen AH, Cook HT, Drachenberg CB, Ferrario F, Fogo AB, Haas M, de Heer E, Joh K, Noel LH, Radhakrishnan J, Seshan SV, Bajema IM, Bruijn JA, Renal Pathology S. Pathologic classification of diabetic nephropathy. J. Am. Soc. Nephrol. 2010;21:556–563. doi: 10.1681/ASN.2010010010. [DOI] [PubMed] [Google Scholar]
- Thallas-Bonke V, Thorpe SR, Coughlan MT, Fukami K, Yap FY, Sourris KC, Penfold SA, Bach LA, Cooper ME, Forbes JM. Inhibition of NADPH oxidase prevents advanced glycation end product-mediated damage in diabetic nephropathy through a protein kinase C-alpha-dependent pathway. Diabetes. 2008;57:460–469. doi: 10.2337/db07-1119. [DOI] [PubMed] [Google Scholar]
- Thomas MC, Brownlee M, Susztak K, Sharma K, Jandeleit-Dahm KA, Zoungas S, Rossing P, Groop PH, Cooper ME. Diabetic kidney disease. Nat. Rev. Dis. Primers. 2015;1:15018. doi: 10.1038/nrdp.2015.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MC, Weekes AJ, Broadley OJ, Cooper ME, Mathew TH. The burden of chronic kidney disease in Australian patients with type 2 diabetes (the NEFRON study) Med. J. Aust. 2006;185:140–144. doi: 10.5694/j.1326-5377.2006.tb00499.x. [DOI] [PubMed] [Google Scholar]
- Ueno N, Takeya R, Miyano K, Kikuchi H, Sumimoto H. The NADPH oxidase Nox3 constitutively produces superoxide in a p22phox-dependent manner: its regulation by oxidase organizers and activators. J. Biol. Chem. 2005;280:23328–23339. doi: 10.1074/jbc.M414548200. [DOI] [PubMed] [Google Scholar]
- Ueyama T, Geiszt M, Leto TL. Involvement of Rac1 in activation of multicomponent Nox1- and Nox3-based NADPH oxidases. Mol. Cell. Biol. 2006;26:2160–2174. doi: 10.1128/MCB.26.6.2160-2174.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinik AI, Nevoret ML, Casellini C, Parson H. Diabetic neuropathy. Endocrinol. Metab. Clin. North Am. 2013;42:747–787. doi: 10.1016/j.ecl.2013.06.001. [DOI] [PubMed] [Google Scholar]
- Wingler K, Wunsch S, Kreutz R, Rothermund L, Paul M, Schmidt HH. Upregulation of the vascular NAD(P)H-oxidase isoforms Nox1 and Nox4 by the renin-angiotensin system in vitro and in vivo. Free Radic. Biol. Med. 2001;31:1456–1464. doi: 10.1016/S0891-5849(01)00727-4. [DOI] [PubMed] [Google Scholar]
- Xu Y, Ruan S, Xie H, Lin J. Role of LOX-1 in Ang II-induced oxidative functional damage in renal tubular epithelial cells. Int. J. Mol. Med. 2010;26:679–690. doi: 10.3892/ijmm_00000514. [DOI] [PubMed] [Google Scholar]
- Yang Y, Zhang Y, Cuevas S, Villar VA, Escano C, L DA, Yu P, Grandy DK, Felder RA, Armando I, Jose PA. Paraoxonase 2 decreases renal reactive oxygen species production, lowers blood pressure, and mediates dopamine D2 receptor-induced inhibition of NADPH oxidase. Free Radic. Biol. Med. 2012;53:437–446. doi: 10.1016/j.freeradbiomed.2012.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu P, Han W, Villar VA, Yang Y, Lu Q, Lee H, Li F, Quinn MT, Gildea JJ, Felder RA, Jose PA. Unique role of NADPH oxidase 5 in oxidative stress in human renal proximal tubule cells. Redox Biol. 2014;2:570–579. doi: 10.1016/j.redox.2014.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]