

Abstract
Although antioxidants promote melanoma metastasis, the role of reactive oxygen species (ROS) in other stages of melanoma progression is controversial. Moreover, genes regulating ROS have not been functionally characterized throughout the entire tumor progression in mouse models of cancer. To address this question, we crossed mice-bearing knock-out of Klf9, an ubiquitous transcriptional regulator of oxidative stress, with two conditional melanocytic mouse models: BrafCA mice, where BrafV600E causes premalignant melanocytic hyperplasia, and BrafCA/Pten−/− mice, where BrafV600E and loss of Pten induce primary melanomas and metastases. Klf9 deficiency inhibited premalignant melanocytic hyperplasia in BrafCA mice but did not affect formation and growth of BrafCA/Pten−/− primary melanomas. It also, as expected, promoted BrafCA/Pten−/− metastasis. Treatment with antioxidant N-acetyl cysteine phenocopied loss of Klf9 including suppression of melanocytic hyperplasia. We were interested in a different role of Klf9 in regulation of cell proliferation in BrafCA and BrafCA/Pten−/− melanocytic cells. Mechanistically, we demonstrated that BRAFV600E signaling transcriptionally upregulated KLF9 and that KLF9-dependent ROS were required for full-scale activation of ERK1/2 and induction of cell proliferation by BRAFV600E. PTEN depletion in BRAFV600E-melanocytes did not further activate ERK1/2 and cell proliferation, but rendered these phenotypes insensitive to KLF9 and ROS. Our data identified an essential role of KLF9-dependent ROS in BRAFV600E signaling in premalignant melanocytes, offered an explanation to variable role of ROS in premalignant and transformed melanocytic cells and suggested a novel mechanism for suppression of premalignant growth by topical antioxidants.
Introduction
Malignant melanoma is a devastating disease with no effective conventional cure [1, 2]. Approximately 60% of melanomas contain activating mutations in BRAF (V600E) which is considered the earliest melanoma driver mutation [3, 4]. It is detected already at premalignant stages such as melanocytic nevi, aggregations of non-proliferative melanocytes that often exhibit senescence-associated features [3, 4]. Activation of BRAF in experimental models in vitro and in vivo initially increases proliferation of melanocytes [5–7], however, in the absence of other genetic events (such as loss of PTEN [7] or overexpression of C-MYC [8]), gradually induces growth arrest which is also accompanied by senescence-like phenotypes [5, 6, 8, 9].
Importantly, ~70–80% of all melanomas originate de novo, i.e. bypassing the “nevus” stage [10, 11]. Therefore, mutations that allow premalignant melanocytes to directly progress to de novo melanomas are likely to occur during BRAFV600E-induced hyperplasia. However, mechanisms regulating BRAFV600E-induced proliferation at that stage are not well studied.
Overproduction of reactive oxygen species (ROS) beyond antioxidant buffer capacity causes oxidative stress [12, 13]. Cancer cells often undergo oxidative stress due to oncogene activation or increased metabolic activity [14]. It has been reported that tumors of multiple origins utilize similar mechanisms of suppression of oxidative stress, the most common of which is accumulation of NRF2 protein (NFE2L2), a key transcriptional activator of antioxidant defense genes [15–17]. It is noteworthy, however, that under certain conditions, an increase in NRF2 protein levels may paradoxically result in amplification of ROS via transactivation of a gene encoding a transcription factor Kruppel-like factor 9 (KLF9) [18, 19]. KLF9 represses multiple anti-oxidant defense genes including mitochondrial thioredoxin reductase (TXNRD2) [20] leading to increased ROS levels [18]. KLF9 is a ubiquitously expressed member of an evolutionary conserved family of KLF transcriptional regulators. It has been implicated in a wide spectrum of processes including differentiation of B cells, maturation of neurons, and differentiation of intestinal cells [19, 21–23]. KLF9 is downregulated in multiple types of human cancers and has been shown to suppress several transformed phenotypes (proliferation, invasion, resistance to apoptosis) in cultured tumor cells [19, 24, 25].
Suppression of oxidative stress appears to be important for advanced melanomagenesis since administration of antioxidants has been shown to promote metastasis in several melanoma mouse models [26, 27]. On the contrary, information is rather scarce on the role of oxidative stress during other stages of melanoma progression. In answer to this question, we interrogated Klf9 deficiency in pre-malignant and transformed melanocytic mouse tissues. We found that Klf9 affects melanoma progression in a stage-specific manner and established a central role of KLF9-dependent ROS in mediation of BRAFV600E signaling in premalignant melanocytes.
Results
Klf9 deficiency or treatment with NAC inhibits BRAFV600E-induced melanocytic proliferation
To identify a functional role of Klf9 in BRAFV600E-expressing premalignant melanocytes, we crossed Klf9−/− null mice with BrafCA/+;Tyr::CreER+/o mice (BC mice), a well-established mouse model of BrafV600E-induced hyperplasia [7]. These mice contain an oncogenic BrafCA allele (BrafV600E) as well as a gene for a tamoxifen-regulated Cre recombinase under the control of melanocyte-specific Tyrosinase (Tyr) promoter. Thus, we generated Klf9−/−; BrafCA/+;Tyr: :CreER+/o mice (KBC mice), which like parental BC mice and Klf9-null mice were viable, healthy and fertile (data not shown).
To induce BrafV600E expression, the backs of 6–8 week-old BC and KBC mice were shaved and topically treated with 5 μl of 1.9 mg/ml (5 mM) 4-HT every other day for 7 days (as previously described [7]). BC and KBC mice started developing skin hyperpigmentation within ~4 weeks after treatment with 4-HT. Punch biopsies were obtained 8 weeks after treatment to assess epidermal hyperpigmentation.
As shown in Fig. 1a, b, KBC mice treated with 4-HT displayed significantly lower levels of pigmentation as compared to 4-HT treated BC mice, suggesting that Klf9 deficiency inhibits melanocytic hyperplasia. To confirm this, immuno-histochemical (IHC) analysis was performed with antibodies against melanocyte lineage-specific marker, S100b [28]. As anticipated, 4-HT-treated KBC mouse skin contained significantly fewer S100b-positive cells than that of 4-HT-treated BC mice (Fig. 1c, d).
Fig. 1.
