

SUMMARY
Group 2 innate lymphoid cells (ILC2s) can initiate pathologic inflammation in allergic asthma by secreting copious amounts of type 2 cytokines, promoting lung eosinophilia and airway hyperreactivity (AHR), a cardinal feature of asthma. We discovered that the TNF/TNFR2 axis is a central immune checkpoint in murine and human ILC2s. ILC2s selectively express TNFR2, and blocking the TNF/TNFR2 axis inhibits survival and cytokine production and reduces ILC2-dependent AHR. The mechanism of action of TNFR2 in ILC2s is through the non-canonical NF-κB pathway as an NF-κB-inducing kinase (NIK) inhibitor blocks the costimulatory effect of TNF-α. Similarly, human ILC2s selectively express TNFR2, and using hILC2s, we show that TNFR2 engagement promotes AHR through a NIK-dependent pathway in alymphoid murine recipients. These findings highlight the role of the TNF/TNFR2 axis in pulmonary ILC2s, suggesting that targeting TNFR2 or relevant signaling is a different strategy for treating patients with ILC2-dependent asthma.
Graphical Abstract
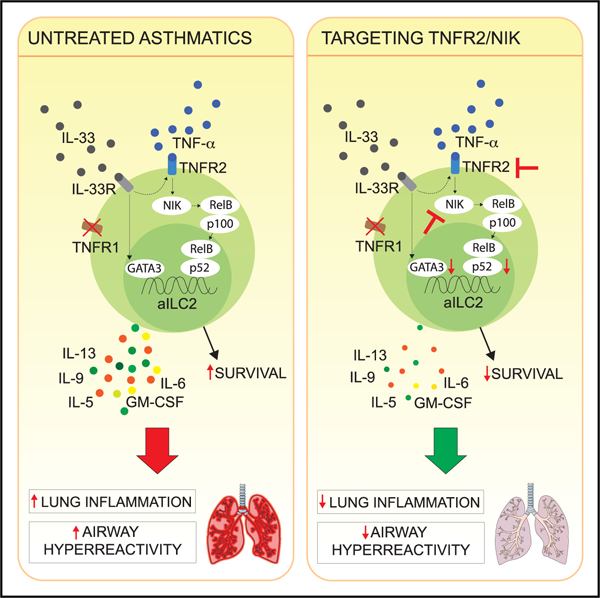
In Brief
TNF-α is highly expressed in the lungs of asthmatic patients. Hurrell et al. show that murine and human ILC2s respond to TNF-α by selectively expressing TNFR2. TNF-α enhances ILC2 survival and cytokine production utilizing the non-canonical NF-κB pathway, leading to increased development of ILC2-dependent AHR.
INTRODUCTION
Asthma is a heterogenous inflammatory disease of the airways characterized by reversible bronchoconstriction and airway hyperreactivity (AHR). Asthma is classically caused by the release of type 2 cytokines such as interleukin-5 (IL-5) and IL-13 from activated T helper 2 (Th2) cells, leading to cardinal symptoms of asthma that include mucus production and smooth muscle cell contraction. Group 2 innate lymphoid cells (ILC2s), however, have been shown recently to rapidly secrete type 2 cytokines in response to alarmins during asthma, and although they lack antigen-specific markers, they can potently induce asthma symptoms in alymphoid mice devoid of T or B cells challenged with IL-33 or IL-25 (Barlow et al., 2013; Kim et al., 2012; Bartemes et al., 2012; Klein Wolterink et al., 2012). Modulation of ILC2 homeostasis and/or activation therefore is a crucial approach to target or alleviate asthma symptoms (Kabata et al., 2018; Maazi and Akbari, 2017; Hurrell et al., 2018).
Members of the tumor necrosis factor superfamilies (TNFSFs) and their receptors (TNFRSFs) provide key communication signals between cells, including T cells and various innate immune cells, such as macrophages, dendritic cells, and natural killer (NK) cells (Ward-Kavanagh et al., 2016; Simhadri et al., 2012; Kobayashi et al., 2015). Recently however, ILC2s have been described to interact with T cells by selectively expressing TNFSF4 (OX40L) in the airways (Halim et al., 2018). Interestingly, ILC2s further express TNFRSF25 (DR3) and TNFRSF18 (GITR), which have been shown to efficiently costimulate ILC2s in disease settings (Nagashima et al., 2018; Galle-Treger et al., 2019; Meylan et al., 2014; Yu et al., 2014; Hurrell et al., 2018). However, in these studies, a better understanding of the availability of membrane-bound ligands in tissues, such as the lungs, and their precise role in induction of AHR remains unclear. TNFSF2, more commonly known as tumor necrosis factor alpha (TNF-α), is a pleiotropic proinflammatory cytokine that plays a significant role in many inflammatory diseases of the lungs, including asthma (Malaviya et al., 2017). TNF-α is elevated in the airways of patients with severe asthma (Howarth et al., 2005; Brightling et al., 2008), and murine studies have confirmed the function of TNF-α in promoting bronchoconstriction and AHR (Kips et al., 1992; Cai et al., 2011). Although TNF-α-neutralizing drugs are commonly used to treat patients with other diseases, their efficacy in humans with asthma is inconclusive, in part because of the heterogeneity of asthma severity (Hasegawa et al., 2001; Raychaudhuri and Raychaudhuri, 2009). Importantly, anti-TNF therapy is generally associated with increased risk of infection and tumor progression because of the existence of two structurally different TNF-α receptors with opposing functions: TNFRSF1a (TNFR1) and TNFRSF1b (TNFR2) (Ali et al., 2013; Baud and Karin, 2001). TNFR1 is constitutively expressed in most cell types and is associated with cytotoxicity because it bears a death domain recruiting death signaling molecules (Sedger and McDermott, 2014). The expression of TNFR2 is, however, restricted and associated with survival and homeostasis because it lacks the death domain observed with TNFR1 (Aggarwal et al., 2012). Interestingly, the TNF/TNFR2 axis in T-regulatory (Treg) cells was recently described as a central checkpoint modulating immune regulation (Cohen and Wood, 2017). Tregs express higher levels of TNFR2 compared with activated conventional T cells and is crucial for Foxp3 stabilization and Treg proliferation and activation through non-canonical nuclear factor κB (NF-κB) pathway activation (Wang et al., 2018; Chen et al., 2013; Yang et al., 2019; Zou et al., 2018; Pfoertner et al., 2006). Interestingly, the expression patterns of TNFR1 and/or TNFR2 on ILC2s and subsequent responsiveness to TNF-α, particularly in the lungs, remain unknown.
In this study, we evaluated the functional requirement and signaling of TNF-α on ILC2s in the context of asthma. We found that murine ILC2s selectively express TNFR2 in response to IL-33. Alveolar macrophages contribute to TNF-α secretion in the lungs, and we discovered that TNF-α signaling through TNFR2 in ILC2s is crucial for their survival and function and induction of AHR. Furthermore, TNFR2 signaling through the non-canonical NF-κB pathway is induced by TNF-α in ILC2s, and selective blocking of NF-κB-inducing kinase (NIK) inhibits the development of ILC2-dependent AHR. Notably, we found that human ILC2s also selectively express TNFR2 and that TNF-α enhances survival and cytokine secretion. Importantly, using a human ILC2 (hILC2) adoptive transfer mouse model, we discovered that the NIK blockade in hILC2s reduces the development of AHR and lung inflammation in response to TNF-α. Our murine and human findings together provide insights into the mechanisms of allergic asthma induction and offer adapted therapeutic strategies by targeting TNFR2 and the relevant signaling pathway.
RESULTS
IL-33 Promotes TNF-α Secretion and TNFR2 Expression on ILC2s
We first addressed whether the proinflammatory cytokine TNF-α was induced in the lung microenvironment in response to IL-33. To this end, we challenged BALB/c mice intranasally (i.n.) with rmIL-33 or PBS on 3 consecutive days. On day 4, the levels of bronchoalveolar lavage (BAL) fluid TNF-α and the number of TNF-α-producing cells were quantified by ELISA and flow cytometry, respectively (Figure 1A). We found that i.n. challenge with IL-33 significantly induced TNF-α levels in the BAL fluid (Figure 1B). We further showed, by intracellular staining, that TNF-α-producing CD45+ immune cells are induced upon IL-33 challenge (Figure 1C). Interestingly, we found that the majority of these cells were alveolar macrophages (AMs) in both naive (Figure 1D) and activated (Figure 1E) lungs (Figure 1F). To confirm our observation, fluorescence-activated cell sorting (FACS)-sorted AMs from activated lungs incubated ex vivo produced significantly more TNF-α in culture compared with naive controls (Figure 1G; Figure S1A). In comparison, SiglecF−CD11b+ cells did not secrete as much TNF-α (Figure S1B), suggesting that AMs are major immune contributors to the TNF-α pool observed in the lungs. We next measured the expression of the TNF-α receptors TNFR1 and TNFR2 on ILC2s. ILC2s were gated on lineage−CD45+CD127+ST2+ viable cells and were GATA-3hi (Figures 1H and 1I; Figures S1D–S1F). We first assessed the expression pattern over time of TNFR1 and TNFR2 by flow cytometry on FACS-sorted naive ILC2s (nILC2s) activated with IL-33 ex vivo (Figure 1J). We observed that neither nILC2s nor IL-33 activation induced TNFR1 expression (Figure 1K). However, notably, we discovered that, although nILC2s expressed little TNFR2, IL-33 significantly induced TNFR2 expression as early as 24 h after activation, reaching a peak after 72 h (Figure 1L). To confirm our observations in an in vivo setting, we challenged mice with PBS or IL-33 i.n. on 3 consecutive days and, on day 4, measured TNFR1 and TNFR2 expression on lung ILC2s (Figure 1M). Consistent with our ex vivo data, we observed that nILC2s and activated ILC2s (aILC2)s expressed little or no TNFR1 (Figure 1N). However, ILC2s selectively and significantly upregulated the expression of TNFR2 in response to IL-33 i.n. challenge (Figure 1O). Of note, we found that the recently claimed ST2− CD127+ population of ILC2s failed to induce TNFR2 in response to IL-33 (Cavagnero et al., 2019; Figures S1G–S1I). Altogether, our data suggest that TNF-α is present in IL-33-activated lungs and that pulmonary aILC2s selectively express TNFR2.
Figure 1. IL-33 Promotes TNF-α Secretion and TNFR2 Expression on ILC2s.

(A) BALB/cByJ mice were challenged i.n. on days 1–3 with 0.5 μg rmIL-33 or PBS.
(B) On day 4, BAL fluid was collected, and supernatant TNF-α was measured by ELISA.
(C) Number of TNF-α-producing CD45+ lung cells on day 4, cultured for 4 h with GolgiPlug.
(D–F) Representative flow cytometry plots in naive (D) and activated (E) lungs of CD45+ TNF-α+ cells backgated for AMs (CD45+ SiglecF+ CD11b−, CD11c+, and F4/80+) and SiglecF−CD11b+ and SiglecF−CD11b− populations and (F) corresponding quantitation presented as mean frequency ± SEM.
(G) AMs were FACS sorted from PBS- or rmIL-33-challenged mice on day 4 and cultured for 24 h in vitro (106 AMs/well), and supernatant TNF-α was measured by ELISA.
(H and I) Gating strategy for murine ILC2s showing (H) lineage− staining and (I) lineage−CD45+ CD127+ ST2+ GATA-3hi ILC2s.
(J) nILC2s were FACS sorted from naive BALB/cByJ mice and cultured (50 × 104/mL) ex vivo in the presence of rmIL-2 (10 ng/mL), rmIL-7 (10 ng/mL), and rmIL-33 (50 ng/mL) for the indicated times. nILC2s were gated as lineage− CD45+ ST2+ CD127+.
