

Abstract
Background:
There have been few reports of mutations in the beta-myosin heavy chain (MYH7) gene in hypertrophic cardiomyopathy (HCM), which is associated with sudden cardiac death caused by HCM. This study aimed to screen the mutation sites in the sarcomeric gene MYH7 in Chinese patients with HCM. We also planned to analyze the pathogenicity of the mutation site as well as its significance in clinical and forensic medicine.
Methods:
From January 2006 to June 2017, autopsy cases were collected from the Department of Pathology, the Affiliated Hospital of Qingdao University. The experiment was to detect MYH7 gene status in formalin-fixed paraffin-embedded tissues from 18 independent autopsy cases who suffered HCM related sudden death (fatal HCM) and 20 cases without cardiomyopathy. Common mutation exon fragments of MYH7 gene were amplified by polymerase chain reaction. The end-of-deoxygenation method and gene cloning method were further performed to analyze the mutation sites. Homologous comparison among mutant sites was conducted using BLAST online database.
Results:
The 1336th nucleotide of MYH7 gene at exon 14 was converted from T to G in one HCM case, resulting in the conversion of threonine (Thr) at position 446 to proline (Pro). In another case, the 1402th nucleotide at exon 14 was converted from T to C, resulting in the conversion of phenylalanine (Phe) at position 468 to leucine (Leu). Homologous comparison results showed that the two amino acid residues of Thr446 and Phe468 are highly conserved among different species.
Conclusions:
Our results showed fatal HCM harbored mutations of Thr446Pro and Phe468Leu in the MYH7 gene. It is significant for clinical and forensic medicine to further explore the functions and detailed mechanisms of these mutations.
Keywords: Hypertrophic cardiomyopathy, MYH7, Gene mutation, Pathology
Introduction
Sudden cardiac death (SCD) is the primary type of sudden death in adults.[1] According to the literature, hereditary cardiomyopathy accounted for 5.9% to 6.2% of SCDs overall, and ranked the third among the causes of SCD.[2] Hypertrophic cardiomyopathy (HCM) is the most common reason for SCD in young adults and is also a major cause of morbidity and mortality in the elderly. It is now believed to affect as many as one in 300 individuals, regardless of race or gender.[3,4]
HCM is one of the most common autosomal dominant single-gene hereditary diseases in cardiomyopathy. It is a primary cardiomyopathy that excludes other myocardial abnormalities, such as hypertensive or valvular heart disease. It is characterized by asymmetrical hypertrophy of the ventricular septum, narrowing of the ventricular cavity, thickening of the ventricular wall, or increasing heart weight, and is the primary cause of SCD in adolescents and athletes.[5,6] Among the adult population in China, the prevalence of HCM is 80/100,000, and approximately 60% of adult patients with HCM will exhibit clear disease-causing gene mutations. China is a populous country, and the total number of patients with HCM is relatively large. The hereditary pattern of HCM appears to involve autosomal dominant inheritance. High heritability of the disease often has a profound impact on the entire family of the patient.[7] Therefore, it is of great significance to screen novel gene mutations in HCM.
More than 1600 pathogenic mutations have been identified in at least 27 genes to date,[8] among which the most frequent mutations were in MYH7 gene.[9,10]MYH7 gene mutations related HCM may lead to insufficiency of ventricular muscle energy supply, thus resulting in ventricular or ventricular septal hypertrophy.[11–14] The pathogenic mutation involved in HCM is divided into two types: familial and sporadic. The prevalence of familial HCM is more concentrated and involves familial aggregation, so it is better to investigate the cause of death.[15,16] The randomization of individuals with sporadic HCM (SHCM) has led to difficulties in determining the cause of death, thus SHCM has become a research hotspot in forensic medicine. SHCM also has substantial research value, since about 30% to 40% of patients are diagnosed as sporadic cases, although the incomplete penetration of some mutations could mean that the percentage of true familial cases is underestimated.[4,17]
In this study, exons of commonly found MYH7 gene mutations in 18 cases of isolated HCM-induced sudden death were sequenced in order to determine the gene mutation sites. The purpose of our study was to identify the phenotypic characteristics of these mutations in the sarcomeric gene MYH7, as observed in patients in China. These findings may be of great significance in determining the molecular pathological features of HCM, and could lead to better prevention and more accurate treatment of the disease in the future.
Methods
Ethical approval
The study was conducted in accordance with the Declaration of Helsinki and was approved by the Ethics Committee of the Affiliated Hospital of Qingdao University. Written informed consent was obtained from all participants.