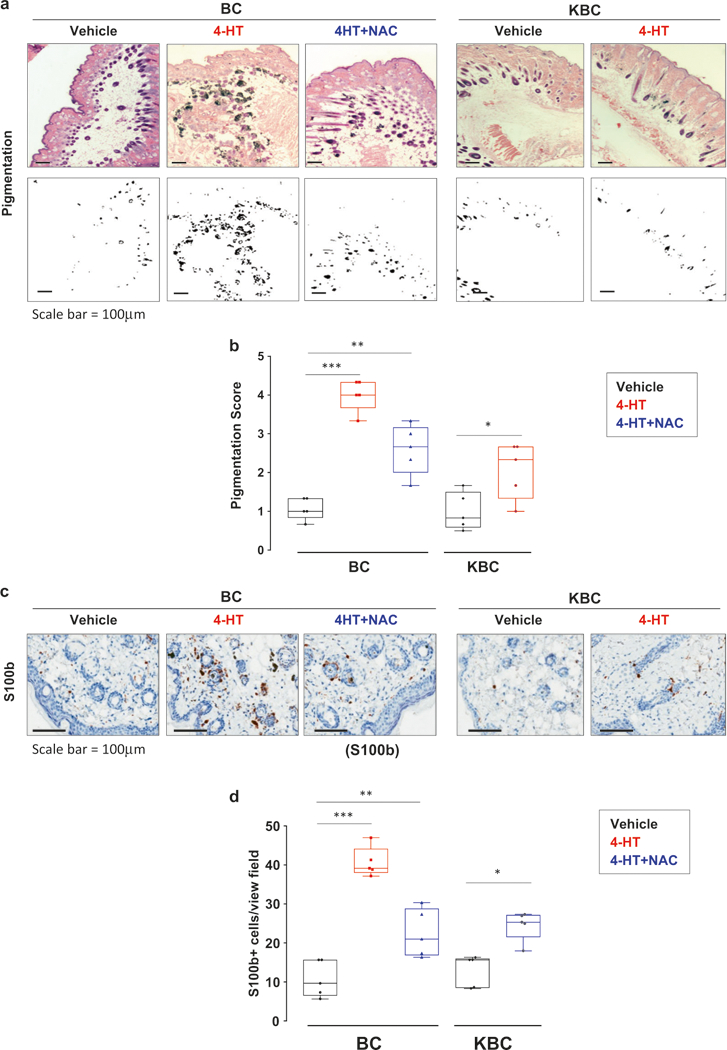
Klf9 deficiency or treatment with NAC inhibits BRAFV600E-induced hyperplasia. a H&E staining of skin sections derived from either BC or KBC mice 8 weeks after treatment with either vehicle (DMSO), 4-HT or 4-HT + NAC as described in the text (upper panel). Representative images shown (scale bar = 100 μm). Phase contrast image of section shown in upper panel highlighting pigmented regions in black (lower panel). Representative images shown. b To quantify the pigmentation in each specimen, three random fields from each H&E section (n = 5 sections per group) were imaged using bright field illumination. The images were then converted to black and white phase contrast images as shown in (a) lower panel using Adobe Photoshop. The area of pigmentation was measured using ImageJ software. To quantify the pigmentation in each specimen, three random fields from each H&E section (n = 5 sections) were imaged using bright field illumination. The images were then converted to black and white phase contrast images as show in (a) lower panel. A pigmentation score was assigned based on the area of pigmentation per total area ranging from (0 no pigmentation, 1 (0–10%), 2 (10–30%, 3 (30–50%), 4 (50–75%), 5 (75–100%) for each field. c Skin sections from mice described in (a) stained with S100b-specific antibodies using IHC. Representative images shown (scale bar = 100 μm). d Quantification of number of S100+ cells shown in panel c. S100b+ cells were counted in three random fields from each H&E section (n = 5 sections per group). Statistical comparisons were made using an unpaired Student’s t-test. (*p <0.05; **p <0.01; ***p <0.001)
We have previously reported that Klf9 is a universal inducer of oxidative stress [18]. However, KLF9 has been implicated in suppression of transformed phenotypes via other mechanisms including for example inhibition of Wnt/beta-catenin/TCF and Notch1 signaling [19, 24, 25]. Therefore, to determine the role of ROS in Klf9-dependent phenotypes, in parallel with the above experiments, we treated BC mice topically with N-acetyl cysteine (NAC), the universal ROS scavenger, for the duration of the experiment starting 2 days after completion of 4-HT treatments. This was followed by collection of punch biopsies 8 weeks after treatment as described above. As expected, treatment with NAC significantly suppressed the intensity of staining with antibodies to 8-hydroxy-2′-deoxyguanosine (8-OHdG, a common biomarker of oxidative stress [29] in the entire skin (Supplementary Figure S1)). Importantly, pigmentation levels and the number of S100b-positive cells in 4-HT-treated BC mice were also decreased by NAC treatment (Fig. 1a-d). We therefore, concluded that BrafV600E-induced premalignant melanocytic proliferation requires Klf9 and ROS.
Klf9 deficiency does not affect primary tumor growth
While BRAFV600E expression in melanocytes promotes an initial phase of proliferation, additional genetic alterations, such as loss of PTEN, are required for progression of these premalignant cells to advanced tumors with robust invasive and metastatic capabilities [7]. We were also interested in identifying the role of Klf9 at more advanced stages of melanoma progression. To this end, we crossed Klf9−/− mice with BrafCA/+;Ptenlox/lox;TYR::CreERT2 mice (BPC mice) [7] to generate Klf9−/−;BrafCA/+;Ptenlox/lox;TYR: CreERT2 mice (KBPC mice).
Localized melanoma induction in BPC and KBPC mice was achieved as described above for BC and KBC mice. Following 4-HT treatments, BPC mice were further divided into two cohorts and treated every other day via i.p. injection with either vehicle or NAC. Intravenous administration has been previously demonstrated as an efficient mean of NAC delivery to both primary tumors and metastatic nodules [26]. As expected, ~7–14 days after administration of 4-HT, all mice displayed expansion of highly pigmented cells and confluent melanocytic proliferation, which progressed to primary tumors ~14 days post-treatment.
Interestingly, the number and size of primary tumors were comparable between KBPC and BPC mouse groups, as well as between BPC mice treated with vehicle or NAC via i.v. injection (Fig. 2a–c). Similar results were obtained in BPC mice topically treated with vehicle or NAC (Supplemental Figure S2). At the same time, the intensity of 8-OHdG (a common biomarker of oxidative stress [29]) was decreased in specimens from KBPC mice vs. BPC mice and in NAC-treated vs. vehicle-treated BPC mice (Fig. 2d, e). These data suggest that Klf9 deficiency or NAC treatment do not affect primary tumor growth in the studied mouse models, although they continue to suppress intracellular ROS.
Fig. 2.