(K and L) Representative flow cytometry plot of TNFR1 (K) and TNFR2 (L) expression and corresponding quantitation, presented as Mean Fluorescence Intensity (MFI) ± SEM.
(M) BALB/cByJ mice were challenged i.n. on days 1–3 with 0.5 μg rmIL-33 or PBS.
(N and O) Representative flow cytometry plot of lung ILC2 expression of TNFR1 (N) and TNFR2 (O) expression on day 4 and corresponding quantitation, presented as MFI ± SEM.
Data are representative of 4 individual experiments (n = 5). *p < 0.05, **p < 0.01, ***p < 0.001; ns, non-significant. See also Figure S1.
aILC2s and TNF-α Promote the Development of AHR
To investigate the functional requirement of TNF-α by ILC2s, we incubated FACS-sorted pulmonary aILC2s with TNF-α and measured cytokine secretion in the culture supernatants by Luminex (Figure 2A). We discovered that TNF-α significantly induced secretion of effector cytokines, including IL-5, IL-6, IL-9, IL-13, and granulocyte-macrophage colony-stimulating factor (GM-CSF), by ILC2s (Figure 2B). We next addressed whether TNF-α is involved in development of AHR and lung inflammation in a model of ILC2-dependent asthma. A cohort of Rag2−/− mice was challenged with IL-33 i.n. and anti-mouse TNF-α (αTNF-α) or a corresponding isotype control intravenously (i.v.) on 3 consecutive days. On day 4, lung function was measured as described in the STAR Methods. Furthermore, BAL eosinophilia and lung ILC2s were measured by flow cytometry (Figure 2C). As expected, control mice challenged with IL-33 developed significantly increased lung resistance compared with PBS-challenged mice. However, notably, the lung resistance in mice treated with aTNF-α was significantly lower than that of controls (Figure 2D). In line with our lung resistance findings, we observed a significantly improved dynamic compliance response in mice that received αTNF-α compared with controls (Figure 2E). Furthermore, we observed a reduced number of BAL eosinophils and lung inflammation (Figures 2F, 2G, and 2J), as well as pulmonary ILC2s (Figures 2H and 2I), in mice treated with αTNF-α compared with controls, suggesting overall decreased airway inflammation. In line with our observations in alymphoid mice, we found similar results in the BALB/c mouse model (Figure S2). We next confirmed our findings in a more physiological setting using Alternaria alternata (A. alternata), a common allergen known to induce ILC2-dependent AHR (Figure 2K). Similar to our previous findings, we first found that i.n. challenge with A. alternata also induced TNF-α levels in the BAL fluid (Figure 2L). We further found that mice treated with αTNF-α antibody developed significantly less lung resistance compared with the controls (Figure 2M), and this observation was associated with decreased BAL eosinophilia (Figures 2N and 2O) and lung ILC2s (Figures 2P and 2Q). Together, our results suggest, using the IL-33 model of airway inflammation as well as the common allergen A. alternata, that TNF-α enhances the development of ILC2-dependent lung inflammation and AHR.
Figure 2. aILC2s and TNF-α Promote Development of AHR.

(A) BALB/cByJ mice were challenged i.n. on days 1–3 with 0.5 μg rmIL-33. On day 4, aILC2s were FACS sorted as lineage− CD45+ ST2+ CD127+ and cultured ex vivo (50 × 104/mL) for 48 h with rmIL-2 (10 ng/mL) and rmIL-7 (10 ng/mL) with or without recombinant mouse (rm) TNF-α (40 ng/mL).
(B) Levels of IL-5, IL-6, IL-9, IL-13, and GM-CSF in culture supernatants.
(C) Rag2−/− mice received i.v. injections of 100 μg αTNF-α or an isotype control and 0.5 αg rmIL-33 or PBS i.n. on days 1–3. On day 4, lung function, BAL eosinophils, lung ILC2s, and histology were analyzed.
(D and E) Lung resistance (D) and dynamic compliance (E) in response to increasing doses of methacholine.
(F and H) Representative FACS plots of BAL eosinophils (F) and lung ILC2s (H). Eosinophils were gated as CD45+, SiglecF+ CD11c− and ILC2s as lineage− CD45+ ST2+ CD127+.
(G and I) Total number of eosinophils in the BAL fluid (G) and of ILC2s in the lungs (I), presented as mean numbers ± SEM.
(J) Lung histology. Scale bars, 50 μm.
(K) Rag2−/− mice received i.v. injections of 100 μg αTNF-α or an isotype control and 100 μg A. alternata or PBS i.n. on days 1–4. On day 5, lung function, BAL eosinophils, and lung ILC2s were analyzed.
(L) BAL fluid was collected, and supernatant TNF-α was measured by ELISA.
(M) Lung resistance in response to increasing doses of methacholine.
(N and P) Representative FACS plots of BAL eosinophils (N) and lung ILC2s (P).
(O and Q) Total number of eosinophils in the BAL fluid (O) and of ILC2s in the lungs (Q), presented as mean numbers ± SEM.
Data are representative of 3 individual experiments (n = 5). *p < 0.05, **p < 0.01, ***p < 0.001. See also Figures S2 and S4.
TNF-α Enhances ILC2 Survival and Activation via TNFR2
To better characterize the function of TNF-α on lung ILC2s, we incubated FACS-sorted pulmonary aILC2s with TNF-α and monitored cell viability, proliferation, and activation by flow cytometry and ELISA (Figure 3A). We found that TNF-α significantly decreased the frequency of late apoptotic and/or necrotic (AnnexinV+ DAPI+) ILC2s after 24 h of ex vivo culture compared with controls, suggesting that the presence of TNF-α increases ILC2 survival (Figures 3B and 3C). Of note, at this time point, aILC2 proliferation was surprisingly not altered by the presence of TNF-α, as evidenced by intranuclear expression of Ki67 by ILC2s (Figure 3D). Because TNF-α induced cytokine secretion by aILC2s ex vivo (Figure 2B), we next investigated whether the observed induction was the result of increased survival alone or a combination of the latter and increased overall activation. We therefore normalized IL-5 and IL-13 secretion of aILC2s in the presence of TNF-α ex vivo to the number of viable cells after culture, as performed previously (Cushnie et al., 2014; Hlushchuk et al., 2017). Interestingly, we found that, on a per-cell basis, aILC2s stimulated with TNF-α secrete significantly more IL-5 and IL-13 compared with controls (Figure 3E). Similarly, aILC2 cultured for 4 h with TNF-α significantly upregulated intracellular IL-5 and IL-13 expression ex vivo (Figures 3F and 3G). Overall, our observations suggest that TNF-α increases ILC2 survival as well as cytokine secretion. Because we observed that ILC2s selectively expressed TNFR2, we next addressed the effects of TNFR2 engagement on ILC2s ex vivo. We incubated FACS-sorted aILC2s in the presence or absence of TNF-α and/or αTNFR2 and measured ILC2 survival by flow cytometry (Figure 3H). Although TNF-α significantly decreased late apoptotic/necrotic ILC2s, blocking TNFR2 in the presence of TNF-α significantly neutralized the observed effects to numbers comparable with the controls (Figures 3I and 3J). Importantly, we measured cytokine secretion in culture supernatants by ELISA. In line with our previous findings, TNF-α significantly increased the IL-5 and IL-13 levels in culture supernatants, but blocking TNFR2 in the presence of TNF-α neutralized this effect (Figure 3K). Of note, we obtained similar results when normalizing cytokine secretion to the number of viable cells in culture (Figure S3A). Overall, our ex vivo findings suggest that TNF-α directly affects ILC2 survival and effector function via TNFR2 signaling.
Figure 3. TNF-α Enhances ILC2 Survival and Activation via TNFR2.

(A) BALB/cByJ mice were challenged i.n. on days 1–3 with 0.5 μg rmIL-33. On day 4, aILC2s were FACS sorted as lineage− CD45+ ST2+ CD127+ and cultured ex vivo (50 × 104/mL) for 24 h with rmIL-2 (10 ng/mL) and rmIL-7 (10 ng/mL) with or without rmTNF-α (40 ng/mL).
(B) Representative flow cytometry plots of AnnexinV and DAPI expression on cultured ILC2s.
(C) Frequency of AnV+DAPI+ ILC2s ± SEM on cultured ILC2s.
(D) Representative flow cytometry plots of intranuclear Ki67-expressing ILC2s among cultured ILC2s and corresponding quantitation, presented as mean frequency ± SEM.
(E) Levels of IL-5 and IL-13 in cultured supernatants, measured by ELISA and normalized to the number of viable cells after culture.
(F) BALB/cByJ mice were challenged i.n. on days 1–3 with 0.5 μg rmIL-33. On day 4, aILC2s were FACS sorted as lineage− CD45+ ST2+ CD127+ and cultured ex vivo (50 × 104/mL) for 4 h with rmIL-2 (10 ng/mL) and rmIL-7 (10 ng/mL) with or without rmTNF-α (40 ng/mL) and GolgiPlug.
(G) Intracellular expression of IL-5 and IL-13 in cultured ILC2s and corresponding quantitation, presented as mean frequency ± SEM.
(H) BALB/cByJ mice were challenged i.n. on days 1–3 with 0.5 μg rmIL-33. On day 4, aILC2s were FACS sorted as lineage− CD45+ ST2+ CD127+ and cultured in vitro for 24 h with rmIL-2 (10 ng/mL) and rmIL-7 (10 ng/mL) with or without rmTNF-α (40 ng/mL) and αTNFR2 (10 μg/mL). αTNFR2 was added 30 min prior to rmTNF-α.
(I) Representative flow cytometry plots of AnnexinV/DAPI expression on cultured ILC2s.
(J) Frequency of AnV+DAPI+ ILC2s ± SEM on cultured ILC2s.
(K) Levels of IL-5 and IL-13 in culture supernatants, measured by ELISA.
Data are representative of 4 individual experiments (n = 5). *p < 0.05, **p < 0.01. See also Figure S3.
TNF-α, via TNFR2 Signaling, Enhances ILC2 Survival and Activation In Vivo
We next investigated the function of TNF-α and TNFR2-expressing ILC2s in an in vivo setting. A cohort of Rag2−/− mice were challenged with IL-33 i.n. and anti-mouse TNFR2 (αTNFR2) or the corresponding isotype control i.v. on 3 consecutive days. On day 4, we measured pulmonary ILC2 numbers, proliferation, and survival by flow cytometry (Figure 4A). Consistent with our previous observations using αTNF-α, we discovered that mice treated with αTNFR2 showed significantly less lung ILC2s (Figures 4B and 4C), as well as BAL eosinophilia (Figures S4A–S4C), compared with control mice. In confirmation of our ex vivo data, we found that, although the frequency of early apoptotic (AnnexinV+ DAPI−) ILC2s was not affected by αTNFR2 treatment (data not shown), the frequency of late apoptotic/necrotic (AnnexinV+ DAPI+) ILC2s was increased in αTNFR2-treated mice compared with controls (Figure 4D). Furthermore, the frequencies of Ki67-positive ILC2s were not affected by TNFR2 signaling (Figure 4E). Our in vivo results therefore support an effect of TNF-α on ILC2 survival via TNFR2 signaling. Because we also observed a direct effect of TNF-α on cytokine secretion, we further characterized the functional requirement of the TNF/TNFR2 axis for ILC2 activation by performing cytokine intracellular staining and measuring the expression of the transcription factor GATA binding protein-3 (GATA-3) by flow cytometry. In support of our previous findings, we found that ILC2s from αTNFR2-treated mice produced significantly less IL-5 and IL-13 compared with controls on a per-cell basis (Figure 4F). The absolute number of ILC2-producing IL-5 and IL-13 was equally lower in αTNFR2-treated mice because of the lower number of ILC2s in these mice (data not shown). Additionally, we found that ILC2s in αTNFR2-treated mice expressed significantly less GATA-3 compared with the controls, suggesting that the GATA-3 pathway is affected by TNF-α in ILC2s (Figure 4G). Interestingly, we found that ILC2s downregulated ST2 expression (Figure 4H) and that ILC2s further downregulated the adhesion molecule ICAM-1 and activation marker CD44, suggesting that they are phenotypically altered when blocking TNFR2 (Figure S5). In confirmation of our results, we observed a similar effect of TNFR2 in mice challenged with a more physiological allergen in A. alternata (Figures S4D–S4L). Together, our in vivo data confirm that TNF-α enhances ILC2 survival and effector function via TNFR2 signaling.