Research subjects
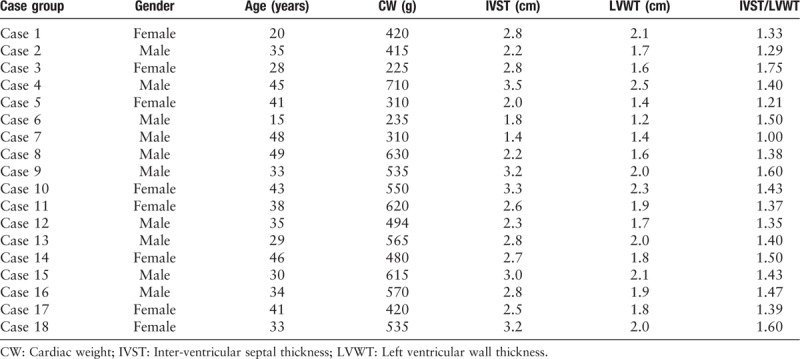
We identified 18 cases of sudden death in patients with pathological diagnosis of HCM (non-hypertensive) as the case group for this study by consulting the autopsy archives from the Department of Pathology, the Affiliated Hospital of Qingdao University from January 2006 to June 2017. The pathological criteria of HCM were as follows: (1) The weight of the heart was increased by 1 to 2 fold, where the average heart weight of the adult was more than 500 g. (2) The inter-ventricular septum was significantly thickened, and the ratio of ventricular septum thickness to left ventricular wall thickness (LVWT) increased from 0.95 (normal) to 1.3 or higher, which is also the main indicator for diagnosis of HCM. (3) The histological changes mainly showed that the myocardial fibers had significant hypertrophy, were arranged disorderly in a whirlpool or cluster, and their nuclei were large and darkly stained, with different karyotypes, which could be pleomorphic. The walls of the small blood vessels in the interstitial space were also thickened, and stenosis was observed in the lumens. The myocardial interstitial tissue often exhibited focal collagen fibrosis proliferation and fibrotic changes.
The control group consisted of 20 cases of violent death autopsies that excluded cardiac hypertrophy and structural changes of the heart.
We collected clinical data from other members of the HCM family. Physical examinations included height, weight, blood pressure, heart rate, and so on. Clinical examination included electrocardiogram, two-dimensional and Doppler echocardiography.
Autopsy and material selection
The histological examinations were performed on major organs such as the heart, liver, spleen, kidney, lung, and brain after systematic autopsy. Genomic deoxyribonucleotide (DNA) was extracted from the tissue of cardiac paraffin specimens.
Genomic DNA extraction
The paraffin-embedded myocardial tissue of the subject was cut with a paraffin microtome (RM2235, Leica, Germany). The genomic DNA was extracted using the TIANamp FFPE DNA Kit (DP331, Tiangen, Shanghai, China).
Polymerase chain reaction (PCR) and cloning of the PCR product
Based on the MYH7 gene sequence from the National Center for Biotechnology Information (NCBI) Genbank, we used Nucleotide BLAST to design exon primers. The designed primers were synthesized by the Shanghai Bio-engineering Company (Shanghai, China). The PCR amplification reagent used for amplification was Premix TaqTM EX Version 2.0 (TaKaRa, Japan). Exon 14 primers were as follows: the upstream sequence: 5′-GTCTCTCCTCCACCTTGCAG-3′; the downstream sequence: 5′-CGAGTGAGTGAT TGTTCTCC-3′. The PCR amplification volume was 25.0 μL, the reaction volume containing the DNA templates was 2.0 μL, the Premix volume was 12.5 μL, the upstream primer concentration was 3 μmol/L, the downstream primer concentration was 3 μmol/L, and the volume of sterile water was 4.5 μL. The amplified product was cloned and sequenced by the Shanghai Bioengineering Company.
Determination of mutation site and inter-species conservative analysis
Based on the genomic sequence of the normal human MYH7 gene provided by NCBI and University of California, Santa Cruz (UCSC) Genome Browser, we used the Chromas program to analyze the sequencing results and determine the mutation sites. The BLAST online database was used to compare the homology between species in the mutation sites and to analyze the conservation among species.
Results
Patients and gross specimen
Among the 18 cases of SHCM deaths, there were ten men and eight women, aged 15 to 49 years (mean age: 35.7 ± 9.3 years), the mean heart weight was 479.9 ± 140.2 g. The post-mortem characteristics of the cardiac specimens in the case group were consistent with the diagnostic criteria of HCM [Table 1]. We found two mutations in the case group. For the eighth case, the weight of the heart was 630 g and the ratio of inter-ventricular septum thickness (IVST) to LVWT was 1.38. For the 12th case, the weight of the heart was 494 g and the ratio of IVST to LVWT was 1.35.
Table 1.
Post-mortem examination of hearts in the case group of hypertrophic cardiomyopathy.