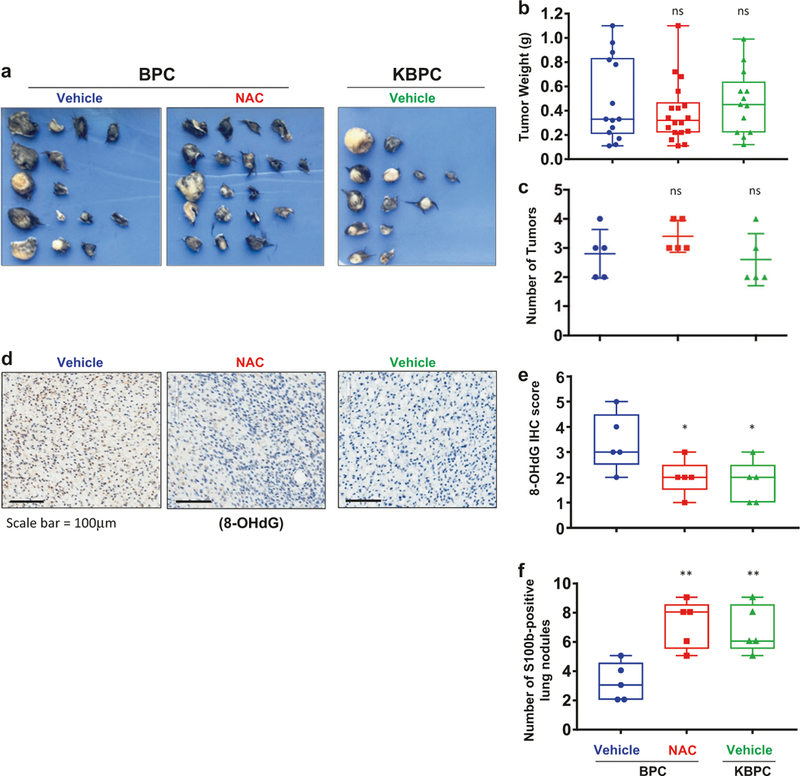
Klf9 deficiency or treatment with NAC does not affect primary tumor growth but promotes metastasis in BrafCA/Pten−/− mice. a Images of primary tumors resected from BPC or KBPC mice (n = 5) 8 weeks after treatment with 4-HT and either vehicle (saline) or NAC (100 mg/kg) as described in the text. b Quantification of weight of the tumors from mice shown in (a) (n = 5, ns = nonsignificant, Student’s t-test). (scale bar = 100 μm). c Quantification of number of tumors from mice shown in (a) (n = 5, ns = nonsignificant, Student’s t-test). d Representative images of primary tumors resected from mice described in (a) and stained with anti-8-OHdG antibodies (scale bar = 100 μm). e Quantification of intensity of staining with anti-8-OHdG antibodies; scored on a scale from 0 (low) to 5 (high) and represented as an average of three fields per specimen (n = 5, *p < 0.05; Student’s t-test). f Lung sections derived from mice from (a) were stained with anti-S100b antibodies using IHC. Shown is quantification of S100b+ positive lung nodules. Lungs of five mice per group were examined, three sections per lungs. **p < 0.01; Student’s t-test). Each experiment was performed at least two times with consistent results
Klf9 deficiency or treatment with NAC promotes melanoma metastasis
It has been demonstrated that treatment with antioxidants suppresses melanoma metastasis in several experimental settings [26, 27]. We were interested in whether loss of Klf9 will too promote metastasis in BrafCA/Pten−/− mice. To this end, we examined lungs of tumor-bearing mice at the time of sacrifice. We identified multiple S100b-positive nodules (Supplementary Figure S3) in lungs of all mouse groups. Importantly, the number of metastases was higher in KBPC and NAC-treated BPC mice compared to BPC mice treated with vehicle (Fig. 2f), suggesting that genetic and pharmacological inhibition of ROS promote BrafCA/Pten−/− melanoma metastasis.
KLF9 levels decrease during melanoma progression
To evaluate the expression pattern of KLF9 in melanocytic cells, we started by assessing KLF9 levels in a panel of metastatic melanoma cells and normal human melanocytes (NHMs). As shown in Fig. 3a, a significant drop in KLF9 protein expression was observed in metastatic melanoma cells compared to NHMs. Next, we measured KLF9 protein expression in human melanoma specimens via immuno-histochemistry. Our analysis revealed that KLF9 levels decrease with high statistical significance in metastatic vs. primary melanomas (Fig. 3b). Furthermore, stage-wise stratification of primary melanoma specimens (T1–T4) revealed a statistically significant drop in KLF9 expression in the thickest (T4) primary melanomas compared to thinner tumors (T1–T3) (Fig. 3c). Accordingly, analysis of the cutaneous melanoma TCGA RNAseq dataset revealed poorer survival outcomes for patients with lower KLF9 levels in melanoma specimens (Fig. 3d). Collectively, these results support our animal data demonstrating that Klf9 tumor suppressor functions are manifested at advanced stages of melanoma progression.
Fig. 3.
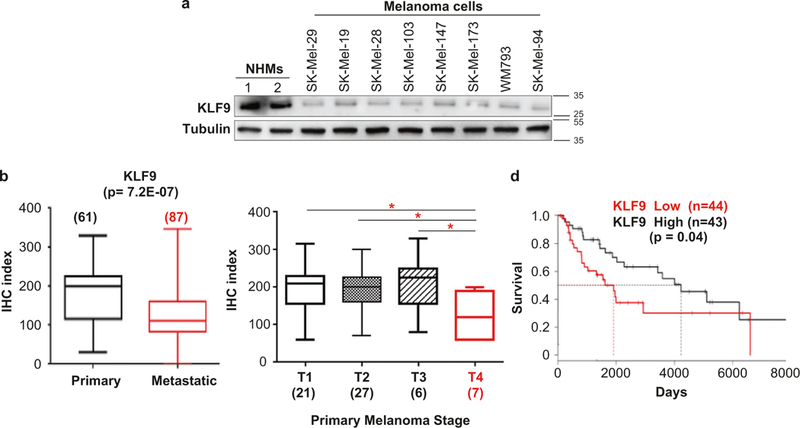
KLF9 levels decrease during melanoma progression in patients. a Two independent populations of NHMS (1 and 2) and indicated melanoma cell lines were probed in immunoblotting with antibodies indicated on the left. Molecular weights are indicated on the right in kDa. b and c The box plots represent the distribution of the IHC index. The number of patient samples (n) is indicated for each cohort. See Materials and methods for IHC description. d Overall survival of melanoma patients with high (highest 15%) vs. low (lowest 15%) KLF9 levels. TCGA skin cutaneous melanoma dataset SKCM, RNAseq was used. Kaplan-Meier survival curves were calculated and compared by log-rank tests (*p < 0.05)
BRAFV600 upregulates KLF9 and requires KLF9-dependent ROS for full-scale activation of ERK and cell proliferation in human melanocytes
Several mechanisms have been described that underline suppression of melanoma metastasis by oxidative stress in mice [26, 27]. On the other hand, the function of ROS in BRAFV600E signaling in premalignant melanocytic cells is largely understudied.
To start addressing this question, we transduced NHMs with an empty vector or BRAFV600E-expressing vector. As previously reported [5, 8], BRAFV600E-expressing NHMs demonstrated increased proliferation during the first several days post-infection compared to the controls (Fig. 4a, b). Activated BRAF increased levels of intracellular ROS, as was evidenced by staining cells with CM-H2DCFDA, general oxidative stress indicator, and monitoring its intensity using flow cytometry (Fig. 4c). Levels of NRF2, the major transcriptional regulator of antioxidant response in the cell [15], were also increased (Fig. 4a). Importantly, levels of KLF9 were also increased, whereas levels of mitochondrial thioredoxin reductase (TXNRD2, a gene repressed by KLF9 [18]) decreased (Fig. 4a).
Fig. 4.