Figure 4. TNF-α, via TNFR2 Signaling, Enhances ILC2 Survival and Activation In Vivo.

(A) Rag2−/− mice received i.v. injections of 100 μg αTNFR2 or an isotype control and 0.5 μg rmIL-33 or PBS i.n. on days 1–3. On day 4, lung ILC2 numbers, proliferation, and apoptosis were measured.
(B) Representative flow cytometry plots of lung ILC2s in isotype control- or αTNFR2-treated mice. ILC2s were gated as lineage− CD45+ ST2+ CD127+.
(C) Total number of lung ILC2s, presented as mean numbers ± SEM.
(D and E) Representative flow cytometry plots of AnnexinV/DAPI (D) and intranuclear Ki67 expression (E) with corresponding quantitation, presented as frequency of Ki67+ ILC2s ± SEM (D) and AnV+DAPI+ ILC2s ± SEM (E).
(F) Representative flow cytometry plots of intracellular IL-5 and IL-13 expression by lung ILC2s cultured for 4 h with phorbol 12-myristate 13-acetate (PMA), ionomycin, and GolgiPlug and corresponding quantitation, presented as mean frequency ± SEM.
(G) Representative flow cytometry plots of intranuclear GATA-3 in lung ILC2s (CD45+Lin−ST2+CD127+) and corresponding quantitation, presented as MFI ± SEM.
(H) Representative flow cytometry plots of ST2 expression in GATA-3+ ILC2s and corresponding quantitation, presented as mean MFI ± SEM.
(I) C57BL/6 and TNFR2−/− mice were challenged i.n. on days 1–3 with 0.5 μg rmIL-33. On day 4, aILC2s were FACS sorted as lineage− CD45+ ST2+ CD127+ and cultured in vitro for 3 days with rmIL-2 (10 ng/mL), rmIL-7 (10 ng/mL), and rmIL-33 (50 μg/mL) prior to transfer of 50 × 103 aILC2s in 2 separate cohorts of Rag−/− Il2rg−/− mice, followed by i.n. administration of 50 ng rmTNF-α on days 1–3. On day 4, lung function, BAL, and lung ILC2s were analyzed.
(J) Lung resistance in response to increasing doses of methacholine.
(K) Total number of eosinophils in the BAL fluid, presented as mean numbers ± SEM.
(L) Total number of ILC2s in the lungs, presented as mean numbers ± SEM.
Data are representative of 3 individual experiments (n = 4–5). *p < 0.05, **p < 0.01. See also Figures S4–S6.
TNF-α Signaling via TNFR2 Induces ILC2-Dependent AHR
We next investigated the functional relevance of the TNF/TNFR2 axis in the context of ILC2-dependent development of AHR and lung inflammation. To remove the effects of type 2 cytokine-producing cells such as T cells, we adoptively transferred FACS-sorted aILC2s from wild-type (WT) or TNFR2−/− mice into Rag2−/− Il2rg−/− mice, as described in the STAR Methods. We then challenged the mice with either TNF-α or PBS i.n. on 3 consecutive days and, on day 4, measured lung function and airway inflammation (Figure 4I). We found that mice receiving WT aILC2s and challenged with TNF-α i.n. had significantly higher lung resistance compared with the PBS-treated controls. However, notably, mice receiving TNFR2−/− aILC2s developed significantly less lung resistance compared with the WT controls (Figure 4J). Furthermore, mice receiving TNFR2−/− ILC2s harbored fewer BAL eosinophils, suggesting that airway inflammation is impaired in the absence of TNFR2 on ILC2s (Figure 4K). In line with our previous findings, we found fewer pulmonary ILC2s in mice receiving TNFR2−/− ILC2s compared with controls, suggesting that TNFR2 is essential for ILC2 maintenance in the context of IL-33-induced airway inflammation (Figure 4L). Of note, TNFR2−/− mice did not show defects in ILC2 numbers at homeostasis but showed a decrease in survival and activation upon IL-33 stimulation (Figure S6). Altogether, our findings suggest that the TNF/TNFR2 axis in ILC2s enhances the development of AHR and lung inflammation.
TNFR2 Engagement on ILC2s Induces Non-canonical NF-κB Pathway
To further elucidate the mechanisms behind the TNFR2-dependent effect of TNF-α on cell survival and cytokine secretion, we cultured FACS-sorted aILC2s with or without TNF-α and performed RNA sequencing analysis (Figure 5A). We discovered that ILC2s treated with TNF-α differentially modulated 692 genes (320 down, 372 up, p < 0.05, 2-fold change [2FC]; Figure 5B). Importantly, we found that TNF-α significantly induced il5, il9, il13, csf2, and tnf mRNA (Figure 5C), confirming that TNF-α enhances ILC2 cytokine production (Figure 2). We next further analyzed the pathways regulated by TNF-α in ILC2s using the Ingenuity Pathway Analysis (IPA) tool. As expected, we found that TNFR2 signaling via non-canonical NF-κB signaling was enriched, with a p value of 2.04 × 10−7 and a Z score of 0.816 (Figure 5D). More specifically, both TNF receptor-associated factor 1 (Traf1) and Traf2 were upregulated by TNF-α treatment. Furthermore, we found that ILC2s upregulated NIK (encoded by Map3k14), a crucial member of the NF-κB pathway and regulator of the immune system (Thu and Richmond, 2010). Notably, the non-canonical NF-κB genes Nfkb2 and Relb were statistically upregulated by TNF-α, whereas the canonical NF-κB genes Nfkb1 and Rela were either downregulated or unchanged (Figure 5D). Our transcriptomic analysis therefore suggests non-canonical NF-κB pathway engagement in response to TNF-α in ILC2s. NIK is essential for inhibitor of nuclear factor-κB kinase-α (IKKa)-induced NF-κB2 p100 processing to p52, which, along with RelB, translocate to the nucleus to activate specific gene transcription (Ling et al., 1998; Sun, 2012). To further confirm non-canonical NF-κB pathway engagement in response to TNF-α, we measured the expression of p65 and p52, members of the canonical and non-canonical NF-κB pathway, respectively. We cultured FACS-sorted aILC2s with or without TNF-α and measured p65 and p52 expression by flow cytometry. Although p65 was expressed in ILC2s but unchanged in response to TNF-α (Figure 5E), ILC2s expressed and induced p52 following treatment with TNF-α (Figure 5F). Altogether, our data demonstrate that TNF-α selectively induces TNFR2-dependent non-canonical NF-κB signaling via NIK in ILC2s.
Figure 5. TNFR2 Engagement on ILC2s Induces the Non-canonical NF-κB Pathway.

(A) BALB/cByJ mice were challenged i.n. on days 1–3 with 0.5 μg rmIL-33. On day 4, aILC2s were FACS sorted as lineage−, CD45+ ST2+ CD127+ and cultured in vitro for 24 h with rmIL-2 (10 ng/mL) and rmIL-7 (10 ng/mL) with or without rmTNFα (40 ng/mL).
(B) Volcano plot comparison of whole-transcriptome gene expression on ILC2s from PBS-versus rmTNF-α—stimulated ILC2s. Differentially downregulated or upregulated genes (p < 0.05 and 2-fold change cutoff) are presented in blue and yellow, respectively.
(C) Heatmap representation of statistically differentially regulated cytokines, chemokines, and their receptors in ILC2s from PBS-versus rmTNF-α-stimulated ILC2s.
(D) Upregulated (red) and downregulated (green) genes in the non-canonical NF-κB pathway and corresponding quantitation, presented as normalized counts ± SEM.
(E and F) Representative flow cytometry plots of NF-κB p65 (E) and NF-κB p52 (F) and corresponding quantitation, presented as MFI ± SEM in rmTNF-α-versus PBS-treated ILC2s.
Data are representative of 3 individual experiments (n = 3–5). *p < 0.05, **p < 0.01, ***p < 0.001.
TNF-α Signaling via NIK Enhances ILC2-Dependent AHR
Specific modulation of NIK activity is a potential therapeutic approach for multiple diseases, including systemic lupus erythematosus (SLE) (Brightbill et al., 2018). We therefore characterized the effects of NIK on ILC2s in response to TNF-α using a highly selective and potent NIK small-molecule inhibitor (Das et al., 2019). We cultured FACS-sorted aILC2s with TNF-α with or without αTNFR2 or a NIK inhibitor and analyzed p52 expression as a measure of non-canonical NF-κB pathway activation. We found that treatment with αTNFR2 or a NIK inhibitor prevented the induction of p52 observed in response to TNF-α with similar efficacy, suggesting that the NIK inhibitor is a potent tool to block TNF-α-induced TNFR2 signaling in ILC2s (Figures 6A and 6B). We next characterized the effects of NIK on ILC2 cytokine secretion. Although TNF-α increased IL-5 and IL-13 levels in culture supernatants, ILC2s cultured in the presence of the NIK inhibitor produced significantly fewer cytokines (Figure 6C; Figure S3B). We next investigated the effects of NIK on ILC2 survival because NIK in human T cells dramatically affects cell survival (Odqvist et al., 2013). Although TNF-α inhibited ILC2 apoptosis, treatment with the NIK inhibitor increased the frequency of late apoptotic/necrotic ILC2s to values comparable with the controls or αTNFR2-treated ILC2s (Figures 6D and 6E). Furthermore, the effects of TNFR2 or NIK did not synergize when blocking both simultaneously, suggesting that they are involved in the same pathway (Figures 6D and 6E). We next investigated the effects of NIK on the development of ILC2-dependent AHR and lung inflammation. We adoptively transferred WT aILC2s to Rag−/− Il2rg−/− mice as described in the STAR Methods, followed by TNF-α i.n. and NIK inhibitor or vehicle intraperitoneally (i.p.) on 3 consecutive days before lung function, BAL eosinophilia, and lung ILC2s were measured (Figure 6F). Although vehicle-treated mice had significantly higher lung resistance compared with controls, NIK inhibition prevented the development of lung resistance in response to TNF-α (Figure 6G). Furthermore, mice treated with the NIK inhibitor harbored significantly fewer BAL eosinophils compared with vehicle-treated mice (Figure 6H). Consistent with our ex vivo findings, we found fewer lung ILC2s in mice treated with NIK inhibitor compared with controls, suggesting that NIK is involved in ILC2 survival (Figure 6I). Altogether, our findings suggest that TNFR2 and the downstream signaling molecule NIK are involved in the development of ILC2-dependent AHR and lung inflammation via modulation of ILC2 survival and effector functions.
Figure 6. TNF-α Signaling via NIK Enhances ILC2-Dependent AHR.