Histopathologic findings
Two mutation sites were screened out from the case group. Hematoxylin and eosin (HE) staining showed that the myocardial fibers had significant hypertrophy, were arranged disorderly in a whirlpool or cluster, and their nuclei were large and darkly stained, with different karyotypes, which could be pleomorphic. Masson staining showed that the myocardial interstitial tissue exhibited focal collagen fibrosis proliferation and fibrotic changes. No significant pathological changes were found in other major organs. Histological findings and general cardiac specimens in the control group were not found to be abnormal [Figure 1].
Figure 1.
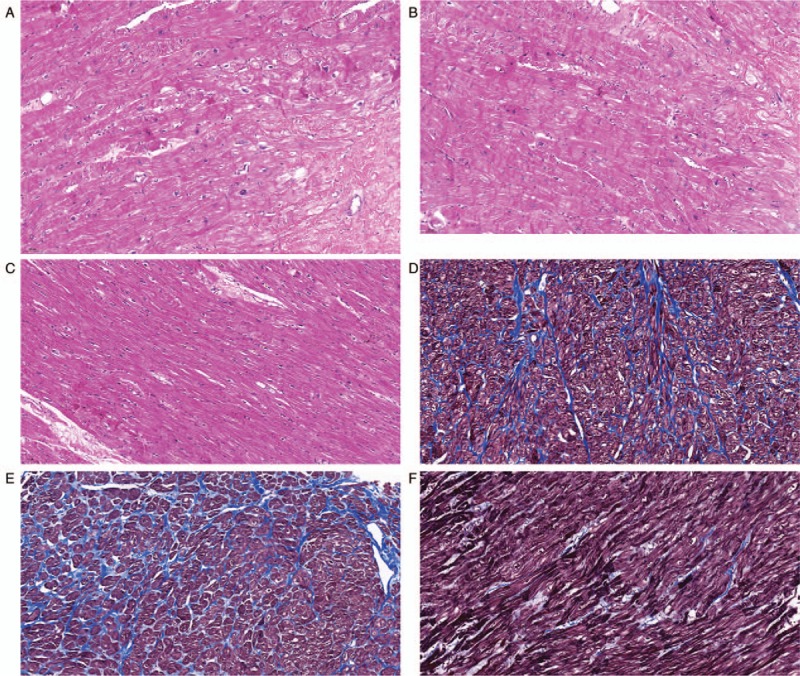
Histopathological characteristics of mutation sites (original magnification ×200). (A) HE staining of myocardial tissue with Thr446Pro mutation. (B) HE staining of myocardial tissue with Phe468Leu mutation. (C) HE staining of normal myocardium. (D) Masson staining of myocardial tissue with Thr446Pro mutation. (E) Masson staining of myocardial tissue with Phe468Leu mutation. (F) Masson staining of normal myocardium. HE: Hematoxylin and eosin.
Genomic DNA sequencing and bioinformatics analysis
Candidate genes were detected in 18 cases of unrelated Chinese SHCM. Thr446Pro mutation (rs796553039) was found in the eighth case, that is, the 1336th base of the human MYH7 genome was converted from T to G, which transformed the genetic code from TGG into GGG, resulting in the conversion of threonine (Thr) at position 446 to proline (Pro). A Phe468Leu mutation (rs727504338) was found in the 12th case, that is, the 1402th base of the human MYH7 genome was converted from T to C, which transformed the genetic code from TTC into CTC, resulting in the conversion of phenylalanine (Phe) at position 468 to leucine (Leu). No mutations were found in the other sporadic cases and the control group. The MYH7 amino acid sequences of human, cattle, orangutan, rat, rabbit, dog, cat, and pig were compared by BLAST online database. The results showed that the two amino acid residues of Thr446 and Phe468 are highly conserved among different species [Figures 2 and 3].
Figure 2.