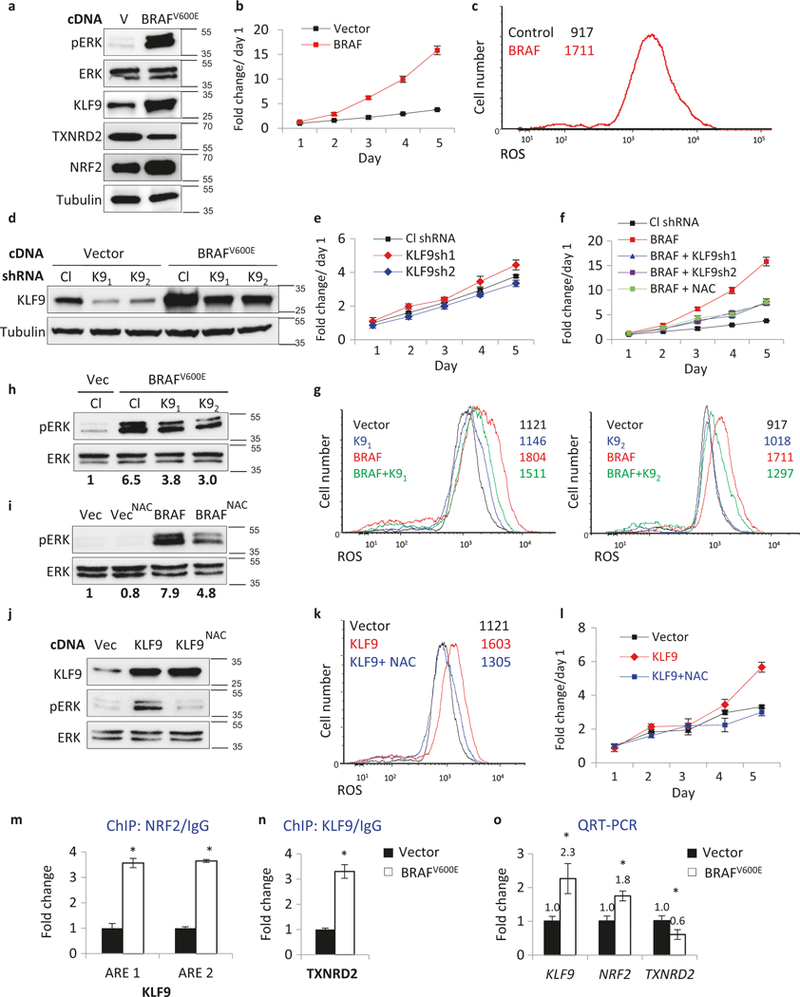
KLF9-dependent ROS regulate BRAFV600E activation of ERK1/2 and proliferation in NHM. a NHMs infected with an empty vector (V) or BRAFV600E-expressing vector (BRAFV600E) were probed in immunoblotting with indicated antibodies (representative immuno-blots shown). Molecular weights are indicated on the right in kDa. b NHMs described in (a) were counted in triplicates on indicated days using trypan blue exclusion assay starting 48 h after infection (*p < 0.05; Student’s t-test). c Cells described in (a) were stained with H2DCFDA (DCF) followed by FACS analysis to determine intracellular ROS. d NHMs were infected with control (Cl) or KLF9 (K9) shRNAs followed by transduction with empty vector (V) or BRAFV600E-expressing vector (BRAFV600E). Cells were probed in immunoblotting with indicated antibodies 48 h after second infection. Molecular weights are indicated on the right in kDa. e, f NHMs described in (d) were counted on indicated days in triplicates using trypan blue exclusion assay starting 48 h after infection (*p <0.05; Student’s t-test). g Cells described in (d) were stained with H2DCFDA (DCF) followed by FACS analysis to determine intracellular ROS. h NHMs were infected with control (Cl) or KLF9 (K9) shRNAs followed by transduction with empty vector (V) or BRAFV600E-expres-sing vector (BRAFV600E). Cells were probed in immunoblotting with indicated antibodies 48 h after infection. pERK and ERK signals were quantified using ImageQuant software. Molecular weights are indicated on the right in kDa. pERK/ERK signal ratio was identified and normalized by that in “Vec/Cl” lane. i NHMs infected with the empty vector (Vec) or BRAFV600E-expressing vector (BRAFV600E) were treated with 0.5 mM NAC for 48 h and probed in immunoblotting with antibodies designated on the left. Molecular weights are indicated on the right in kDa. pERK and ERK signals were quantified using ImageQuant software. pERK/ERK signal ratio was calculated and normalized to that of vector control (“Vec” lane). j NHMs infected with empty vector (Vec) or KLF9-expressing vector (KLF9) were treated or not with 0.5 mM NAC for 48 h and probed in immunoblotting with indicated antibodies. Molecular weights are indicated on the right in kDa. k Cells described in (j) were stained with H2DCFDA (DCF) followed by FACS analysis to determine intracellular ROS. 1 Cells described in (j) were cultured in media containing 0.5 mM NAC and were counted in triplicates on indicated days using to trypan blue exclusion assay starting 48 h after infection (*p <0.05; Student’s t-test). m, n ChIP assay. QPCR signals in reactions with DNA that was precipitated with NRF2, KLF9, or control IgG antibodies from NHMs expressing Vector or BRAFV600E. QPCR was performed with primers encompassing NRF2 binding site in KLF9 promoter (ARE1) (ARE2) or KLF9-binding site in TXNRD2 promoter. All QPCR signals were normalized by those obtained from IgG-precipitated DNA in Vector cells. Representative experiment shown (*n = 3, technical replicas, p <0.05; Student’s t-test). o NHMs expressing Vector or BRAFV600E were probed in QRT-PCR with primers corresponding to indicated genes. QRT-PCR signals were normalized by β-actin signal and by the corresponding signals in Vector cells. Representative experiment shown, (*n = 3, technical replicas, p <0.05; Student’s t-test). Each experiment was performed at least two times with consistent results
Interestingly, although shRNA-mediated depletion of KLF9 did not affect proliferation of NHM-vector cells (Fig. 4d, e), it did suppress proliferation rates and levels of total ROS in BRAFV600E-expressing cells (Fig. 4f, g). Levels of mitochondrial superoxide measured with Mito-SOX™ Red reagent via flow cytometry were also decreased (Supplemental Figure S4a). Accordingly, mitochondrial membrane potential (MMP) determined using MitoProbe™ DiIC1[5] dye also via flow cytometry was decreased in BRAFV600E NHM compared to Vector-NHM. Depletion of KLF9 in BRAFV600E NHM partially restored MMP (Supplemental Figure S4b). These data are in general agreement with those obtained in TXNRD2-deficient or TXNRD2-inhibited cells [30, 31]. Proliferation rates of BRAFV600E-expressing NHMs were also decreased by treatment with 0.5 mM NAC (Fig. 4f) suggesting that KLF9-induced ROS are required for BRAFV600E-dependent proliferation in NHMs.
Activation of ERK1/2 kinases is the major mechanism of BRAFV600E-induced proliferation. Induction of ERK1/2 by BRAFV600E was substantially (~50%) decreased in KLF9-depleted NHMs or NHMs treated with NAC compared to Vector-treated or untreated cells, respectively (Fig. 4h, i). Reciprocally, overexpression of KLF9 in NHMs (Fig. 4j) increased intracellular ROS levels (Fig. 4k), levels of p-ERK1/2 (Fig. 4j), and cell proliferation (although less robustly than overexpression of BRAFV600E, Fig. 4l) which were reverted by treatment with NAC (Fig. 4j–l). Collectively, these data suggest that full-scale activation of ERK1/2 by BRAFV600E requires KLF9 or KLF9-dependent ROS in NHMs.