(A–E) BALB/cByJ mice were challenged i.n. on days 1–3 with 0.5 μg rmIL-33. On day 4, aILC2s were FACS sorted as lineage− CD45+ ST2+ CD127+ and cultured ex vivo for 24 h with rmIL-2 (10 ng/mL) and rmIL-7 (10 ng/mL) with or without rmTNF-α (40 ng/mL), αTNFR2 (10 μg/mL), or NIK inhibitor (10 μM). αTNFR2 and the NIK inhibitor were added 30 min prior to rmTNF-α.
(A and B) NF-κB p52 representative flow cytometry plots in TNFR2 (A) and NIK (B) blocking experiments on ILC2s and corresponding quantitation, presented as MFI ± SEM.
(C) Levels of IL-5 and IL-13 secretion in culture supernatants, measured by ELISA.
(D) Frequency of AnnexinV+DAPI+ late apoptotic/necrotic ILC2s, presented as mean frequency ± SEM.
(E) Representative flow cytometry plots of AnnexinV+DAPI+ late apoptotic/necrotic ILC2s.
(F) C57BL/6 mice were challenged i.n. on days 1–3 with 0.5 μg rmIL-33. On day 4, aILC2s were FACS sorted as lineage−, CD45+ ST2+ CD127+ and cultured ex vivo for 3 days with rmIL-2 (10 ng/mL), rmIL-7 (10 ng/mL), and rmIL-33 (50 μg/mL) prior to transfer of 50 × 103 aILC2s in 2 separate cohorts of Rag−/− Il2rg−/− mice, followed by i.n. administration of 50 ng rmTNF-α and i.p. injection of 250 μg NIK inhibitor or vehicle on days 1–3. On day 4, lung function, BAL, and lung ILC2s were analyzed.
(G) Lung resistance in response to increasing doses of methacholine.
(H) Total number of eosinophils in the BAL fluid, presented as mean numbers ± SEM.
(I) Total number of ILC2s in the lungs, presented as mean numbers ± SEM.
Data are representative of 3 individual experiments (n = 5). *p < 0.05, **p < 0.01. See also Figure S3.
TNFR2 and NIK Signaling Enhance the Induction of AHR in hILC2 Recipient Mice
We next investigated whether the observations from our murine studies translate to hILC2s. Peripheral blood ILC2s from healthy donors were FACS-sorted as described in the STAR Methods and cultured ex vivo with IL-33 to measure TNFR1 and TNFR2 expression by flow cytometry (Figure 7A). hILC2s were gated as CD45+, lineage−, CRTH2+, and CD127+ and sorted to a purity of more than 95% (Figure 7B). Consistent with our murine observations, we discovered that hILC2s did not express TNFR1, neither at steady state nor upon IL-33 stimulation (Figure 7C). However, notably, they expressed basal levels of TNFR2, which was efficiently induced by IL-33 stimulation (Figure 7D). To evaluate the function of the TNF/TNFR2 axis in hILC2s, we cultured FACS-sorted hILC2s with TNF-α in the presence of anti-human TNFR2 and measured survival and cytokine production in the culture supernatants (Figure 7E). In confirmation of our murine studies, we found that TNF-α decreased the frequency of late apoptotic/necrotic hILC2s, an effect that was neutralized by blocking TNFR2 (Figure 7F–G). We next found that hILC2s secreted higher levels of IL-5 and IL-13 under TNF-α stimulation, which was inhibited when blocking TNFR2 (Figure 7H; Figure S3C). Interestingly, we found a similar effect on IL-6, IL-9, and GM-CSF (data not shown). We next assessed whether the TNF/TNFR2 axis was involved in the development of AHR and lung inflammation in hILC2-transferred mice. We therefore adoptively transferred FACS-sorted hILC2s to Rag−/− Il2rg−/− mice, as described in the STAR Methods, followed by 4 consecutive days of TNF-α i.n. challenge. Because we previously observed that the NIK inhibitor efficiently suppressed TNFα-induced TNFR2 signaling, mice received either vehicle or the NIK inhibitor i.p., and on day 5, lung function, BAL eosinophilia, and lung ILC2s were measured (Figure 7I). We found that mice treated with the vehicle developed significantly higher lung resistance compared with the controls. However, notably, treatment of mice with the NIK inhibitor prevented the development of lung resistance to levels similar to the controls (Figure 7J). Moreover, the number of BAL eosinophils was lower in mice that received the NIK inhibitor (Figure 7K). Finally, we discovered that treatment with the NIK inhibitor decreased the number of pulmonary ILC2s in response to TNF-α challenge, suggesting that TNF-α and TNFR2 signaling modulate hILC2 survival (Figure 7L). Taken together, these results indicate that human peripheral ILC2s selectively express TNFR2 and that the TNF/TNFR2 axis plays a crucial role in hILC2 survival and function in the context of asthma.
Figure 7. TNFR2 Signaling, via NIK Signaling, Enhances AHR in hILC2 Recipient Mice.

(A) Human peripheral blood ILC2s were FACS sorted from human peripheral blood mononuclear cells (PBMCs) and cultured (5 × 104/mL) for 72 h with rhIL-2 (10 ng/mL) and rhIL-7 (10 ng/mL) with or without rhIL-33 (50 ng/mL).
(B) hILC2s were gated as CD45+ lineage— CD127+ CRTH2+.
(C and D) Representative flow cytometry plots of hTNFR1 (C) and hTNFR2 (D) expression in hILC2s and corresponding quantitation, presented as MFI ± SEM.
(E) FACS-sorted hILC2s were cultured (5 × 104/mL) for 72 h with rhIL-2 (10 ng/mL) and rhIL-7 (10 ng/mL) with or without rhIL-33 (50 ng/mL) or αTNFR2 (10 μg/mL). αTNFR2 was added 30 min prior to rhTNF-α.
(F) Representative flow cytometry plots of AnnexinV/DAPI expression on cultured hILC2s.
(G) Frequency of AnV+DAPI+ ILC2s ± SEM on cultured hILC2s.
(H) Levels of IL-5 and IL-13 secretion in culture supernatants, measured by ELISA.
(I) FACS-sorted hILC2s were cultured (5 × 104/mL) for 72 h with rhIL-2 (10 ng/mL), rhIL-7 (10 ng/mL), and rhIL-33 (50 ng/mL) in vitro for 3 days prior to transfer of 50 × 103 ILC2s in 2 separate cohorts of Rag−/− Il2rg−/− mice, followed by i.n. administration of 50 ng rhTNF-α and i.p. injection of 250 μg NIK inhibitor or vehicle on days 1–4. On day 5, lung function, BAL, and lung ILC2s were analyzed.
(J) Lung resistance in response to increasing doses of methacholine.
(K) Total number of eosinophils in the BAL, presented as mean numbers ± SEM.
(L) Total number of ILC2s in the lungs, presented as mean numbers ± SEM.
Data are representative of 4 individual donors. *p < 0.05, **p < 0.01, ***p < 0.001. See also Figure S3.
DISCUSSION
Elevated levels of TNF-α were found in the airways of asthmatics, suggesting that TNF-α is involved in the development of asthma (Bradding et al., 1994). Notably, TNF-α is rapidly induced in the sputum of allergic asthma patients upon allergen exposure, and TNF-α inhalation induces lung resistance in healthy volunteers (Keatings et al., 1997; Thomas, 2001). TNF-α is known to trigger smooth muscle cell contractility, epithelial cell proliferation, and neutrophil and eosinophil chemotaxis, suggesting a direct effect on the development of asthma symptoms (Vieira et al., 2009; Anticevich et al., 1995; Sasaki et al., 2000). The sources of TNF-α following airway allergen exposure include lung epithelial cells, AMs, monocytes, and mast cells (Berry et al., 2006; Bradding et al., 1994). Using the model of IL-33-induced airway contraction, we show that IL-33 induces TNF-α secretion by AMs, suggesting that AMs contribute to the total TNF-α pool in the lungs. We observed that freshly FACS-sorted AMs from IL-33-challenged airways produced significantly more TNF-α compared with naive mice ex vivo; our observations are in line with previous findings suggesting that IL-33 drives AM activation in a model of chronic asthma (Bunting et al., 2013).
Anti-TNF therapy consists of antagonizing TNF-α, a pleiotropic cytokine with a wide range of biological functions that include proliferation, apoptosis, and tissue remodeling (Malaviya et al., 2017). The various activities of TNF-α are mediated by two structurally distinct receptors with opposing functions, TNFR1 and TNFR2, suggesting that targeting such cytokines may cause unwanted adverse effects linked to the fact that signaling through both receptors is affected (Aggarwal, 2003). Although TNF-blocking agents have caused improvements of asthma symptoms in both steroid-resistant and -dependent patients, there have been some major concerns about increased infection risks and tumor development in patients (Taillè et al., 2013; Ali et al., 2013). Common side effects include bacterial, fungal, viral, and parasitic infections that can, in some cases, lead to treatment withdrawal. A better understanding of the TNF receptor signature expressed by the cells involved in diseases such as asthma will therefore provide valuable information for the development of an adapted treatment to target TNF-α in patients.
We observed in our study that pulmonary ILC2s selectively induced TNFR2 expression following IL-33 stimulation ex vivo and in vivo, and, notably, they did not express or induce TNFR1. aILC2s stimulated with TNF-α secreted significantly higher levels of effector cytokines, including IL-5, IL-6, IL-9, IL-13, and GM-CSF, suggesting a beneficial role of TNF-α in ILC2 effector functions. In such settings, we discovered that blocking TNF-α in vivo using anti-mouse TNF-α efficiently ameliorated the development of ILC2-dependent AHR using IL-33 and A. alternata models of airway inflammation. We observed overall decreased lung inflammation with reduced numbers of eosinophils in the BAL fluid and notably fewer pulmonary ILC2s compared with controls. Our results suggest that TNF-α plays a pivotal role in lung inflammation and eosinophilia in ILC2-dependent asthma.
Our ex vivo and in vivo studies suggest that TNF-α acts on ILC2 survival and effector functions. Specifically, ex vivo, TNF-α enhanced ILC2 survival and also notably increased cytokine secretion; this effect was TNFR2-dependent. In confirmation of our observations, we found that blocking TNFR2 upon IL-33 i.n. challenge affected pulmonary ILC2-dependent airway inflammation in vivo. Most notably, we found that blocking TNFR2 signaling in vivo dramatically affected ILC2 survival because mice treated with TNFR2-blocking antibody harbored significantly fewer pulmonary ILC2s, and apoptotic markers were increased in ILC2s of these mice. In addition to being crucial for ILC2 survival, we observed that TNF-α substantially contributed to cytokine production by ILC2s; we found that the production of IL-5 and IL-13 by ILC2s was significantly reduced in the absence of TNFR2 in vivo. In agreement with these findings, we observed that TNFR2−/− ILC2s induced significantly lower AHR and lung inflammation compared with WT ILC2s transferred to Rag2−/− Il2−/− mice challenged i.n. with TNF-α. Together, our findings suggest that TNFR2 on ILC2s positively modulates their survival and effector function in response to exogenous TNF-α and that it promotes the development of ILC2-dependent AHR. A role of the TNF/TNFR2 axis in cell survival and activation has been observed previously on T cells (Cohen and Wood, 2017). TNFR2 is predominantly expressed on a subset of highly suppressive human and mouse CD4+ CD25+ FoxP3+ Tregs and is associated with FoxP3 stabilization as well as Treg proliferation and cytokine secretion (Chen et al., 2007, 2008, 2013; Chopra et al., 2016; Yang et al., 2019; Aggarwal, 2003). Blocking the TNF/TNFR2 axis by targeting TNFR2 rather than TNF-α may therefore provide a different approach for treating diseases such as asthma, where TNF-α plays a pathologic role.