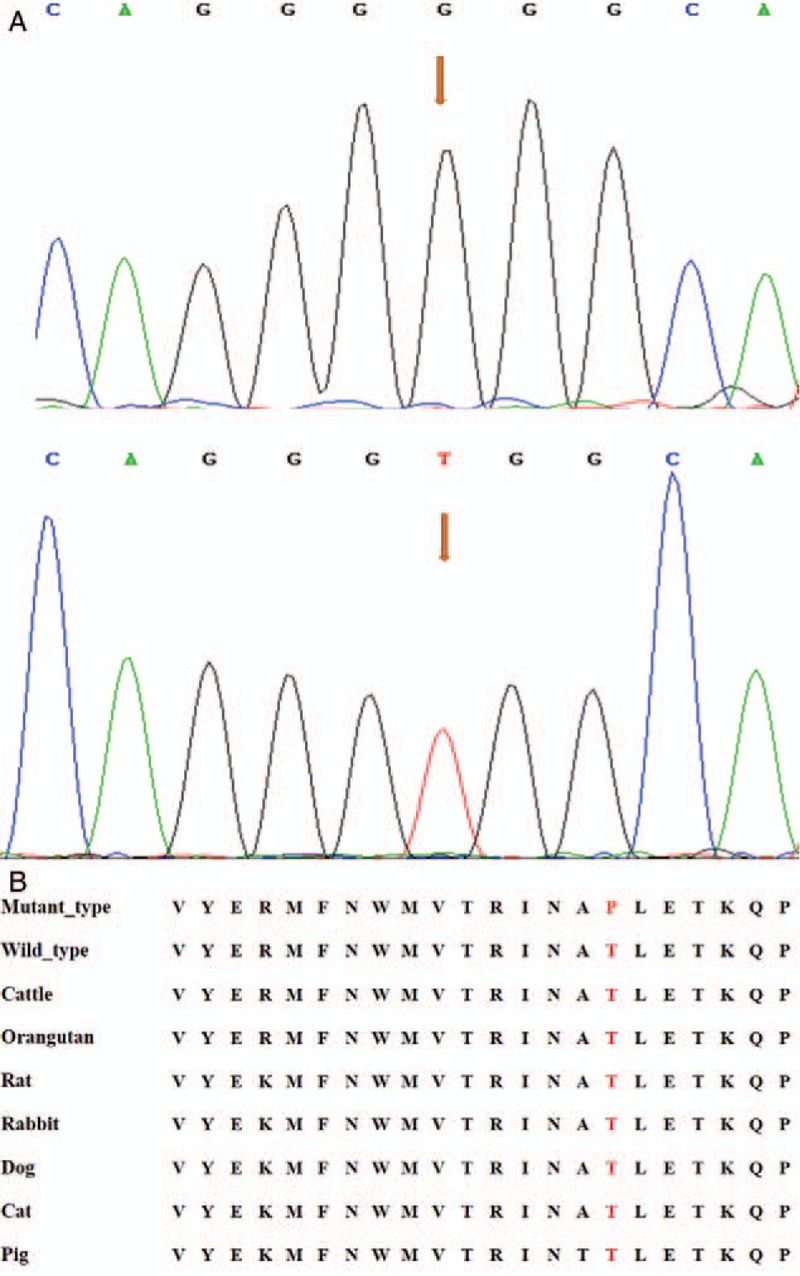
The Thr446Pro mutation sequencing and inter-species conserved analysis of MYH7 gene. (A) The upper frame is the mutation, and the arrow shows the mutation site; the lower frame is the normal wild type, and the arrow shows the normal site, base 1336 of exon 14 was converted from T to G, resulting in the conversion of Thr at position 446 to Pro. (B) The results of the inter-species homology comparison showed that the amino acid residues of Thr446 are highly conserved among different species. Pro: Proline; Thr: Threonine.
Figure 3.
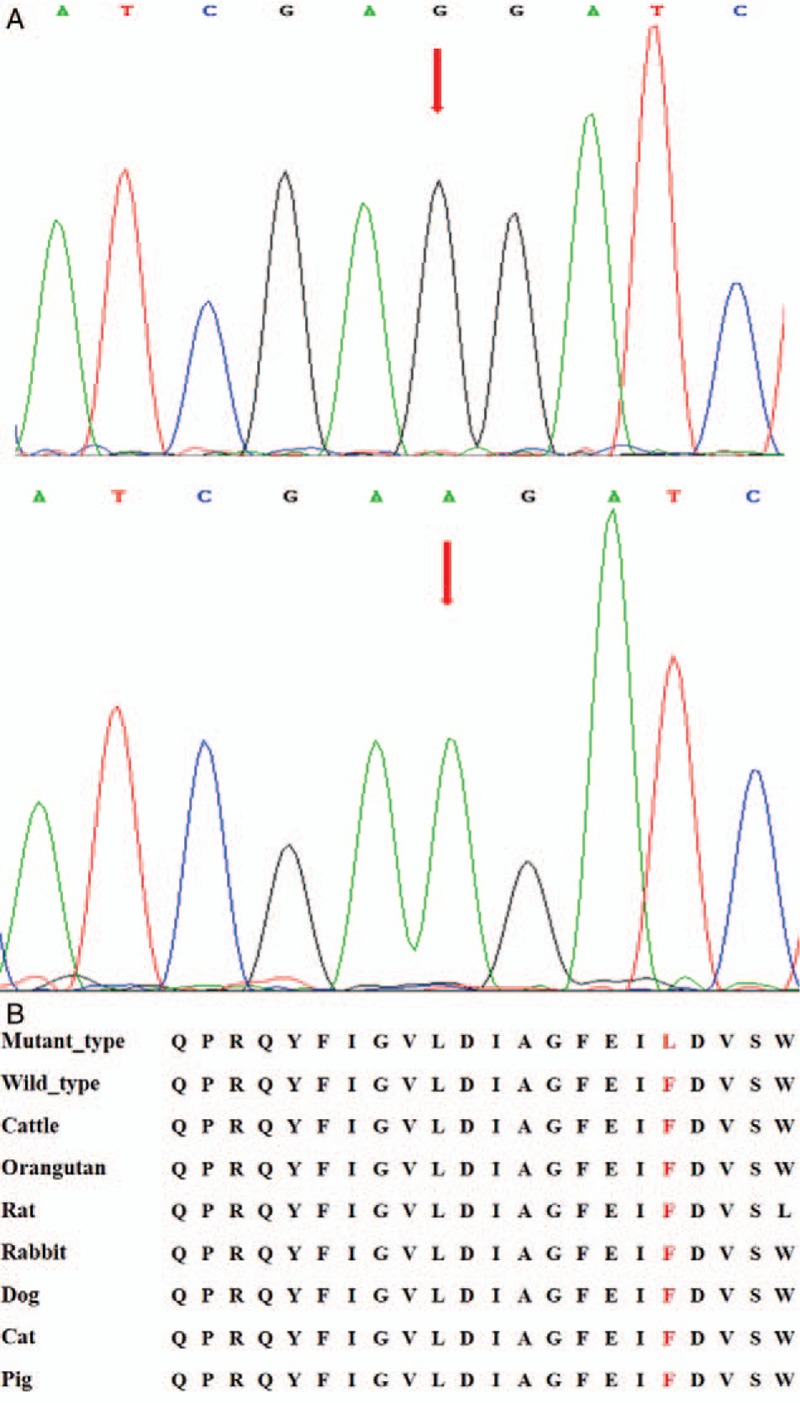
The Phe468Leu mutation sequencing and inter-species conserved analysis of MYH7 gene. (A) The upper frame is the mutation, and the arrow shows the mutation site; the lower frame is the normal wild type, and the arrow shows the normal site, base 1402 of exon 14 was converted from T to C (Base pair: A and T, G and C), resulting in the conversion of threonine (Phe) at position 468 to proline (Leu). (B) Results of the inter-species homology comparison showed that the amino acid residues of Phe468 are highly conserved among different species. Leu: Leucine; Phe: Phenylalanine.
Discussion
This study discovered the Thr446Pro and Phe468Leu mutations of the MYH7 gene. This information would be of vital significance for patients at early stages of the disease, and could affect risk assessment, prognosis, corresponding preventative measures, improvement of quality of life, and other specific treatments.[18] Furthermore, such precise diagnosis and treatment methods and personalized medicine research on the basis of gene sequencing can prevent medical waste and help clinicians in achieving individual precision treatment, pushing medical treatment to new heights.[19] In terms of forensic medicine, continuous development of molecular testing could allow us to clarify the identity of the dead as well as determine specific reasons of death.[20] Thus, the analysis of genetic mutations could play an important role in the identification of unknown individuals, which has been complicated by the increasing mobility of the population.
Among patients with MYH7 mutations, 30% to 40% were sporadic cases. Higher penetration has been reported for MYH7 mutations, which would increase the probability that the disease will manifest in the carriers of each family.[21–23] Patients with MYH7 mutations can manifest the disease at an earlier age and have a higher degree of hypertrophy, a more malignant phenotype, and a poorer prognosis.[24] According to the encoding MYH7 gene information in the database, it is located in chromosome 14q12, it contains 40 exons, and its overall exon length is 5808 bp. In addition, the head of the MYH7 gene is made up of 3 to 21 exons, the neck consists of 21 to 25 exons, also known as the head-rod joints, and 25 to 40 exons form the valve stem. The mutation of the MYH7 gene involves aggregation, which can be found in the head and head-rod joints.[25–27]