We have recently reported that the KLF9 gene is a direct transcriptional target of NRF2, however, in comparison to other NRF2 targets, activation of KLF9 regulatory regions require higher amounts of NRF2 [18]. By activating KLF9, NRF2 paradoxically leads to upregulation of ROS [18]. Thus, we were interested in whether NRF2 mediates BRAFV600E-dependent increase in KLF9 levels in NHMs. To this end, we assessed the binding of NRF2 to the regulatory regions of KLF9 gene in Vector-and BRAFV600E-NHMs using chromatin immunoprecipitation (ChIP) assay with NRF2-specific, KLF9-specific, or control (IgG) antibodies. The precipitated materials were probed in Q-PCR with primers flanking the previously identified NRF2-binding sites in the KLF9 regulatory regions [18]. As shown in Fig. 4m, NRF2 more efficiently interacts with KLF9 regulatory regions in BRAFV600E-expressing NHMs compared to Vector-NHMs, suggesting that BRAFV600E-induced NRF2 is important for KLF9 transcriptional activation. Accordingly, KLF9 binding to the promoter of its major target TXNRD2 [18] was also increased in BRAFV600E-expressing compared to Vector NHMs (Fig. 4n), which is in good agreement with the decreased levels of TXNRD2 observed in BRAFV600E-cells (Fig. 4a).
Although NRF2 is usually regulated at posttranslational level [15], NRF2 gene can be activated transcriptionally by BRAFV600E-c-Jun or BRAFV600E-Myc axes at least in mouse embryonic fibroblasts [17]. We demonstrated that NRF2 mRNA levels were indeed increased in NHM-BRAFV600E-compared to Vector-NHMs (Fig. 4o), further supporting the existence of a BRAFV600E → NRF2 → KLF9 axis in NHMs. Collectively, our data demonstrate that KLF9-dependent ROS mediate BRAFV600E-induced melanocytic proliferation in vitro and in vivo.
Our animal data show that growth of primary melanoma in BrafCA/Pten−/− mice is insensitive to deficiency of Klf9 or treatment with NAC (Fig. 2). We were interested in whether this feature can be recapitulated in cultured human melanocytes. To this end, we depleted endogenous PTEN in BRAFV600E-expressing NHMs using previously evaluated PTEN shRNA (BRAFV600E/PTENsh melanocytes). PTEN depletion did not change levels of KLF9 or p-ERK1/2 in BRAFV600E-expressing melanocytes (Fig. 5a, b). Next, BRAFV600E/PTENsh melanocytes were superinfected with control shRNA or KLF9 shRNAs (Fig. 5b). Although depletion of KLF9 in BRAFV600E/PTENsh melanocytes decreased ROS levels, it did not change levels of p-ERK1/2 and proliferation rates (Fig. 5c, d).
Fig. 5.
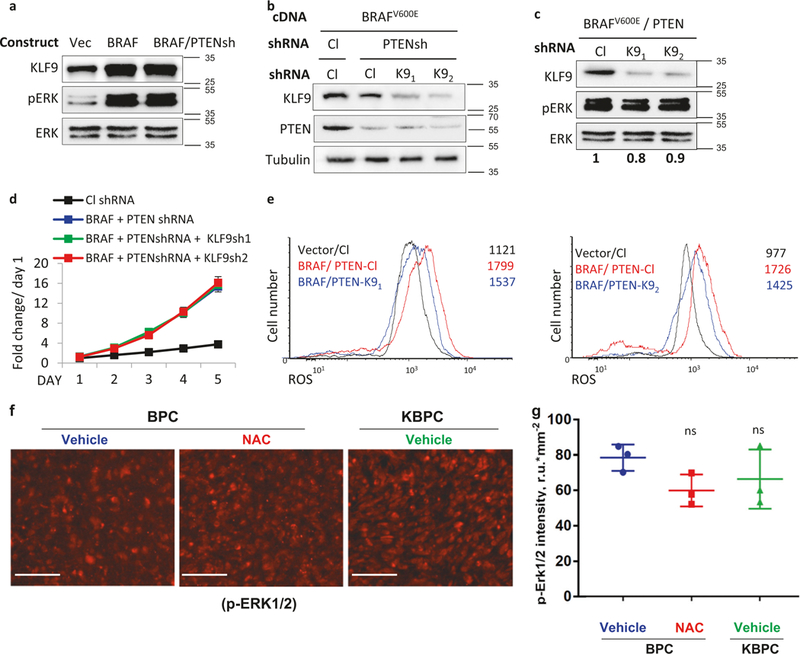
Depletion of PTEN renders proliferation and ERK1/2 activation in BRAFV600E melanocytes insensitive to KLF9-dependent ROS. a NHMs infected with an empty vector (V) or BRAFV600E-expressing vector (BRAFV600E) or BRAFV600E-expressing vector together with PTEN shRNA (BRAF/PTENsh) were probed in immunoblotting with antibodies designated on the left. Molecular weights are indicated on the right in kDa. b BRAFV600E-expressing NHMs were infected with control (Cl) or KLF9 (K9) shRNAs followed by infection with control shRNA or PTEN shRNA (PTENsh). Cells were probed in immuno-blotting with indicated antibodies 48 h after second infection. Molecular weights are indicated on the right in kDa. c NHMs expressing BRAFV600E and PTEN shRNA were infected with control (Cl) or KLF9 (K9) shRNAs. Cells were probed in immunoblotting with indicated antibodies 48 h after second infection. pERK and ERK signals were quantified using ImageQuant software. pERK/ERK signal ratio was identified and normalized to that in “Cl” lane. Molecular weights are indicated on the right in kDa. d NHMs described in (b) were counted on indicated days in triplicates using trypan blue exclusion assay starting 48 h after last infection. e Cells described in (d) were stained with H2DCFDA (DCF) followed by FACS analysis to determine intracellular ROS. f Representative images of primary tumors resected from BPC mice treated i.v. with vehicle or NAC or KBPC mice treated with vehicle. Tumors were stained with anti phospho-ERK1/ERK2 antibodies (scale bar = 50 μm). g Quantification of intensity of staining with anti phospho-ERK1/ERK2 antibodies was performed using ImageJ software. Each point represents a mean fluorescence of seven fields of view (n = 3 tumors, ns = non-significant, Student’s t-test). Each experiment was performed at least two times with consistent results
The fact that KLF9 depletion did not affect p-ERK in BRAFV600E/PTENsh melanocytes prompted us to evaluate ERK1/2 activation in melanomas in BPC and KBP mice. As shown in Fig. 5f, the levels of p-ERK1/2 did not vary with statistical significance among BPC and KBPC mice or BPC mice treated or not with NAC. Taken together our results suggest that PTEN inhibition renders BRAFV600E-induced ERK activation and proliferation of melanocytic cells insensitive to KLF9-depenent ROS in vivo and in vitro.
Discussion
Function of ROS in premalignant melanocytic cells is largely understudied, although processes regulating proliferation of these cells are important for de novo melanomagenesis. In the current manuscript, we focused on the role of KLF9-depedent ROS in proliferation induced by the activating (V600E) mutation in BRAF gene which is considered the earliest melanoma driver mutation [3, 4]. In addition to increasing ROS, KLF9 has been implicated in regulation of several pathways mostly as a tumor suppressor [19, 24, 25]. However, in our experiments treatment with ROS scavenger NAC largely phenocopied KLF9 deficiency or depletion, suggesting that ROS play the major role in the studied KLF9-dependent phenotypes.