The transcription factor GATA-3 is expressed by ILC2s and plays a pivotal role in the regulation of ILC2 effector functions (Mjösberg et al., 2012; Lei et al., 2018). Notably, we found that GATA-3 expression was impaired in the absence of TNFR2 signaling in both IL-33 and A. alternata models of airway inflammation. This suggests that the TNF/TNR2 axis may directly control the secretion of effector cytokines such as IL-5 and IL-13 by affecting GATA-3 expression, as observed in other studies (Lei et al., 2018). This effect on GATA-3 may explain the observed decrease in IL-33 receptor (ST2) expression, supporting previous reports that GATA-3 controls ST2 expression in ILC2s (Zhu, 2017; Lewis et al., 2019). Interestingly, impaired responsiveness to IL-33 has been shown previously to further affect ILC2 effector functions (Kearley et al., 2015). Together, our data suggest that the TNF/TNFR2 axis controls ILC2 effector functions by modulating GATA-3 expression.
Although also involved in the canonical NF-κB pathway, NIK is a serine/threonine kinase involved in phosphorylation of IKKa and p100, generation of p52, and translocation to the nucleus (Xiao et al., 2001). Interestingly, NIK activity is associated with multiple malignancies, and studies have shown that knockdown of NIK leads to anti-tumor effects (Thu et al., 2012; Saitoh et al., 2008). Importantly, NIK is linked to T and B cell survival in tumors by targeting non-canonical NF-κB pathway activation (Sasaki et al., 2008; Odqvist et al., 2013). Furthermore, targeting the non-canonical NF-κB pathway using small-molecule NIK inhibitors in cancer or experimental lupus showed promising results regarding pathology scores (Brightbill et al., 2018; Das et al., 2019; Mortier et al., 2010). In our study, we observed that ILC2s treated with TNF-α ex vivo selectively activated the non-canonical NF-κB pathway. Our transcriptomic analysis showed that Nfkb2 and RelB were upregulated, whereas Nfkb1 and Rela were either unaffected or downregulated. Furthermore, treatment of ILC2s with TNF-α did not affect p65 expression in ILC2s but efficiently upregulated p52. Importantly, NIK, encoded by the Map3k14 gene, was induced by TNF-α treatment, and blocking TNFR2 or NIK efficiently reduced p52 expression in response to TNF-α. As a result, we showed that inhibiting NIK ex vivo efficiently neutralized the beneficial effects of TNF-α on ILC2 survival and activation. In agreement with our findings, Rag2−/− Il2−/− mice receiving WT ILC2s and treated with the NIK inhibitor in vivo developed less lung resistance and airway inflammation compared with controls. Importantly, these mice also harbored fewer pulmonary ILC2s, suggesting that, similar to TNFR2 blocking, NIK inhibition affects ILC2 survival in vivo. Together, our observations suggest that the TNF/TNFR2 axis, via NIK, enhances ILC2-dependent development of AHR.
In addition, we found that hILC2s selectively expressed and induced TNFR2 in response to IL-33 stimulation. Similar to our murine findings, we observed that TNF-α increased hILC2 survival and cytokine secretion in culture. We further found that the increased IL-5 and IL-13 secretion in ILC2s treated with TNF-α ex vivo was abrogated when TNFR2 was blocked. We next validated our murine observations in a hILC2 transfer mouse model, as described previously (Maazi et al., 2015). This model is a platform to study the function of hILC2s in the context of asthma and to test potential therapeutic strategies. We showed, using this model, that treatment of mice with the NIK inhibitor efficiently inhibited the development of lung resistance and airway inflammation in response to TNF-α. Furthermore, mice that received the NIK inhibitor harbored fewer pulmonary ILC2s compared with controls, suggesting that TNF-α is involved in hILC2 survival. Together, these findings highlight the importance of the TNF/TNFR2 axis in hILC2 survival and activation and provide valuable evidence that our findings on murine ILC2s can translate to a clinical approach.
In conclusion, we demonstrate in this study that the TNF/TNFR2 axis enhances ILC2-dependent development of AHR and lung inflammation in murine models and in mice reconstituted with hILC2s. We show that both mouse and human aILC2s selectively express TNFR2 and that exogenous TNF-α provides essential modulatory signals to ILC2s, positively affecting survival and activation. Importantly, we show that inhibiting TNFR2-dependent NF-κB non-canonical signaling using a NIK inhibitor efficiently affected murine and hILC2 survival as well as effector function, preventing development of AHR and lung inflammation. Although inhibition of TNF-α had similar effects, systemic toxicity of such treatment cannot be excluded because signaling through TNFR1 and TNFR2 is affected. Targeting specific TNF-α receptors or, more specifically, the downstream signaling molecule NIK therefore is a more targeted approach to monitor the effects of TNF-α. With the discovery of a different pathway regulating ILC2 survival and effector functions, we believe that our findings set the stage for the development of adapted therapeutic strategies against diseases such as asthma.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Omid Akbari (akbari@usc.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse experiments
Experimental protocols were approved by the USC institutional Animal Care and Use Committee (IACUC) and conducted in accordance with the USC Department of Animal Resources’ guidelines. 5–10 week old age and sex matched mice were used in the studies. C57BL/6J, BALB/cByJ, TNFR2 deficient (B6.129S7-Tnfrsf1btm1lmx/J), RAG2 deficient (C.B6(Cg)-Rag2tm1.1Cgn/J), RAG2-, IL-2Rg-deficient (C;129S4-Rag2tm1.1Flv Il2rgtm1.1Flv/J) mice were bred in our animal facility at the Keck School of Medicine, University of Southern California (USC).
Human subjects
Experimental protocols were approved by the USC Institutional Review Board (IRB) and conducted in accordance to the principles of the Declaration of Helsinki. Studies were performed on blood from adult unidentified healthy donors collected at Children’s Hospital in Los Angeles. A total of 8 donors were required to perform experiments.
METHOD DETAILS
In vivo experiments and tissue preparation
When indicated, mice were challenged on 3 consecutive days with 0.5μg/mouse in 50μL of carrier-free rmIL-33 or vehicle. For A. alternata experiments, mice were challenged on 4 consecutive days with 100μg/mouse in 50μL of A. alternata or vehicle. In studies investigating the effect of TNFα or TNFR2, mice were intravenously injected on 3 consecutive days with 100μg anti-mouse TNFα (clone XT3.11) or anti-mouse TNFR2 (clone TR75–54.7) or appropriate isotype controls. On day 4 (rmIL33) or 5 (A. alternata), lungs were collected and processed for the indicated readout. Following transcardial perfusion with PBS 1X to clear lungs of red blood cells, collected lungs were then digested in collagenase Type IV (400U/mL) at 37°C for one hour and then processed to single cell suspension through a 70μm nylon cell strainer (Falcon®) as described previously (Maazi et al., 2015).
Flow Cytometry
The following murine antibodies were used: biotinylated anti-mouse lineage CD3ε (145–2C11), CD5 (53–7.3), TCRβ (H57–597), TCR-γδ (eBioGL3), TCRb (H57–597), CD45R (RA3–6B2), Gr-1 (RB6–8C5), CD11c (N418), CD11b (M1/70), Ter119 (TER-119), FcεRIα (MAR-1), Streptavidin-FITC, PE-Cy7 anti-mouse CD127 (A7R34), APCCy7 anti-mouse CD45 (30-F11), PECy7 anti-mouse CD45 (30-F11), APC anti-mouse CD120a (55R-286), PerCPCy5.5 anti-mouse CD11c (N418), APCCy7 anti-mouse CD11c (N418), FITC anti-mouse CD19 (6D5), APC anti-mouse Gr-1 (RB6–8C5), PerCPCy5.5 anti-mouse CD3 (17A2), APC anti-mouse SiglecF (S17007L), BV421 anti-mouse CD135 (Flt3, AFF10), BV510 anti-mouse CD25 (PC61) and APC anti-mouse LPAM-1 (DATK32), PECy7 anti-mouse F4/80 (BM8), PerCP-eFluor710 anti-mouse ST2 (RMST2–2), eFluor450 anti-mouse CD11b (M1/70), PE anti-mouse SiglecF (E50–2440), PE anti-mouse CD120b, Streptavidin Alexa Fluor 647. Intranuclear staining was performed using the Foxp3 Transcription Factor Staining Kit according to the manufacturer’s instructions and APC anti-mouse Ki67 (SolA15) and PE anti-GATA-3 (TWAJ) were used. Intracellular staining was performed using the BD Cytofix/Cytoperm kit. When indicated, cytokine production was measured following 4 hours in vitro stimulation with 50μg/mL PMA, 500μg/mL ionomycin and/or 1μg/mL Golgi plug. PE anti-mouse/human IL-5 (TRFK5), PE anti-mouse TNFα (MP6-XT22) and eFluor450 anti-mouse IL-13 (eBio13A) were used. Intracellular straining of NFκB p65 and p52 was performed according to the manufacturer’s instructions and PE anti-human/mouse RelA NFκB p65 (IC5078P) and Alexa Fluor 647 anti-human/mouse NFκB p52 (C-5) were used. For apoptosis staining, AnnexinV-PE and DAPI were used according to the manufacturer’s instructions. The following human antibodies were used: FITC anti-human lineage cocktail including CD3 (clone UCHT1), CD14 (clone HCD14), CD16 (clone 3G8), CD19 (clone HIB19), CD20 (clone 2H7), CD56 (clone HCD56). Additional lineage markers were added: FITC anti-human CD235a (Clone HI264), FITC anti-human FCεRIα (clone AER-37), FITC anti-human CD1a (clone HI149), FITC anti-human CD123 (clone 6H6) and FITC anti-human CD5 (clone L17F12). APCCy7 anti-human CD45 (HI30), PECy7 anti-human CD127 (A019D5), APC anti-human CD120a (W15099A), BV421 anti-human CD120b (hTNFR-M1 RUO) were further used. Live/dead fixable violet or aqua dead cell stain kits were used to exclude dead cells and CountBright absolute counting beads to calculate absolute cell numbers when indicated. Stained cells were analyzed on FACSCanto II and/or FACSARIA III systems and the data was analyzed with FlowJo version 9 or 10 software.
Murine ILC2 or macrophage isolation and in vitro culture
Murine ILC2s and alveolar macrophages were FACS-sorted to a purity of > 95% on a FACSARIA III system. nILC2s and naive AM macrophages were purified from the lungs of naive mice, whereas aILC2s and activated AM macrophages were purified from the lungs of mice challenged on 3 consecutive days with 0.5μg/mouse in 50μL of carrier-free rmIL-33. ILC2s were gated as lineage (CD3ε, CD5, CD45R, Gr-1, CD11c, CD11b, Ter119, TCRγδ, TCRβ and FCεRIα) negative CD45+, ST2+, CD117+ cells. Isolated ILC2s were cultured at 37°C (5x104/mL) with rmIL-2 (10ng/mL) and rmIL-7 (10ng/mL) in complete RPMi (cRPMi). For cRPMi, RPMI (GIBCO) was supplemented with 10% heat-inactivated FBS (Omega Scientific), 100 units/mL penicillin and 100mg/mL streptomycin (GenClone). In TNFR1 and TNFR2 expression kinetics experiments, nILC2s were activated with 50ng/mL rmIL-33 for the indicated times. When mentioned, ILC2s were treated with 40ng/mL rmTNFα for the indicated times. In TNFR2 and NIK blocking experiments, 10μg/mL anti-TNFR2 or corresponding isotype, and 10μM NIK inhibitor 1, 3[2H, 4H]-Isoquinolinedione or vehicle were added 30 minutes prior rmTNFα. AM macrophages were gated as CD45+, SiglecF+, CD11b-, CD11c+, F4/80+. Naive or activated AM macrophages were cultured (106/mL) in complete DMEM (GIBCO) supplemented with 10% heat-inactivated FBS (Omega Scientific), 100 units/mL penicillin and 100mcg/mL streptomycin (GenClone) for the indicated times at 37°C.