The exons selected in this study are common mutant exons reported in the literature. The exons measured have high frequency of mutation and most of them are malignant mutations, which has research value and needs to be further studied and improved. Through Sanger sequence analysis of common mutations exons 3, 8, 14, 16, 17, 18, 19, 20, 22, 23, 26, and 27 of the MYH7 gene we found that exon 14 in the MYH7 gene clearly exhibited the missense mutation Thr446Pro and Phe468Leu. Interestingly, two mutations were detected previously in China, but were not reported in the ESP, Online Mendelian Inheritance in Man, UCSC, and NCBI databases. The death of Thr446Pro mutation was 48 years old, and the death of Phe468Leu mutation was 35 years old. Both of the deceased died of unexplained sudden fall to the ground and that the age of sudden death was younger. In these two cases of mutated myocardial tissue, under the microscope, typical changes of HCM can be seen under HE staining, and myocardial fibrosis can be seen under Masson staining. Myocardial fibrosis is a cardiac interstitial remodeling characterized by excessive proliferation, collagen deposition, and abnormal distribution of cardiac interstitial fibroblasts, which is a potential risk factor for SCD. Cardiac myocytes are non-renewable cells. Once fibrous tissue proliferates, the inter-ventricular septum becomes thicker and harder, which limits ventricular filling and decreases diastolic capacity, leading to dysfunction of cardiac blood supply and eventually sudden death.[28]
The Thr446Pro and Phe468Leu mutations of MYH7 gene are located in exon 14, as we all know. No. 14 exon coding corresponds to a link between two homocysteines in the adjacent hinge region of the β myosin heavy chain gene, which is an important functional area of the spherical head. Mutations in the head region of the MYH7 gene also include Arg453Ser, Gly425Arg, and Thr441Met, which have high clinically extraneous rates.[29–31] Research has shown that mutation in this region can enhance the adenosine triphosphate enzyme activity of myosin S1, which may prevent changes in the conformation of myosin or alter its interaction with actin and other molecules. This ultimately can cause HCM, which is characterized by high penetrance, rapid progression, heart failure, and other malignant clinical manifestations.[32] In addition, homologous comparison results show that the two amino acid residues of Thr446 and Phe468 are highly conserved among different species, suggesting that once the mutation occurs, it will play an important role in life activities. To sum up, these two mutations are malignant mutations that can lead to sudden death.