We identified that BRAFV600E-induced proliferation requires KLF9-dependent ROS in cultured melanocytes and melanocytes in the skin. On the contrary, it has been reported that conditional activation of endogenous BrafV619E (equivalent to human BRAFV600E) in mouse embryonic fibroblasts led to suppression of ROS [17]. The discrepancy between these results and our findings is most likely due to a difference between studied cell types, as melanocytic cells display unique regulation of ROS [32]. Unlike many other primary cells, epidermal melanocytes continuously undergo increased oxidative stress due to melanin synthesis that produces superoxide anion and H2O2 [33]. As a result, reaction of melanocyte to changes in ROS generated by endogenous or exogenous stimuli differs from that of other cell types. For example, shRNA-mediated depletion of p16INK4A in melanocytes, keratinocytes, and fibroblasts generated from the skin of the same donor led to significantly higher ROS levels (both basal and H2O2-induced) in melanocytes compared to other skin cell types [34].
Oxidative stress caused by external sources, most commonly UV-A part of solar radiation, has been considered a major risk factor in human skin cancers including melanoma [35]. Indirect DNA damage induced by UV-A exposure is an important driver of genetic instability causing cutaneous neoplasms in several animal models [36]. As a result, topical preparations containing antioxidants, such as vitamins C and E, are currently being used to protect skin from UV-induced damage [37, 38]. To mimic such preparations, we used topical treatment with NAC and demonstrated for the first time that it suppressed BRAFV600E-dependent hyperplasia. These data suggest that endogenous oxidative stress caused by BRAF activation is essential for proliferation of premalignant melanocytic cells which in turn is important for the acquisition of additional genetic lesions (such as loss of Pten). Thus, our data suggest that topical antioxidants could prevent de novo melanomagenesis via suppression of BRAFV600E-induced proliferation independently from suppressing consequences of UV exposure.
On the other hand, treatment with NAC failed to inhibit formation of primary BrafCA/Pten−/− melanomas. This could be due to the fact that in BPC mice, activation of BrafV600E and deletion of both Pten alleles occurs simultaneously, resulting in a very fast progression from pre-malignant hyperplasia to malignant lesions. With this respect, this mouse model does not completely reflect the situation in human skin where progression from premalignant to malignant stages might take considerable amount of time.
We found that BRAFV600E upregulates levels of NRF2 and KLF9 proteins (Fig. 4a). An increase in NRF2 levels in BRAFV600E melanocytes (Fig. 4a) could be achieved via several mechanisms including a rather unconventional transcriptional upregulation of NRF2 gene via BrafV600E-Jun or BrafV600E-Myc axis described in MEFs by DeNicola et al. [17]. In support of this possibility, we detected an increase in NRF2 mRNA levels in BRAFV600E-expressing melanocytes compared to control cells (Fig. 4o). It is also possible that other BRAFV600E-dependent but KLF9-independent pathways induce ROS and, subsequently NRF2 since depletion of KLF9 did not completely eliminate BRAFV600E-induced ROS (Fig. 4g).
We determined that a full-scale activation of ERK1/2 by BRAFV600E in melanocytes requires KLF9-dependent ROS. Treatment of cells with oxidative agents increased levels of p-ERK1/2 in multiple cell lines, although in the vast majority of cases ROS effectors (e.g. receptor tyrosine kinases, PKC, members of Src-or RAS-families [39, 40]) functioned via activation of CRAF which is unlikely to cooperate with BRAFV600E for ERK1/2 activation in melanocytes [41]. A more plausible mechanism may involve ROS-mediated suppression of ERK-inhibitory dual-specificity phosphatases [42, 43].
Depletion of KLF9 decreased ROS in BRAFV600E/PTENshRNA melanocytes but did not significantly affect pERK levels or cell proliferation suggesting that PTEN depletion triggered ROS-independent mechanism of ERK1/2 activation. Loss of PTEN has been reported to activate ERK independently of RAF via PI3K-RAC1/CDC42-PAK1-MEK pathway [44, 45]. It is conceivable that PTEN depletion produces more robust activation of ERK1/2 compared to KLF9-dependent ROS, and as a result, p-ERK1/2 levels in BRAFV600E-PTENshRNA melanocytes become insensitive to changes in KLF9 and ROS.
In summary (Supplemental Table S1), our data identify KLF9 as an important mediator of BRAFV600E-dependent signaling in premalignant melanocytes in vitro and in mouse skin, and provide an explanation to a variable role of ROS in premalignant and transformed melanocytic cells.
Materials and methods
Mice
Klf9-/– mice were described previously [46]. Tyr-CreER, Ptenlox, and BrafCA, alleles have also been described [7] and were acquired from Jackson Labs (B6.129P2(Cg)-Braftm1Mmcm/J, 017837 and B6.Cg-Braftm1Mmcm Ptentm1Hwu Tg(Tyr-cre/ERT2)13Bos/BosJ, 013590). All mice were genotyped using quantitative real-time PCR by Transnetyx genotyping services using recommended primers.
Klf9−/− mice were crossed with BrafCA Tyr-CreER (BC mice) and separately with BrafCA Ptenlox Tyr-CreER (BPC mice) to generate Klf9−/− BrafCA Tyr-CreER (KBC mice) and Klf9−/− BrafCA Ptenlox Tyr-CreER (KBPC mice). All strains were maintained on a C57BL/6 background. All animal experiments were performed in compliance with ‘Care and Use of Animals’, and were approved by the Roswell Park Comprehensive Cancer Center IACUC.
To induce BrafV600E expression, 6–8-week-old mice were treated topically with 5 μl of 1.9 mg/ml (5 mM) 4-HT every other day for 4 days for 1 week. BC and KBC mice developed skin hyperpigmentation within 4–6 weeks after treatment with tamoxifen. After 1 week of treatment with 4-HT, mice were divided into two groups and treated daily with either vehicle (Saline) or 100 mg/kg NAC intraperitoneally until experimental endpoint.
For induction of melanoma on the back skin, 6–8-week-old mice were treated topically with 5 μl of 1.9 mg/ml (5 mM) 4-HT every other day for 4 days for 1 week. For treatment with NAC, mice were injected intraperitoneally daily with either vehicle (saline) or NAC (100 mg/kg) until experimental endpoint. Tumor size was measured using a vernier caliper. Volume (mm3) = (L × W2)/2, where L is the longest and W the shortest radius of the tumor (mm).
For topical administration of NAC, a 20% N-acetyl-L-cysteine (NAC) (Sigma-Aldrich, A7250) solution was freshly prepared using water as the solvent. Two hundred microliters of this solution was applied daily directly to a localized region of mouse skin for the entire course of the experiment.
Immunohistochemistry
Positive and negative control slides were included with every immunochemistry run. The S100b (Abcam, ab62642) and 8-OHdG (Biorbyt, orb10011) antibodies were visualized with the Novocastra (Newcastle, UK) PowerVision kit, followed by Fast Red (Thermo Scientific). Phospho-ERK1/ERK2 (Thr185, Tyr187) (ThermoFisher Scientific (#44–680G)) antibody signal was visualized with Alexa Fluor 594 goat anti-rabbit secondary antibodies (Invitrogen).