Human ILC2 isolation and in vitro culture
Human peripheral blood ILC2s were isolated from total peripheral blood mononuclear cells (PBMCs) to a purity of > 95% on a FACSARIA III system as described previously (Maazi et al., 2015). Briefly, human fresh blood was first diluted 1:1 in PBS 1X and transferred to SepMateTM-50 separation tubes (STEMCELL Technologies) filled with 12mL Lymphoprep™. Samples were centrifuged for 10 minutes and PBMCs were collected. CRTH2+ cells were then isolated using the CRTH2 MicroBead Kit, used according to the manufacturer’s conditions. Samples were then stained and ILC2s were isolated based on the absence of common lineage markers (CD3, CD5, CD14, CD16, CD19, CD20, CD56, CD235a, CD1a, CD123), and the expression of CD45, CRTH2 and CD127. Isolated ILC2s were cultured at 37°C (5x104/mL) with rhIL-2 (10ng/mL) and rhIL-7 (10ng/mL) in cRPMi. When indicated, human ILC2s were activated with 50ng/mL rhIL-33 for the indicated times. In TNFR2 blocking experiments, 10μg/mL anti-human TNFR2 (3G7A02) was added 30 minutes prior adding 40ng/mL rhTNFα.
Murine adoptive transfer, humanized mice and measurement of lung function
Murine C57BL/6 or TNFR2 deficient aILC2s were isolated as described above and cultured with rmIL-2, rmIL-7 and rmIL-33 for 3 days prior transfer. 50 × 103 aILC2s were intravenously transferred to sex and age matched Rag2−/− Il2rg−/− mice followed by intranasal administration of 50ng in 50μL rmTNFα. For humanized mice, peripheral blood ILC2s were isolated as described above and cultured with rhIL-2, rhIL-7 and rhIL-33 for 3 days prior transfer. 50 × 103 ILC2s were intravenously transferred to sex and age matched Rag2−/− Il2rg−/− mice followed by intranasal administration of 50ng in 50μL rhTNFα. In experiments studying the effect of NIK, 250μg of NIK inhibitor 1, 3[2H, 4H]-Isoquinolinedione or vehicle were i.p. injected for the indicated times. On day 4 or 5, lung function was measured as described previously using the FinePointe RC system (Buxco Research Systems) (Maazi et al., 2013, 2015). Briefly, mice were surgically tracheotomized under deep anesthesia and placed on the mechanically ventilated system where increasing doses of methacholine are sequentially nebulized. For each dose, lung resistance and dynamic compliance were measured and computed over a 3 minutes period.
Collection of BAL fluid and histology
BAL fluid was collected as previously described (Maazi et al., 2015). Briefly, tracheotomized mice were cannulated and lungs washed 3 times with 1mL PBS 1X. Harvested cells were centrifuged to collect BAL supernatant and cells were further stained for flow cytometry analysis. When stated, one lobe per lung was collected and stored in PFA 4% for histology, as described previously (Maazi et al., 2015). Briefly, lungs were embedded in paraffin and sections of 4μm prepared for hematoxylin and eosin (H&E) staining. Histology pictures were acquired on a Leica DME microscope and Leica ICC50HD camera (Leica) and analyzed with ImageJ.
Cytokine measurements
The amounts of cytokines in BAL or culture supernatants were measured by ELISA. Murine IL-5, IL-13, TNFα and human IL-5 and IL-13 ELISA kits were used according to the manufacturer’s instructions. Other cytokines were measured by multiplexed fluorescent bead-based immunoassay detection (MILLIPLEX® MAP system, Millipore Corporation) according to the manufacturer’s instructions, using a combination of 32-plex (MCYTMAG70KPX32) kit. When indicated, ELISA results were normalized to cell viability in culture, using CountBright absolute counting beads, used according to the manufacturer’s instructions
RNA sequencing and data analysis
Purified aILC2s were cultured with or without rmTNFα for 24 hours as described above. Following culture, ILC2s were recovered in RLT buffer and total RNA was extracted using the MicroRNeasy kit. For each sample, a total of 10ng of RNA was used to generate cDNA (SMARTer Ultra Low Input RNA v3 kit, Clontech) for library preparation. Samples were then amplified and sequenced on a NextSeq 500 system (Illumina) where on average 30 million reads were generated from each sample. Raw reads were then further processed on Partek® Genomics Suite® software, version 7.0 Copyright ©; Partek Inc. Briefly, raw reads were aligned by STAR – 2.6.1d with mouse reference index mm10 and GENECODE M20 annotations. Aligned reads were further quantified and normalized using the upper quartile method and differential analysis by GSA. QIAGEN Ingenuity Pathway Analysis (IPA) software was used for pathway analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
Experiments were repeated at least three times (n = 4–8 each) and data are shown as the representative of > 2 independent experiments. A Student’s t test for unpaired data was used for comparisons between each group using Prism Software (GraphPad Software Inc.). The degree of significance was indicated as: *p < 0.05, **p < 0.01, ***p < 0.001.
DATA AND CODE AVAILABILITY
The accession number for the data generated by RNAseq reported in this paper is GEO: GSE133672.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Biotin anti-mouse CD3ε (clone 145-2C11) | BioLegend | Cat # 100304; RRID: AB_312669 |
| Biotin anti-mouse CD5 (clone 53-7.3) | BioLegend | Cat # 100604; RRID: AB_312733 |
| Biotin anti-mouse TCR-β (clone H57-597) | BioLegend | Cat # 109204; RRID: AB_313427 |
| Biotin anti-mouse/human CD45R (clone RA3-6B2) | BioLegend | Cat # 103204; RRID: AB_312989 |
| Biotin anti-mouse Gr-1 (clone RB6-8C5) | BioLegend | Cat # 108404; RRID: AB_313369 |
| Biotin anti-mouse CD11c (clone N418) | BioLegend | Cat # 117304; RRID: AB_313773 |
| Biotin anti-mouse/human CD11b (clone M1/70) | BioLegend | Cat # 101204; RRID: AB_312787 |
| Biotin anti-mouse TER-119 (clone TER-119) | BioLegend | Cat # 116204; RRID: AB_313705 |
| Biotin anti-mouse FCεRIα (clone MAR-1) | BioLegend | Cat # 134304; RRID: AB_1626106 |
| FITC Streptavidin | BioLegend | Cat # 405202 |
| PE/Cyanine7 anti-mouse CD127 (clone A7R34) | BioLegend | Cat # 135014; RRID: AB_1937265 |
| APC/Cyanine7 anti-mouse CD45 (clone 30-F11) | BioLegend | Cat # 103116; RRID: AB_312981 |
| PE/Cyanine7 anti-mouse CD45 (clone 30-F11) | BioLegend | Cat # 103114; RRID: AB_312979 |
| APC anti-mouse CD120a (clone 55R-286) | BioLegend | Cat # 113006; RRID: AB_2208779 |
| PerCP/Cyanine5.5 anti-mouse CD11c (clone N418) | BioLegend | Cat # 117328; RRID: AB_2129641 |
| APC/Cyanine7 anti-mouse CD11c (clone N418) | BioLegend | Cat # 117324; RRID: AB_830649 |
| FITC anti-mouse CD19 (clone 6D5) | BioLegend | Cat # 115506; RRID: AB_313641 |
| APC anti-mouse Gr-1 (clone RB6-8C5) | BioLegend | Cat # 108412; RRID: AB_313377 |
| PerCP/Cyanine5.5 anti-mouse CD3 (clone 17A2) | BioLegend | Cat # 100218; RRID: AB_1595492 |
| APC anti-mouse CD170/SiglecF (clone S17007L) | BioLegend | Cat # 155508; RRID: AB_2750237 |
| Brilliant Violet 421 anti-mouse CD135 (clone A2F10) | BioLegend | Cat # 135314; RRID: AB_2562339 |
| Brilliant Violet 510 anti-mouse CD25 (clone PC61) | BioLegend | Cat # 102042; RRID: AB_2562270 |
| APC anti-mouse LPAM-1 (clone DATK32) | BioLegend | Cat # 120608; RRID: AB_10730607 |
| PE anti-mouse TNF-α (clone MP6-XT22) | BioLegend | Cat # 506306; RRID: AB_315427 |
| Biotin anti-mouse TCR-γδ (clone eBioGL3) | Thermofisher | Cat # 13-5711-85; RRID: AB_466669 |
| PE/Cyanine7 anti-mouse F4/80 (clone BM8) | Thermofisher | Cat # 25-4801-82; RRID: AB_469653 |
| PerCP/eFluor710 anti-mouse ST2 (clone RMST2-2) | Thermofisher | Cat # 46-9335-82; RRID: AB_2573883 |
| eFluor 450 anti-mouse CD11b (clone M1/70) | Thermofisher | Cat # 48-0112-82; RRID: AB_1582236 |
| APC anti-mouse/human Ki-67 (clone SolA15) | Thermofisher | Cat # 17-5698-82; RRID: AB_2688057 |
| PE anti-mouse/human GATA-3 (clone TWAJ) | Thermofisher | Cat # 12-9966-42; RRID: AB_1963600 |
| eFluor 450 anti-mouse IL-13 (clone eBio13A) | Thermofisher | Cat # 48-7133-82; RRID: AB_11219690 |
| PE anti-mouse/human IL-5 (clone TRFK5) | Biolegend | Cat # 504304; RRID: AB_315328 |
| PE anti-mouse CD170/SiglecF (clone E50-2440) | BD Biosciences | Cat # 552126; RRID: AB_394341 |
| PE anti-mouse CD120b (clone TR75-89) | BD Biosciences | Cat # 550086; RRID: AB_393556 |
| APC Streptavidin | BioLegend | Cat # 405207 |
| FITC anti-human lineage CD3 (clone UCHT1), CD14 (clone HCD14), CD16 (clone 3G8), CD19 (clone HIB19), CD20 (clone 2H7), CD56 (clone HCD56) | BioLegend | Cat # 348801; RRID: AB_10612570 |
| APC/Cyanine7 anti-human CD45 (clone HI30) | BioLegend | Cat # 304014; RRID: AB_314402 |
| PE/Cyanine anti-human CD127 (clone A019D5) | BioLegend | Cat # 351320; RRID: AB_10897098 |
| FITC anti-human CD235a (clone HI264) | BioLegend | Cat # 349104, RRID: AB_10613463 |
| FITC anti-human FCεRIα (clone AER-37) | BioLegend | Cat # 334608; RRID: AB_1227653 |
| FITC anti-human CD1a (clone HI149) | BioLegend | Cat # 300104; RRID: AB_314018 |
| FITC anti-human CD123 (clone 6H6) | BioLegend | Cat # 306014; RRID: AB_2124259 |
| FITC anti-human CD5 (clone L17F12) | BioLegend | Cat # 364022; RRID: AB_2566248 |
| APC anti-human CD120a (W15099A) | BioLegend | Cat # 369906; RRID: AB_2650764 |
| BV421 anti-human CD120b (hTNFR-M1) | BD Biosciences | Cat # 742984; RRID: AB_2741188 |
| PE anti-mouse/human RelA/NFκB p65 (clone 532301) | R&D Systems | Cat # IC5078P |
| Alexa Fluor 647 anti-mouse/human NFκB p52 (clone C-5) | Santa Cruz Biotechnology | Cat # sc-7386 AF647 |
| Anti-mouse CD120b (clone TR75-54.7) | BioXcell | Cat # BE0247; RRID: AB_2687728 |
| Anti-mouse TNF-a (clone XT3.11) | BioXcell | Cat # BP0058; RRID: AB_1107764 |
| Anti-human CD120b (clone 3G7A02) | BioLegend | Cat # 358408; RRID: AB_2563224 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Collagenase Type 4 | Worthington Biochemical | Cat # LS004189 |
| Recombinant mouse IL-2 | BioLegend | Cat # 575408 |
| Recombinant mouse IL-7 | BioLegend | Cat # 577808 |
| Recombinant mouse TNF-a | BioLegend | Cat # 575204 |
| Recombinant mouse IL-33 | BioLegend | Cat # 580508 |
| Recombinant human IL-2 | BioLegend | Cat # 589108 |