In addition, there are various reasons that can influence the occurrence and development of HCM, including genetic differences, insertion, and deletion of modified genes, geographical and environmental factors, and ethnicity.[33] Some experiments have studied the patients with SHCM and screened the mutation sites of HCM pathogenic genes. Five cases of cardiac myosin binding protein C (MYBPC3) gene mutation, two cases of cardiac troponin I (TNNI3) gene mutation and 1 case of MYH7 gene mutation have been found. It shows that familial and SHCM have the same pathogenic genes.[34] Therefore, SHCM patients should be alert to the risk of hereditary offspring. Because the two positive cases in this study were sudden death, no clinical data and no family members were found in autopsy records, it is impossible to do family analysis, but it can provide reference for other cases to make these two positive mutations again, which is still of great significance. Sometimes the propositus may carry two disease-causing mutations, but genetic screening tests can only find one, thus affecting the results of genetic testing. In this study, only common mutant exons were detected, and the exons that were not detected had the possibility of mutation. However, due to a large amount of literature reading, no mutation sites of other exons were reported. In addition, because of the experimental method, only exons that are prone to mutation are studied, and deletion mutation is possible in undetected exons. These are the reasons for the low detection rate. Therefore, it is necessary to carry out genetic testing analysis and clinical phenotyping of large samples, in order to provide sufficient basis for the formulation of HCM treatments and prevention strategies. Further research studies are needed on the distribution of pathogenic genes in HCM and analysis of its pathological parameters, especially with regards to the relationship between severe clinical phenotypes and mutation genotypes as well as related factors that can affect the phenotype.[32]
In conclusion, this study discovered that the Thr446Pro and Phe468Leu mutations of the MYH7 gene had been found earlier in Chinese patients, and that it could cause HCM. Furthermore, the risk of passing mutations involved in SHCM to the next generation is high, so it will be crucial to conduct family history screenings for patients with the disease. Detailed investigation and analysis of gene mutations related to HCM will not only have value in forensic medicine teaching and research services, but can also lead to medical determination of HCM through genetic testing and prevention of sudden deaths associated with the disease.[20]
Funding
The study was supported by the grants from the “Clinical Medicine + X” Project of Qingdao University (No. 2017Q12) and the Postdoctoral Applied Research Foundation of Qingdao City, the China Postdoctoral.
Conflicts of interest
None.