Patient selection and statistical analysis
The study was approved by the Roswell Park Cancer Institute (RPCI) Institutional Review Board. Formalin fixed and paraffin-embedded skin and melanoma specimens were processed at the Pathology Core Facility (RPCI). Patients included in the study (total 149) were diagnosed with melanoma between July 1970 and April 2005 at RPCI or Ohio State University (OSU). Selection of patients included all patients in this time period with adequate material in the RPCI or OSU archival bank for whole section use or tissue microarray (TMA) construction. Patients in the primary cutaneous cohorts were assembled from patients at RPCI. All specimens in the primary cutaneous cohort were studied using whole sections to evaluate the full architecture of the tumor. Specimens in the metastatic melanoma cohort were assembled from patients at the Ohio State University and were evaluated using TMAs, which is deemed representative since the overall architecture of the tumor is no longer a part of the diagnostic evaluation and is not a requirement for accurate assessment. There was no overlap between the primary cutaneous cohorts and the metastatic cohort to avoid bias of selection of patients with altered KLF9 expression in the primary disease. The 61 patients in the primary cutaneous melanoma cohort had a median patient age at first diagnosis of 60 years (average 59; range 21–86) with approximately even distribution between males and females. For the 87 patients in the metastatic melanoma cohort, specimens represented metastatic melanoma of various sites, including lymph node and visceral metastases. The expression IHC-based staining index for KLF9 was calculated as the product of the intensity of the stained cells (0–3) and the percentage of cells stained. For statistical analysis of IHC scores, an unpaired Student’s t-test was used and a value of p ≤ 0.05 was considered statistically significant.
Cell culture
Populations of NHMs were purchased from Invitrogen and maintained in Medium 254 (Invitrogen) supplemented with human melanocyte growth supplement (Invitrogen). Cells were routinely tested for mycoplasma contamination.
Cell proliferation analysis
Cell proliferation was assessed by counting cells daily using trypan blue exclusion assay.
Flow cytometry assays
Approximately 3 × 105 cells were collected after trypsinization, washed twice with PBS and subjected to the following assays according to the manufacturer’s recommendations. Total ROS levels were measured using CM-H2DFCDA reagent (Invitrogen (C6827)). Levels of mitochondrial superoxide were measured using MitoSOX™ Red reagent (Thermo Fisher Scientific (M36008)). MMP was measured using MitoProbe™ DiICl [5] dye (Thermo Fisher Scientific (M34151)). Fluorescence was measured via flow cytometry using LSRIIA (BECTON DICKINSON). Detection was based on mean fluorescence intensity of at least 10,000 cells.
Reverse transcription-PCR analysis
Total RNA was isolated from cells using the RNeasy Mini Kit (Qiagen, Valencia, CA, USA). cDNA was prepared using cDNA reverse transcription kit (Invitrogen). Quantitative reverse transcription-PCR was performed using 7900HT fast real-time PCR system (Applied Biosystems, Carlsbad, CA, USA) using SYBr GreenMaster Mix (Invitrogen). See Supplemental Table S2 for primer sequences.
Immunoblotting
PVDF membranes were developed using alkaline phosphatase-conjugated secondary antibodies and signals were detected and visualized using the Alpha-Innotech FluorChem HD2 imaging system (Alpha Innotech) and quantified using ImageQuant software (GE Healthcare Life Sciences). The following antibodies have been utilized KLF9 sc-376422, TXNRD2 sc-46279, NRF2 sc-365949 (All from Santa Cruz Biotechnology), Tubulin HRP-66031 (Proteintech Biotechnology), p-ERK1/2 9101, ERK1/2 9102 (both from Cell Signaling Technology).
Statistical analysis
Each experiment was performed at least two times with consistent results. For in vitro studies, statistical significance was determined using Student’s t-test. A two-tailed p-value was considered significant for all analyses. For animal studies, sample size was determined as a function of effect size ((difference in means)/(standard deviation) = 2.0) for a two-sample t-test comparison assuming a significance level of 5%, a power of 90% and a two-sided t-test. Normal distribution was confirmed using normal probability plot (GraphPad Prism 6.0, Graphpad Software, Inc., San Diego, CA, USA), variance was also assessed using GraphPad Prism 6.0 both within and between groups and were approximately the same. For statistically analysis of immunohistological scores, an unpaired Student’s t-test was used and a value of p ≤ 0.05 was considered statistically significant.
Supplementary Material
Acknowledegements
We are grateful to the Pathology Resource Network, the Clinical Data Network, and the transgenic shared core facility.
Funding This work has been supported by the following NCI grants to MAN: CA190533, CA193981, CA220096, CA224434, by NCI P30CA16056 to Roswell Park Cancer Institute and partially supported by the Program for Basic Research of Russian State Academies of Sciences for 2013–2020, and by the Russian Foundation for Basic Research (grant No. 17–04-00716).
Footnotes
Compliance with ethical standards
Conflict of interest The authors declare that they have no conflict of interest.
Supplementary information The online version of this article (https://doi.org/10.1038/s41388–019-0689–6) contains supplementary material, which is available to authorized users.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Ekwueme DU, Guy GP Jr, Li C, Rim SH, Parelkar P, Chen SC. The health burden and economic costs of cutaneous melanoma mortality by race/ethnicity-United States, 2000 to 2006. J Am Acad Dermatol. 2011;65:S133–143. [DOI] [PubMed] [Google Scholar]
- 2.Linos E, Swetter SM, Cockburn MG, Colditz GA, Clarke CA. Increasing burden of melanoma in the United States. J Invest Dermatol. 2009;129:1666–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pollock PM, Harper UL, Hansen KS, Yudt LM, Stark M, Robbins CM, et al. High frequency of BRAF mutations in nevi. Nat Genet. 2003;33:19–20. [DOI] [PubMed] [Google Scholar]
- 4.Curtin JA, Fridlyand J, Kageshita T, Patel HN, Busam KJ, Kutzner H, et al. Distinct sets of genetic alterations in melanoma. N Engl J Med. 2005;353:2135–47. [DOI] [PubMed] [Google Scholar]
- 5.Denoyelle C, Abou-Rjaily G, Bezrookove V, Verhaegen M, Johnson TM, Fullen DR, et al. Anti-oncogenic role of the endoplasmic reticulum differentially activated by mutations in the MAPK pathway. Nat Cell Biol. 2006;8:1053–63. [DOI] [PubMed] [Google Scholar]
- 6.Dhomen N, Reis-Filho JS, da Rocha Dias S, Hayward R, Savage K, Delmas V, et al. Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer Cell. 2009;15:294–303. [DOI] [PubMed] [Google Scholar]
- 7.Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE, et al. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet. 2009;41:544–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhuang D, Mannava S, Grachtchouk V, Tang WH, Patil S, Wawrzyniak JA, et al. C-MYC overexpression is required for continuous suppression of oncogene-induced senescence in melanoma cells. Oncogene. 2008;27:6623–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–4. [DOI] [PubMed] [Google Scholar]