| Recombinant human IL-7 | BioLegend | Cat # 581908 |
| Recombinant human TNF-a | BioLegend | Cat # 570104 |
| Recombinant human IL-33 | BioLegend | Cat # 581808 |
| 1,3[2H,4H]-Isoquinolinedione | AK Scientific | Cat # W6338 |
| Phorbol 12-myristate 13-acetate | Sigma-Aldrich | Cat # P1585-1MG |
| Ionomycin calcium salt | Sigma-Aldrich | Cat # I0634-1MG |
| DAPI | Sigma-Aldrich | Cat # D9542-1MG |
| Lymphoprep | Axis-Shield | Cat # AXS-1114544 |
| Acetyl-β-methylcholine chloride | Sigma-Aldrich | Cat # A2251-25G |
| Alternaria alternata | Greer Laboratories | Cat # XPM1D3A25 |
| Critical Commercial Assays | ||
| Mouse IL-5 Uncoated ELISA Kit | Thermofisher | Cat # 88-7054-88 |
| Mouse IL-13 Uncoated ELISA Kit | Thermofisher | Cat # 88-7137-88 |
| Mouse TNFα Uncoated ELISA Kit | Thermofisher | Cat # 88-7324-88 |
| Human IL-5 Uncoated ELISA Kit | Thermofisher | Cat # 88-7056-88 |
| Human IL-13 Uncoated ELISA Kit | Thermofisher | Cat # 88-7439-88 |
| RNeasy Micro kit | QIAGEN | Cat # 74004 |
| Annexin V Apoptosis Detection Kit-PE | Thermofisher | Cat # 88-8102-74 |
| FOXP3/Transcription Factor Staining Buffer Set | Thermofisher | Cat # 00-5523-00 |
| BD Cytofix/Cytoperm Plus | BD Biosciences | Cat # 555028 |
| Deposited Data | ||
| RNaseq data | This paper | GEO: GSE133672 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | Jackson laboratories | Cat # 000664 |
| Mouse: BALB/cByJ | Jackson laboratories | Cat # 000651 |
| Mouse: B6.129S7-Tnfrsf1btm1lmx/J | Jackson laboratories | Cat # 003246 |
| Mouse: C.B6(Cg)-Rag2tm1.1Cgn/J | Jackson laboratories | Cat # 008448 |
| Mouse: C;129S4-Rag2tm1.1Flv Il2rgtm1.1Flv/J | Jackson laboratories | Cat # 017707 |
| Software and Algorithms | ||
| FlowJo analysis software version 9 and 10 | TreeStar | N/A |
| Prism analysis software version 5-8 | GraphPad | N/A |
| Partek Flow Genomic Analysis Software | Partek | N/A |
| Ingenuity Pathway Analysis (IPA) | QIAGEN | N/A |
| ImageJ | NIH | N/A |
| Other | ||
| LIVE/DEAD Fixable Aqua Dead Cell Stain Kit | Thermofisher | Cat # L34957 |
| LIVE/DEAD Fixable Violet Dead Cell Stain Kit | Thermofisher | Cat # L34955 |
| CD294 (CRTH2) Microbead Kit human | Miltenti Biotec | Cat # 130-091-274 |
| CountBright absolute counting beads | Thermofisher | Cat # C36950 |
Highlights.
Murine activated lung ILC2s lack TNFR1 but selectively express TNFR2
TNF-α enhances ILC2 survival, activation, and induction of AHR using NIK signaling
Inhibition of NIK blocks the costimulatory effects of the TNF/TNFR2 axis in ILC2s
Human ILC2s express TNFR2, and a NIK inhibitor reduces AHR in humanized ILC2 mice
ACKNOWLEDGMENTS
B.P.H. is supported by the Swiss National Science Foundation early postdoctoral mobility grant 181286. This article was financially supported by NIH Public Health Service grants R01 ES025786, R01 ES021801, R01 HL144790, and R21 AI109059 (to O.A.). We thank the Bioinformatics Service from USC, in particular Dr. Yibu Chen and Meng Li, for technical support.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.11.102.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Aggarwal BB (2003). Signalling pathways of the TNF superfamily: a doubleedged sword. Nat. Rev. Immunol 3, 745–756. [DOI] [PubMed] [Google Scholar]
- Aggarwal BB, Gupta SC, and Kim JH (2012). Historical perspectives on tumor necrosis factor and its superfamily: 25 years later, a golden journey. Blood 119, 651–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali T, Kaitha S, Mahmood S, Ftesi A, Stone J, and Bronze MS (2013). Clinical use of anti-TNF therapy and increased risk of infections. Drug Healthc. Patient Saf 5, 79–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anticevich SZ, Hughes JM, Black JL, and Armour CL (1995). Induction of human airway hyperresponsiveness by tumour necrosis factor-alpha. Eur. J. Pharmacol 284, 221–225. [DOI] [PubMed] [Google Scholar]
- Barlow JL, Peel S, Fox J, Panova V, Hardman CS, Camelo A, Bucks C, Wu X, Kane CM, Neill DR, et al. (2013). IL-33 is more potent than IL-25 in provoking IL-13-producing nuocytes (type 2 innate lymphoid cells) and airway contraction. J. Allergy Clin. Immunol 132, 933–941. [DOI] [PubMed] [Google Scholar]
- Bartemes KR, Iijima K, Kobayashi T, Kephart GM, McKenzie AN, and Kita H (2012). IL-33-responsive lineage-CD25+ CD44(hi) lymphoid cells mediate innate type 2 immunity and allergic inflammation in the lungs. J. Immunol 188, 1503–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baud V, and Karin M (2001). Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol 11, 372–377. [DOI] [PubMed] [Google Scholar]
- Berry MA, Hargadon B, Shelley M, Parker D, Shaw DE, Green RH, Bradding P, Brightling CE, Wardlaw AJ, and Pavord ID (2006). Evidence of a role of tumor necrosis factor alpha in refractory asthma. N. Engl. J. Med 354, 697–708. [DOI] [PubMed] [Google Scholar]
- Bradding P, Roberts JA, Britten KM, Montefort S, Djukanovic R, Mueller R, Heusser CH, Howarth PH, and Holgate ST (1994). Interleukin-4, −5, and −6 and tumor necrosis factor-alpha in normal and asthmatic airways: evidence for the human mast cell as a source of these cytokines. Am. J. Respir. Cell Mol. Biol. 10, 471–480. [DOI] [PubMed] [Google Scholar]
- Brightbill HD, Suto E, Blaquiere N, Ramamoorthi N, Sujatha-Bhaskar S, Gogol EB, Castanedo GM, Jackson BT, Kwon YC, Haller S, et al. (2018). NF-κB inducing kinase is a therapeutic target for systemic lupus erythematosus. Nat. Commun 9, 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brightling C, Berry M, and Amrani Y (2008). Targeting TNF-alpha: a novel therapeutic approach for asthma. J. Allergy Clin. Immunol 121, 5–10, quiz 11–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunting MM, Shadie AM, Flesher RP, Nikiforova V, Garthwaite L, Tedla N, Herbert C, and Kumar RK (2013). Interleukin-33 drives activation of alveolar macrophages and airway inflammation in a mouse model of acute exacerbation of chronic asthma. BioMed Res. Int 2013, 250938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Y, Cao YX, Lu SM, Xu CB, and Cardell LO (2011). Infliximab alleviates inflammation and ex vivo airway hyperreactivity in asthmatic E3 rats. Int. Immunol 23, 443–451. [DOI] [PubMed] [Google Scholar]
- Cavagnero KJ, Badrani JH, Naji LH, Amadeo MB, Shah VS, Gasparian S, Pham A, Wang AW, Seumois G, Croft M, et al. (2019). Unconventional ST2- and CD127-negative lung ILC2 populations are induced by the fungal allergen Alternaria alternata. J. Allergy Clin. Immunol 144, 1432–1435.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Bäumel M, Männel DN, Howard OM, and Oppenheim JJ (2007). Interaction of TNF with TNF receptor type 2 promotes expansion and function of mouse CD4+CD25+ T regulatory cells. J. Immunol 179, 154–161. [DOI] [PubMed] [Google Scholar]
- Chen X, Subleski JJ, Kopf H, Howard OM, Männel DN, and Oppenheim JJ (2008). Cutting edge: expression of TNFR2 defines a maximally suppressive subset of mouse CD4+CD25+FoxP3+ T regulatory cells: applicability to tumor-infiltrating T regulatory cells. J. Immunol 180, 6467–6471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Wu X, Zhou Q, Howard OM, Netea MG, and Oppenheim JJ (2013). TNFR2 is critical for the stabilization of the CD4+Foxp3+ regulatory T. cell phenotype in the inflammatory environment. J. Immunol 190, 1076–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chopra M, Biehl M, Steinfatt T, Brandl A, Kums J, Amich J, Vaeth M, Kuen J, Holtappels R, Podlech J, et al. (2016). Exogenous TNFR2 activation protects from acute GvHD via host T reg cell expansion. J. Exp. Med 213, 1881–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JL, and Wood KJ (2017). TNFR2: The new Treg switch? OncoImmunology 7, e1373236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cushnie EK, Ulery BD, Nelson SJ, Deng M, Sethuraman S, Doty SB, Lo KW, Khan YM, and Laurencin CT (2014). Simple signaling molecules for inductive bone regenerative engineering. PLoS ONE 9, e101627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das R, Coupar J, Clavijo PE, Saleh A, Cheng TF, Yang X, Chen J, Van Waes C, and Chen Z (2019). Lymphotoxin-β receptor-NIK signaling induces alternative RELB/NF-κB2 activation to promote metastatic gene expression and cell migration in head and neck cancer. Mol. Carcinog 58, 411–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galle-Treger L, Sankaranarayanan I, Hurrell BP, Howard E, Lo R, Maazi H, Lewis G, Banie H, Epstein AL, Hu P, et al. (2019). Costimulation of type-2 innate lymphoid cells by GITR promotes effector function and ameliorates type 2 diabetes. Nat. Commun 10, 713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halim TYF, Rana BMJ, Walker JA, Kerscher B, Knolle MD, Jolin HE, Serrao EM, Haim-Vilmovsky L, Teichmann SA, Rodewald HR, et al. (2018). Tissue-Restricted Adaptive Type 2 Immunity Is Orchestrated by Expression of the Costimulatory Molecule OX40L on Group 2 Innate Lymphoid Cells. Immunity 48, 1195–1207.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa A, Takasaki W, Greene MI, and Murali R (2001). Modifying TNFalpha for therapeutic use: a perspective on the TNF receptor system. Mini Rev. Med. Chem 1, 5–16. [DOI] [PubMed] [Google Scholar]
- Hlushchuk R, Styp-Rekowska B, Dzambazi J, Wnuk M, Huynh-Do U, Makanya A, and Djonov V (2017). Endoglin inhibition leads to intussusceptive angiogenesis via activation of factors related to COUP-TFII signaling pathway. PLoS ONE 12, e0182813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howarth PH, Babu KS, Arshad HS, Lau L, Buckley M, McConnell W, Beckett P, Al Ali M, Chauhan A, Wilson SJ, et al. (2005). Tumour necrosis factor (TNFalpha) as a novel therapeutic target in symptomatic corticosteroid dependent asthma. Thorax 60, 1012–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurrell BP, Shafiei Jahani P, and Akbari O (2018). Social Networking of Group Two Innate Lymphoid Cells in Allergy and Asthma. Front. Immunol 9, 2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabata H, Moro K, and Koyasu S (2018). The group 2 innate lymphoid cell (ILC2) regulatory network and its underlying mechanisms. Immunol. Rev 286, 37–52. [DOI] [PubMed] [Google Scholar]