Footnotes
How to cite this article: Liu HT, Ji FF, Wei L, Zuo AJ, Gao YX, Qi L, Jin B, Wang JG, Zhao P. Screening of MYH7 gene mutation sites in hypertrophic cardiomyopathy and its significance. Chin Med J 2019;132:2835–2841. doi: 10.1097/CM9.0000000000000428
References
- 1.Rahimtoola SH. Sudden cardiac death. Foreword. Curr Probl Cardiol 2015; 40:131.doi: 10.1016/j.cpcardiol.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 2.Shah LL, Daack-Hirsch S. Family communication about genetic risk of hereditary cardiomyopathies and arrhythmias: an integrative review. J Genet Couns 2018; 27:1022–1039. doi: 10.1007/s10897-018-0225-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA 2002; 287:1308–1320. doi: 10.1001/jama.287.10.1308. [DOI] [PubMed] [Google Scholar]
- 4.Garcia-Castro M, Coto E, Reguero JR, Berrazueta JR, Alvarez V, Alonso B, et al. Mutations in sarcomeric genes MYH7, MYBPC3, TNNT2, TNNI3, and TPM1 in patients with hypertrophic cardiomyopathy. Rev Esp Cardiol 2009; 62:48–56. doi: 10.1016/S0300-8932(09)70020-X. [PubMed] [Google Scholar]
- 5.Schmied C, Borjesson M. Sudden cardiac death in athletes. J Intern Med 2014; 275:93–103. doi: 10.1111/joim.12184. [DOI] [PubMed] [Google Scholar]
- 6.Marian A. Recent advances in genetics and treatment of hypertrophic cardiomyopathy. Future Cardiol 2005; 1:341–353. doi: 10.1517/14796678.1.3.341. [DOI] [PubMed] [Google Scholar]
- 7.Zou Y, Song L, Wang Z, Ma A, Liu T, Gu H, et al. Prevalence of idiopathic hypertrophic cardiomyopathy in china: a population-based echocardiographic analysis of 8080 adults. Am J Med 2004; 116:14–18. doi: 10.1016/j.amjmed.2003.05.009. [DOI] [PubMed] [Google Scholar]
- 8.Wang B, Guo RQ, Wang J, Yang F, Zuo L, Liu Y, et al. The cumulative effects of the myh7-v878a and cacna1c-a1594v mutations in a Chinese family with hypertrophic cardiomyopathy. Cardiology 2017; 138:228–237. doi: 10.1159/000478900. [DOI] [PubMed] [Google Scholar]
- 9.Maron BJ, Maron MS, Semsarian C. Genetics of hypertrophic cardiomyopathy after 20 years: clinical perspectives. J Am Coll Cardiol 2012; 60:705–715. doi: 10.1016/j.jacc.2012.02.068. [DOI] [PubMed] [Google Scholar]
- 10.Zhao P, Cui HL, He TT, Wang JG, Wang D, Feng XX, et al. Familial hypertrophic cardiomyopathy caused by a de novo gly716arg mutation of the beta-myosin heavy chain. Cardiol Young 2017; 27:467–472. doi: 10.1017/S1047951116000731. [DOI] [PubMed] [Google Scholar]
- 11.Jarcho JA, McKenna W, Pare JA, Solomon SD, Holcombe RF, Dickie S, et al. Mapping a gene for familial hypertrophic cardiomyopathy to chromosome 14q1. N Engl J Med 1989; 321:1772–1378. doi: 10.1056/NEJM198911163212005. [DOI] [PubMed] [Google Scholar]
- 12.Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg HP, McKenna W, Seidman CE, et al. A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell 1990; 62:999–1006. doi: 10.1016/0092-8674(90)90274-I. [DOI] [PubMed] [Google Scholar]
- 13.Rosenzweig A, Watkins H, Hwang DS, Miri M, McKenna W, Traill TA, et al. Preclinical diagnosis of familial hypertrophic cardiomyopathy by genetic analysis of blood lymphocytes. N Engl J Med 1991; 325:1753–1760. doi: 10.1056/NEJM199112193252501. [DOI] [PubMed] [Google Scholar]
- 14.Watkins H, Rosenzweig A, Hwang DS, Levi T, McKenna W, Seidman CE, et al. Characteristics and prognostic implications of myosin missense mutations in familial hypertrophic cardiomyopathy. N Engl J Med 1992; 326:1108–1114. doi: 10.1056/NEJM199204233261703. [DOI] [PubMed] [Google Scholar]
- 15.Seidman CE, Seidman JG. Molecular genetic studies of familial hypertrophic cardiomyopathy. Basic Res Cardiol 1998; 93: Suppl 3: 13–16. doi: 10.1007/s003950050196. [DOI] [PubMed] [Google Scholar]
- 16.Marian AJ, Roberts R. Recent advances in the molecular genetics of hypertrophic cardiomyopathy. Circulation 1995; 92:1336–1347. doi: 10.1161/01.CIR.92.5.1336. [DOI] [PubMed] [Google Scholar]
- 17.Erdmann J, Daehmlow S, Wischke S, Senyuva M, Werner U, Raible J, et al. Mutation spectrum in a large cohort of unrelated consecutive patients with hypertrophic cardiomyopathy. Clin Genet 2003; 64:339–349. doi: 10.1034/j.1399-0004.2003.00151.x. [DOI] [PubMed] [Google Scholar]