- 10.Tucker MA, Halpern A, Holly EA, Hartge P, Elder DE, Sagebiel RW, et al. Clinically recognized dysplastic nevi. A central risk factor for cutaneous melanoma. JAMA. 1997;277:1439–44. [PubMed] [Google Scholar]
- 11.Cymerman RM, Shao Y, Wang K, Zhang Y, Murzaku EC, Penn LA. et al. De novo vs nevus-associated melanomas: differences in associations with prognostic indicators and survival. J Natl Cancer Inst. 2016;108:djw121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol. 2014;24:R453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zuo L, Zhou T, Pannell BK, Ziegler AC, Best TM. Biological and physiological role of reactive oxygen species—the good, the bad and the ugly. Acta Physiol. 2015;214:329–48. [DOI] [PubMed] [Google Scholar]
- 14.Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11:85–95. [DOI] [PubMed] [Google Scholar]
- 15.Kansanen E, Kuosmanen SM, Leinonen H, Levonen AL. The Keap1-Nrf2 pathway: mechanisms of activation and dysregulation in cancer. Redox Biol. 2013;1:45-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leinonen HM, Kansanen E, Pölönen P, Heinäniemi M, Levonen AL. Role of the Keap1-Nrf2 pathway in cancer. Adv Cancer Res. 2014;122:281–320. [DOI] [PubMed] [Google Scholar]
- 17.DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zucker SN, Fink EE, Bagati A, Mannava S, Bianchi-Smiraglia A, Bogner PN, et al. Nrf2 amplifies oxidative stress via induction of Klf9. Mol Cell. 2014;53:916–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tetreault MP, Yang Y, Katz JP. Krüppel-like factors in cancer. Nat Rev Cancer. 2013;13:701–13. [DOI] [PubMed] [Google Scholar]
- 20.Arner ES. Focus on mammalian thioredoxin reductases--important selenoproteins with versatile functions. Biochim Biophys Acta. 2009;1790:495–526. [DOI] [PubMed] [Google Scholar]
- 21.Good KL, Tangye SG. Decreased expression of Kruppel-like factors in memory B cells induces the rapid response typical of secondary antibody responses. Proc Natl Acad Sci USA. 2007;104:13420–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bonett RM, Hu F, Bagamasbad P, Denver RJ. Stressor and glucocorticoid-dependent induction of the immediate early gene kruppel-like factor 9: implications for neural development and plasticity. Endocrinology. 2009;150:1757–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simmen FA, Xiao R, Velarde MC, Nicholson RD, Bowman MT, Fujii-Kuriyama Y, et al. Dysregulation of intestinal crypt cell proliferation and villus cell migration in mice lacking Kruppel-like factor 9. Am J Physiol Gastrointest Liver Physiol. 2007;292: G1757–1769. [DOI] [PubMed] [Google Scholar]
- 24.Qiao F, Yao F, Chen L, Lu C, Ni Y, Fang W, et al. Krüppel-like factor 9 was down-regulated in esophageal squamous cell carcinoma and negatively regulated beta-catenin/TCF signaling. Mol Carcinog. 2016;55:280–91. [DOI] [PubMed] [Google Scholar]
- 25.Ying M, Sang Y, Li Y, Guerrero-Cazares H, Quinones-Hinojosa A, Vescovi AL, et al. Krüppel-like family of transcription factor 9, a differentiation-associated transcription factor, suppresses Notch1 signaling and inhibits glioblastoma-initiating stem cells. Stem Cells. 2011;29:20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Le Gal K, Ibrahim MX, Wiel C, Sayin VI, Akula MK, Karlsson C, et al. Antioxidants can increase melanoma metastasis in mice. Sci Transl Med. 2015;7:308re8. [DOI] [PubMed] [Google Scholar]
- 27.Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. 2015;527:186–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gaynor R, Irie R, Morton D, Herschman HR. S100 protein is present in cultured human malignant melanomas. Nature. 1980;286:400–1. [DOI] [PubMed] [Google Scholar]
- 29.Kaneko T, Tahara S, Matsuo M. Non-linear accumulation of 8-hydroxy-2’-deoxyguanosine, a marker of oxidized DNA damage, during aging. Mutat Res. 1996;316:277–85. [DOI] [PubMed] [Google Scholar]
- 30.Chen Y, Cai J, Jones DP. Mitochondrial thioredoxin in regulation of oxidant-induced cell death. FEBS Lett. 2006;580:6596–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kirsch J, Schneider H, Pagel JI, Rehberg M, Singer M, Hellfritsch J, et al. Endothelial dysfunction, and a prothrombotic, proin-flammatory phenotype is caused by loss of mitochondrial thior-edoxin reductase in endothelium. Arterioscler Thromb Vasc Biol. 2016;36:1891–9. [DOI] [PubMed] [Google Scholar]
- 32.Denat L, Kadekaro AL, Marrot L, Leachman SA, Abdel-Malek ZA. Melanocytes as instigators and victims of oxidative stress. J Invest Dermatol. 2014;134:1512–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Simon JD, Peles D, Wakamatsu K, Ito S. Current challenges in understanding melanogenesis: bridging chemistry, biological control, morphology, and function. Pigment Cell Melanoma Res. 2009;22:563–79. [DOI] [PubMed] [Google Scholar]
- 34.Jenkins NC, Liu T, Cassidy P, Leachman SA, Boucher KM, Goodson AG. et al. Thep16(INK4A) tumor suppressor regulates cellular oxidative stress. Oncogene. 2011;30:265–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sander CS, Hamm F, Elsner P, Thiele JJ. Oxidative stress in malignant melanoma and non-melanoma skin cancer. Br J Dermatol. 2003;148:913–22. [DOI] [PubMed] [Google Scholar]
- 36.Day CP, Marchalik R, Merlino G, Michael H. Mouse models of UV-induced melanoma: genetics, pathology, and clinical relevance. Lab Invest. 2017;97:698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lin JY, Selim MA, Shea CR, Grichnik JM, Omar MM, Monteiro-Riviere NA, et al. UV photoprotection by combination topical antioxidants vitamin C and vitamin. E. J Am Acad Dermatol. 2003;48:866–74. [DOI] [PubMed] [Google Scholar]
- 38.Matsui MS, Hsia A, Miller JD, Hanneman K, Scull H, Cooper KD, et al. Non-sunscreen photoprotection: antioxidants add value to a sunscreen. J Investig Dermatol Symp Proc. 2009;14:56–59. [DOI] [PubMed] [Google Scholar]
- 39.Son Y, Cheong YK, Kim NH, Chung HT, Kang DG, Pae HO. Mitogen-activated protein kinases and reactive oxygen species: how can ROS activate MAPK pathways? J Signal Transduct. 2011;2011:792639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McCubrey JA, Lahair MM, Franklin RA. Reactive oxygen species-induced activation of the MAP kinase signaling pathways. Antioxid Redox Signal. 2006;8:1775–89. [DOI] [PubMed] [Google Scholar]
- 41.Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kidger AM, Keyse SM. The regulation of oncogenic Ras/ERK signalling by dual-specificity mitogen activated protein kinase phosphatases (MKPs). Semin Cell Dev Biol. 2016;50:125–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bonham CA, Vacratsis PO. Redox regulation of the human dual specificity phosphatase YVH1 through disulfide bond formation. J Biol Chem. 2009;284:22853–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiang K, Zhong B, Gilvary DL, Corliss BC, Hong-Geller E, Wei S, et al. Pivotal role of phosphoinositide-3 kinase in regulation of cytotoxicity in natural killer cells. Nat Immunol. 2000;1:419–25. [DOI] [PubMed] [Google Scholar]
- 45.Chu JY, Dransfield I, Rossi AG, Vermeren S. Non-canonical PI3K-Cdc42-Pak-Mek-Erk signaling promotes immune-complex-induced apoptosis in human neutrophils. Cell Rep. 2016;17:374–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Morita M, Kobayashi A, Yamashita T, Shimanuki T, Nakajima O, Takahashi S, et al. Functional analysis of basic transcription element binding protein by gene targeting technology. Mol Cell Biol. 2003;7:2489–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.