- Kearley J, Silver JS, Sanden C, Liu Z, Berlin AA, White N, Mori M, Pham TH, Ward CK, Criner GJ, et al. (2015). Cigarette smoke silences innate lymphoid cell function and facilitates an exacerbated type I interleukin-33-dependent response to infection. Immunity 42, 566–579. [DOI] [PubMed] [Google Scholar]
- Keatings VM, O’Connor BJ, Wright LG, Huston DP, Corrigan CJ, and Barnes PJ (1997). Late response to allergen is associated with increased concentrations of tumor necrosis factor-alpha and IL-5 in induced sputum. J. Allergy Clin. Immunol 99, 693–698. [DOI] [PubMed] [Google Scholar]
- Kim HY, Chang YJ, Subramanian S, Lee HH, Albacker LA, Matangkasombut P, Savage PB, McKenzie AN, Smith DE, Rottman JB, et al. (2012). Innate lymphoid cells responding to IL-33 mediate airway hyperreactivity independently of adaptive immunity. J. Allergy Clin. Immunol 129, 216–27.e1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kips JC, Tavernier J, and Pauwels RA (1992). Tumor necrosis factor causes bronchial hyperresponsiveness in rats. Am. Rev. Respir. Dis 145, 332–336. [DOI] [PubMed] [Google Scholar]
- Klein Wolterink RG, Kleinjan A, van Nimwegen M, Bergen I, de Bruijn M, Levani Y, and Hendriks RW (2012). Pulmonary innate lymphoid cells are major producers of IL-5 and IL-13 in murine models of allergic asthma. Eur. J. Immunol 42, 1106–1116. [DOI] [PubMed] [Google Scholar]
- Kobayashi Y, Kiguchi N, Fukazawa Y, Saika F, Maeda T, and Kishioka S (2015). Macrophage-T cell interactions mediate neuropathic pain through the glucocorticoid-induced tumor necrosis factor ligand system. J. Biol. Chem 290, 12603–12613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei AH, Xiao Q, Liu GY, Shi K, Yang Q, Li X, Liu YF, Wang HK, Cai WP, Guan YJ, et al. (2018). ICAM-1 controls development and function of ILC2. J. Exp. Med 215, 2157–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis G, Wang B, Shafiei Jahani P, Hurrell BP, Banie H, Aleman Muench GR, Maazi H, Helou DG, Howard E, Galle-Treger L, et al. (2019). Dietary Fiber-Induced Microbial Short Chain Fatty Acids Suppress ILC2-Dependent Airway Inflammation. Front. Immunol 10, 2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling L, Cao Z, and Goeddel DV (1998). NF-kappaB-inducing kinase activates IKK-alpha by phosphorylation of Ser-176. Proc. Natl. Acad. Sci. USA 95, 3792–3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maazi H, and Akbari O (2017). Type two innate lymphoid cells: the Janus cells in health and disease. Immunol. Rev 278, 192–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maazi H, Singh AK, Speak AO, Lombardi V, Lam J, Khoo B, Inn KS, Sharpe AH, Jung JU, and Akbari O (2013). Lack of PD-L1 expression by iNKT cells improves the course of influenza A infection. PLoS ONE 8, e59599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maazi H, Patel N, Sankaranarayanan I, Suzuki Y, Rigas D, Soroosh P, Freeman GJ, Sharpe AH, and Akbari O (2015). ICOS:ICOS-ligand interaction is required for type 2 innate lymphoid cell function, homeostasis, and induction of airway hyperreactivity. Immunity 42, 538–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaviya R, Laskin JD, and Laskin DL (2017). Anti-TNFα therapy in inflammatory lung diseases. Pharmacol. Ther 180, 90–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meylan F, Hawley ET, Barron L, Barlow JL, Penumetcha P, Pelletier M, Sciumè G, Richard AC, Hayes ET, Gomez-Rodriguez J, et al. (2014). The TNF-family cytokine TL1A promotes allergic immunopathology through group 2 innate lymphoid cells. Mucosal Immunol 7, 958–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mjösberg J, Bernink J, Golebski K, Karrich JJ, Peters CP, Blom B, te Velde AA, Fokkens WJ, van Drunen CM, and Spits H (2012). The transcription factor GATA3 is essential for the function of human type 2 innate lymphoid cells. Immunity 37, 649–659. [DOI] [PubMed] [Google Scholar]
- Mortier J, Masereel B, Remouchamps C, Ganeff C, Piette J, and Frederick R (2010). NF-kappaB inducing kinase (NIK) inhibitors: identification of new scaffolds using virtual screening. Bioorg. Med. Chem. Lett 20, 4515–4520. [DOI] [PubMed] [Google Scholar]
- Nagashima H, Okuyama Y, Fujita T, Takeda T, Motomura Y, Moro K, Hidaka T, Omori K, Sakurai T, Machiyama T, et al. (2018). GITR cosignal in ILC2s controls allergic lung inflammation. J. Allergy Clin. Immunol 141, 1939–1943.e8. [DOI] [PubMed] [Google Scholar]
- Odqvist L, Sánchez-Beato M, Montes-Moreno S, Martín-Sánchez E, Pajares R, Sánchez-Verde L, Ortiz-Romero PL, Rodriguez J, Rodríguez-Pinilla SM, Iniesta-Martínez F, et al. (2013). NIK controls classical and alternative NF-κB activation and is necessary for the survival of human T-cell lymphoma cells. Clin. Cancer Res 19, 2319–2330. [DOI] [PubMed] [Google Scholar]
- Pfoertner S, Jeron A, Probst-Kepper M, Guzman CA, Hansen W, Westendorf AM, Toepfer T, Schrader AJ, Franzke A, Buer J, and Geffers R (2006). Signatures of human regulatory T cells: an encounter with old friends and new players. Genome Biol 7, R54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raychaudhuri SP, and Raychaudhuri SK (2009). Biologics: target-specific treatment of systemic and cutaneous autoimmune diseases. Indian J. Dermatol 54, 100–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh Y, Yamamoto N, Dewan MZ, Sugimoto H, Martinez Bruyn VJ, Iwasaki Y, Matsubara K, Qi X, Saitoh T, Imoto I, et al. (2008). Overexpressed NF-kappaB-inducing kinase contributes to the tumorigenesis of adult T-cell leukemia and Hodgkin Reed-Sternberg cells. Blood 111, 5118–5129. [DOI] [PubMed] [Google Scholar]
- Sasaki M, Kashima M, Ito T, Watanabe A, Izumiyama N, Sano M, Kagaya M, Shioya T, and Miura M (2000). Differential regulation of metalloproteinase production, proliferation and chemotaxis of human lung fibroblasts by PDGF, interleukin-1beta and TNF-alpha. Mediators Inflamm 9, 155–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki Y, Calado DP, Derudder E, Zhang B, Shimizu Y, Mackay F, Nishikawa S, Rajewsky K, and Schmidt-Supprian M (2008). NIK overexpression amplifies, whereas ablation of its TRAF3-binding domain replaces BAFF:BAFF-R-mediated survival signals in B cells. Proc. Natl. Acad. Sci. USA 105, 10883–10888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedger LM, and McDermott MF (2014). TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants - past, present and future. Cytokine Growth Factor Rev 25, 453–472. [DOI] [PubMed] [Google Scholar]
- Simhadri VL, Hansen HP, Simhadri VR, Reiners KS, Bessler M, Engert A, and von Strandmann EP (2012). A novel role for reciprocal CD30-CD30L signaling in the cross-talk between natural killer and dendritic cells. Biol. Chem 393, 101–106. [DOI] [PubMed] [Google Scholar]
- Sun SC (2012). The noncanonical NF-κB pathway. Immunol. Rev 246, 125–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taillè C, Poulet C, Marchand-Adam S, Borie R, Dombret MC, Crestani B, and Aubier M (2013). Monoclonal Anti-TNF-α Antibodies for Severe Steroid-Dependent Asthma: A Case Series. Open Respir. Med. J 7, 21–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas PS (2001). Tumour necrosis factor-alpha: the role of this multifunctional cytokine in asthma. Immunol. Cell Biol 79, 132–140. [DOI] [PubMed] [Google Scholar]
- Thu YM, and Richmond A (2010). NF-κB inducing kinase: a key regulator in the immune system and in cancer. Cytokine Growth Factor Rev 21, 213–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thu YM, Su Y, Yang J, Splittgerber R, Na S, Boyd A, Mosse C, Simons C, and Richmond A (2012). NF-κB inducing kinase (NIK) modulates melanoma tumorigenesis by regulating expression of pro-survival factors through the β-catenin pathway. Oncogene 31, 2580–2592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira SM, Lemos HP, Grespan R, Napimoga MH, Dal-Secco D, Freitas A, Cunha TM, Verri WA Jr., Souza-Junior DA, Jamur MC, et al. (2009). A crucial role for TNF-alpha in mediating neutrophil influx induced by endogenously generated or exogenous chemokines, KC/CXCL1 and LIX/CXCL5. Br. J. Pharmacol 158, 779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Ferreira R, Lu W, Farrow S, Downes K, Jermutus L, Minter R, Al-Lamki RS, Pober JS, and Bradley JR (2018). TNFR2 ligation in human T regulatory cells enhances IL2-induced cell proliferation through the non-canonical NF-κB pathway. Sci. Rep 8, 12079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward-Kavanagh LK, Lin WW, Šedý JR, and Ware CF (2016). The TNF Receptor Superfamily in Co-stimulating and Co-inhibitory Responses. Immunity 44, 1005–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao G, Harhaj EW, and Sun SC (2001). NF-kappaB-inducing kinase regulates the processing of NF-kappaB2 p100. Mol. Cell 7, 401–409. [DOI] [PubMed] [Google Scholar]
- Yang S, Xie C, Chen Y, Wang J, Chen X, Lu Z, June RR, and Zheng SG (2019). Differential roles of TNFα-TNFR1 and TNFα-TNFR2 in the differentiation and function of CD4+Foxp3+ induced Treg cells in vitro and in vivo periphery in autoimmune diseases. Cell Death Dis 10, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Pappu R, Ramirez-Carrozzi V, Ota N, Caplazi P, Zhang J, Yan D, Xu M, Lee WP, and Grogan JL (2014). TNF superfamily member TL1A elicits type 2 innate lymphoid cells at mucosal barriers. Mucosal Immunol 7, 730–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J (2017). GATA3 Regulates the Development and Functions of Innate Lymphoid Cell Subsets at Multiple Stages. Front. Immunol 8, 1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou H, Li R, Hu H, Hu Y, and Chen X (2018). Modulation of Regulatory T Cell Activity by TNF Receptor Type II-Targeting Pharmacological Agents. Front. Immunol 9, 594. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the data generated by RNAseq reported in this paper is GEO: GSE133672.