- 18.Waldmuller S, Muller M, Rackebrandt K, Binner P, Poths S, Bonin M, et al. Array-based resequencing assay for mutations causing hypertrophic cardiomyopathy. Clin Chem 2008; 54:682–687. doi: 10.1373/clinchem.2007.099119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang J, Wang Y, Zou Y, Sun K, Wang Z, Ding H, et al. Malignant effects of multiple rare variants in sarcomere genes on the prognosis of patients with hypertrophic cardiomyopathy. Eur J Heart Fail 2014; 16:950–957. doi: 10.1002/ejhf.144. [DOI] [PubMed] [Google Scholar]
- 20.Zhao P, Wang JG, Gao P, Li X, Brewer R. Sudden unexpected death from natural diseases: fifteen years’ experience with 484 cases in seychelles. J Forensic Leg Med 2016; 37:33–38. doi: 10.1016/j.jflm.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 21.Arad M, Seidman JG, Seidman CE. Phenotypic diversity in hypertrophic cardiomyopathy. Hum Mol Genet 2002; 11:2499–2506. doi: 10.1093/hmg/11.20.2499. [DOI] [PubMed] [Google Scholar]
- 22.Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 2003; 107:2227–2232. doi: 10.1161/01.CIR.0000066323.15244.54. [DOI] [PubMed] [Google Scholar]
- 23.Feng X, He T, Wang JG, Zhao P. Asn391thr mutation of beta-myosin heavy chain in a hypertrophic cardiomyopathy family. Int Heart J 2018; 59:596–600. doi: 10.1536/ihj.17-250. [DOI] [PubMed] [Google Scholar]
- 24.Xu CC, Bai YZ, Xu XS, Lu GL, Lai XP, Chen R, et al. Gene analysis for the sudden death of hypertrophic cardiomyopathy by whole exome sequencing (in Chinese). J Foren Med 2017; 33:339–343. doi: 10.3969/j.issn.1004-5619.2017.04.001. [DOI] [PubMed] [Google Scholar]
- 25.Epstein ND, Cohn GM, Cyran F, Fananapazir L. Differences in clinical expression of hypertrophic cardiomyopathy associated with two distinct mutations in the beta-myosin heavy chain gene. A 908leu----val mutation and a 403arg----gln mutation. Circulation 1992; 86:345–352. doi: 10.1161/01.CIR.86.2.345. [DOI] [PubMed] [Google Scholar]
- 26.Oldfors A. Hereditary myosin myopathies. Neuromuscul Disord 2007; 17:355–367. doi: 10.1016/j.nmd.2007.02.008. [DOI] [PubMed] [Google Scholar]
- 27.Walsh R, Rutland C, Thomas R, Loughna S. Cardiomyopathy: a systematic review of disease-causing mutations in myosin heavy chain 7 and their phenotypic manifestations. Cardiology 2010; 115:49–60. doi: 10.1159/000252808. [DOI] [PubMed] [Google Scholar]
- 28.Rakowski H, Hoss S, Williams LK. Echocardiography in the diagnosis and management of hypertrophic cardiomyopathy. Cardiol Clin 2019; 37:11–26. doi: 10.1016/j.ccl.2018.09.001. [DOI] [PubMed] [Google Scholar]
- 29.Perrot A, Schmidt-Traub H, Hoffmann B, Prager M, Bit-Avragim N, Rudenko RI, et al. Prevalence of cardiac beta-myosin heavy chain gene mutations in patients with hypertrophic cardiomyopathy. J Mol Med (Berl) 2005; 83:468–477. doi: 10.1007/s00109-005-0635-7. [DOI] [PubMed] [Google Scholar]
- 30.Stockler S, Corvera S, Lambright D, Fogarty K, Nosova E, Leonard D, et al. Single point mutation in rabenosyn-5 in a female with intractable seizures and evidence of defective endocytotic trafficking. Orphanet J Rare Dis 2014; 9:141.doi: 10.1186/s13023-014-0141-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fan XP, Yang ZW, Feng XL, Yang FH, Xiao B, Liang Y. Mutation analysis of beta myosin heavy chain gene in hypertrophic cardiomyopathy families (in Chinese). Chin J Med Genet 2011; 28:387–392. doi: 10.3760/cma.j.issn.1003-9406.2011.04.006. [DOI] [PubMed] [Google Scholar]
- 32.Baxi AJ, Restrepo CS, Vargas D, Marmol-Velez A, Ocazionez D, Murillo H. Hypertrophic cardiomyopathy from A to Z: genetics, pathophysiology, imaging, and management. Radiographics 2016; 36:335–354. doi: 10.1148/rg.2016150137. [DOI] [PubMed] [Google Scholar]
- 33.Semsarian C, Ingles J, Maron MS, Maron BJ. New perspectives on the prevalence of hypertrophic cardiomyopathy. J Am Coll Cardiol 2015; 65:1249–1254. doi: 10.1016/j.jacc.2015.01.019. [DOI] [PubMed] [Google Scholar]
- 34.Niimura H, Patton KK, McKenna WJ, Soults J, Maron BJ, Seidman JG. Sarcomere protein gene mutations in hypertrophic cardiomyopathy of the elderly. Circulation 2002; 105:446–451. doi: 10.1161/hc0402.102990. [DOI] [PubMed] [Google Scholar]